

Abstract
Using spike-timing-dependent plasticity (STDP) protocols that consist of pairing an EPSP and a postsynaptic backpropagating action potential (BAP), we investigated the contribution of the changes in EPSP waveform induced by the slow Ca2+-dependent K+-mediated afterhyperpolarization (sAHP) in the regulation of long-term potentiation (LTP). The “temporal window” between Schaffer collateral EPSPs and BAPs in CA1 pyramidal neurons required to induce LTP was narrowed by a reduction of the amplitude and decay time constant of the EPSP, which could be reversed with cyclothiazide. The EPSP changes were caused by the increased conductance induced by activation of the sAHP. Therefore, the EPSP waveform and its regulation by the sAHP are central in determining the duration of the temporal window for STDP, thus providing a possible dynamic regulatory mechanism for the encoding of cognitive processes.
Keywords: temporal window, LTP regulation, increased conductance, EPSP profile, cyclothiazide, temporal coding
Introduction
Activity-dependent modifications of synaptic efficacy are thought to underlie learning and memory and to be essential for the maturation of the nervous system (Bliss and Collingridge, 1993; Malinow et al., 2000; Malinow and Malenka, 2002; Malenka and Bear, 2004; Dan and Poo, 2006). Different patterns of activity can increase or decrease synaptic efficacy, effects termed long-term potentiation (LTP) and long-term depression (LTD), respectively (Bliss and Lomo, 1973; Bliss and Collingridge, 1993; Malinow et al., 2000; Malinow and Malenka, 2002). At glutamatergic Schaffer collateral (SC) synapses of CA3 with CA1 hippocampal pyramidal neurons LTP is thought to originate postsynaptically and require the activation of the NMDA receptors (NMDARs) and a rise in spine Ca2+ caused by Ca2+ influx through NMDARs (Lynch et al., 1983; Malenka et al., 1989; Regehr and Tank, 1990). The induction of LTP classically involves presynaptic stimulation at high frequency, but low-frequency stimulation paired with postsynaptic depolarization has also been commonly used (Bekkers and Stevens, 1990; Bliss and Collingridge, 1993; Malenka and Bear, 2004). An LTP induction protocol that appears to be closer to natural physiological conditions consists of the pairing of a presynaptic and a postsynaptic spike (Magee and Johnston, 1997; Markram et al., 1997; Bi and Poo, 1998). This LTP induction procedure, termed spike-timing-dependent plasticity (STDP) (Song et al., 2000), requires the generation of a subthreshold EPSP followed at short delays (∼0–30 ms) by a backpropagating action potential (BAP). This pairing protocol must be repeated to induce the sustained synaptic plasticity that characterizes LTP (Magee and Johnston, 1997; Markram et al., 1997; Bi and Poo, 1998; Abbott and Nelson, 2000; Feldman, 2000; Song et al., 2000; Sjöström et al., 2001).
The slow Ca2+-dependent K+-mediated afterhyperpolarization (sAHP) regulates synaptic efficacy (Sah and Bekkers, 1996; Borde et al., 1999; Le Ray et al., 2004) and the threshold for LTP induction (Sah and Bekkers, 1996; Le Ray et al., 2004). The sAHP also regulates the spread and integration of EPSPs and BAPs at apical dendrites of pyramidal neurons (Sah and Bekkers, 1996; Borde et al., 1999; Le Ray et al., 2004; Fernandez de Sevilla et al., 2007). Although these effects of the sAHP are expected to contribute to the regulation of STDP, their involvement is unknown.
Using somatic whole-cell in vitro recordings in CA1 pyramidal cells, we analyzed the role of sAHP in the regulation of the “temporal window” during which the association of presynaptic and postsynaptic activity induces LTP with STDP protocols. We show that the temporal window is narrowed by the reduction of the amplitude and decay time constant of the EPSP caused by the increased membrane conductance (gm) induced by the activation of the Ca2+-dependent K+ channels that mediate the sAHP. Therefore, the sAHP effectively regulates the induction threshold and the magnitude of the LTP evoked by physiological stimulation protocols such as the STDP.
Materials and Methods
Procedures of animal care, surgery, and slice preparation were in accordance with the guidelines laid down by the European Communities Council. The procedures will be described briefly because they have been extensively detailed previously (Borde et al., 1995, 1999; Le Ray et al., 2004).
Slice preparation.
Young Wistar rats (14–16 d old) were decapitated, and the brain was removed and submerged in cold (∼4°C) artificial CSF (ACSF) (in mm): 124.00 NaCl, 2.69 KCl, 1.25 KH2PO4, 2.00 Mg2SO4, 26.00 NaHCO3, 2.00 CaCl2, and 10.00 glucose. The pH was stabilized at 7.4 by bubbling the ACSF with carbogen (95% O2, 5% CO2). Transverse hippocampal slices (300–350 μm) were cut with a Vibratome (3000; Pelco, St. Louis, MO) and incubated in the ACSF (>1 h at room temperature, 20–22°C). Slices were transferred to a 2 ml chamber fixed to an upright microscope stage (BX51WI; Olympus, Tokyo, Japan) equipped with infrared differential interference contrast video microscopy and a 40× water-immersion objective. Slices were superfused with carbogen-bubbled ACSF (2 ml/min) and maintained at room temperature. All recordings were made under picrotoxin (50 μm), whereas 2-amino-5-phosphonopentanoic acid (APV) (50 μm), nimodipine (10 μm), thapsigargin, (1 μm), 4-ethylphenylamino-1,2-dimethyl-6-methylaminopyrimidinium chloride (ZD7288) (50 μm), apamin (100 nm), isoproterenol (10 μm), and cyclothiazide (CTZ) (100 μm) were added to the ACSF as needed.
Electrophysiology.
Whole-cell recordings from soma of CA1 pyramidal cells were performed with patch pipettes (4–8 MΩ) filled with an internal solution that contained the following (in mm): 135 K-MeSO4, 10 HEPES, 0.5 EGTA, 2 Na2-ATP, and 0.4 Na3-GTP, buffered to pH 7.2–7.3 with KOH. In some experiments, the intracellular solutions contained heparin (5 mg/ml), ruthenium red (100 μm), or BAPTA (50 mm). The BAPTA solution contained the following (in mm): 60 K-MeSO4, 10 HEPES, 50 BAPTA-K4, 2 Na2-ATP, and 0.4 Na3-GTP, buffered to pH 7.2–7.3 with KOH. Chemicals were purchased from Sigma-Aldrich Química (Madrid, Spain), Tocris (Biogen Científica, Madrid, Spain), and Alomone Labs (Jerusalem, Israel).
Recordings were performed both in the current- and voltage-clamp modes using an Axoclamp-2B amplifier (Molecular Devices, Union City, CA). In voltage-clamp experiments, the holding potential (Vh) was adjusted to −65 mV. In current-clamp conditions, the membrane potential (Vm) was set to the same value by injecting DC current as needed, except when indicated otherwise. In the voltage-clamp configuration, the series resistance was compensated to ∼70%, and neurons were accepted only when the seal resistance was >1 GΩ and the series resistance (7–14 MΩ) did not change >10% during the experiment. Current-clamp recordings were rejected if the resting Vm depolarized to values more than −50 mV. The liquid junction potential was measured (∼6 mV) but was not corrected. Voltage- and current-clamp data were high-pass filtered at 3.0 kHz and sampled at rates between 6.0 and 10.0 kHz, through a Digidata 1322A (Molecular Devices). The pClamp programs (Molecular Devices) were used to generate stimulus timing signals and transmembrane current pulses and to record and analyze data. The sAHPs were activated by a depolarizing current pulse (intensity, 0.5 nA; duration, 200 ms) applied through the somatic recording electrode. The depolarizing pulse induced a burst of spikes (7.4 ± 2.3; n = 41) that was followed by the Ca2+-activated K+-mediated medium AHP (mAHP) and sAHP with faster and slower kinetics, respectively. EPSPs were evoked by stimulation of SCs with a bipolar extracellular electrode (diameter, 60 μm; tip separation, ∼100 μm) placed in the stratum radiatum ∼150 μm from the recorded neuron. To allow comparisons between experiments, the amplitude of the EPSP was set to similar values (4–6 mV) ∼50% under the threshold that could evoke spikes. A Grass S88 and stimulus isolation unit (SIU; Grass Instruments, Quincy, MA) generated the simulation pulse protocols. In some experiments, a separate group of SC fibers was stimulated with a second electrode (as above) connected to a Cibertec SIU (Cibertec, Madrid, Spain) driven by the Clampex program. Paired-pulse response (PPR) tests between the two stimulation sites were used to determine that separate groups of fibers were being stimulated.
Ca2+ imaging.
Simultaneous electrophysiology recordings and intracellular Ca2+ imaging were obtained. The latter was performed by fluorescence microscopy with the Ca2+ indicator fluo-3 (Invitrogen, Eugene, OR). Patch pipettes were filled with the internal solution containing 50–100 μm Fluo-3. Imaging experiments were performed after a 10–15 min stabilization period that allowed the equilibration of the dye in the soma and apical dendritic shaft. Cells were illuminated every 200 ms at 490 nm during 40 ms with a monochromator (Polychrome IV; TILL Photonics, Planegg, Germany), and successive images were obtained at 5 s−1 with a cooled monochrome CCD camera (4920; Cohu, San Diego, CA) attached to the Olympus microscope, which was equipped with a filter cube optimized for Fluo-3 (Chroma Technology, Rockingham, VT). Camera control, synchronization with electrophysiological measurements, and quantitative epifluorescence measurements were made with the Imaging Workbench software (INDEC BioSystems, Santa Clara, CA). Changes in fluorescence signals were expressed as the proportion (percentage) of relative change in fluorescence (ΔF/F0), where F0 is the prestimulus fluorescence level when the cell is at rest, and ΔF is the change in fluorescence during activity. Plots of Ca2+ signal variations versus time were obtained off-line at specified regions of interest from stored image stacks and expressed as ΔF/F0. Corrections were made for indicator bleaching during trials by subtracting the signal measured under the same conditions when the cell was not stimulated.
A baseline control EPSC was evoked under voltage clamp by repeated stimulation at 0.3 Hz with the same intensity as EPSPs. The recording was then switched to the current-clamp mode, and a single EPSP was paired with a single postsynaptic action potential evoked by a transmembrane current pulse (3 ms, 0.5–2 nA) applied at delays of 2–40 ms through the somatic recording electrode. This LTP induction protocol was repeated 60 times at 1 s−1 (Bi and Poo, 1998). The magnitude of the LTP was measured under voltage clamp 25–35 min after the end of induction protocol and expressed as a proportion (percentage) of the baseline control EPSC amplitude. The presynaptic or postsynaptic origin of the changes in EPSC amplitude was tested using the PPR test (Bekkers and Stevens, 1990; Fernandez de Sevilla et al., 2002).
Data analysis.
Data were analyzed with the pClamp programs (Molecular Devices). Statistical analysis was performed using Origin 7.0 (OriginLab, Northampton, MA). Results are given as mean ± SEM (n = number of cells), and percentages are presented as percentage of control. Statistical analysis was performed using Student's two-tailed t test, the difference was considered statistically significant at p < 0.05, and p < 0.01 and p < 0.001 are also indicated.
Results
The sAHP raises the threshold for LTP induction
The STDP protocol at an EPSP–BAP delay of 10 ms induced a strong, long-lasting increase of EPSC peak amplitude or LTP (218.0 ± 22.2%; p < 0.05; n = 20) (Fig. 1A, top records, B, control, C, open circles). In contrast, no LTP was induced when the pre-postsynaptic pairing interval was increased to 30 ms (98.5 ± 2.5%; p > 0.05; n = 10) (Fig. 2B). However, reducing the pairing interval to 5 ms resulted in a larger LTP (see below), indicating that within a temporal window the ability of the STDP protocol to induce LTP decreases rapidly with the time delay between the EPSP and BAP (Bi and Poo, 1998; Abbott and Nelson, 2000; Song et al., 2000; Dan and Poo, 2006) (see below).
Figure 1.
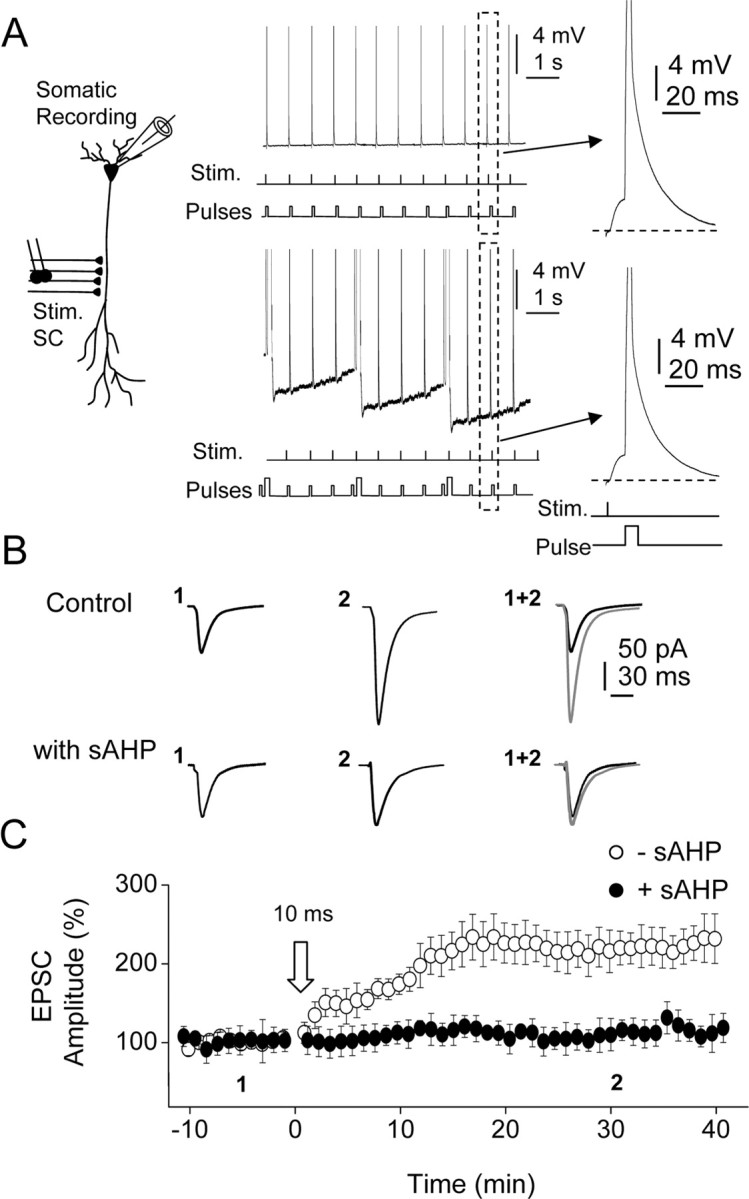
The sAHP suppresses the induction of LTP. A, Left, Diagram showing recording and stimulating electrode locations. Top and bottom right, Representative current-clamp records showing the STDP protocol (10 ms) delivered in the control and during the sAHP. The sAHP was activated continuously by repeated depolarizing pulses (0.5 nA, 200 ms, at 0.25 s−1) that persistently hyperpolarized the cell during the STDP protocol. Insets, Expanded versions of the pairing between EPSP and BAP at a 10 ms delay in the control and during sAHP. Stim., Stimulation. B, Averaged EPSCs (n = 10 as in other cases) recorded before (1) and 30 min after (2) the STDP protocol delivered in control condition (top records) and during the sAHP (bottom records). C, Time course of the changes in EPSC amplitude induced by the 10 ms pairing interval STDP protocol (open arrow, as in all other figures) in the control (−sAHP, open circles) and during the sAHP (+sAHP, filled circles).
Figure 2.

The sAHP narrows the temporal window for LTP. A, The LTP is suppressed by the sAHP induced with a 10 ms EPSP–BAP interval. In contrast, a 5 ms interval induced LTP during the sAHP in the same cells (n = 6). The insets show averaged EPSC recorded before (1) and 5 min after (2) the first STDP (10 ms) protocol and 20 min after the second STDP (5 ms) protocol (3). B, Summary data showing the functional relationship between the EPSC amplitude (%) and the EPSP–BAP interval in control conditions (open circles) and during the sAHP (filled circles). The control single exponential fit showed τ = 9.3 ± 0.5 ms and R2 = 0.97 (n = 36), and during the sAHP, τ = 2.5 ± 0.5 ms and R2 = 0.99 (n = 32). *p < 0.05; **p < 0.01. C, Representative current-clamp records showing averaged EPSPs recorded before and during the sAHP (gray bars). The inset shows superimposed EPSPs recorded before (black trace) and during (gray trace) the sAHP. D, Left, Diagram showing somatic and dendritic (150 μm) recording electrode locations. Right, Representative somatic and dendritic recordings showing EPSPs and APs before and during the sAHP (black and gray traces, respectively).
The sAHP decreases excitability and may regulate the induction of LTP by the STDP by both (1) controlling action potential generation (Madison and Nicoll, 1984; Lancaster and Adams, 1986; Le Ray et al., 2004) and (2) reducing synaptic efficacy by means of modifying the amplitude, shape, and propagation of EPSPs at the apical dendrites of CA1 pyramidal cells (Sah and Bekkers, 1996; Fernandez de Sevilla et al., 2007). Therefore, we designed experiments to investigate whether the sAHP could control the induction threshold of the LTP evoked by STDP protocols.
The sAHP was evoked by a depolarizing current pulse applied through the somatic electrode at a rate of 0.25 s−1 (see Materials and Methods), a repetition rate at which the sAHP is permanently activated (Borde at al., 1995) (Fig. 1A, bottom records, B, with sAHP). In these conditions, the sAHP attained amplitudes between 5.9 and 9.8 mV (7.5 ± 0.3 mV; n = 10) from the resting Vm (−58.8 ± 0.8 mV; same cells). We first applied the STDP protocol at EPSP–BAP delays of 10 ms. The 10 ms delay STDP protocol failed to induce LTP during the sAHP, and the control and post-STDP EPSCs were essentially identical (p > 0.5; n = 12) (Fig. 1C, filled circles).
The sAHP narrows the critical temporal window for LTP induction
The above results indicate that sAHP may prevent the induction of LTP with an EPSP–BAP delay of 10 ms, but the ability of the STDP protocol to induce LTP increases with the reduction of the EPSP–BAP delay (Bi and Poo, 1998; Song et al., 2000; Dan and Poo, 2006), suggesting that the sAHP may not inhibit LTPs induced with briefer EPSP–BAP delays. The sAHP reduces the amplitude and τ of EPSPs at CA1 pyramidal neuron dendrites to 77.0 ± 0.5% and τ d 50.8 ± 11.7%, respectively (n = 8; p < 0.05 in both cases) (Fig. 2C) (Sah and Bekkers, 1996; Fernandez de Sevilla et al., 2007), and the briefer EPSPs could effectively narrow the temporal window of coincidence between EPSP and BAP required to induce the LTP. We examined the changes in the waveform of the EPSP and the BAP before and during sAHP. During the sAHP, the EPSP amplitude decreased (from 3.4 ± 0.2 to 2.4 ± 0.2 mV, a 29.7 ± 5.4% reduction; p < 0.05; n = 10), and the EPSP decayed with a faster τ than in the control (from 62.1 ± 3.8 ms to 33.2 ± 2.4 ms, a 53.2 ± 3.1% reduction; p < 0.01; n = 10) (Fig. 2C). In contrast, neither the amplitude nor the waveform of the BAP were affected by the sAHP, and this was true both for somatic and dendritic recordings at the sites where the LTP was induced ∼150 μm from the soma (p > 0.05; n = 10) (Fig. 2D).
Therefore, we tested whether the LTP could be induced during the sAHP when the time interval between the EPSP and BAP was reduced below 10 ms. Although with an EPSP–BAP interval of 10 ms the sAHP suppressed the LTP (p > 0.05; n = 6) (Fig. 2A), a second STDP protocol at a briefer delay of 5 ms delivered in the same cells ∼10 min later was able to induce the LTP during the sAHP (160.8 ± 9.7%; p < 0.05; n = 6), suggesting that the sAHP acted by narrowing the temporal window for coincidence detection required to induce the LTP with STDP protocols (Fig. 2A).
To quantify the modifications of the temporal window induced by the sAHP, we performed experiments with STDP protocols at different EPSP–BAP delays in control conditions (i.e., in the absence of sAHP activation). The LTP was consistently generated (166.6 ± 10.2%; p < 0.05; n = 6) at delays of 15 ms, and the LTP increased in magnitude (231.6 ± 5.7%; p < 0.001; n = 6) when the EPSP–BAP interval was reduced to 5 ms. With EPSP–BAP intervals of 30 ms and above, the LTP was not induced (Fig. 2B). In contrast, during the sAHP, the STDP protocols induced the LTP only if the EPSP–BAP interval was 5 ms (129.1 ± 4.1%; p < 0.05; n = 10) or briefer (at 2 ms, the LTP was 201.6 ± 15.0%; p < 0.05; n = 6). With EPSP–BAP delays of 10 ms or longer, the sAHP suppressed the LTP (p > 0.05; n = 6) (Fig. 2B). Therefore, the sAHP reduced the magnitude of the LTP and narrowed the temporal window of coincidence. We plotted the functional relationships between the EPSP–BAP interval and the magnitude of the LTP (% EPSC amplitude), which were well fitted by single exponential functions both in the control condition and with the sAHP (Fig. 2B). Exponential fits were significantly different (p < 0.05) with τ = 9.3 ± 0.5 ms and a R2 = 0.97 in the control (n = 36) and τ = 2.5 ± 0.5 ms and a R2 = 0.99 (n = 32) during the sAHP, respectively, confirming that the sAHP effectively narrowed the temporal window of coincidence required to induce the LTP.
LTP induction is regulated by the raise in gm underlying the sAHP
We have previously shown that the modifications in EPSP amplitude and duration were induced by the gm raise that parallels the activation of the sAHP (Fernandez de Sevilla et al., 2007). Consequently, we tested whether a relationship existed between the EPSP properties and the changes in gm. The “shunting” of the EPSP by the increased gm may be caused by the AP itself (Haüsser et al., 2001) rather than by the rise in gm that parallels the sAHP activation. However, pairing the EPSP with the BAP at 10 ms in the control condition revealed that the peak amplitude of the EPSP was unaffected by the AP (p > 0.05; n = 8), suggesting that the rise in gm associated with the activation of the sAHP was the key factor in modifying the EPSP. We estimated the gm modifications by measuring the changes in the amplitude of the voltage responses evoked by depolarizing current pulses without and with sAHPs of different magnitudes from different experiments (n = 30) (Fig. 3A,B). We measured the changes in EPSP amplitude and decay τ as a function of the gm raise and found that both functional relationships were well described by linear functions with negative slopes, indicating an inverse relationship of both amplitude and decay τ of the EPSPs with the increased gm (Fig. 3B), suggesting that the change in gm was the central factor underlying the effects of the sAHP. We also measured the relationship between the gm change caused by the activation of the sAHP and the magnitude (percentage) of the LTP. There was an inverse relationship of the gm with the magnitude of the LTP evoked by pairing at EPSP–BAP delays of 10 ms, indicating that the LTP was gradually smaller with larger gm values (Fig. 3C). In addition, when the rise in gm induced by the sAHP activation was ≥140% of the control value, the LTP was suppressed (p > 0.05; n = 30), indicating a threshold value of the gm over which the LTP was prevented.
Figure 3.
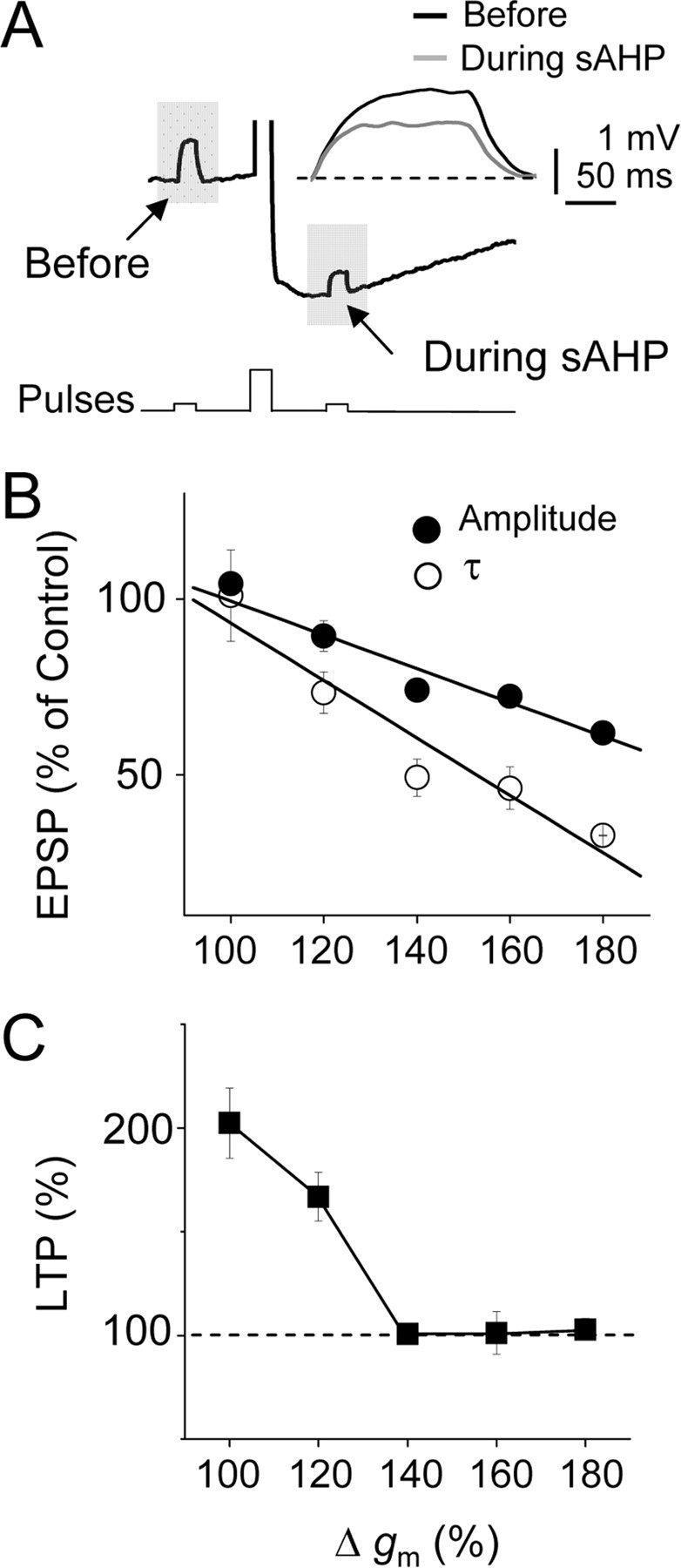
LTP induction is regulated by the rise in gm underlying the sAHP. A, Representative current-clamp records showing the changes of gm during the sAHP estimated from responses evoked by brief depolarizing current pulses (10 pA, 200 ms). The inset shows superimposed responses before (black trace) and during (gray trace) the sAHP. B, Summary data showing the relationship between the gm (%) and the amplitude (filled circles) and decay τ (open circles) of the EPSP during the sAHP (n = 30). C, Summary data illustrating the relationship between gm and magnitude of the LTP (%). Note that when the gm increased >140%, the LTP was suppressed (same cells).
Together, these results suggest that the increased gm caused by the activation of the sAHP regulated both the induction threshold and the magnitude of the LTP. The results also suggest that the gm reduces the temporal window of coincidence between the EPSP and BAP required to induce the LTP by decreasing the amplitude and decay τ of the EPSP. Therefore, the properties of the EPSP are a key factor regulating the temporal window of coincidence.
Hyperpolarization and reblock of NMDARs do not contribute to the effects of the sAHP
Membrane hyperpolarization during the sAHP and Mg2+ reblock of NMDARs could effectively contribute to the regulation of the LTP by the sAHP by diminishing the influx of Ca2+. To distinguish between those possibilities, we performed two different tests. First, we delivered the 10 ms EPSP–BAP delay protocol during a “simulated” sAHP that was generated by applying a current waveform obtained from an averaged “real” sAHP through the somatic electrode (Fig. 4A). The STDP protocol delivered during the simulated sAHP induced a robust LTP (185.2 ± 7.2%; p < 0.05; n = 4), indicating that hyperpolarization caused by the simulated sAHP did not affect the LTP (Fig. 4A,B). We also applied the STDP protocol during a real sAHP generated with depolarizing pulses while holding the cell at −85 mV (close to the K+ reversal potential) by continuous injection of hyperpolarizing current (Fig. 4C,D). In this condition the sAHP effectively suppressed the LTP (104.6 ± 10.6%; p > 0.05; n = 6), although the hyperpolarization associated with the sAHP activation was absent (Fig. 4C,D). However, when the STDP protocol was delivered at the same hyperpolarized Vm of −85 mV in the absence of sAHP activation, a robust LTP was induced (201.1 ± 22.6%; p < 0.05; n = 4).
Figure 4.

Hyperpolarization and reblock of NMDARs do not contribute to the effects of the sAHP. A, Records showing the STDP protocol (10 ms) delivered during the simulated sAHP. B, Summary data showing that the sustained hyperpolarization during the simulated sAHP failed to suppress the LTP. The insets are averaged EPSCs recorded before (1) and 30 min after (2) the STDP protocol. C, STDP protocol (10 ms) delivered during the sAHP while holding the Vm at −85 mV by continuous current injection. D, Summary data illustrating the suppression of the LTP by the sAHP when the STDP protocol (10 ms) was delivered at −85 mV (filled circles). In the absence of sAHP, the same STDP protocol delivered at −85 mV induced a robust LTP. The insets show EPSC averages recorded before (1) and 30 min after (2) the STDP protocol with the sAHP. Stim., Stimulation.
The above results are consistent with a scenario in which the hyperpolarization associated with the sAHP activation is not essential, but in contrast, the gm change underlying the sAHP is vital in terms of blocking the LTP induction by the STDP protocol (Fig. 4D).
To confirm that the increased gm during the sAHP was the key factor in the suppression of the LTP, we applied brief test depolarizing current pulses (10 pA, 200 ms) during the simulated and real sAHP. The amplitude and time constant (τ) of the membrane depolarization evoked by the brief test pulses were essentially identical before and during the simulated sAHP (p > 0.05 in both cases; n = 6), consistent with the absence of modifications in gm caused by the possible activation of the hyperpolarization-activated cationic H-current (Ih) or by reblock of NMDARs by extracellular Mg2+. In addition, the gm increments associated with the real sAHP were similar at both resting and hyperpolarized Vms (at −60 mV, 161.2 ± 11.1% and −85 mV, 154.5 ± 8.3%, respectively; p < 0.05 in both cases; n = 6), although membrane hyperpolarization was present at −60 mV and absent at −85 mV, respectively (data not shown).
The above results are consistent with the suppression of the LTP being caused by the increase in gm induced by the opening of the Ca2+-dependent K+ channels that mediate the sAHP. In contrast, the results also imply that in the present conditions, membrane hyperpolarization per se had no effect in the induction threshold of LTP.
Blockade of the sAHP prevented the suppression of the LTP
When the sAHP was inhibited (by 80.2 ± 10.2%; p < 0.05; n = 8) by perfusion with the β-adrenergic receptors agonist isoproterenol (10 μm) (Fig. 5A), the STDP protocol (10 ms) induced a robust LTP (Fig. 5B,D). Isoprotorenol blocked the sAHP without modifying the spike burst induced by the depolarizing pulse (data not shown), suggesting that the absence of a contribution of the initial spike response in the suppression of the LTP. Although activation of β-adrenergic receptors can enhance the homosynaptic (Yang et al., 2002) and associative LTP (Lin et al., 2003), we could not detect changes in the magnitude of the LTP (p > 0.05; n = 8) nor modification of the control EPSC under isoproterenol (p > 0.05; n = 4). In addition, blockade of the sAHP with isoproterenol did not significantly modify the intracellular Ca2+ signal induced by the depolarizing pulse used to trigger the sAHP (in control, the Ca2+ signal was 15.8 ± 1.4% and with isoproterenol, 14.5 ± 1.2%; n = 5; p > 0.05) (Fig. 5A), suggesting that the intracellular Ca2+ raise was not involved in the suppression of the LTP. The above results are consistent with the notion that the sAHP specifically modifies the induction threshold and the temporal window of the LTP. However, we could not record the Ca2+ signal in the spines, which would have provided conclusive information on the interactions between the depolarization provided by the modified EPSP and the BAP in control solution and with isoproterenol.
Figure 5.

The LTP was induced after blockade of the sAHP. A, Top left, Superfusion with 10 μm isoproterenol markedly reduced the sAHP (n = 8; p < 0.01). Bottom left, Representative records of the intracellular Ca2+ signal recorded at the soma during the sAHP and under inhibition of sAHP with 10 μm Isoproterenol (black and gray traces, respectively). Right, Images showing changes in intracellular Ca2+ signals before (1), during the sAHP (2), and after (3) the sAHP was inhibited with 10 μm isoproterenol. B, Pooled data showing that under blockade of the sAHP with 10 μm isoproterenol (Iso), the 10 ms STDP protocol induced a robust LTP (n = 8). Top, Insets show averaged EPSCs recorded before (1) and 30 min after (2) STDP protocol. C, Inhibition of the mAHP with apamin (100 nm) did not alter the ability of the sAHP to suppress the LTP (filled circles; n = 6) and had no effect on the LTP induced in the absence of sAHP activation (open circles; n = 6). D, Summary data showing the effects of Iso (10 μm) and apamin (Apa; 100 nm), as well as the inhibition of Ih with ZD7288 (50 μm), on the LTP (% EPSC amplitude) during the sAHP. P-AHP, Pairing during AHP. ***p < 0.001. The numbers in the bars represent sample size (n).
The mAHP is also activated by the pulse protocol and may contribute to the described effects. We tested the blockade of the mAHP with apamin (100 nm), which did not prevent the sAHP, to suppress the LTP (Fig. 5C,D). In addition, Ih could be activated by the hyperpolarization induced by the sAHP and modify the induction threshold of the LTP by the STDP protocol. The specific blocker of Ih channels, ZD7288 (50 μm), did not modify the inhibition of the STDP induced by activation of the sAHP (Fig. 5D). The above results are consistent with the lack of contribution of both the mAHP and Ih to the blockade of the LTP and suggest that the suppression of the LTP is exclusively mediated by the sAHP.
The amplitude and waveform of the EPSP regulates the temporal window
The above results suggest that the sAHP narrows the temporal window by reducing the amplitude and decay τ of the EPSP. Therefore, manipulations that increase EPSP amplitude and duration should broaden the temporal window both in control conditions and during the sAHP, thus possibly counteracting the effects of the sAHP on the LTP. We tested the effects of CTZ (100 μm), which inhibits AMPA receptor desensitization and increases the amplitude and duration of EPSPs (Arai et al., 1994; Pelletier and Hablitz, 1994) (Fig. 6C).
Figure 6.

The EPSP waveform regulates the temporal windows of STDP/LTP. A, Under superfusion with 100 μm CTZ, the STDP protocol (10 ms) delivered during the sAHP induced a robust LTP. The inset shows EPSC averages before (1) and 30 min after (2) the STDP protocol. B, Summary data illustrating the functional relationship between the EPSC amplitude (%) and the EPSP–BAP pairing interval under CTZ (100 μm) (open circles), during the sAHP in control ACSF (gray circles), and with 100 μm CTZ (filled circles). The exponential fits show as follows: in control ACSF with the sAHP, τ = 2.5 ± 0.4 ms and R2 = 0.99 (n = 32); with the sAHP under CTZ, τ = 9.3 ± 1.3 ms and R2 = 0.99; and in control ACSF with CTZ, τ = 13.7 ± 2.1 ms and R2 = 0.99. Note that CTZ broadened the temporal window both in the control and with the sAHP. *p < 0.05. C, Representative EPSP averages in control ACSF and under CTZ (black and gray traces, respectively) before and during the sAHP. D, Left, Representative voltage-clamp recordings showing EPSCs in control conditions and with CTZ (black and gray traces, respectively). D, Right, Same as left but scaled to the peak amplitude.
In the absence of sAHP activation, the functional relationship between the EPSP–BAP intervals and the magnitude of the LTP (% EPSC amplitude) revealed that under the CTZ, the temporal window was widened to 30 ms, and larger LTPs (than control) were induced by briefer EPSP–BAP intervals (Fig. 6B). In the presence of sAHP activation, robust LTPs (larger than control) were generated under 100 μm CTZ with a 10 ms pairing interval (191.3 ± 5.1%; p < 0.05; n = 6) (Fig. 6A,B). Larger LTPs were also induced by pairing intervals between 2 and 20 ms during sAHP (Fig. 6B), suggesting that CTZ counteracted the effects of the sAHP (Fig. 2B). Under CTZ, the functional relationships between the EPSP–BAP pairing intervals and the LTP (% EPSC amplitude) were well fitted by single exponential functions (Fig. 6B). The fit had a τ of 13.7 ± 2.1 ms (R2 = 0.99) with the sAHP and a τ of 9.3 ± 1.3 ms (R2 = 0.99) in the control, respectively (p < 0.05; n = 34 and n = 30, respectively), confirming that the sAHP effectively narrowed the temporal window of coincidence required to induce the LTP. Therefore, CTZ counteracted the effects of the sAHP on LTP because the control τ = 9.3 ± 0.6 ms (Fig. 2B) and τ = 9.3 ± 1.3 ms with the sAHP under CTZ were essentially identical (p < 0.05) (Fig. 6C).
We also compared the amplitude and decay τ of EPSPs in control conditions and during the sAHP both under 100 μm CTZ and in control ACSF. CTZ increased the amplitude and decay τ of EPSPs relative to the corresponding controls in the absence of sAHP activation (by 145.7 ± 0.5% and 159.4 ± 7.7%, respectively; p < 0.05 in both cases; n = 10) (Fig. 6C). The EPSP amplitude and τ were also increased by CTZ during the sAHP (59.0 ± 2.4% and 118.1 ± 9.5%, respectively, in both cases; p < 0.05; n = 10) (Fig. 6C). In addition, CTZ did not modify BAPs (p > 0.05; n = 10), the input resistance (p > 0.05; same cells), nor the sAHP (p > 0.05; n = 10). The changes induced by CTZ on the EPSC waveform clearly illustrate that the EPSP modification was caused by the inhibition of AMPA receptor desensitization (Fig. 6D).
Therefore, CTZ can effectively widen the temporal window both in the absence of sAHP activation and with the sAHP, in the latter case counteracting the effects of the sAHP on the induction of the LTP. The above results are consistent with the notion that the amplitude and waveform of the EPSPs are key factors controlling the duration of the temporal window for STDP both in control conditions and during the sAHP.
The LTP in the CA1 region is input specific (Bliss and Collingridge, 1993; Malinow and Malenka, 2002; Malenka and Bear, 2004; Dan and Poo, 2006). However, it is not known whether the suppression of the LTP by the sAHP is restricted to particular regions or is widespread. We tested the input specificity of the sAHP by stimulating different groups of SC fibers in the apical dendrite close to and far from the soma (proximal, ∼150 μm and distal, ∼250 μm, respectively). The absence of common fibers in proximal and distal inputs was confirmed by the lack of cross-facilitation. The LTP was induced at the input that received the STDP protocol at a 10 ms pairing interval (proximal, 215.1 ± 14.9% and distal, 147.7 ± 8.9%; p > 0.05; n = 8) but not the other input that was used as a control (p < 0.05; same cells). The STDP protocol delivered during the sAHP failed to induce LTP in both inputs (p > 0.05; same cells).
The LTP induced by high-frequency stimulation of SC synapses is thought to be mediated postsynaptically by Ca2+ influx through NMDARs (Lynch et al., 1983; Malenka et al., 1989; Regehr and Tank, 1990; Bliss and Collingridge, 1993). We tested the effects of blockade of NMDARs with APV (50 μm; n = 8) and of chelation of intracellular Ca2+ with BAPTA (50 mm; n = 4), which inhibited the LTP induced by STDP protocols. We also examined the effect of nimodipine (10 μm), which selectively blocks L-type Ca2+ channels that play a major role in depolarization-mediated Ca2+ influx in CA1 pyramidal cells (Raymond and Redman, 2002, 2006; Zucker, 2002) and could contribute to the LTP (Yasuda et al., 2003; Moosmang et al., 2005). However, nimodipine (10 μm) did not modify the LTP induced by the STDP protocol (p < 0.05; n = 10). Endoplasmic reticulum (ER) internal stores are another potential source of Ca2+ that could contribute to the LTP (Schiegg et al., 1995; Raymond and Redman, 2002, 2006). However, the IP3 receptor antagonist heparin (5 mg/ml) and the ryanodine receptor antagonist ruthenium red (100 μm) in the pipette solution, as well as depletion of internal Ca2+ stores with thapsigargin (1 μm), respectively, were unable to prevent the induction of LTP.
We analyzed the modifications of paired-pulse responses to test for presynaptic changes in release probability paralleling the LTP. All of the EPSCs tested showed paired-pulse facilitation that did not change with the induction of the LTP (p > 0.05; n = 10), consistent with the LTP induced by STDP protocols being exclusively mediated by postsynaptic mechanisms.
Discussion
We present original evidence indicating that activation of the sAHP narrows the temporal window between EPSPs and BAP required to induce the LTP with STDP protocols. Our results suggest that changes in the amplitude and decay kinetics of SC EPSPs, caused by the rise in gm associated with the opening of the K+ channels that mediate the sAHP (Sah and Bekkers, 1996; Fernandez de Sevilla et al., 2007), are the key factors in determining the width of the temporal window in STDP.
The timing requirement for induction of associative LTP with STDP protocols has been described both for hippocampal (Magee and Johnston, 1997; Bi and Poo, 1998) and neocortical pyramidal neuron synapses (Markram et al., 1997; Feldman, 2000). The induction of LTP requires an EPSP followed by a BAP at delays under 10–20 ms (Magee and Johnston, 1997; Markram et al., 1997; Bi and Poo, 1998; Feldman, 2000; Song et al., 2000; Sjöström et al., 2001). The magnitude of LTP depends on the EPSP–BAP interval, with briefer intervals inducing larger LTPs (Bi and Poo, 1998; Song et al., 2000; Sjöström et al., 2001; Dan and Poo, 2006). In agreement with previous reports in hippocampal SC–CA1 synapses in slices (Lin et al., 2003), we show that the temporal window for the induction of LTP is ∼20 ms and that reductions of the EPSP–BAP interval increase the LTP.
The pre-postsynaptic timing of activity, which determines the temporal window, depends mainly on the dendritic morphology and the interaction of dendritic ionic channels with EPSPs and BAPs. The activation of dendritic ionic conductances may profoundly change the temporal interaction between EPSPs and BAPs in pyramidal cells and may modulate the induction of LTP by dynamically regulating the dendritic spread and waveform of EPSPs and BAPs (Migliore et al., 1999; Shouval et al., 2002; Watanabe et al., 2002). These effects on EPSPs and BAPs may modify the temporal window for LTP (Bi and Poo, 1998; Song et al., 2000; Sjöström et al., 2001; Dan and Poo, 2006). Indeed, the density of dendritic potassium conductance [i.e., IA and Ca2+-dependent K+ channels (large conductance and small conductance)] in pyramidal neuron dendrites may modify STDP (Shouval et al., 2002; Watanabe et al., 2002; Dan and Poo, 2006), and the EPSP-dependent inactivation of transient IA in the dendrites contributes to the induction of LTP by STDP protocols in CA1 pyramidal cells by boosting BAPs (Watanabe et al., 2002). In addition, activation of β-adrenergic receptors (Foehring et al., 1989; Fernandez de Sevilla et al., 2007), muscarinic receptors (Huerta and Lisman, 1993; Segal and Auerbach; 1997), or metabotropic glutamate receptors (Melyan et al., 2002) and hormones (Carrer et al., 2003), which exert a robust, although unspecific inhibition of the sAHP, can regulate the threshold for LTP induction in CA1 pyramidal neurons. Indeed, neurotransmitters that reduce the amplitude of the sAHP, and therefore increase dendritic excitability, facilitate the spread of EPSPs and favor BAPs, crucial factors required to reach a critical dendritic depolarization and Ca2+ elevation for LTP induction (Kampa et al., 2004, 2006; Le Ray et al., 2004).
We propose that the reduced amplitude and duration of the EPSP induced by the sAHP narrows the temporal window of coincidence between the EPSP and BAP by decreasing the spine depolarization required to activate NMDARs and induce the LTP. Consistent with this view, we show that the sAHP reduces the amplitude and duration of EPSPs and that the effects of the sAHP are counteracted by increasing the amplitude and duration of the EPSP via inhibiting AMPA receptor desensitization with CTZ. Indeed, CTZ effectively widens the temporal window for LTP induction not only when the sAHP is activated but also in control conditions. The contrasting effects of the sAHP and CTZ on the temporal window provide novel evidence consistent with a key importance of the EPSP profile in STDP and specially in establishing the timing requirement for induction of associative LTP. Recent results support the above view because both in the cortex (Feldman, 2000; Sjöström et al., 2001) and hippocampal cultures (Bi and Poo, 1998), the induction of LTP demands large EPSPs suggesting that a critical depolarization must be provided by the EPSP for the generation of the LTP by the STDP protocol. The contribution of the EPSP properties to STDP should be analyzed in detail especially because it has been reported that a positive regulation of AMPA receptors may facilitate synaptic plasticity and learning (Staubli et al., 1994; Arai et al., 2004). We show that the BAP was not modified by the sAHP in the soma and the apical dendrite at sites where the LTP was induced. These results are consistent with an insignificant contribution of the modifications induced by the sAHP on the BAP.
Although the cellular mechanisms that mediate the LTP induction by the STDP are not well understood, it has been established that it relies on the activation of NMDARs and is mediated by Ca2+ influx through NMDARs after the relief of the Mg2+ block caused by the combined depolarization induced by the EPSP and the BAP (Bi and Poo, 1998; Kampa et al., 2004, 2006; Froemke et al., 2005). Blockade of NMDARs with APV and intracellular Ca2+ chelation with BAPTA loading suppressed the LTP, confirming that activation of NMDARs and the resulting intracellular Ca2+ elevation are both necessary to induce the LTP (present results). In addition, experimental evidence indicates that the cooperative interplay between the activation of the EPSP and the BAP boosts Ca2+ influx and that the temporal window of STDP is dependent on the supralinear summation of voltage and Ca2+ signals in the spine produced by the interaction of the EPSP and the BAP (Koester and Sakmann, 1998; Schiller et al., 1998; Nevian and Sakmann, 2006). Other mechanisms that increase Ca2+ concentration, such as activation of voltage-gated Ca2+ channels or release from intracellular Ca2+ ER stores either by the mechanism of Ca2+-induced Ca2+ release via activation of Ca2+-sensitive ryanodine receptors or by the production of IP3 and activation of IP3 receptors, can increase Ca2+ in spine and dendrite and could contribute to the LTP (Schiegg et al., 1995; Raymond and Redman, 2002, 2006). However, blockade of L-type Ca2+ channels with nimodipine, inhibition of ryanodine, and IP3 receptors with ruthenium red and heparin in the pipette solution, respectively, did not modify the LTP (present results). Therefore, in our experimental conditions, the LTP induced by the STDP protocols is entirely independent both on Ca2+ influx through L-type channels and Ca2+ release from ER stores.
In conclusion, the activation of the Ca2+-dependent K+ channels that mediate the sAHP may act as a dynamic cellular switch that modifies the narrow critical interval between presynaptic and postsynaptic activity necessary for the induction of LTP. Changes in the temporal window may also function as a gain control mechanism that regulates the magnitude of the LTP. Our results also point to a crucial functional role of the amplitude and duration of the EPSP in controlling the temporal window of the STDP, thus providing yet another dynamic regulatory mechanism that may be decisive for the encoding of cognitive processes. It has been proposed that the sAHP level might determine the degree of synaptic plasticity and learning (Disterhoft and Oh, 2006), and this function of the sAHP could be operative in STDP. Indeed, the EPSP waveform modifications that parallel the sAHP activation may function in physiological conditions when action potential bursts are induced in CA1 pyramidal neurons by excitatory synaptic inputs as may occur during the theta rhythm in vivo (Nuñez et al., 1987; Kamondi et al., 1998) and under activation of NMDARs in vitro in which the sAHP plays a key role (Bonansco and Buño, 2003).
Footnotes
This work was supported by Dirección General de Investigación Científica and Tecnológica, Ministerio de Ciencia and Tecnología Grants BFI2002-01107 and BFU2005-07486, and Comunidad Autónoma de Madrid Grant GR/SAL/0877/2004 (W.B.). M.F. (on leave from Centro de Neurobiología y Plasticidad del Desarrollo, Facultad de Ciencias, Universidad de Valparaíso, Valparaíso, Chile) is a doctoral fellow supported by a Ministerio de Relaciones Exteriores–AGCI Grant. D.F.d.S. is a postdoctoral fellow supported by Comunidad Autónoma de Madrid Grant GR/SAL/0877/2004 and Ministerio de Ciencia and Tecnología Grant BFU2005-07486. We thank Dr. A. Araque for his valuable suggestions on the final version of this manuscript.
References
- Abbott LF, Nelson SB. Synaptic plasticity: taming the beast. Nat Neurosci. 2000;3:1178–1183. doi: 10.1038/81453. [DOI] [PubMed] [Google Scholar]
- Arai A, Kessler M, Xiao P, Ambros-Ingerson J, Rogers G, Lynch G. A centrally active drug that modulates AMPA receptor gated currents. Brain Res. 1994;638:343–346. doi: 10.1016/0006-8993(94)90669-6. [DOI] [PubMed] [Google Scholar]
- Arai AC, Xia YF, Suzuki E. Modulation of AMPA receptor kinetics differentially influences synaptic plasticity in the hippocampus. Neuroscience. 2004;123:1011–1024. doi: 10.1016/j.neuroscience.2003.10.033. [DOI] [PubMed] [Google Scholar]
- Bekkers JM, Stevens CF. Presynaptic mechanism for long-term potentiation in the hippocampus. Nature. 1990;346:724–729. doi: 10.1038/346724a0. [DOI] [PubMed] [Google Scholar]
- Bi GQ, Poo MM. Synaptic modifications in cultured hippocampal neurons: dependence on spike timing, synaptic strength, and postsynaptic cell type. J Neurosci. 1998;18:10464–10472. doi: 10.1523/JNEUROSCI.18-24-10464.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol (Lond) 1973;232:331–356. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonansco C, Buño W. Cellular mechanisms underlying the rhythmic bursts induced by NMDA microiontophoresis at the apical dendrites of CA1 pyramidal neurons. Hippocampus. 2003;13:150–153. doi: 10.1002/hipo.10067. [DOI] [PubMed] [Google Scholar]
- Borde M, Cazalets JR, Buño W. Activity-dependent response depression in rat hippocampal CA1 pyramidal neurons in vitro. J Neurophysiol. 1995;74:1714–1729. doi: 10.1152/jn.1995.74.4.1714. [DOI] [PubMed] [Google Scholar]
- Borde M, Bonansco C, Buño W. The activity-dependent potentiation of the slow Ca2+-activated K+ current regulates synaptic efficacy in rat CA1 pyramidal neurons. Pflügers Arch. 1999;437:261–266. doi: 10.1007/s004240050778. [DOI] [PubMed] [Google Scholar]
- Carrer HF, Araque A, Buño W. Estradiol regulates the slow Ca2+-activated K+ current in hippocampal pyramidal neurons. J Neurosci. 2003;23:6338–6344. doi: 10.1523/JNEUROSCI.23-15-06338.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dan Y, Poo MM. Spike timing-dependent plasticity: from synapse to perception. Physiol Rev. 2006;86:1033–1048. doi: 10.1152/physrev.00030.2005. [DOI] [PubMed] [Google Scholar]
- Disterhoft JF, Oh MM. Learning, aging and intrinsic neuronal plasticity [review] Trends Neurosci. 2006;29:587–589. doi: 10.1016/j.tins.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Feldman DE. Timing-based LTP and LTD at vertical inputs to layer II/III pyramidal cells in rat barrel cortex. Neuron. 2000;27:45–56. doi: 10.1016/s0896-6273(00)00008-8. [DOI] [PubMed] [Google Scholar]
- Fernandez de Sevilla D, Cabezas C, de Prada AN, Sanchez-Jimenez A, Buño W. Selective muscarinic regulation of functional glutamatergic Schaffer collateral synapses in rat CA1 pyramidal neurons. J Physiol (Lond) 2002;545:51–63. doi: 10.1113/jphysiol.2002.029165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez de Sevilla D, Fuenzalida Marco, Porto Pazos Ana, Buño W. Selective shunting of the NMDA EPSP component by the slow after hyperpolarization in rat CA1 pyramidal neurons. J Neurophysiol. 2007;97:3242–3255. doi: 10.1152/jn.00422.2006. [DOI] [PubMed] [Google Scholar]
- Foehring RC, Schwindt PC, Crill WE. Norepinephrine selectively reduces slow Ca2+- and Na+-mediated K+ currents in cat neocortical neurons. J Neurophysiol. 1989;61:245–256. doi: 10.1152/jn.1989.61.2.245. [DOI] [PubMed] [Google Scholar]
- Froemke RC, Poo MM, Dan Y. Spike-timing-dependent synaptic plasticity depends on dendritic location. Nature. 2005;434:221–225. doi: 10.1038/nature03366. [DOI] [PubMed] [Google Scholar]
- Haüsser M, Major G, Stuart GJ. Differential shunting of EPSPs by action potentials. Science. 2001;291:138–141. doi: 10.1126/science.291.5501.138. [DOI] [PubMed] [Google Scholar]
- Huerta PT, Lisman JE. Heightened synaptic plasticity of hippocampal CA1 neurons during a cholinergically induced rhythmic state. Nature. 1993;364:723–725. doi: 10.1038/364723a0. [DOI] [PubMed] [Google Scholar]
- Kamondi A, Acsády L, Wang X-I, Buzsaki G. Theta oscillations in somata and dendrites of hippocampal pyramidal cell in vivo: activity dependent phase precession of action potentials. Hippocampus. 1998;8:244–251. doi: 10.1002/(SICI)1098-1063(1998)8:3<244::AID-HIPO7>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Kampa BM, Clements J, Jonas P, Stuart G. Kinetics of Mg2+ unblock of NMDA receptors: implications for spike-timing dependent synaptic plasticity. J Physiol (Lond) 2004;556:337–345. doi: 10.1113/jphysiol.2003.058842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampa BM, Letzkus JJ, Stuart GJ. Requirement of dendritic calcium spikes for induction of spike-timing-dependent synaptic plasticity. J Physiol (Lond) 2006;574:283–290. doi: 10.1113/jphysiol.2006.111062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koester HJ, Sakmann B. Calcium dynamics in single spines during coincident pre- and postsynaptic activity depend on relative timing of back-propagating action potentials and subthreshold excitatory postsynaptic potentials. Proc Natl Acad Sci USA. 1998;95:9596–9601. doi: 10.1073/pnas.95.16.9596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster B, Adams PR. Calcium-dependent current generating the afterhyperpolarization of hippocampal neurons. J Neurophysiol. 1986;55:1268–1282. doi: 10.1152/jn.1986.55.6.1268. [DOI] [PubMed] [Google Scholar]
- Le Ray D, Fernandez de Sevilla D, Porto AB, Fuenzalida M, Buño W. Heterosynaptic metaplastic regulation of synaptic efficacy in CA1 pyramidal neurons of rat hippocampus. Hippocampus. 2004;14:1011–1025. doi: 10.1002/hipo.20021. [DOI] [PubMed] [Google Scholar]
- Lin YW, Min MY, Chiu TH, Yang HW. Enhancement of associative long-term potentiation by activation of beta-adrenergic receptors at CA1 synapses in rat hippocampal slices. J Neurosci. 2003;23:4173–4181. doi: 10.1523/JNEUROSCI.23-10-04173.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch G, Larson J, Kelso S, Barrionuevo G, Schottler F. Intracellular injections of EGTA block induction of hippocampal long-term potentiation. Nature. 1983;305:719–721. doi: 10.1038/305719a0. [DOI] [PubMed] [Google Scholar]
- Madison DV, Nicoll RA. Control of the repetitive discharge of rat CA 1 pyramidal neurones in vitro. J Physiol (Lond) 1984;354:319–331. doi: 10.1113/jphysiol.1984.sp015378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC, Johnston D. A synaptically controlled, associative signal for Hebbian plasticity in hippocampal neurons. Science. 1997;275:209–213. doi: 10.1126/science.275.5297.209. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Kauer JA, Perkel DJ, Nicoll RA. The impact of postsynaptic calcium on synaptic transmission–its role in long-term potentiation. Trends Neurosci. 1989;12:444–450. doi: 10.1016/0166-2236(89)90094-5. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Malinow R, Mainen ZF, Hayashi Y. LTP mechanisms: from silence to four-lane traffic. Curr Opin Neurobiol. 2000;10:352–357. doi: 10.1016/s0959-4388(00)00099-4. [DOI] [PubMed] [Google Scholar]
- Markram H, Lubke J, Frotscher M, Sakmann B. Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science. 1997;275:213–215. doi: 10.1126/science.275.5297.213. [DOI] [PubMed] [Google Scholar]
- Melyan Z, Wheal HV, Lancaster B. Metabotropic-mediated kainate receptor regulation of IsAHP and excitability in pyramidal cells. Neuron. 2002;34:107–114. doi: 10.1016/s0896-6273(02)00624-4. [DOI] [PubMed] [Google Scholar]
- Migliore M, Hoffman DA, Magee JC, Johnston D. Role of an A-type K+ conductance in the back-propagation of action potentials in the dendrites of hippocampal pyramidal neurons. J Comput Neurosci. 1999;7:5–15. doi: 10.1023/a:1008906225285. [DOI] [PubMed] [Google Scholar]
- Moosmang S, Haider N, Klugbauer N, Adelsberger H, Langwieser N, Muller J, Stiess M, Marais E, Schulla V, Lacinova L, Goebbels S, Nave KA, Storm DR, Hofmann F, Kleppisch T. Role of hippocampal Cav1.2 Ca2+ channels in NMDA receptor-independent synaptic plasticity and spatial memory. J Neurosci. 2005;25:9883–9892. doi: 10.1523/JNEUROSCI.1531-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevian T, Sakmann B. Spine Ca2+ signaling in spike-timing-dependent plasticity. J Neurosci. 2006;26:11001–11013. doi: 10.1523/JNEUROSCI.1749-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuñez A, García-Austt E, Buño W. Intracellular θ-rhythm generation in identified hippocampal pyramids. Brain Res. 1987;416:289–300. doi: 10.1016/0006-8993(87)90909-7. [DOI] [PubMed] [Google Scholar]
- Pelletier MR, Hablitz JJ. Altered desensitization produces enhancement of EPSPs in neocortical neurons. J Neurophysiol. 1994;72:1032–1036. doi: 10.1152/jn.1994.72.2.1032. [DOI] [PubMed] [Google Scholar]
- Raymond CR, Redman SJ. Different calcium sources are narrowly tuned to the induction of different forms of LTP. J Neurophysiol. 2002;88:249–255. doi: 10.1152/jn.2002.88.1.249. [DOI] [PubMed] [Google Scholar]
- Raymond CR, Redman SJ. Spatial segregation of neuronal calcium signals encodes different forms of LTP in rat hippocampus. J Physiol (Lond) 2006;570:97–111. doi: 10.1113/jphysiol.2005.098947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regehr WG, Tank DW. Postsynaptic NMDA receptor-mediated calcium accumulation in hippocampal CA1 pyramidal cell dendrites. Nature. 1990;345:807–810. doi: 10.1038/345807a0. [DOI] [PubMed] [Google Scholar]
- Sah P, Bekkers JM. Apical dendritic location of slow afterhyperpolarization current in hippocampal pyramidal neurons: implications for the integration of long-term potentiation. J Neurosci. 1996;16:4537–4542. doi: 10.1523/JNEUROSCI.16-15-04537.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiegg A, Gerstner W, Ritz R, van Hemmen JL. Intracellular Ca2+ stores can account for the time course of LTP induction: a model of Ca2+ dynamics in dendritic spines. J Neurophysiol. 1995;74:1046–1055. doi: 10.1152/jn.1995.74.3.1046. [DOI] [PubMed] [Google Scholar]
- Schiller J, Schiller Y, Clapham DE. NMDA receptors amplify calcium influx into dendritic spines during associative pre- and postsynaptic activation. Nat Neurosci. 1998;1:114–118. doi: 10.1038/363. [DOI] [PubMed] [Google Scholar]
- Segal M, Auerbach JM. Muscarinic receptors involved in hippocampal plasticity. Life Sci. 1997;60:1085–1091. doi: 10.1016/s0024-3205(97)00051-9. [DOI] [PubMed] [Google Scholar]
- Shouval HZ, Bear MF, Cooper LN. A unified model of NMDA receptor-dependent bidirectional synaptic plasticity. Proc Natl Acad Sci USA. 2002;99:10831–10836. doi: 10.1073/pnas.152343099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjöström PJ, Turrigiano GG, Nelson SB. Rate, timing, and cooperativity jointly determine cortical synaptic plasticity. Neuron. 2001;32:1149–1164. doi: 10.1016/s0896-6273(01)00542-6. [DOI] [PubMed] [Google Scholar]
- Song S, Miller KD, Abbott LF. Competitive Hebbian learning through spike-timing-dependent synaptic plasticity. Nat Neurosci. 2000;3:919–926. doi: 10.1038/78829. [DOI] [PubMed] [Google Scholar]
- Staubli U, Rogers G, Lynch G. Facilitation of glutamate receptors enhances memory. Proc Natl Acad Sci USA. 1994;91:777–781. doi: 10.1073/pnas.91.2.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Hoffman DA, Migliore M, Johnston D. Dendritic K+ channels contribute to spike-timing dependent long-term potentiation in hippocampal pyramidal neurons. Proc Natl Acad Sci USA. 2002;99:8366–8371. doi: 10.1073/pnas.122210599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H-W, Lin Y-W, Yen C-D, Min M-Y. Change in bidirectional plasticity at CA1 synapses in hippocampal slices taken from 6-hydroxydopamine treated rats: the role of endogenous norepinephrine. Eur J Neurosci. 2002;16:1117–1128. doi: 10.1046/j.1460-9568.2002.02165.x. [DOI] [PubMed] [Google Scholar]
- Yasuda R, Sabatini BL, Svoboda K. Plasticity of calcium channels in dendritic spines. Nat Neurosci. 2003;6:948–955. doi: 10.1038/nn1112. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]