

Abstract
Circadian rhythmicity in the primary mammalian circadian pacemaker, the suprachiasmatic nucleus (SCN) of the hypothalamus, is maintained by transcriptional and translational feedback loops among circadian clock genes. Photic resetting of the SCN pacemaker involves induction of the clock genes Period1 (Per1) and Period2 (Per2) and communication among distinct cell populations. Gastrin-releasing peptide (GRP) is localized to the SCN ventral retinorecipient zone, from where it may communicate photic resetting signals within the SCN network. Here, we tested the putative role of GRP as an intra-SCN light signal at the behavioral and cellular levels, and we also tested whether GRP actions are dependent on activation of the cAMP response element-binding protein (CREB) pathway and Per1. In vivo microinjections of GRP to the SCN regions of Per1::green fluorescent protein (GFP) mice during the late night induced Per1::GFP throughout the SCN, including a limited population of arginine vasopressin-immunoreactive (AVP-IR) neurons. Blocking spike-mediated communication with tetrodotoxin did not disrupt overall Per1::GFP induction but did reduce induction within AVP-IR neurons. In vitro GRP application resulted in persistent increases in the spike frequency of Per1::GFP-induced neurons. Blocking endogenous Per1 with antisense oligodeoxynucleotides inhibited GRP-induced increases in spike frequency. Furthermore, inhibition of CREB-mediated gene activation with decoy oligonucleotides blocked GRP-induced phase shifts of PER2::luciferase rhythms in SCN slices. Altogether, these results indicate that GRP communicates phase resetting signals within the SCN network via both spike-dependent and spike-independent mechanisms, and that activation of the CREB pathway and Per1 are key steps in mediating downstream events in GRP resetting of SCN neurons.
Keywords: GRP, cAMP, tetrodotoxin, oligodeoxynucleotides, phase shift, Per2
Introduction
Daily, physiological and behavioral rhythms persist in the absence of environmental time cues with a near 24 h period. In an exact 24 h light/dark (LD) cycle, endogenous circadian rhythms are reset or entrained daily by light, which can advance or delay the phase of molecular, electrical, and behavioral activity. At a molecular level, light upregulates the mammalian circadian clock genes Period1 (Per1) and Period 2 (Per2) (Shearman et al., 1997; Shigeyoshi et al., 1997; Kuhlman et al., 2000; Moriya et al., 2000), which are key participants in the transcriptional/translational feedback loops that maintain the rhythm-generating mechanism of the primary mammalian circadian clock (for review, see Reppert and Weaver, 2001), the suprachiasmatic nucleus (SCN) of the hypothalamus. The SCN receives photic information directly from the retina through the retinohypothalamic tract (RHT) (Pickard, 1985; Johnson et al., 1988). The RHT terminals synapse onto the ventral or “core” region of the SCN, which is characterized by light-responsive cells that produce the peptides vasoactive intestinal polypeptide (VIP) and gastrin-releasing peptide (GRP) (Abrahamson and Moore, 2001). After light exposure, upregulation of the Per clock genes is first apparent in ventral VIP- or GRP-containing cells and later follows in the dorsal or “shell” region of the SCN, which is characterized by rhythmic expression of Per1 and immunoreactivity for arginine vasopressin (AVP) (Hamada et al., 2001; Dardente et al., 2002; Moore et al., 2002; Yan and Silver, 2002; Kawamoto et al., 2003; Kuhlman et al., 2003; Karatsoreos et al., 2004). It remains unknown how photic information is conveyed from retinorecipient cells throughout the SCN network.
A potential candidate for intra-SCN mediation of photic information is GRP. Light induces c-Fos and Per1 mRNA in most of the GRP-producing SCN neurons (Earnest et al., 1993; Romijn et al., 1996; Aida et al., 2002; Kawamoto et al., 2003; Karatsoreos et al., 2004; Antle et al., 2005), and application of GRP phase shifts the circadian clock in vivo and in vitro (Albers et al., 1995; Piggins et al., 1995; McArthur et al., 2000; Kallingal and Mintz, 2006). GRP is a bombesin-like peptide that activates BB2 receptors in the dorsal and medial regions of the mouse SCN (Aida et al., 2002; Karatsoreos et al., 2006). Activation of these receptors initiates the phospholipase C pathway, resulting in intracellular Ca2+ release and activation of mitogen-activated protein kinase (MAPK) (Woodruff et al., 1996; Moody and Merali, 2004).
The functional role and transduction pathway of GRP in mediating photic information remains to be elucidated. Here, we infused GRP into the SCN regions of transgenic mice in which a dynamic, short half-life green fluorescent protein (GFP) reports Per1 gene activation (Kuhlman et al., 2000). After measurement of behavioral phase advances, we used double-labeled immunocytochemistry to determine the phenotype and distribution of Per1-induced SCN cells after GRP infusion with and without spike-mediated communication. We further examined whether GRP induces persistent neuronal activity and whether Per1 induction is necessary for GRP-induced increases in spike rate. Finally, we determined whether GRP application induces phase shifts in a cAMP response element-binding protein (CREB)-dependent manner using SCN slice cultures from PER2::luciferase (PER2::LUC) reporter mice.
Materials and Methods
Animals and housing.
Adult, Per1::GFP mice (∼3 months of age) hemizygous for the mPer1::d2EGFP transgene on a B6C3H hybrid background (Kuhlman et al., 2000) were used for behavioral and immunocytochemistry experiments, whereas 16- to 28-d-old Per1::GFP mice were used for electrophysiology experiments. Adult, ∼30-d-old, mPER2::LUC knock-in mice (Yoo et al., 2004) (generously donated by J. Takahashi, Northwestern University, Evanston, IL) were backcrossed to C57BL/6J for 10 generations and crossed with B6C3H hybrid mice and were used to measure PER2-driven luminescence. All mice were housed in a 12:12 LD cycle for a minimum of 2 weeks before the start of the experiment and had food and water provided ad libitum. All handling of animals was done in accordance with the Vanderbilt University Institutional Animal Care and Use Committee guidelines.
Behavioral analyses.
To reduce interindividual variability among mice and to allow comparison between behavior and immunocytochemistry results, the behavioral phase shifts were determined using a modified Aschoff type II procedure (Aschoff, 1965), in which mice were maintained on a 12:12 LD cycle, and, after treatment at Zeitgeber time 21 (ZT 21) (ZT 12 refers to lights off), the lights remained off for 10 d. Wheel-running activity was recorded and analyzed using ClockLab software (Actimetrics, Wilmette, IL). Behavioral phase shifts were quantified using the linear regression method (Daan and Pittendrigh, 1976). A regression line was determined for the daily activity onsets for days 3–9 after the injection and compared with the average activity onset for the 7 d preceding the injection. Phase shifts were calculated as the difference between the onsets of activity predicted by the regression line and the average activity onset for the preceding days. Activity onsets were defined as the first of at least three consecutive 5 min periods of >10 wheel revolutions. If the SE of the regression line was >30 min, the injection was not included in the analysis. The average circadian period was determined for each animal by averaging the day-to-day differences in activity onsets for days 3–9 under constant dark conditions. The average circadian period was similar for both GRP- and vehicle-injected animals (23.76 ± 0.03 and 23.67 ± 0.11 h, respectively), as well as similar to what was reported previously for this line of mice (Kuhlman et al., 2000).
Surgery.
For stereotaxic surgery, animals were anesthetized with a mixture of ketamine (60 mg/kg) and xylazine (5 mg/kg). Stainless steel guide cannulas (4.0 mm, 26 gauge; Plastics One, Roanoke, VA) were fixed to the skull at a 9° angle using cyanoacrylate gel (Plastics One) and dental cement (OrthoJet; Lang Dental, Wheeling, IL). The following coordinates from bregma were used: +0.1 mm anterior, +0.8 mm lateral, and a final depth from the skull of 4.1 mm. Animals were allowed a 1–2 week recovery before experimentation.
Microinjections.
Unilateral microinjections were given in a 200 nl volume using a 33 gauge needle attached by polyethylene tubing to a 1 μl Hamilton syringe in a dark room by use of a dim red light (<3 lux) and night vision goggles. Animals were gently restrained for ∼30 s. For immunocytochemistry, injections of colchicine (5 mm; Sigma, St. Louis, MO) were given on the day before treatment at ZT 24 to visualize VIP immunoreactivity. Two additional animals also received GRP or vehicle without colchicine to ensure that colchicine did not have any effects or disrupt the effects of GRP (data not shown), in agreement with Kuhlman et al. (2003). All drugs were purchased from Sigma and dissolved in a vehicle of saline, and the respective concentrations were as follows: GRP (0.15 mm; 84 ng in 200 nl), tetrodotoxin (TTX; 1 μm) (De Groote et al., 2003), and a mixture of GRP (0.15 mm, final) plus TTX (1 μm, final).
Perfusion and tissue preparation for immunocytochemistry.
Three hours after microinjection (ZT 24 or ZT 19), all animals were killed with an overdose of sodium pentobarbital (200 mg/kg, i.p.) anesthesia in a completely dark room with the aid of night vision goggles. Once unconscious, the animal's head was covered with a hood, and the animal was perfused intracardially [50 ml of 0.9% saline and 100 ml of 4% paraformaldehyde in 0.1 m phosphate buffer (Kuhlman et al., 2003)]. Brains were cryoprotected in 20% sucrose and sectioned (40 μm) through the anterior hypothalamus on a cryostat.
Immunocytochemistry.
Alternate sections were double labeled for AVP–GFP or VIP–GFP, using guinea pig polyclonal antisera against AVP (1:5000) or VIP (1:2500) (DiaSorin, Stillwater, MN) and rabbit polyclonal antisera against GFP (1:20,000; Invitrogen, Eugene, OR). For supplemental Figure 2 (available at www.jneurosci.org as supplemental material), sections were labeled with rabbit polyclonal antisera against PER1 (1:500; Abcam, Cambridge, MA). For visualization, sections were rinsed in secondary antibodies (1:500): Alexa Fluor 594 goat anti-guinea pig IgG and Alexa Fluor 488 goat anti-rabbit IgG (Invitrogen). Sections were examined under a confocal microscope (LSM5 PASCAL; Zeiss, Thornwood, NY) at excitation wavelengths of 488 nm for Alexa Fluor 488 and 543 nm for Alexa Fluor 594. Several animals that did not receive an injection were run as controls.
Image analysis.
Images were digitized using software from the LSM5. Because unilateral microinjections within 0.5 mm of the SCN border results in strong labeling of both SCN (Rea et al., 1993), the analysis only included animals with injection traces that were within 300 μm of the SCN border but did not damage the SCN or penetrate the third ventricle (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). The number of single- and double-labeled cells was bilaterally counted using the Cell Count macro in NIH Image by an experimenter who was blind to the injection type. For each brain, three VIP/GFP-IR and three AVP/GFP-IR sections best representing rostral, mid, and caudal SCN regions were used to ensure that an equal number of sections were counted for each animal. The percentage of double-labeled cells was determined for each slice individually, and then an average of the three slices for that animal was used in the analysis. For each section, V++ 4.0 software (Digital Optics, Auckland, New Zealand) was used to define bright cells as having twice the intensity of the average of three dim cells. Dim cells were defined as having a round shape of a similar size as the brighter cells but having the lowest detectable intensity above background in the slice. In supplemental Figure 2 (available at www.jneurosci.org as supplemental material), GFP intensity was measured in MetaMorph 7.1.2.0 software (Molecular Devices, Downingtown, PA) by drawing using a circular region with a 30 μm diameter in the middle of one SCN and subtracting the intensity of the same size region placed in the anterior hypothalamus just outside the SCN.
Slice preparation and electrophysiological recordings.
For electrophysiology experiments, Per1::GFP mice were killed by cervical dislocation, and brains were removed and blocked in cold, HBSS (Invitrogen). Coronal, hypothalamic slices (200 μm) containing the SCN were cut on a vibroslicer (World Precision Instruments, Sarasota, FL) at 4–10°C, trimmed to ∼4 × 4 mm squares, and transferred directly to culture membranes (Millipore, Billerica, MA) in 35 mm culture dishes with 1.0 ml of MEM (Invitrogen) containing 30 mm HEPES, 20 mm d-glucose, 5% B27, 5.0 mm l-glutamine, and 25 U/ml streptomycin/penicillin (Han et al., 2006) and maintained at 37°C and 5% CO2.
The following day, at projected ZT 21, an 8 μl volume of 37.5 μm GRP (final concentration, 0.3 μm; 841 ng in 8 μl, diluted into 1.0 ml of culture media) or vehicle (sterile culture water) was added directly to the culture dish medium. For Per1 gene knockdown, either antisense oligodeoxynucleotide (ODN) sequence against the 5′ translational start site of Per1 (taggggaccactcatgtct; IDT, Coralville, IA) or a random ODN with equivalent GC content (ccgttagtactgagctgac) (as used previously by Akiyama et al., 1999; Tischkau et al., 2003) was applied at a final concentration of 10 μm at projected ZT 20. Importantly, the promoter sequences in the Per1::GFP transgene do not contain the 5′ start site sequence targeted by these antisense ODNs (Gekakis et al., 1998; Kuhlman et al., 2000). At projected ZT 22, slices were transferred to an open recording chamber (Warner Instruments, Hamden, CT) that was continuously perfused at a rate of 2.0 ml/min with extracellular solution (in mm: 130.5 NaCl, 26.0 NaHCO3, 1.25 Na2HPO4-7H20, 1.2 MgSO4-7H20, 10 glucose, 3.5 KCl, and 2.5 CaCl2), bubbled with 5% CO2/95% O2 and heated to 34 ± 0.5°C. Antisense ODN inhibition of Per1 expression was verified by immunocytochemistry for PER1 (see above) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Briefly, coronal brain slices (200 μm) containing the SCN were established and maintained on culture membranes and treated with anti-Per1 ODN or random Per1 ODN at ZT 20 followed by GRP at ZT 21 using identical, aforementioned methods. After two rinses and a medium change at ZT 22, the cultures were fixed at ZT 24 with 1.0 ml of 4% paraformaldehyde in 0.1 m phosphate buffer for 45 min and then processed for immunocytochemistry as above.
Neurons were visualized with an Axioskop microscope (Zeiss) equipped for near-infrared differential interference contrast (DIC) and epifluorescence. Recordings were made between projected ZT 24 and ZT 1 (3–4 h after GRP application and 2–3 h after GRP washout). After recording, fluorescent time-lapse images were captured every 30 min from projected ZT 1 to ZT 3 to observe the circadian increase in Per1::GFP and ensure the health of the slice. Each experimental treatment contained data from three to five animals. Recordings were made from SCN neurons located in the dorsal and medial regions of the SCN because the majority of BB2 receptors in the SCN lie within this region (Aida et al., 2002; Karatsoreos et al., 2006). All recordings were confirmed to be from GFP-positive (GFP+) neurons by aligning digital images of the same neuron under near-infrared differential interference contrast and GFP fluorescence illumination. Extracellular patch electrodes with a pipette resistance of 3–5 MΩ were filled with filtered, normal HEPES solution (in mm: 150 NaCl, 10 HEPES, and 10 glucose). Firing rate frequency was measured as the average of a 100 s record. Electrophysiological signals were processed and controlled by an Axopatch 1D amplifier, and pClamp 10.0 software (Molecular Devices, Palo Alto, CA) in gap-free mode. Recordings were sampled at 10 kHz and filtered at 1 kHz.
Tissue preparation and treatment for PER2::LUC cultures.
At ∼4 h after lights on (approximately ZT 4), PER2::LUC mice were killed by cervical dislocation. Brains were removed and blocked in cold HBSS. Hypothalamic coronal slices (250 μm) containing the SCN were cut on a vibroslicer (World Precision Instruments) at 4–10°C, trimmed to ∼1.5 × 1.5 mm squares, and transferred directly to culture membranes (Millipore) in 35 mm culture dishes with 1.0 ml of DMEM (D-2902; Sigma) supplemented with 3.5 g/liter d-glucose, 10 mm HEPES, 25 U/ml penicillin/streptomycin, 2% B27, and 0.1 mm beetle luciferin (Promega, Madison, WI). Slice cultures containing the SCN were maintained at 36.8°C, and, after stable rhythmicity was established, they were treated with new media containing either antisense decoy ODN to cAMP-response element (CRE) (100 μm; tgacgtcatgacgtcatgacgtca; IDT) or a 100 μm concentration of a mismatched sequence of the same nucleotide bases (CRE-mis) [tgtggtcatgtggtcatgtggtca (as by Tischkau et al., 2003)], in addition to either 0.3 μm GRP or vehicle. Cultures were treated for 1 h beginning at circadian time 15 (CT 15) to CT 16 (CT 12 refers to peak luminescence) (Field et al., 2000; Yoo et al., 2004) and then transferred back into the original media. After two or more full cycles, each culture received a media change and two rinses and was treated for a second time with vehicle, if GRP was given for the first treatment and vice versa. The order of GRP and control treatments was randomized and counterbalanced.
PER2::LUC bioluminescence analysis.
Bioluminescence was measured with a LumiCycle (Actimetrics). Data were analyzed by using Lumicycle data analysis software (Actimetrics). For each selection of data (two to three cycles before treatment or two cycles after treatment), baseline drift was removed by fitting a polynomial curve with an order of one less than the number of cycles. The baseline-subtracted data were then used to estimate the circadian period by fitting a sine wave multiplied by an exponential decay. A goodness of fit was computed, and only data with at least 80% of the variance accounted for by the curve was used for analysis. Two predictions of the peak of the first cycle after treatment were made: one based on the curve that best fit the cycles preceding treatment, and a second based on the curve that best fit the cycles after treatment. The difference between these two predictions was determined to be the phase shift.
Statistical analysis.
For behavioral phase-shifting analysis and quantitative real-time PCR, independent samples t tests (two-tailed) were used. When appropriate, one-way ANOVA and Fisher's least significant difference were used for comparison of more than two groups, except for when the variances were not homogeneous, as indicated by a significant Levene's test. In these cases, a nonparametric Kruskal–Wallis omnibus test was used. For the bioluminescence analysis, a mixed factorial ANOVA was used, with repeated measures on the GRP application variable. Significance was ascribed at p < 0.05.
Results
Injection of GRP into the SCN region induces phase shifts and Per1 gene activity
Our first step in examining the effects of GRP within the SCN was to verify that microinjection specifically to the SCN regions of mice during the late night would induce phase shifts in wheel-running behavior. We chose to focus on the late night phase to allow comparison with a previous study that used the same animal model to investigate light-induced GFP immunoreactivity, AVP immunoreactivity, and VIP immunoreactivity in the SCN (Kuhlman et al., 2003). Although injection of GRP into the SCN region has been shown to induce phase shifts in hamsters (Piggins et al., 1995), only ventricular injection of GRP has been examined in the mouse, specifically during the early night (Aida et al., 2002). Therefore, the first part of this study was designed to test whether GRP injection into the SCN regions of mice can successfully induce phase advances during the late night. Behavioral phase-shifting analysis revealed that microinjection of 0.15 mm GRP into the SCN regions of Per1::GFP mice at ZT 21 elicited significantly larger phase advances (53.1 ± 12.5 min; n = 4) compared with vehicle (2.2 ± 13.8 min; n = 5; t(7) = −2.61; p < 0.05) (Fig. 1).
Figure 1.
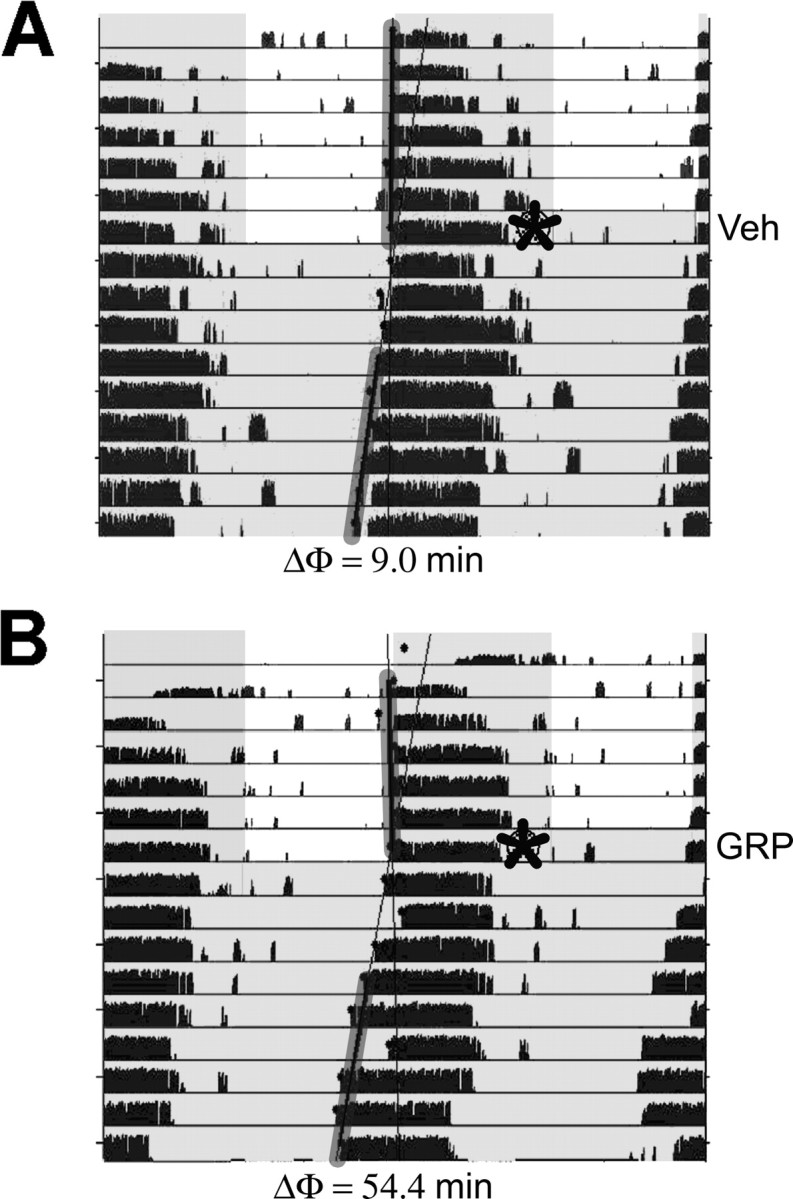
Representative actograms of the wheel-running activity of mice injected with vehicle (A; Veh) or GRP (B) at ZT 21, as indicated by the asterisks. Black tick marks represent the number of wheel revolutions, and the shaded areas indicate lights off. The data are double plotted so that each line represents 48 h. The phase shift (ΔΦ) for each animal is indicated under each actogram and was determined from the difference between the average activity onset for the days before injection and a regression line calculated from the daily activity onsets for days 3–9 after the injection. The activity onsets for the days included in the analysis are indicated by the gray-shaded lines.
After measuring phase shifts, we examined the pattern of Per1 induction after in vivo GRP application at both early and late night phases. The animals from the behavioral experiment, along with additional mice, were reentrained to an LD cycle for 2 weeks and then were used to examine immunoreactivity for GFP, VIP, and AVP after microinjection of vehicle, GRP, GRP and TTX together, or TTX at ZT 21. A separate cohort of mice received microinjections of either GRP or vehicle at ZT 16 for comparison. GRP injection elicited a significantly greater number of GFP+ cells than vehicle-injected controls at ZT 21 (F(3,23) = 4.58; p < 0.05) (Figs. 2, 3) but not at ZT 16 (F(1,9) = 1.41; p > 0.05) (Table 1). In addition, the percentage of double-labeled GFP–AVP and GFP–VIP cells during the early night did not differ significantly between GRP- and vehicle-injected groups (F(1,9) = 0.79 and 0.00, respectively; p > 0.05) (Table 1). This lack of GRP effect that we observed during the early night is in agreement with findings that light pulses at this time do not significantly induce Per1 in the dorsal and medial SCN regions in which BB2 receptors are most densely localized (Yan and Silver, 2002; Karatsoreos et al., 2006) but differs from the findings of Aida et al. (2002) who did report significant Per1 mRNA induction after intraventricular injection of GRP in the early night.
Figure 2.
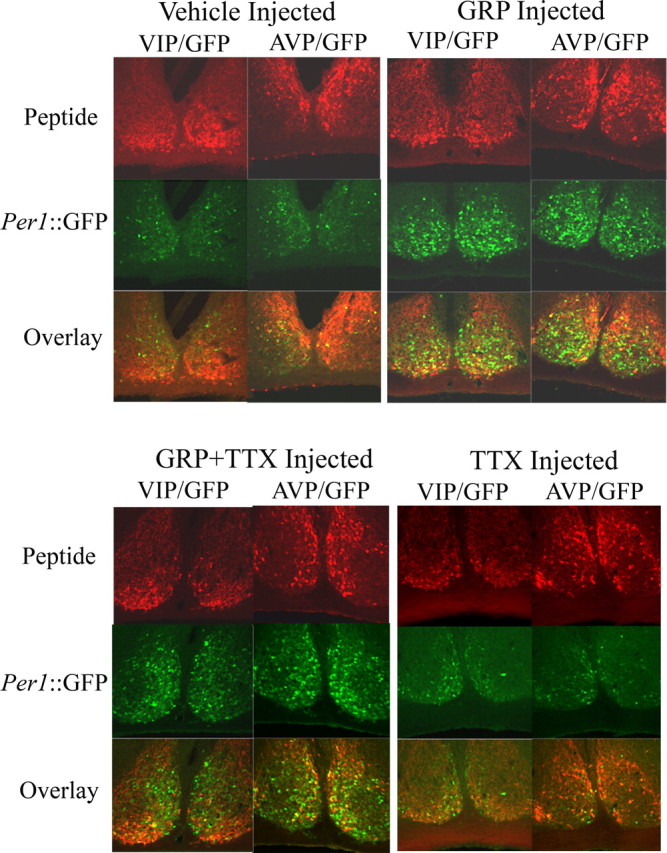
Representative confocal images of Per1::GFP mice injected with vehicle or GRP (top) and GRP plus TTX or TTX (bottom) into the SCN region at ZT 21. Mice were perfused at ZT 24. Alternating sections (40 μm thick) were double labeled for VIP–GFP (left column of each treatment group) or AVP–GFP (right column of each treatment group). Top row, Single-labeled images for AVP immunoreactivity or VIP immunoreactivity (red); middle row, single-labeled images for GFP immunoreactivity (green); bottom row, double-labeled composite of AVP–GFP or VIP–GFP (yellow cells are double labeled).
Figure 3.

Microinjection of GRP into the mouse SCN region at ZT 21 induces Per1::GFP expression. Mean number of GFP-IR cells per animal (A), bright GFP-IR cells per animal (B), and percentage AVP–GFP double-labeled cells (as a percentage of AVP-IR cells) per animal (C) after microinjection of vehicle, GRP, a mixture of GRP plus TTX, or TTX; *p < 0.05.
Table 1.
Average cell counts and percentage double-labeled cells
| GFP+ | AVP+ | VIP+ | GFP-AVP (%AVP) | GFP-VIP (%VIP) | |
|---|---|---|---|---|---|
| ZT 21 | |||||
| Vehicle | 445.9 (60) | 265.9 (28) | 159.7 (30) | 7.6 (1.8) | 7.3 (2.0) |
| GRP | 678.3 (71) | 270.6 (32) | 130.9 (25) | 16.6 (4.2) | 7.0 (2.7) |
| GRP + TTX | 704.8 (102) | 238.6 (30) | 174.4 (21) | 6.0 (1.6) | 3.6 (1.1) |
| TTX | 372.0 (12) | 242.5 (39) | 185.8 (15) | 3.0 (1.1) | 1.8 (0.9) |
| ZT 16 | |||||
| Vehicle | 442.0 (40) | 228.4 (21) | 64.2 (13) | 12.2 (2.1) | 6.0 (2.0) |
| GRP | 475.5 (24) | 236.8 (21) | 50.3 (8) | 16.5 (4.0) | 6.2 (1.9) |
Mean (SEM) after microinjection at ZT 21 of vehicle (n = 10), GRP (n = 8), GRP plus TTX (n = 5), or TTX (n = 4) and microinjection at ZT 16 of vehicle (n = 5) and GRP (n = 6). Bold, Significant difference from vehicle and TTX, p < 0.05; bold italics, significant difference from all other groups, p < 0.05.
Interestingly, widespread GRP induction of Per1::GFP at the late night phase was not reduced by coapplication of TTX (means and SEMs are shown in Fig. 3A and Table 1). Approximately 16% of the GFP+ cells were also AVP-IR, which was more than twice the percentage of GFP–AVP+ double-labeled cells after either vehicle or TTX injection (F(3,23) = 3.78; p < 0.05) (Fig. 3B). Surprisingly, when GRP was injected with TTX, only ∼6% of the GFP+ cells were AVP-IR (p < 0.05).
To determine whether GRP increases the level of Per1 activity, as well as the number of Per1::GFP neurons, we quantified the number of bright cells (defined as twice the intensity of the average of three dim cells in each section). Because this reporter mouse harbors a short half-life version of GFP, fluorescence intensity at the cellular level reflects Per1 promoter activity. Animals injected with GRP or GRP combined with TTX had significantly more bright GFP+ cells than those injected with vehicle or TTX alone (Kruskal–Wallis, χ2(3) = 15.1, p < 0.01; Dunnett's T3 post hoc test for unequal variances, p < 0.05) (Fig. 3C). The number of double-labeled GFP–AVP+ cells were also counted; however, the variance was too large for any differences to be determined statistically (mean ± SEM; vehicle, 17.3 ± 4.3; GRP, 21.7 ± 3.1; GRP plus TTX, 16.2 ± 1.3; and TTX, 17.6 ± 5.3). Because Per1 activation, as reflected by Per1::GFP intensity, is significantly correlated with SCN neuronal spike frequency (Kuhlman et al., 2003), these data suggest that GRP application elevates spike frequencies in SCN neurons as part of phase resetting.
GRP persistently increases spike frequency in SCN neurons
Phase-resetting photic stimulation increases the frequency of spontaneous action potentials, even hours later, in ventral SCN neurons, and this increase is correlated with the induction of Per1, as reflected by Per1::GFP (Kuhlman et al., 2003). Thus, we sought to investigate whether GRP, potentially released from activated ventral SCN neurons, subsequently increases the neuronal activity of dorsal SCN neurons in a similar manner. Although the acute effects of GRP application to SCN neurons have been investigated (Piggins and Rusak, 1993; Pinnock et al., 1994), we tested the longer-term effects of GRP associated with Per1 induction and phase resetting. Specifically, we compared the frequency of spontaneous action potentials of SCN neurons 3–4 h after either a pulse of GRP or vehicle application at projected ZT 21. Hypothalamic slices were prepared during lights on (ZT 1–ZT 4) and cultured (Han et al., 2006) until the following day to avoid the phase-shifting effects of slice preparation during the night (Gillette, 1986). At projected ZT 21 of the following day, 0.3 μm GRP was applied to the culture medium and washed out 1 h later (projected ZT 22) as the slice was transferred to an open recording chamber that was continuously perfused with extracellular solution. Two to 3 h after the termination of GRP application (projected ZT 0–ZT 1), loose patch extracellular recordings were made from Per1::GFP-induced, SCN neurons. The mean firing rate of Per1 fluorescent neurons of GRP-treated slices was nearly double the mean firing rate of Per1 fluorescent neurons from vehicle-treated slices (Fig. 4A,B). The mean firing rate for GRP-treated neurons was 6.7 ± 1.0 Hz (mean ± SEM; n = 10), which was significantly greater than the mean firing rate for vehicle-treated neurons (3.6 ± 0.9 Hz; n = 9; t(17) = −2.3; p < 0.05, two-tailed).
Figure 4.

Per1 gene activity is necessary for GRP-induced increases in neuronal activity of Per1-expressing neurons. A, Examples of individual, extracellular recordings from slices that were treated with either vehicle (top trace) or GRP (bottom trace) at projected ZT 21. Calibration: 40 pA, 1.0 s. B, Bar graph indicating the mean spontaneous action potential (A.P.) frequency of Per1 fluorescent neurons after treatment with either vehicle or GRP at projected ZT 21. *p < 0.05. C, Examples of individual extracellular recordings from slices that were treated with either random ODN followed by GRP (top trace) or mPer1 antisense ODN followed by GRP (bottom trace) at projected ZT 21. Calibration: 40 pA, 1.0 s. D, Bar graph indicating the mean spontaneous action potential frequency of Per1 fluorescent neurons after treatment with mPer1 antisense ODN followed by GRP (AntiPer1 + GRP), random ODN followed by GRP (RandPer1 + GRP), or mPer1 antisense ODN followed by vehicle (AntiPer1 + Veh). Brain slices were prepared during the day (ZT 1–ZT 4), cultured overnight, and treated with ODN at projected ZT 20 (C, D) and GRP or vehicle at projected ZT 21. Slices were transferred to the recording chamber at projected ZT 22, and recordings were made from projected ZT 0–ZT 1. *p < 0.05, significantly different from the other two groups.
GRP-induced spike frequency elevation is through Per1
Our TTX experiments showed that spike-mediated mechanisms are not necessary for Per1::GFP induction by GRP in the majority of SCN neurons, suggesting that Per1 induction may be upstream or independent of GRP-induced increases in spike rate. Therefore, we sought to determine whether Per1 induction is necessary for GRP-induced increases in neuronal activity. Although the reporter transgene contains E-box and CRE sequences from the mPer1 promoter so that it accurately reports transcriptional activation of Per1, sequence differences surrounding the translation start site of the Per1::GFP transgene versus the native mPer1 gene (Gekakis et al., 1998; Kuhlman et al., 2000) enabled a unique experimental strategy. Here, we used Per1::GFP to identify SCN neurons transcriptionally activated by GRP, whereas translation of native Per1 and subsequent downstream events were blocked with antisense ODNs shown previously to disrupt mPer1 expression and light- and glutamate-induced phase resetting (Akiyama et al., 1999; Tischkau et al., 2003). We used this strategy to identify SCN neurons that had transduced the GRP signal, as shown by increased Per1::GFP, and then to determine whether GRP induction of elevated spike frequency persists in the presence of anti-Per1 ODN.
Hypothalamic slice cultures were treated with anti-Per1 ODN or a control ODN of the same GC content as the anti-Per1 ODN but of randomized sequence (random ODN) at projected ZT 20. At projected ZT 21, either 0.3 μm GRP or vehicle was added and subsequently washed out during transfer to the recording chamber at projected ZT 22. As before, we compared the frequency of spontaneous action potentials of SCN neurons at projected ZT 0–ZT 1, which was 3–4 h after GRP or vehicle application. In contrast to the experiments described above, GRP failed to persistently increase spiking in SCN neurons when applied in the presence of anti-Per1 ODN (Fig. 4C,D). After GRP and anti-Per1 ODN application, Per1::GFP neurons exhibited mean spike frequencies (3.7 ± 0.4 Hz; n = 7) that were essentially identical to the vehicle-treated SCN neurons shown in Figure 4, A and B (3.6 ± 0.9 Hz). However, when GRP was applied in the presence of a random ODN, Per1::GFP SCN neurons did show the expected significant increase in spike frequency (5.9 ± 0.6 Hz; n = 8; χ2(2) = 7.0; p < 0.05, Kruskal–Wallis test). Application of anti-Per1 ODN with vehicle did not affect spike frequency (3.5 ± 0.8 Hz; n = 8). Immunocytochemistry of coronal brain slices containing the SCN confirmed ODN inhibition of GRP-induced PER1 expression. The intensity of PER1 immunoreactivity after anti-Per1 ODN treatment was reduced to 35.1% of that after random ODN treatment (mean ± SEM; random Per1 ODN, 53.6 ± 5.3; anti-Per1 ODN, 18.8 ± 7.7; t(7) = 3.85; p < 0.01) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). These results indicate that persistent elevation of spike frequency in SCN neurons by GRP is downstream of mPer1 gene induction.
GRP phase shifts SCN gene expression rhythms in a CREB-dependent manner
Stimulation of BB2 receptors by GRP activates the MAPK pathway (Woodruff et al., 1996; Moody and Merali, 2004), which can affect SCN clock gene transcription through a CREB-dependent mechanism (Butcher et al., 2005). To determine whether GRP stimulates SCN gene transcription and phase resetting through the CREB pathway, we tested whether inhibiting CREB protein binding to CRE elements with CRE-decoy ODNs blocked GRP-induced phase resetting of SCN rhythms. CRE decoy ODNs compete with native CRE sites in gene promoters (e.g., Per1 and Per2) for binding of the phospho-CREB transcription factor (Tischkau and Gillette, 2005) and have been shown to block light-induced phase shifts in the rodent SCN (Tischkau et al., 2003). In these experiments, we used SCN from PER2::LUC reporter mice and luminescence recording to take advantage of the increased throughput of the multiwell Lumicycle luminometer for pharmacological experiments. After two to three stable cycles of luminescence were established, coronal hypothalamic slice cultures containing the SCN from PER2::LUC mice were transferred to new media treated with either 100 μm CRE-decoy ODN or 100 μm mismatched sequence of the same nucleotide bases (CRE-mis), in addition to either 0.3 μm GRP or vehicle. Because an increase in Per2 transcription is more robust after a phase shifting light stimulus presented in the early rather than late night (Yan and Silver, 2002), cultures received treatment for 1 h beginning at CT 15–CT 16 (CT 12 refers to peak PER2::LUC luminescence) (Field et al., 2000; Yoo et al., 2004) and then transferred back into the original media. After two or more full cycles, each culture received a medium change and two rinses and was then treated for a second time with vehicle, if GRP was given for the first treatment and vice versa. The results showed that application of GRP and CRE-mis for 1 h at CT 15–CT 16 resulted in a phase delay of ∼2 h (Fig. 5A,C). Treatment of the cultures with CRE-mis alone resulted in a phase delay of ∼0.7 h. In contrast, the GRP-induced phase delays in PER2::LUC luminescence rhythms were completely blocked when GRP was applied in the presence of CRE-decoy, and CRE-decoy alone resulted in minimal phase delays (Fig. 5B,C) (F(1,6) = 7.72; p < 0.05). Thus, activation of CRE elements is a necessary step in GRP-mediated phase resetting of the SCN.
Figure 5.
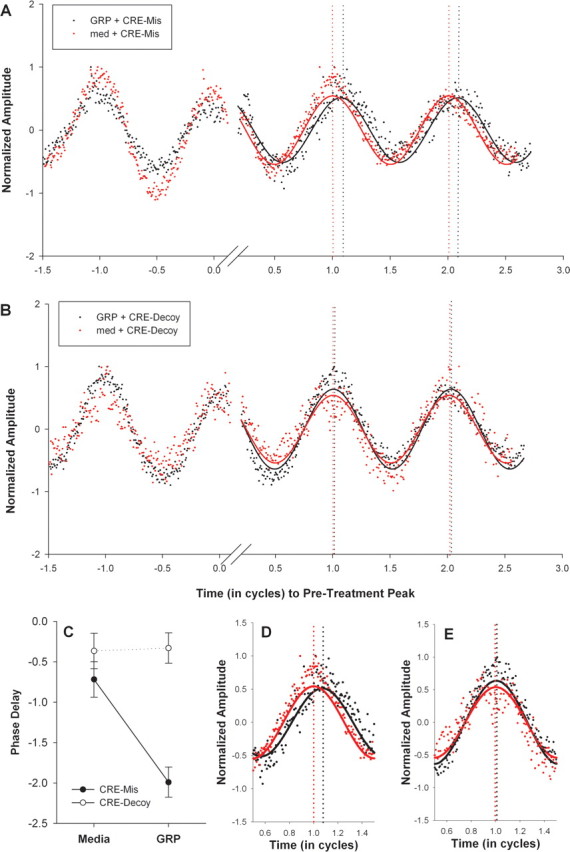
A, B, Bioluminescence recorded from SCN explants cultured from PER2::LUC mice after treatment with CRE-mis (A) or CRE-decoy (B) at CT 15–CT 16. Baseline-subtracted traces (see Materials and Methods) within each graph are from the same culture treated with GRP (black) or culture medium (med; red). The luminescence amplitude (y-axis) was normalized to the maximum luminescence for that trace. The x-axis represents time in cycles (where 1 cycle = 24/free-running period). The two traces in each graph were aligned to the peak just before treatment (defined as time 0) to allow phase-shift comparison. The solid lines indicate the fitted sine wave for the luminescence data points from cultures treated with GRP (black) or media (red). For the CRE-mis culture (A), the phase difference between GRP and medium is 0.098 cycles, which equals 2.34 h. For the CRE-decoy culture (B), the phase difference between GRP and medium is 0.014 cycles, which equals 0.34 h. C, Line graph of average ± SEM phase delays: filled circles represent cultures that received CRE-mis, and open circles represent cultures that received CRE-decoy. Each culture was treated with GRP or medium for 1 h at CT 15–CT 16 (in which CT 12 is defined as peak PER2::LUC luminescence). Every culture received both conditions (GRP and medium in a counterbalanced order), which were separated by two rinses and a medium change. D, E, Enlarged bioluminescence traces of the first cycle after GRP (black) or culture medium (red) treatment at CT 15–CT 16 for the CRE-mis-treated (D) and a CRE-decoy-treated (E) cultures in A and B above.
Discussion
During photic entrainment, a series of neural signals originating with the transduction of environmental light by the retina resets the rhythmic neural network of the SCN. In the presence of a phase-shifting light stimulus, the retinal signal to the retinorecipient zone of the SCN must be transformed into a neuropeptide/neurochemical signal that communicates photic cues throughout the SCN network. Initially, light-induced glutamate and pituitary adenylate cyclase-activating polypeptide release from retinal afferents stimulates Per1 gene activity (Nielsen et al., 2001) and high levels of spontaneous spike activity in neurons of the retinorecipient core SCN region (Kuhlman et al., 2003) in which VIP-IR and GRP-IR populations are located (Abrahamson and Moore, 2001), and thus VIP and GRP are proposed to be signaling peptides for intra-SCN communication of light information. The present data add to this model, providing three novel findings that further our understanding of the role of GRP in the phase-shifting process of SCN circadian rhythms. First, GRP receptor activation results in widespread transmission of the light signal as Per1 induction throughout the SCN via a spike-independent mechanism and in some AVP-expressing SCN neurons via a spike-dependent mechanism. Second, persistent increases in SCN neuronal spike rate frequency induced by GRP are dependent on Per1 activation, suggesting that activation of the Per1 gene occurs upstream from the resulting change in neuronal activity. Third, GRP mediates phase resetting via a CREB-dependent pathway, providing a link between GRP modulation of circadian phase and CREB-mediated transcription.
GRP mediates SCN gene induction through spike-dependent and spike-independent mechanisms
Using the Per1::GFP reporter for Per1 gene activity that closely overlaps the spatial pattern of PER1 protein (LeSauter et al., 2003), the present study showed that GRP injection into the SCN regions of Per1::GFP mice during the late night induced phase advances that were also accompanied by Per1 gene activity throughout the SCN, including a subpopulation of AVP-producing cells. One proposed function of GRP is to directly mediate photic information from the retinorecipient region to the AVP-producing cells in the dorsomedial or shell region of the SCN (Hamada et al., 2001; Aida et al., 2002; Karatsoreos et al., 2004). Our results do not support this proposed mechanism of GRP action because only a minority of AVP-producing cells responded to GRP application. Of these responsive cells, spike-mediated mechanisms were necessary, indicating the possibility of an indirect mode of activation by GRP. Alternatively, membrane depolarization may be necessary for a GRP-induced increase in Per1 activity in this small population of AVP-producing cells. In general, our results and those of Antle et al. (2005) in the hamster demonstrate that GRP directly induces gene activation in non-AVP SCN neurons. Specifically, ventricular injection of GRP into the hamster SCN in the early night does not increase Per1 and Per2 mRNA levels in the AVP region of the hamster SCN but rather in a small region in the dorsolateral SCN that additionally expresses the phosphorylated form of extracellular signal-regulated kinases 1/2 (ERK1/2) (Antle et al., 2005). Despite the notable difference in the circadian time of GRP administration (early vs late night), both studies point to the same conclusion, that GRP-containing neurons do not transmit photic information by activating Per1 directly in the majority of AVP-producing cells within the SCN.
In the present study, GRP injection induced Per1 reporter activity throughout the SCN even when Na+-dependent action potentials were blocked with TTX. This spike-independent Per1 activation by GRP is presumably attributable to the direct action of GRP on BB2 receptors and occurred in many cells that were neither AVP- nor VIP-expressing neurons (‘80% of the Per1-induced neurons). This pattern of Per1 gene induction is different from the initial transduction of the photic signal, in which light stimulation induces Per1 gene activity in many VIP-expressing cells (Kuhlman et al., 2003). The widespread Per1 gene induction by GRP, however, mimics the widespread expression of BB2 receptors, although they are more highly concentrated in the dorsal and medial regions of the SCN (Karatsoreos et al., 2006).
During the early night, GRP application to the SCN region did not increase the number of Per1::GFP-labeled cells. This result is in agreement with the finding of Yan and Silver (2004) demonstrating that PER1 protein induction rapidly returns to control levels in the shell region of the SCN after a phase-delaying light stimulus but is in contrast with the findings of Aida et al. (2002), in which ventricular injection of GRP during the early night upregulates Per1 and Per2 mRNA levels in the dorsal SCN. It is possible that GRP-induced upregulation of Per1 mRNA does not translate into a significant increase in PER1 protein. In addition, induction of PER1 protein in the SCN shell region may not be critical for phase delays (Yan and Silver, 2004), which may explain the lack of GRP-induced Per1::GFP in the early night in the present study.
GRP induces persistent spiking of SCN neurons through Per1 activation
Initial transduction of photic input in the SCN induces Per1 gene activity that is significantly correlated with persistent increases in the spike rates of gene-induced neurons in the ventral region of the SCN (Kuhlman et al., 2003), in which GRP-IR and VIP-IR cells are located (Abrahamson and Moore, 2001). Although upregulated Per1 gene activity later spreads to include the dorsal shell region as well, it was previously unclear whether stimulation of GRP receptors, which are concentrated in the dorsal and medial regions of the SCN (Karatsoreos et al., 2006), would also result in induced Per1 gene activity along with a corresponding persistent increase in spike rate. Here, the results of the present study show that GRP induced Per1 reporter gene activity and a persistent increase in spike rate that is presumably the initial phase advance of the electrical activity rhythm. More importantly, our data show that this persistent increase in spike rate observed hours after GRP application is linked to upregulation of the native Per1 gene.
We now show that the induction of Per1 gene activity is a key step required for GRP to communicate spike frequency changes induced throughout the SCN network (Fig. 6). It remains to be shown how induction of Per1 results in high levels of spontaneous spike activity in the GRP-induced SCN cell population. One possibility is that a reduction in a hyperpolarizing K+ current may account for the increase in spontaneous firing rate, similar to the neural mechanisms mediating transduction of the retinal signal to the core SCN (Kuhlman et al., 2003). Although Per1 gene activity could alter many cellular processes, including transcriptional regulation of membrane channel abundance and/or channel function, it is interesting to note that both PER1 and some voltage-dependent K+ channels contain PAS (Period–Arnt–Singleminded) structural domains, which could allow direct protein–protein regulating interactions (Morais Cabral et al., 1998; Reppert and Weaver, 2001).
Figure 6.

Model of GRP mediation of photic phase resetting in the SCN. During photic phase resetting, we propose that GRP cells and GRP are involved in the following ways. “Transduction of the photic signal in the SCN,” Ventral SCN cells in the night phase expressing GRP and/or VIP (top cell) show a persistent increase in the frequency of spontaneous action potentials (far right, top), as well as the induction of Per1 in the presence of light (represented by the light bulb) (Kuhlman et al., 2003). “Communication of the intra-SCN photic signal,” GRP is released from ventral cells and activates BB2 receptors (BB2R) on cells concentrated in the dorsal and medial SCN regions (Karatsoreos et al., 2006). One should note that other neurotransmitters and peptides such as VIP may also be released and involved in this process. “Initiation of SCN phase resetting,” MAPK, ERK, and CREB intracellular pathways are activated (Butcher et al., 2002; Antle et al., 2005; this study), resulting in activation of Per1 that is necessary for a persistent increase in the frequency of spontaneous action potentials to be induced (this study). “Phase shift of SCN rhythms and behavior,” Phase advance of the electrical activity rhythm, behavior, and gene expression.
GRP induces SCN phase shifts through CREB activation
Several lines of evidence suggest that light induction of Per1 during phase shifting occurs through activation of the MAPK and CRE pathways. For example, activation of the MAPK pathway in the SCN is observed after nighttime photic stimulation (Obrietan et al., 1998) and is necessary for light-induced immediate-early gene expression and phase shifts (Butcher et al., 2002; Dziema et al., 2003). Likewise, phosphorylation of CREB occurs after nighttime photic stimulation and in vitro glutamate application (Ding et al., 1997) and is necessary for light-induced and glutamate-induced phase shifts, as well as glutamate-induced Per1 induction (Tischkau et al., 2003). Both the MAPK and cAMP pathways synergistically activate the Per1 promoter (Travnickova-Bendova et al., 2002), and activation of CREB is necessary for the MAPK pathway to induce Per1 expression (Butcher et al., 2005). As part of the photic signaling pathway within the SCN, GRP-induced phase shifts and Per induction also require activation of the MAPK pathway (Woodruff et al., 1996; Moody and Merali, 2004; Antle et al., 2005). We now show that coapplication of GRP with CRE-decoy ODN blocked GRP-induced phase delays of PER2::LUC rhythms. Altogether, the present and previous findings support the notion that GRP phase shifts SCN neurons via activation of MAPK, CREB, and Per gene activation (Fig. 6).
Summary
The results of this study support the hypothesis that GRP mediates photic modulation of circadian phase, although it appears that GRP is not solely responsible for mediating photic information to the AVP-rich region of the SCN. For example, VIP is likely to be involved the photic entrainment process within the SCN because it has been shown to phase shift activity rhythms (Albers et al., 1991, 1995), and mice that are deficient for VIP or its receptor have reduced phase-shifting responses to light (Harmar et al., 2002; Colwell et al., 2003). In addition to VIP, recent evidence has shown that NMDA receptor activation is required for GRP-induced phase shifts in behavior (Kallingal and Mintz, 2006), indicating the complexity of this pathway. The present findings suggest that GRP communicates phase-resetting signals throughout the SCN network via spike-independent mechanisms and to a small population of AVP-containing neurons via spike-dependent mechanisms. Our results further imply that activation of the CREB pathway and Per1 are key steps in mediating downstream physiological events in GRP-resetting of SCN neurons. Future studies will be necessary to identify the link between GRP-stimulated gene induction and persistent increases in neural activity.
Footnotes
This work was supported by National Institutes of Health Grants MH63341 (D.G.M.), NS051183 (K.L.G.), and T32 MH65215 (Vanderbilt Neurogenomics Training Program) and the Vanderbilt Kennedy Center. We thank Guoxiang Ruan, Kevin McMahon, Shin Yamazaki, Earnestine Jideama, and Shi Meng for technical assistance.
References
- Abrahamson EE, Moore RY. Suprachiasmatic nucleus in the mouse: retinal innervation, intrinsic organization and efferent projections. Brain Res. 2001;916:172–191. doi: 10.1016/s0006-8993(01)02890-6. [DOI] [PubMed] [Google Scholar]
- Aida R, Moriya T, Araki M, Akiyama M, Wada K, Wada E, Shibata S. Gastrin-releasing peptide mediates photic entrainable signals to dorsal subsets of suprachiasmatic nucleus via induction of Period gene in mice. Mol Pharmacol. 2002;61:26–34. doi: 10.1124/mol.61.1.26. [DOI] [PubMed] [Google Scholar]
- Akiyama M, Kouzu Y, Takahashi S, Wakamatsu H, Moriya T, Maetani M, Watanabe S, Tei H, Sakaki Y, Shibata S. Inhibition of light- or glutamate-induced mPer1 expression represses the phase shifts into the mouse circadian locomotor and suprachiasmatic firing rhythms. J Neurosci. 1999;19:1115–1121. doi: 10.1523/JNEUROSCI.19-03-01115.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albers HE, Liou SY, Stopa EG, Zoeller RT. Interaction of colocalized neuropeptides: functional significance in the circadian timing system. J Neurosci. 1991;11:846–851. doi: 10.1523/JNEUROSCI.11-03-00846.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albers HE, Gillespie CF, Babagbemi TO, Huhman KL. Analysis of the phase shifting effects of gastrin releasing peptide when microinjected into the suprachiasmatic region. Neurosci Lett. 1995;191:63–66. doi: 10.1016/0304-3940(95)11559-1. [DOI] [PubMed] [Google Scholar]
- Antle MC, Kriegsfeld LJ, Silver R. Signaling within the master clock of the brain: localized activation of mitogen-activated protein kinase by gastrin-releasing peptide. J Neurosci. 2005;25:2447–2454. doi: 10.1523/JNEUROSCI.4696-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschoff J. Circadian clocks. Amsterdam: North-Holland; 1965. Response curves in circadian periodicity; pp. 95–111. [Google Scholar]
- Butcher GQ, Dziema H, Collamore M, Burgoon PW, Obrietan K. The p42/44 mitogen-activated protein kinase pathway couples photic input to circadian clock entrainment. J Biol Chem. 2002;277:29519–29525. doi: 10.1074/jbc.M203301200. [DOI] [PubMed] [Google Scholar]
- Butcher GQ, Lee B, Cheng HY, Obrietan K. Light stimulates MSK1 activation in the suprachiasmatic nucleus via a PACAP-ERK/MAP kinase-dependent mechanism. J Neurosci. 2005;25:5305–5313. doi: 10.1523/JNEUROSCI.4361-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colwell CS, Michel S, Itri J, Rodriguez W, Tam J, Lelievre V, Hu Z, Liu X, Waschek JA. Disrupted circadian rhythms in VIP- and PHI-deficient mice. Am J Physiol Regul Integr Comp Physiol. 2003;285:R939–R949. doi: 10.1152/ajpregu.00200.2003. [DOI] [PubMed] [Google Scholar]
- Daan S, Pittendrigh CS. A functional analysis of circadian pacemakers in nocturnal rodents. II. The variability of phase response curves. J Comp Physiol [A] 1976;106:253–266. [Google Scholar]
- Dardente H, Poirel VJ, Klosen P, Pevet P, Masson-Pevet M. Per and neuropeptide expression in the rat suprachiasmatic nuclei: compartmentalization and differential cellular induction by light. Brain Res. 2002;958:261–271. doi: 10.1016/s0006-8993(02)03563-1. [DOI] [PubMed] [Google Scholar]
- De Groote L, Olivier B, Westenberg HG. Role of 5-HT1B receptors in the regulation of extracellular serotonin and dopamine in the dorsal striatum of mice. Eur J Pharmacol. 2003;476:71–77. doi: 10.1016/s0014-2999(03)02154-x. [DOI] [PubMed] [Google Scholar]
- Ding JM, Faiman LE, Hurst WJ, Kuriashkina LR, Gillette MU. Resetting the biological clock: mediation of nocturnal CREB phosphorylation via light, glutamate, and nitric oxide. J Neurosci. 1997;17:667–675. doi: 10.1523/JNEUROSCI.17-02-00667.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziema H, Oatis B, Butcher GQ, Yates R, Hoyt KR, Obrietan K. The ERK/MAP kinase pathway couples light to immediate-early gene expression in the suprachiasmatic nucleus. Eur J Neurosci. 2003;17:1617–1627. doi: 10.1046/j.1460-9568.2003.02592.x. [DOI] [PubMed] [Google Scholar]
- Earnest DJ, DiGiorgio S, Olschowka JA. Light induces expression of fos-related proteins within gastrin-releasing peptide neurons in the rat suprachiasmatic nucleus. Brain Res. 1993;627:205–209. doi: 10.1016/0006-8993(93)90322-e. [DOI] [PubMed] [Google Scholar]
- Field MD, Maywood ES, O'Brien JA, Weaver DR, Reppert SM, Hastings MH. Analysis of clock proteins in mouse SCN demonstrates phylogenetic divergence of the circadian clockwork and resetting mechanisms. Neuron. 2000;25:437–447. doi: 10.1016/s0896-6273(00)80906-x. [DOI] [PubMed] [Google Scholar]
- Gekakis N, Staknis D, Nguyen HB, Davis FC, Wilsbacher LD, King DP, Takahashi JS, Weitz CJ. Role of the CLOCK protein in the mammalian circadian mechanism. Science. 1998;280:1564–1569. doi: 10.1126/science.280.5369.1564. [DOI] [PubMed] [Google Scholar]
- Gillette MU. The suprachiasmatic nuclei: circadian phase-shifts induced at the time of hypothalamic slice preparation are preserved in vitro. Brain Res. 1986;379:176–181. doi: 10.1016/0006-8993(86)90273-8. [DOI] [PubMed] [Google Scholar]
- Hamada T, LeSauter J, Venuti JM, Silver R. Expression of Period genes: rhythmic and nonrhythmic compartments of the suprachiasmatic nucleus pacemaker. J Neurosci. 2001;21:7742–7750. doi: 10.1523/JNEUROSCI.21-19-07742.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han MH, Bolanos CA, Green TA, Olson VG, Neve RL, Liu RJ, Aghajanian GK, Nestler EJ. Role of cAMP response element-binding protein in the rat locus ceruleus: regulation of neuronal activity and opiate withdrawal behaviors. J Neurosci. 2006;26:4624–4629. doi: 10.1523/JNEUROSCI.4701-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmar AJ, Marston HM, Shen S, Spratt C, West KM, Sheward WJ, Morrison CF, Dorin JR, Piggins HD, Reubi JC, Kelly JS, Maywood ES, Hastings MH. The VPAC(2) receptor is essential for circadian function in the mouse suprachiasmatic nuclei. Cell. 2002;109:497–508. doi: 10.1016/s0092-8674(02)00736-5. [DOI] [PubMed] [Google Scholar]
- Johnson RF, Morin LP, Moore RY. Retinohypothalamic projections in the hamster and rat demonstrated using cholera toxin. Brain Res. 1988;462:301–312. doi: 10.1016/0006-8993(88)90558-6. [DOI] [PubMed] [Google Scholar]
- Kallingal GJ, Mintz EM. Glutamatergic activity modulates the phase-shifting effects of gastrin-releasing peptide and light. Eur J Neurosci. 2006;24:2853–2858. doi: 10.1111/j.1460-9568.2006.05165.x. [DOI] [PubMed] [Google Scholar]
- Karatsoreos IN, Yan L, LeSauter J, Silver R. Phenotype matters: identification of light-responsive cells in the mouse suprachiasmatic nucleus. J Neurosci. 2004;24:68–75. doi: 10.1523/JNEUROSCI.1666-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karatsoreos IN, Romeo RD, McEwen BS, Silver R. Diurnal regulation of the gastrin-releasing peptide receptor in the mouse circadian clock. Eur J Neurosci. 2006;23:1047–1053. doi: 10.1111/j.1460-9568.2006.04633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamoto K, Nagano M, Kanda F, Chihara K, Shigeyoshi Y, Okamura H. Two types of VIP neuronal components in rat suprachiasmatic nucleus. J Neurosci Res. 2003;74:852–857. doi: 10.1002/jnr.10751. [DOI] [PubMed] [Google Scholar]
- Kuhlman SJ, Quintero JE, McMahon DG. GFP fluorescence reports Period 1 circadian gene regulation in the mammalian biological clock. NeuroReport. 2000;11:1479–1482. [PubMed] [Google Scholar]
- Kuhlman SJ, Silver R, Le Sauter J, Bult-Ito A, McMahon DG. Phase resetting light pulses induce Per1 and persistent spike activity in a subpopulation of biological clock neurons. J Neurosci. 2003;23:1441–1450. doi: 10.1523/JNEUROSCI.23-04-01441.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeSauter J, Yan L, Vishnubhotla B, Quintero JE, Kuhlman SJ, McMahon DG, Silver R. A short half-life GFP mouse model for analysis of suprachiasmatic nucleus organization. Brain Res. 2003;964:279–287. doi: 10.1016/s0006-8993(02)04084-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArthur AJ, Coogan AN, Ajpru S, Sugden D, Biello SM, Piggins HD. Gastrin-releasing peptide phase-shifts suprachiasmatic nuclei neuronal rhythms in vitro. J Neurosci. 2000;20:5496–5502. doi: 10.1523/JNEUROSCI.20-14-05496.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moody TW, Merali Z. Bombesin-like peptides and associated receptors within the brain: distribution and behavioral implications. Peptides. 2004;25:511–520. doi: 10.1016/j.peptides.2004.02.012. [DOI] [PubMed] [Google Scholar]
- Moore RY, Speh JC, Leak RK. Suprachiasmatic nucleus organization. Cell Tissue Res. 2002;309:89–98. doi: 10.1007/s00441-002-0575-2. [DOI] [PubMed] [Google Scholar]
- Morais Cabral JH, Lee A, Cohen SL, Chait BT, Li M, Mackinnon R. Crystal structure and functional analysis of the HERG potassium channel N terminus: a eukaryotic PAS domain. Cell. 1998;95:649–655. doi: 10.1016/s0092-8674(00)81635-9. [DOI] [PubMed] [Google Scholar]
- Moriya T, Horikawa K, Akiyama M, Shibata S. Correlative association between N-methyl-d-aspartate receptor-mediated expression of period genes in the suprachiasmatic nucleus and phase shifts in behavior with photic entrainment of clock in hamsters. Mol Pharmacol. 2000;58:1554–1562. doi: 10.1124/mol.58.6.1554. [DOI] [PubMed] [Google Scholar]
- Nielsen HS, Hannibal J, Knudsen SM, Fahrenkrug J. Pituitary adenylate cyclase-activating polypeptide induces period1 and period2 gene expression in the rat suprachiasmatic nucleus during late night. Neuroscience. 2001;103:433–441. doi: 10.1016/s0306-4522(00)00563-7. [DOI] [PubMed] [Google Scholar]
- Obrietan K, Impey S, Storm DR. Light and circadian rhythmicity regulate MAP kinase activation in the suprachiasmatic nuclei. Nat Neurosci. 1998;1:693–700. doi: 10.1038/3695. [DOI] [PubMed] [Google Scholar]
- Pickard GE. Bifurcating axons of retinal ganglion cells terminate in the hypothalamic suprachiasmatic nucleus and the intergeniculate leaflet of the thalamus. Neurosci Lett. 1985;55:211–217. doi: 10.1016/0304-3940(85)90022-9. [DOI] [PubMed] [Google Scholar]
- Piggins HD, Rusak B. Electrophysiological effects of pressure-ejected bombesin-like peptides on hamster suprachiasmatic nucleus neurons in vitro. J Neuroendocrinol. 1993;5:575–581. doi: 10.1111/j.1365-2826.1993.tb00524.x. [DOI] [PubMed] [Google Scholar]
- Piggins HD, Antle MC, Rusak B. Neuropeptides phase shift the mammalian circadian pacemaker. J Neurosci. 1995;15:5612–5622. doi: 10.1523/JNEUROSCI.15-08-05612.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinnock RD, Reynolds T, Woodruff GN. Different types of bombesin receptors on neurons in the dorsal raphe nucleus and the rostral hypothalamus in rat brain slices in vitro. Brain Res. 1994;653:119–124. doi: 10.1016/0006-8993(94)90379-4. [DOI] [PubMed] [Google Scholar]
- Rea MA, Buckley B, Lutton LM. Local administration of EAA antagonists blocks light-induced phase shifts and c-fos expression in hamster SCN. Am J Physiol. 1993;265:R1191–R1198. doi: 10.1152/ajpregu.1993.265.5.R1191. [DOI] [PubMed] [Google Scholar]
- Reppert SM, Weaver DR. Molecular analysis of mammalian circadian rhythms. Annu Rev Physiol. 2001;63:647–676. doi: 10.1146/annurev.physiol.63.1.647. [DOI] [PubMed] [Google Scholar]
- Romijn HJ, Sluiter AA, Pool CW, Wortel J, Buijs RM. Differences in colocalization between Fos and PHI, GRP, VIP and VP in neurons of the rat suprachiasmatic nucleus after a light stimulus during the phase delay versus the phase advance period of the night. J Comp Neurol. 1996;372:1–8. doi: 10.1002/(SICI)1096-9861(19960812)372:1<1::AID-CNE1>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Shearman LP, Zylka MJ, Weaver DR, Kolakowski LF, Jr, Reppert SM. Two period homologs: circadian expression and photic regulation in the suprachiasmatic nuclei. Neuron. 1997;19:1261–1269. doi: 10.1016/s0896-6273(00)80417-1. [DOI] [PubMed] [Google Scholar]
- Shigeyoshi Y, Taguchi K, Yamamoto S, Takekida S, Yan L, Tei H, Moriya T, Shibata S, Loros JJ, Dunlap JC, Okamura H. Light-induced resetting of a mammalian circadian clock is associated with rapid induction of the mPer1 transcript. Cell. 1997;91:1043–1053. doi: 10.1016/s0092-8674(00)80494-8. [DOI] [PubMed] [Google Scholar]
- Tischkau SA, Gillette MU. Oligodeoxynucleotide methods for analyzing the circadian clock in the suprachiasmatic nucleus. Methods Enzymol. 2005;393:593–610. doi: 10.1016/S0076-6879(05)93031-0. [DOI] [PubMed] [Google Scholar]
- Tischkau SA, Mitchell JW, Tyan SH, Buchanan GF, Gillette MU. Ca2+/cAMP response element-binding protein (CREB)-dependent activation of Per1 is required for light-induced signaling in the suprachiasmatic nucleus circadian clock. J Biol Chem. 2003;278:718–723. doi: 10.1074/jbc.M209241200. [DOI] [PubMed] [Google Scholar]
- Travnickova-Bendova Z, Cermakian N, Reppert SM, Sassone-Corsi P. Bimodal regulation of mPeriod promoters by CREB-dependent signaling and CLOCK/BMALI activity. Proc Natl Acad Sci USA. 2002;99:7728–7733. doi: 10.1073/pnas.102075599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff GN, Hall MD, Reynolds T, Pinnock RD. Bombesin receptors in the brain. Ann NY Acad Sci. 1996;780:223–243. doi: 10.1111/j.1749-6632.1996.tb15126.x. [DOI] [PubMed] [Google Scholar]
- Yan L, Silver R. Differential induction and localization of mPer1 and mPer2 during advancing and delaying phase shifts. Eur J Neurosci. 2002;16:1531–1540. doi: 10.1046/j.1460-9568.2002.02224.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L, Silver R. Resetting the brain clock: time course and localization of mPER1 and mPER2 protein expression in suprachiasmatic nuclei during phase shifts. Eur J Neurosci. 2004;19:1105–1109. doi: 10.1111/j.1460-9568.2004.03189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo SH, Yamazaki S, Lowrey PL, Shimomura K, Ko CH, Buhr ED, Siepka SM, Hong HK, Oh WJ, Yoo OJ, Menaker M, Takahashi JS. PERIOD2::LUCIFERASE real-time reporting of circadian dynamics reveals persistent circadian oscillations in mouse peripheral tissues. Proc Natl Acad Sci USA. 2004;101:5339–5346. doi: 10.1073/pnas.0308709101. [DOI] [PMC free article] [PubMed] [Google Scholar]