

Abstract
The Kiss1 gene codes for kisspeptin, which binds to GPR54, a G-protein-coupled receptor. Kisspeptin and GPR54 are expressed in discrete regions of the forebrain, and they have been implicated in the neuroendocrine regulation of reproduction. Kiss1-expressing neurons are thought to regulate the secretion of gonadotropin-releasing hormone (GnRH) and thus coordinate the estrous cycle in rodents; however, the precise role of kisspeptin–GPR54 signaling in the regulation of gonadotropin secretion is unknown. In this study, we used female mice with deletions in the GPR54 gene [GPR54 knock-outs (KOs)] to test the hypothesis that kisspeptin–GPR54 signaling provides the drive necessary for tonic GnRH/luteinizing hormone (LH) release. We predicted that tonic GnRH/LH secretion would be disrupted in GPR54 KOs and that such animals would be incapable of showing a compensatory rise in LH secretion after ovariectomy. As predicted, we found that GPR54 KO mice do not exhibit a postovariectomy rise in LH, suggesting that tonic GnRH secretion is disrupted in the absence of kisspeptin–GPR54 signaling. We also postulated that kisspeptin–GPR54 signaling is critical for the generation of the estradiol (E)-induced GnRH/LH surge and thus E should be incapable of inducing an LH surge in the absence of GPR54. However, we found that E induced Fos expression in GnRH neurons and produced a GnRH-dependent LH surge in GPR54 KOs. Thus, in mice, kisspeptin–GPR54 signaling is required for the tonic stimulation of GnRH/LH secretion but is not required for generating the E-induced GnRH/LH surge.
Keywords: arcuate nucleus, kisspeptin, Kiss1, LH surge, AVPV, Kiss1R
Introduction
Gonadal steroids provide feedback signals to the brain that coordinate the ovulatory cycle in mammals. These signals are relayed to gonadotropin-releasing hormone (GnRH)-producing neurons in the forebrain that control luteinizing hormone (LH) secretion. In rodents, estradiol (E) exerts an inhibitory (negative feedback) effect on GnRH/LH secretion throughout the estrous cycle, except during the afternoon of proestrus, when a rising tide of E initiates a surge of GnRH/LH release (positive feedback) that causes ovulation (Freeman, 2006). Both negative and positive feedback effects of E appear to be mediated by estrogen receptor α (ERα) (Scully et al., 1997; Wintermantel et al., 2006), which is not expressed in GnRH neurons (Hrabovszky et al., 2000). However, ERα is expressed in the arcuate nucleus (Arc) and anteroventral periventricular nucleus (AVPV), which contain neurons that project directly to GnRH neurons (Gu and Simerly, 1997; Clarkson and Herbison, 2006). These unidentified cells in the AVPV and Arc may be Kiss1 neurons.
Neurons in both the Arc and AVPV express the Kiss1 gene (Gottsch et al., 2004). Kiss1 codes for a family of RF-amide proteins, collectively called kisspeptins, which bind to the G-protein-coupled receptor GPR54 (Lee et al., 1999; Kotani et al., 2001; Ohtaki et al., 2001). Mounting evidence suggests kisspeptin–GPR54 signaling activates the neuroendocrine reproductive axis. Humans and mice with disabling mutations in GPR54 remain sexually infantile and are infertile as adults (de Roux et al., 2003; Funes et al., 2003; Seminara et al., 2003). These animals have low circulating levels of gonadotropins, reflecting diminished GnRH secretion, which may be attributable to a lack of trophic activation from kisspeptin. Indeed, kisspeptin stimulates GnRH secretion by a direct action on GnRH neurons, virtually all of which express GPR54 (Gottsch et al., 2004; Dhillo et al., 2005; Irwig et al., 2005; Messager et al., 2005; Navarro et al., 2005; Ramaswamy et al., 2007). E exerts dramatic, but opposite, effects on Kiss1 mRNA expression in the AVPV and Arc (in the Arc, E inhibits the expression of Kiss1, whereas in the AVPV, E induces the expression of Kiss1) (Smith et al., 2005). Because Kiss1 neurons in the Arc and AVPV express ERα (Smith et al., 2005), they represent a possible conduit for mediating the bimodal actions of E on GnRH secretion.
Based on these observations, we have proposed a model whereby Kiss1 neurons in the Arc relay the negative feedback effects of E on GnRH secretion and Kiss1 neurons in the AVPV, when stimulated by E, generate the preovulatory GnRH/LH surge. If this model were valid, we would expect both the tonic and surge release of GnRH/LH to depend on an intact kisspeptin–GPR54 signaling pathway. To test the hypothesis that GPR54 is necessary for tonic GnRH/LH secretion, we evaluated the ability of mice lacking GPR54 to maintain high levels of GnRH/LH secretion after ovariectomy. Likewise, to test the hypothesis that GPR54 plays an essential role in E-positive feedback, we investigated the ability of E to induce a GnRH/LH surge in female mice lacking GPR54.
Materials and Methods
Generation of the GPR54 knock-out strain and animal care
GPR54 knock-out (KO) mice were generated by Omeros Corporation via retroviral mutagenesis as described previously (Krasnow et al., 2004). Briefly, an embryonic stem (ES) cell library was constructed by infecting 129/Sv ES cells with a retroviral vector containing a transcription termination site [Gatanaris GA (2001) U.S. Patent US6228639B1]. Mutations in the GPR54 gene were found in the library by PCR analysis of genomic DNA by using vector-specific and gene-specific primers. Mutant clones isolated from the library were used for animal production, with the use of standard injection methods. Chimeric mice were bred with 129S1/SvImJ mice to generate heterozygotes on an inbred background. The resulting progeny were genotyped by PCR of tail DNA to identify pups containing a disruption in the GPR54 gene. For phenotypic studies, heterozygous males in 129S1/SvImJ inbred background were bred with C57BL/6J females to obtain 129/B6 F1 hybrid heterozygous mice, which were then bred with each other to obtain homozygous GPR54 KO mice and wild-type (WT) control littermates in 129/B6 F2 hybrid background.
All animals were housed in groups of two or three with ad libitum to water and standard rodent chow. The light cycle was set for 14/10 h light/dark, with lights on at 4:00 A.M. and lights off at 6:00 P.M. All animal care and techniques were conducted in accordance with the National Institutes of Health animal care and use guidelines and with the approval of the Animal Care Committee of the University of Washington.
Experimental design
Experiment 1: confirmation and behavioral characterization of GPR54 KO strain.
The purpose of this experiment was to confirm that GPR54 KO mice do not express GPR54 mRNA in GnRH neurons and to examine the LH response of GPR54 KO mice to kisspeptin treatment. For the former, brains were collected from castrated male WT and GPR54 KO mice (n = 5 per group) and subjected to double in situ hybridization (ISH) for GnRH and GPR54 as described above. For the latter, adult female WT and GPR54 KO mice (n = 5 per group) were primed with two intraperitoneal injections of GnRH given 7 and 3 d before kisspeptin challenge. After GnRH priming, mice received intraperitoneal injections of either the vehicle alone (100 μl of sterile 0.9% saline) or 0.1 nmol of kisspeptin-54 (Phoenix Pharmaceuticals, Phoenix, AZ) suspended in vehicle. Serum was collected 30 min after injection and assayed for LH as described below.
Behavioral tests were performed on intact adult female GPR54 KO mice and WT littermates. These tests, including the hot-plate test, light/dark box test, tail suspension test, and contextual fear conditioning test, were performed as described in the supplemental Methods (available at www.jneurosci.org as supplemental material).
Experiment 2: LH response to ovariectomy and E treatment.
The purpose of this experiment was to test whether GPR54 signaling is necessary for maintaining tonic GnRH/LH secretion. To accomplish this, we eliminated negative feedback by removing the ovaries and examined serum LH levels 7 d later, when serum levels of LH would normally become elevated. Female WT and GPR54 KO mice were ovariectomized (OVX) and implanted with empty (sham) or E-filled capsules to induce supraphysiological serum E levels as described previously (Smith et al., 2005) (n = 4–5 per group). After 7 d, blood was collected from the orbital sinus in the morning (8:00–10:00 A.M.), while keeping the animals anesthetized under inhaled isoflurane anesthesia; subsequently, the mice were killed by cervical dislocation. Brains were immediately collected, frozen, and prepared for ISH to measure Kiss1 mRNA. Serum was assayed for LH by radioimmunoassay (RIA), as described below.
Experiment 3: E-induced LH surge.
The purpose of this experiment was to test the model that GPR54 signaling mediates the positive feedback effects of E on LH secretion. To accomplish this, we used a protocol that was previously shown to induce a daily, evening LH surge in mice (Christian et al., 2005). Adult female WT and GPR54 KO mice were subjected to the LH surge protocol as described below. WT mice were divided into two groups for blood collection at the A.M. or P.M. time point (A.M., n = 8; P.M., n = 7). KO mice were likewise divided into two groups for blood collection in the A.M. or P.M. (A.M., n = 7; P.M., n = 6). Brains were also collected and processed for ISH as described below.
Experiment 4: acyline blockade of E-induced LH surge.
The purpose of this experiment was to determine whether the P.M. rise in LH observed in GPR54 KO mice in experiment 3 is mediated either by GnRH or through a direct action of E on the pituitary gonadotropes. Adult female WT and GPR54 KO mice were subjected to the LH surge protocol described below. Both WT and KO mice were divided into three groups. The first group was collected at the A.M. time point, with no additional treatments (A.M.: WT, n = 8; KO, n = 7). The second group received two subcutaneous injections of 50 μg of the GnRH antagonist acyline (Rivier et al., 1995; Herbst et al., 2002) dissolved in 100 μl of water, one injection 26 h before and one injection 1 h before the mice were killed (P.M. plus acyline: WT, n = 5; KO, n = 8). Acyline was provided as a gift from Dr. W. Bremner (University of Washington, Seattle, WA). The third group received injections of 100 μl of water (P.M. plus vehicle: WT, n = 9; KO, n = 10). Brains were collected and processed for double-label immunohistochemistry (IHC) in experiment 5. All injections were given while maintaining the animals under brief isofluorane anesthesia.
Experiment 5: Fos expression in GnRH neurons during the E-induced LH surge.
The purpose of this experiment was to determine whether GnRH neurons coexpress Fos at the time of the E-induced LH surge. If the E-induced LH surge we had observed in experiment 3 is dependent on GnRH secretion, then we would expect to see more GnRH neurons coexpressing Fos in the P.M. than in the A.M. in both genotypes. The brains collected in experiment 4 were subjected to double IHC for GnRH and Fos protein as described below.
RIAs
Serum levels of E and LH were measured at the University of Virginia Center for Research in Reproduction Ligand Assay and Analysis core (Charlottesville, VA). E was measured with a double-antibody kit (Diagnostics Systems Laboratories, Webster, TX), which had a sensitivity of 10 pg/ml, an intraassay coefficient of variation of 5.7%, and an interassay coefficient of variation of 11.6%. LH was measured using modified two-site sandwich immunoassay using two monoclonal antibodies against bovine and human LH. The sensitivity of the LH assay was 0.04 ng/ml, the intraassay coefficient of variation was 3.6%, and the interassay coefficient of variation was 9.2%. LH values in experiments 3 and 4 were measured in separate assays.
Induction of daily LH surge
We used the protocol devised by Christian et al. (2005) to induce a predictably timed LH surge in adult female mice. Briefly, on the morning of day 0, mice were anesthetized by inhaled isofluorane, and the ovaries were removed through bilateral incisions. A SILASTIC (Dow-Corning, Midland, MI) capsule containing 0.652 μg of E suspended in sesame seed oil was immediately implanted under the skin in the mid-scapular region (1.5 cm, 1.0 mm inner diameter × 2.2 mm outer diameter). Each animal was given an intramuscular injection of 0.05 ng of ketoprofen, a long-acting analgesic (Ketofen; Fort Dodge Animal Health, Fort Dodge, IA), and was allowed to recover for several hours in a warmed cage, under supervision, before being returned to central animal housing. Animals were assigned to a morning (A.M.) or evening (P.M.) group. For animals in the A.M. group, on the morning of day 3, between 7:00 and 8:00 A.M., blood was collected from the orbital sinus, while the animals were anesthetized under isofluorane anesthesia. All animals in the P.M. group were anesthetized with isofluorane between 6:30 and 7:30 P.M. (30–90 min after lights out) on the evening of day 3, and blood was collected from the orbital sinus. Immediately after blood collection, all animals were killed by cervical dislocation, and brains were collected. All animal procedures were done in accordance with the National Institutes of Health animal care and use guidelines and with the approval of the Institutional Animal Care and Use Committee of the University of Washington.
ISH
Brain tissue was prepared for ISH by freezing on dry ice immediately after collection and storage at −80°C. Single-label ISH for Kiss1 was performed as described previously (Gottsch et al., 2004). Briefly, radiolabeled (33P) antisense Kiss1 riboprobe was denatured, diluted in hybridization buffer at a concentration of 0.03 pmol/ml along with tRNA (2 mg/ml), and applied to slides (100 μl per slide). After hybridization, slides were treated with RNase (29 μg/ml), washed, and dehydrated as reported previously (Cunningham et al., 2002). Slides were then dipped in liquid emulsion type NTB (Eastman Kodak, Rochester, NY) and stored at 4°C. Slides were developed 4 d later. After developing, slides were dehydrated with a series of ethanol washes, followed by two 10 min washes in Citrisolv (Fisher Scientific, Baltimore, MD) and coverslipping with Permaslip mounting medium (Alban Scientific, St. Louis, MO). Slides were coded and randomized to obscure group identification and examined with dark-field microscopy.
Double-label ISH for GnRH and GPR54 mRNA was performed as described previously (Irwig et al., 2005). Radiolabeled antisense GPR54 (0.1 pmol/ml) and digoxigenin (DIG)-labeled GnRH riboprobes (concentration determined empirically) were denatured, dissolved in the same hybridization buffer along with tRNA (2 mg/ml), and applied to slides. Slides were allowed to hybridize overnight. After hybridization, slides were treated with RNase (29 μg/ml), washed in conditions of increasing stringency, and transferred to blocking buffer. Slides were then washed in Tris buffer containing anti-DIG fragments conjugated to alkaline phosphatase (diluted 1:300; Roche, Indianapolis, IN). Vector Red substrate (SK-5100; Vector Laboratories, Burlingame, CA) was used to visualize DIG-containing cells, following the directions of the manufacturer. Slides were dipped in 70% ethanol, air dried, and dipped in NTB liquid emulsion (Eastman Kodak). Slides were developed 10 d later.
GnRH mRNA-containing cells were identified under fluorescent illumination, and silver grains (corresponding to radiolabeled GPR54 mRNA over each cell) were quantified. The number of grains over each labeled GnRH cell was counted and averaged for each animal. The grains per cell for each animal were then averaged across the group.
Double-label IHC for Fos and GnRH
Fresh whole-brain tissue was prepared for IHC with a series of three washes in 10% phosphate-buffered formalin (Fisher Scientific). Brains were immersed in formalin in sterile glass vials immediately after collection and shaken for 30 min. Then, brains were blocked by slicing off the olfactory bulbs and the brainstem and returned to a fresh formalin wash. Brains from the A.M. groups were then shaken in formalin for 2 h, placed into fresh formalin for another 2 h, and immersed overnight in phosphate-buffered 10% formalin/10% sucrose overnight at 4°C. The next day, brains that were collected in the A.M. were immersed in phosphate-buffered 20% sucrose until the tissue sunk to the bottom, then they were frozen on dry ice and stored at −80°C. Brains from the P.M. groups were treated the same as A.M. brains but were left in the second formalin wash overnight. The next day, brains were shaken in fresh formalin for 2 h and immersed in 10% formalin/10% sucrose at 4°C for up to 4 h. They were then immersed in 20% sucrose overnight and frozen the next day. Four parallel series of 35-μm-thick coronal sections were cut on a cryostat and stored in 0.9% PBS with 0.5% sodium azide at 4°C until processed.
Double IHC for Fos and GnRH protein was performed as described previously (Suzuki and Handa, 2005). Briefly, tissue was washed three times in PBS with 0.1% Triton X-100 (PBST) and blocked for 15 min in 0.3% H2O2. Tissue was then washed twice more in PBST and blocked for 2 h in 6% normal horse serum (NHS). Tissue was shaken at 4°C for 48 h in a solution of PBST with 2% NHS and primary Fos antibody SC-52G (Santa Cruz Biotechnology, Santa Cruz, CA) at a concentration of 1:5000. After primary antibody incubation, tissue was washed three times in PBST and shaken at room temperature for 2 h in a solution of PBST, with 2% NHS and secondary antibody, at a concentration of 1:500 (biotinylated horse anti-goat; Vector Laboratories). After incubation with a secondary antibody, tissue was washed three times in PBST, incubated in avidin/biotin complex (ABC) reagent, and developed with nickel-intensified diaminobenzidine (DAB), according to the manufacturer's instructions (Vectastain ABC kit and DAB kit; Vector Laboratories).
Subsequently, tissue was processed for IHC for GnRH by using the same protocol as above with modifications. The tissue was incubated overnight at 4°C in PBST with GnRH antiserum (1:5000; Affinity Bioreagents, Golden CO). Normal goat serum was used as a blocking agent, and at the DAB staining step, nickel was excluded from reaction. After staining, tissue sections were mounted onto SuperFrost Plus glass slides (VWR, Westchester, PA) and coverslipped. All slides were scored blindly, and immunoreactive cells in the medial septum and diagonal band of Broca (MS/DBB) and the medial preoptic area (MPA) were counted and analyzed separately. Cells stained in brown were counted as GnRH-expressing cells, and brown-stained cells with gray/black stained nuclei were scored as cells coexpressing GnRH and Fos. Any cells that were ambiguous in staining were excluded from analysis. Data are expressed as the percentage of GnRH neurons that also express Fos.
Statistical analysis
Data are expressed at mean ± SEM for each group. One-way ANOVA was used to assess variation of LH levels and Kiss1 mRNA among treatment groups. When the F test for the ANOVA reached statistical significance (p < 0.05), differences among means were assessed by least significant difference tests. All analyses were performed with Statview 5.0.1 for Macintosh (SAS Institute, Cary, NC).
Results
Experiment 1: confirmation of GPR54 KO
Inspection of gross morphology revealed that both genders of adult GPR54 KO mice have smaller gonads and accessory reproductive organs than WT mice (A. S. Kauffman and our own unpublished observations). In the first part of experiment 1, we used double-label ISH to examine the expression of GPR54 mRNA in GnRH neurons. We found that GnRH neurons (identified by red staining) in WT mice have an average of 33.7 ± 10.8 silver grains (denoting GPR54 mRNA) over them (Fig. 1A,B). However, no silver grains are detectable over GnRH neurons in GPR54 KO mice (p < 0.01). We detected no difference between the number of GnRH neurons observed in WT compared with GPR54 KO mice; we counted an average of 29 neurons in GPR54 KO mice and an average of 35 neurons in WT mice.
Figure 1.

A, Photomicrographs of double-label ISH in WT (n = 5) and GPR54 KO (n = 5) brains. White arrows indicate GnRH neurons; black arrows with white outline indicate GnRH cells coexpressing GPR54 mRNA. Scale bars, 50 μm. B, Quantification of silver grains (GPR54 mRNA) on GnRH neurons. C, Serum LH levels in female WT and GPR54 KO mice after treatment with vehicle or kisspeptin (n = 4–5 per group). Double asterisks indicate significant difference at p < 0.01. Error bars indicate SEM.
In the second part of experiment 1, we found that kisspeptin treatment significantly increased serum LH concentrations in GnRH-primed WT mice compared with vehicle treatment (p < 0.05). However, in GPR54 KO mice, treatment had no effect on serum LH levels, which were at the bottom range of detectability (Fig. 1C). Behavioral tests revealed no significant differences between genotypes in indices of learning, anxiety, and nociception (supplemental Figs. 1–4, available at www.jneurosci.org as supplemental material).
Experiment 2: Kiss1 mRNA expression and LH response to OVX
In this experiment, we examined tonic LH secretion in GPR54KO and WT mice after OVX and E replacement. Mice were OVX and implanted with either an oil-filled capsule as a sham control or an E-filled capsule to replace circulating E. In the Arc, OVX and sham replacement (OVX plus sham) resulted in a dramatic increase in the number of Kiss1 mRNA-expressing cells of both genotypes compared with mice given E implants (OVX plus E) (Fig. 2A,B). The difference was significant by treatment (p < 0.05) but not by genotype. In the AVPV, E replacement increased the number of Kiss1 cells compared with sham replacement for both genotypes (p < 0.05). Curiously, we found that, regardless of treatment, GPR54 KO mice had fewer Kiss1 cells than WT mice (p < 0.05 by genotype) (Fig. 2A,C). Mice of both genotypes in the OVX plus E group had low levels of LH. OVX plus sham WT mice had high serum levels of LH. In contrast, OVX plus sham GPR54 KO mice had significantly lower LH levels (p < 0.01) (Fig. 3).
Figure 2.

A, Representative photomicrographs of ISH for Kiss1 mRNA in the Arc and AVPV of OVX sham (OVX + Sham) and E-replaced (OVX + E) mice. Scale bars, 100 μm. B, Quantification of Kiss1-expressing cells in the Arc. Different letters represent significant difference (p < 0.05). C, Quantification of Kiss1-expressing cells in the AVPV. Different letters represent significant difference (p < 0.05; E WT, n = 5; E KO, n = 5; sham WT, n = 5; sham KO, n = 4). Error bars indicate SEM.
Figure 3.

Serum LH levels in OVX E and sham-replaced WT and GPR54 KO mice. Double asterisks indicate significant difference (p < 0.01; E WT, n = 5; E KO, n = 5; sham WT, n = 5; sham KO, n = 4). Error bars indicate SEM.
Experiment 3: E-induced LH surge
In this experiment, we used a paradigm of ovariectomy and E replacement to induce daily LH surges in female mice. We compared LH levels of WT and GPR54 KO mice in the morning and evening of day 3 after the surgeries. As expected, all mice sampled on the morning of day 3 (A.M.) had low levels of LH (0.19 ± 0.1 ng/ml). We used a value of the A.M. mean for both groups of mice plus two times the SD as a threshold for identifying a surge of LH (0.99 ng/ml). All WT mice sampled at the P.M. time point (seven of seven) had serum LH levels above the threshold. The mean LH level for the P.M. WT group was significantly higher than the A.M. LH levels (p < 0.01) (Fig. 4). Among the GPR54 KO mice sampled in the P.M., four of six had serum LH levels above the threshold, and the mean for all six of those animals was significantly higher than the A.M. mean (p < 0.01). Serum E levels were within the physiological range (15.5 ± 1.3 pg/ml).
Figure 4.

Serum LH levels in WT and GPR54 KO mice in the A.M. and P.M.. Different letters indicate significant difference (p < 0.05; A.M. WT, n = 8; A.M. KO, n = 7; P.M. WT, n = 7; P.M. KO, n = 6). Error bars indicate SEM.
Experiment 4: acyline-induced blockade of the LH surge
To determine whether GnRH signaling was required to mediate the LH surge, we repeated the surge-inducing paradigm of ovariectomy and E replacement and added groups that received injections of the GnRH antagonist acyline. As in the previous experiment, LH levels of the A.M. groups were low (0.16 ± 0.05 ng/ml). Again, we used the A.M. mean plus two times the SD to determine a threshold for an LH surge (0.53 ng/ml). The mean LH levels in vehicle-treated animals sampled in the P.M. were significantly higher than animals sampled in the A.M., regardless of genotype (p < 0.05) (Fig. 5). In the P.M., 5 of 9 WT and 3 of 10 KO mice treated with vehicle had LH levels above the threshold. Among the acyline-treated mice sampled in the P.M., 0 of 5 WT and 0 of 8 KO animals had LH levels above the surge threshold, with means at the bottom limit of detection (Fig. 5). Serum levels of E were in the physiological range (13.5 ± 2.8 pg/ml).
Figure 5.

Serum LH levels in WT and GPR54 KO mice in the A.M. and P.M. All mice in the P.M. groups were treated with either vehicle or acyline. Different letters indicate significant difference (p < 0.05; A.M. WT, n = 7; A.M. KO, n = 7; P.M. WT + Veh., n = 9; P.M. KO + Veh., n = 10; P.M. WT + acyline, n = 5; P.M. KO + acyline, n = 8). Error bars indicate SEM. Veh., Vehicle.
Experiment 5: Activation of GnRH neurons at the time of the surge
In this experiment, we used the brains collected in experiment 4 for GnRH/Fos double-label IHC assay. Because no significant difference in the percentage of GnRH neurons expressing Fos was found between the acyline and vehicle-treated groups (data not shown), tissue from the P.M. groups treated with acyline and vehicle were analyzed together. As with LH, we used the A.M. mean plus two times the SD of the percentage of GnRH neurons expressing Fos (47%) as a threshold to determine whether each individual exhibited a significant activation of Fos in its GnRH neurons. By this criterion, Fos was activated in 6 of 14 WT and 6 of 18 KO mice. Likewise, the mean percentage of GnRH neurons coexpressing Fos was higher in the P.M. than in the A.M., regardless of genotype (p < 0.01) (Fig. 6). Analysis of the data by hypothalamic region revealed a nonsignificant trend of increased coexpression in the MPA compared with the MS/DBB of the P.M. groups but not the A.M. groups (data not shown).
Figure 6.
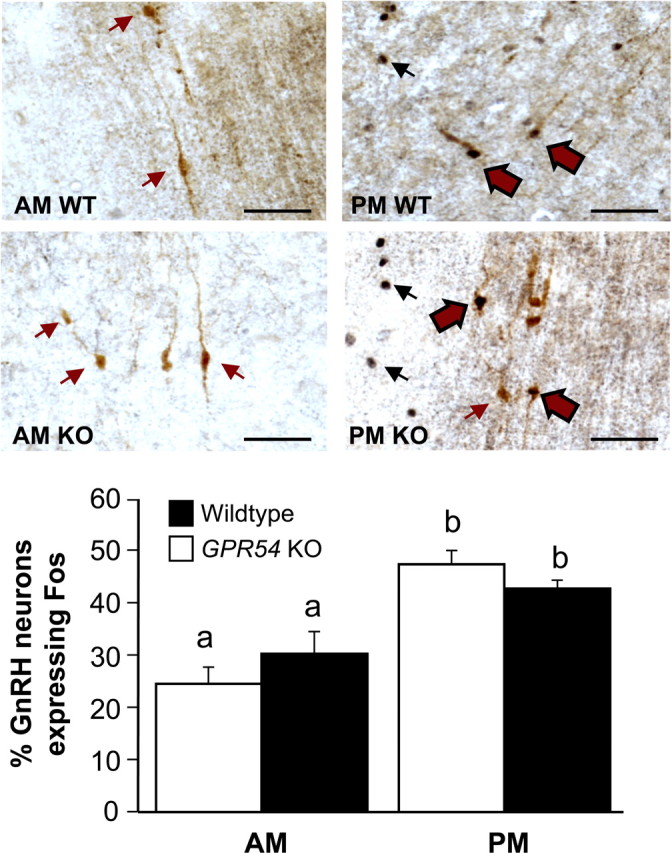
A, Representative photomicrographs of GnRH and Fos double-label IHC in the rostral hypothalamus. Black arrows indicate Fos-stained nuclei. Red arrows indicate GnRH-stained cytoplasm. Red arrows with black outlines indicate cells stained for both GnRH and Fos. B, Percentage of GnRH neurons coexpressing Fos protein in the A.M. and P.M. Different letters indicate significant difference (p < 0.01; A.M. WT, n = 7; A.M. KO, n = 7; P.M. WT plus vehicle or acyline, n = 14; P.M. KO vehicle or acyline, n = 18). Scale bars, 50 μm. Error bars indicate SEM.
Discussion
The results presented here confirm previous observations that deletions of the GPR54 gene produce hypogonadotropic hypogonadism, without any other obvious phenotypic abnormalities (de Roux et al., 2003; Funes et al., 2003; Seminara et al., 2003). Our GPR54 KO mice were hypogonadal but demonstrated no abnormalities in indices of pain, anxiety, or memory compared with WT littermates. These GPR54 KO mice retain the ability to exhibit positive feedback effects of E on GnRH secretion but lack the ability to show an appropriate response to ovariectomy. These observations suggest that the primary defect in the GPR54 KOs is attributable to the loss of tonic support of gonadotropin secretion and hence ovarian function. However, the ability of E to act on the brain and evoke a GnRH/LH surge remains intact, despite the complete loss of kisspeptin–GPR54 signaling.
During the first part of the female reproductive cycle, the negative feedback effects of E keep gonadotropin secretion within the physiological range necessary to support folliculogenesis (Freeman, 2006). Ovariectomy reduces circulating levels of E, which decreases the negative feedback inhibition of GnRH secretion and thus causes serum LH to rise. Because animals lacking a functional GPR54 gene are “hypogonadotropic,” it might be expected that they would be incapable of displaying a postovariectomy rise in LH. However, alternative mechanisms could explain low gonadotropin secretion in GPR54-deficient animals. For example, GPR54 KOs could be hypersensitive to the negative feedback effects of gonadal steroids, such that even small quantities of sex steroids produced by their immature gonads would lead to chronic low levels of gonadotropins, as has been suggested to be the case in normal prepubertal rodents (Ojeda et al., 1983). Nevertheless, our results indicate that even in the absence of any gonadal hormones, LH levels in the GPR54 KO remain low. This finding highlights the importance of kisspeptin–GPR54 input to GnRH neurons for providing tonic stimulation of GnRH/LH.
Although the present study does not directly test the idea that Kiss1 neurons in the Arc mediate negative feedback by E, our results are consistent with this hypothesis. The negative feedback effects of E on GnRH/LH secretion depend on ERα (Hewitt and Korach, 2003), which GnRH neurons do not appear to express (Hrabovszky et al., 2000). Thus, E is thought to exert its negative feedback effects on GnRH neurons through intermediaries that express ERα, and Kiss1 neurons in the Arc may serve this function, based on several lines of indirect evidence. First, neurons with cell bodies that are located in the Arc project to regions of the forebrain containing GnRH neurons, and some of these cells express ERα (Hahn and Coen, 2006; Wintermantel et al., 2006). Second, Kiss1 neurons reside within the Arc, and they express ERα. Third, the expression of Kiss1 mRNA in the Arc is negatively regulated by E (Smith et al., 2005). Finally, in the sheep, GnRH neurons receive direct synaptic input from dynorphin-containing fibers (Goodman et al., 2004), and recent evidence (again in the sheep) reveals that virtually all kisspeptin neurons in the Arc coexpress dynorphin (Goodman et al., 2007). However, it remains to be determined whether the dynorphin fibers that innervate GnRH neurons in the sheep originate from kisspeptin neurons in the Arc and whether this is the case in other species. Thus, despite compelling circumstantial evidence, additional work with careful retrograde and anterograde tract tracing will be required to establish unequivocally that Kiss1 neurons in the Arc connect directly to GnRH neurons.
In addition to the Arc, Kiss1 neurons also reside in the AVPV. These two populations of Kiss1 neurons represent phenotypically distinct cell groups, with only the population in the AVPV coexpressing tyrosine hydroxylase (in the mouse) (Lee et al., 2005). Furthermore, E inhibits the expression of Kiss1 in the Arc, but in the AVPV, E stimulates Kiss1 expression (Smith et al., 2005). Thus, although both groups of neurons express Kiss1, it is likely that they serve different physiological functions. The AVPV is thought to contain neural circuits involved in generating GnRH/LH surges (Wiegand et al., 1978; Simerly, 1998). Kiss1 neurons in the AVPV express ERα, and at the time of the preovulatory LH surge (in the rat), those same neurons express Fos and increase their synthesis of Kiss1 mRNA (Smith et al., 2006; Adachi et al., 2007). Together, these observations implicate Kiss1 neurons in the AVPV for an important role in the positive feedback regulation of GnRH/LH secretion.
Despite strong evidence that kisspeptin normally plays an important role in E positive feedback, we have shown that LH surges can occur in the absence of GPR54. We also demonstrated that Fos is induced in GnRH neurons in temporal association with the E-induced LH surge (even in GPR54 KOs) and that the LH surge in GPR54 KOs can be blocked by the administration of a GnRH antagonist. These experiments demonstrate that the LH surge requires the activation GnRH neurons but does not require kisspeptin-GPR54 signaling. In the Fos experiment, some animals of both genotypes failed to exhibit a robust increase in LH, and the overall amplitude of the surge appeared to be lower in experiment 4 than experiment 3. The apparent attenuated response in experiment 4 could simply reflect the normal variation in the timing/amplitude of the LH surge, compounded by the fact that we could only sample once to characterize the entire surge event. It is also possible that a delay in the onset of the surge, perhaps because of the stress of an additional injection, produced lower LH values that were interpreted as a “failure” of the surge event.
Our observation that the surge mechanism remains unabated in the GPR54 KO mouse appears to conflict with data from the rat showing a blockade of the LH surge with kisspeptin antiserum (Kinoshita et al., 2005; Adachi et al., 2007). There are several possible explanations for this apparent discrepancy. First, it is conceivable that kisspeptin can signal through another receptor besides GPR54. This seems unlikely, because it has been reported that kisspeptin cannot stimulate LH secretion in GPR54 KO mice (Messager et al., 2005), and we have confirmed those observations in the present study. In addition, Kauffman et al. (2006) have demonstrated that although kisspeptin can induce Fos in GnRH neurons of WT mice, it does not do so in GPR54 KOs (Kauffman et al., 2006). Thus, even if kisspeptin were to bind another receptor (besides GPR54), it cannot directly activate Fos in GnRH neurons. Second, the kisspeptin antiserum studies were performed in rats, whereas our experiments were conducted in mice. Perhaps the GnRH/LH surge mechanism in the mouse contains additional parallel (or redundant) circuits that are not present in the rat. Third, kisspeptin–GPR54 signaling was acutely blocked by antiserum in rats but is congenitally absent in GPR54 KOs. Thus, it remains possible that in the congenital absence of GPR54, alternative pathways develop or become activated as a mechanism of compensation. This appears to be the case for other neurotransmitter systems thought to be involved in the generation of GnRH/LH surges. For example, the pharmacological blockade of norepinephrine has been shown to block the LH surge in rats. However, transections that sever noradrenergic input to the hypothalamus only temporarily block LH surges (Clifton and Sawyer, 1979). Several weeks after such transections, LH surges return and can no longer be blocked by anti-noradrenergic agents (Clifton and Sawyer, 1980). Determining which alternative pathway is active in the absence of GPR54 will require further investigation.
Our investigation of Kiss1 mRNA expression in the AVPV revealed a difference in the number of Kiss1 cells between OVX WT and GPR54 KO mice. In both genotypes, E treatment increased the number of Kiss1 cells in the AVPV; however, even in the presence of E, GPR54 KO mice had only about half as many Kiss1 cells as WT mice. The mechanisms responsible for this phenomenon are unclear. Development of Kiss1 cells in the AVPV is thought to be determined by the steroid environment during the neonatal critical period, with exposure to androgens during early life causing Kiss1 cells to disappear (Kauffman et al., 2007). However, it is unlikely that female GPR54 KOs had high androgen levels during the critical period, because a lack of kisspeptin–GPR54 signaling would be expected to diminish androgen production. This suggests that another GPR54-dependent process contributes to the development of Kiss1 cells in the AVPV during maturation. Alternatively, Kiss1 neurons in GPR54 KO mice, which have never been exposed to elevated E levels, require longer than 7 d to respond to E treatment.
In summary, we report that GPR54 signaling is critical for the maintenance of tonic LH secretion. In contrast, we also show that GPR54 is not required for E to induce a GnRH/LH surge and that alternative (or redundant) signaling pathways may compensate for the loss of kisspeptin–GPR54 signaling. Together, these results suggest that infertility in females lacking a functional GPR54 gene is not attributable to an inability to generate a preovulatory LH surge. Instead, it reflects a lack of tonic gonadotropin secretion that normally stimulates follicular development, leading to the high levels of circulating E necessary for the production of a GnRH/LH surge and subsequently ovulation.
Footnotes
This work was supported by National Institutes of Health Grant R01 HD27142 and through Cooperative Agreement U54 HD12629 as part of the Specialized Cooperative Centers Program in Reproduction and Infertility Research. We thank Dr. Alexander Kauffman, Dr. Patience Browne, Sonya Jakawich, Janessa Lawhorn, Alisa Byquist, and Maile Parker for technical assistance. All hormone measurements were performed by the Specialized Cooperative Centers Program in Reproduction and Infertility Research Ligand Assay and Analysis Core at the University of Virginia (Charlottesville, VA).
References
- Adachi S, Yamada S, Takatsu Y, Matsui H, Kinoshita M, Takase K, Sugiura H, Ohtaki T, Matsumoto H, Uenoyama Y, Tsukamura H, Inoue K, Maeda K. Involvement of anteroventral periventricular metastin/kisspeptin neurons in estrogen positive feedback action on luteinizing hormone release in female rats. J Reprod Dev. 2007;53:367–378. doi: 10.1262/jrd.18146. [DOI] [PubMed] [Google Scholar]
- Christian CA, Mobley JL, Moenter SM. Diurnal and estradiol-dependent changes in gonadotropin-releasing hormone neuron firing activity. Proc Natl Acad Sci USA. 2005;102:15682–15687. doi: 10.1073/pnas.0504270102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson J, Herbison AE. Postnatal development of kisspeptin neurons in mouse hypothalamus; sexual dimorphism and projections to gonadotropin-releasing hormone neurons. Endocrinology. 2006;147:5817–5825. doi: 10.1210/en.2006-0787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifton DK, Sawyer CH. LH release and ovulation in the rat following depletion of hypothalamic norepinephrine: chronic vs. acute effects. Neuroendocrinology. 1979;28:442–449. doi: 10.1159/000122893. [DOI] [PubMed] [Google Scholar]
- Clifton DK, Sawyer CH. Positive and negative feedback effects of ovarian steroids on luteinizing hormone release in ovariectomized rats following chronic depletion of hypothalamic norepinephrine. Endocrinology. 1980;106:1099–1102. doi: 10.1210/endo-106-4-1099. [DOI] [PubMed] [Google Scholar]
- Cunningham MJ, Scarlett JM, Steiner RA. Cloning and distribution of galanin-like peptide mRNA in the hypothalamus and pituitary of the macaque. Endocrinology. 2002;143:755–763. doi: 10.1210/endo.143.3.8661. [DOI] [PubMed] [Google Scholar]
- de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc Natl Acad Sci USA. 2003;100:10972–10976. doi: 10.1073/pnas.1834399100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhillo WS, Chaudhri OB, Patterson M, Thompson EL, Murphy KG, Badman MK, McGowan BM, Amber V, Patel S, Ghatei MA, Bloom SR. Kisspeptin-54 stimulates the hypothalamic-pituitary gonadal axis in human males. J Clin Endocrinol Metab. 2005;90:6609–6615. doi: 10.1210/jc.2005-1468. [DOI] [PubMed] [Google Scholar]
- Freeman ME. The neuroendocrine control of the ovarian cycle of the rat. In: Neill JD, editor. Knobil and Neill's physiology of reproduction. Vol 2. Philadelphia: Elsevier; 2006. pp. 2327–2388. [Google Scholar]
- Funes S, Hedrick JA, Vassileva G, Markowitz L, Abbondanzo S, Golovko A, Yang S, Monsma FJ, Gustafson EL. The KiSS-1 receptor GPR54 is essential for the development of the murine reproductive system. Biochem Biophys Res Commun. 2003;312:1357–1363. doi: 10.1016/j.bbrc.2003.11.066. [DOI] [PubMed] [Google Scholar]
- Goodman RL, Coolen LM, Anderson GM, Hardy SL, Valent M, Connors JM, Fitzgerald ME, Lehman MN. Evidence that dynorphin plays a major role in mediating progesterone negative feedback on gonadotropin-releasing hormone neurons in sheep. Endocrinology. 2004;145:2959–2967. doi: 10.1210/en.2003-1305. [DOI] [PubMed] [Google Scholar]
- Goodman RL, Lehman MN, Smith JT, Coolen LM, de Oliveira CV, Jafarzadehshirazi MR, Pereira A, Iqbal J, Caraty A, Ciofi P, Clarke IJ. Kisspeptin neurons in the arcuate nucleus of the ewe express both dynorphin A and neurokinin B. Endocrinology. 2007 doi: 10.1210/en.2007-0961. in press. [DOI] [PubMed] [Google Scholar]
- Gottsch ML, Cunningham MJ, Smith JT, Popa SM, Acohido BV, Crowley WF, Seminara S, Clifton DK, Steiner RA. A role for kisspeptins in the regulation of gonadotropin secretion in the mouse. Endocrinology. 2004;145:4073–4077. doi: 10.1210/en.2004-0431. [DOI] [PubMed] [Google Scholar]
- Gu GB, Simerly RB. Projections of the sexually dimorphic anteroventral periventricular nucleus in the female rat. J Comp Neurol. 1997;384:142–164. [PubMed] [Google Scholar]
- Hahn JD, Coen CW. Comparative study of the sources of neuronal projections to the site of gonadotrophin-releasing hormone perikarya and to the anteroventral periventricular nucleus in female rats. J Comp Neurol. 2006;494:190–214. doi: 10.1002/cne.20803. [DOI] [PubMed] [Google Scholar]
- Herbst KL, Anawalt BD, Amory JK, Bremner WJ. Acyline: the first study in humans of a potent, new gonadotropin-releasing hormone antagonist. J Clin Endocrinol Metab. 2002;87:3215–3220. doi: 10.1210/jcem.87.7.8675. [DOI] [PubMed] [Google Scholar]
- Hewitt SC, Korach KS. Oestrogen receptor knockout mice: roles for oestrogen receptors alpha and beta in reproductive tissues. Reproduction. 2003;125:143–149. doi: 10.1530/rep.0.1250143. [DOI] [PubMed] [Google Scholar]
- Hrabovszky E, Shughrue PJ, Merchenthaler I, Hajszan T, Carpenter CD, Liposits Z, Petersen SL. Detection of estrogen receptor-beta messenger ribonucleic acid and 125I-estrogen binding sites in luteinizing hormone-releasing hormone neurons of the rat brain. Endocrinology. 2000;141:3506–3509. doi: 10.1210/endo.141.9.7788. [DOI] [PubMed] [Google Scholar]
- Irwig MS, Fraley GS, Smith JT, Acohido BV, Popa SM, Cunningham MJ, Gottsch ML, Clifton DK, Steiner RA. Kisspeptin Activation of gonadotropin releasing hormone neurons and regulation of KiSS-1 mRNA in the male rat. Neuroendocrinology. 2005;80:264–272. doi: 10.1159/000083140. [DOI] [PubMed] [Google Scholar]
- Kauffman A, Crown A, Suzuki S, Gottsch M, Hohmann J, Clifton D, Steiner R. Analysis of Fos induction in GnRH neurons of wild-type and GPR54 knock-out mice. Soc Neurosci Abstr. 2006;36:658–11. [Google Scholar]
- Kauffman AS, Gottsch ML, Roa J, Byquist AC, Crown A, Clifton DK, Hoffman GE, Steiner RA, Tena-Sempere M. Sexual differentiation of Kiss1 gene expression in the brain of the rat. Endocrinology. 2007;148:1774–1783. doi: 10.1210/en.2006-1540. [DOI] [PubMed] [Google Scholar]
- Kinoshita M, Tsukamura H, Adachi S, Matsui H, Uenoyama Y, Iwata K, Yamada S, Inoue K, Ohtaki T, Matsumoto H, Maeda K. Involvement of central metastin in the regulation of preovulatory luteinizing hormone surge and estrous cyclicity in female rats. Endocrinology. 2005;146:4431–4436. doi: 10.1210/en.2005-0195. [DOI] [PubMed] [Google Scholar]
- Kotani M, Detheux M, Vandenbogaerde A, Communi D, Vanderwinden JM, Le Poul E, Brezillon S, Tyldesley R, Suarez-Huerta N, Vandeput F, Blanpain C, Schiffmann SN, Vassart G, Parmentier M. The metastasis suppressor gene KiSS-1 encodes kisspeptins, the natural ligands of the orphan G protein-coupled receptor GPR54. J Biol Chem. 2001;276:34631–34636. doi: 10.1074/jbc.M104847200. [DOI] [PubMed] [Google Scholar]
- Krasnow SM, Hohmann JG, Gragerov A, Clifton DK, Steiner RA. Analysis of the contribution of galanin receptors 1 and 2 to the central actions of galanin-like peptide. Neuroendocrinology. 2004;79:268–277. doi: 10.1159/000079632. [DOI] [PubMed] [Google Scholar]
- Lee DK, Nguyen T, O'Neill GP, Cheng R, Liu Y, Howard AD, Coulombe N, Tan CP, Tang-Nguyen AT, George SR, O'Dowd BF. Discovery of a receptor related to the galanin receptors. FEBS Lett. 1999;446:103–107. doi: 10.1016/s0014-5793(99)00009-5. [DOI] [PubMed] [Google Scholar]
- Lee KJ, Maizlin II, Clifton DK, Steiner RA. Coexpression of tyrosine hydroxylase and KiSS-1 mRNA in the anteroventral periventricular nucleus of the female mouse. Soc Neurosci Abstr. 2005;35:758–19. [Google Scholar]
- Messager S, Chatzidaki EE, Ma D, Hendrick AG, Zahn D, Dixon J, Thresher RR, Malinge I, Lomet D, Carlton MB, Colledge WH, Caraty A, Aparicio SA. Kisspeptin directly stimulates gonadotropin-releasing hormone release via G protein-coupled receptor 54. Proc Natl Acad Sci USA. 2005;102:1761–1766. doi: 10.1073/pnas.0409330102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro VM, Castellano JM, Fernandez-Fernandez R, Tovar S, Roa J, Mayen A, Nogueiras R, Vazquez MJ, Barreiro ML, Magni P, Aguilar E, Dieguez C, Pinilla L, Tena-Sempere M. Characterization of the potent luteinizing hormone-releasing activity of KiSS-1 peptide, the natural ligand of GPR54. Endocrinology. 2005;146:156–163. doi: 10.1210/en.2004-0836. [DOI] [PubMed] [Google Scholar]
- Ohtaki T, Shintani Y, Honda S, Matsumoto H, Hori A, Kanehashi K, Terao Y, Kumano S, Takatsu Y, Masuda Y, Ishibashi Y, Watanabe T, Asada M, Yamada T, Suenaga M, Kitada C, Usuki S, Kurokawa T, Onda H, Nishimura O, et al. Metastasis suppressor gene KiSS-1 encodes peptide ligand of a G-protein-coupled receptor. Nature. 2001;411:613–617. doi: 10.1038/35079135. [DOI] [PubMed] [Google Scholar]
- Ojeda SR, Aguado LI, Smith S. Neuroendocrine mechanisms controlling the onset of female puberty: the rat as a model. Neuroendocrinology. 1983;37:306–313. doi: 10.1159/000123565. [DOI] [PubMed] [Google Scholar]
- Ramaswamy S, Seminara SB, Pohl CR, Dipietro MJ, Crowley WFJ, Plant TM. Effect of continuous intravenous administration of human metastin 45-54 on the neuroendocrine activity of the hypothalamic-pituitary-testicular axis in the adult male rhesus monkey (Macaca mulatta) Endocrinology. 2007;148:3364–3370. doi: 10.1210/en.2007-0207. [DOI] [PubMed] [Google Scholar]
- Rivier JE, Jiang G, Porter J, Hoeger CA, Craig AG, Corrigan A, Vale W, Rivier CL. Gonadotropin-releasing hormone antagonists: novel members of the azaline B family. J Med Chem. 1995;38:2649–2662. doi: 10.1021/jm00014a017. [DOI] [PubMed] [Google Scholar]
- Scully KM, Gleiberman AS, Lindzey J, Lubahn DB, Korach KS, Rosenfeld MG. Role of estrogen receptor-alpha in the anterior pituitary gland. Mol Endocrinol. 1997;11:674–681. doi: 10.1210/mend.11.6.0019. [DOI] [PubMed] [Google Scholar]
- Seminara SB, Messager S, Chatzidaki EE, Thresher RR, Acierno JS, Jr, Shagoury JK, Bo-Abbas Y, Kuohung W, Schwinof KM, Hendrick AG, Zahn D, Dixon J, Kaiser UB, Slaugenhaupt SA, Gusella JF, O'Rahilly S, Carlton MB, Crowley WF, Jr, Aparicio SA, Colledge WH. The GPR54 gene as a regulator of puberty. N Engl J Med. 2003;349:1614–1627. doi: 10.1056/NEJMoa035322. [DOI] [PubMed] [Google Scholar]
- Simerly RB. Organization and regulation of sexually dimorphic neuroendocrine pathways. Behav Brain Res. 1998;92:195–203. doi: 10.1016/s0166-4328(97)00191-5. [DOI] [PubMed] [Google Scholar]
- Smith JT, Cunningham MJ, Rissman EF, Clifton DK, Steiner RA. Regulation of Kiss1 gene expression in the brain of the female mouse. Endocrinology. 2005;146:3686–3692. doi: 10.1210/en.2005-0488. [DOI] [PubMed] [Google Scholar]
- Smith JT, Popa SM, Clifton DK, Hoffman GE, Steiner RA. Kiss1 neurons in the forebrain as central processors for generating the preovulatory luteinizing hormone surge. J Neurosci. 2006;26:6687–6694. doi: 10.1523/JNEUROSCI.1618-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S, Handa RJ. Estrogen receptor-beta, but not estrogen receptor-alpha, is expressed in prolactin neurons of the female rat paraventricular and supraoptic nuclei: comparison with other neuropeptides. J Comp Neurol. 2005;484:28–42. doi: 10.1002/cne.20457. [DOI] [PubMed] [Google Scholar]
- Wiegand SJ, Terasawa E, Bridson WE. Persistent estrus and blockade of progesterone-induced LH release follows lesions which do not damage the suprachiasmatic nucleus. Endocrinology. 1978;102:1645–1648. doi: 10.1210/endo-102-5-1645. [DOI] [PubMed] [Google Scholar]
- Wintermantel TM, Campbell RE, Porteous R, Bock D, Grone HJ, Todman MG, Korach KS, Greiner E, Perez CA, Schutz G, Herbison AE. Definition of estrogen receptor pathway critical for estrogen positive feedback to gonadotropin-releasing hormone neurons and fertility. Neuron. 2006;52:271–280. doi: 10.1016/j.neuron.2006.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]