

Abstract
Estrogen depletion in postmenopausal women is a significant risk factor for the development of Alzheimer's disease (AD), and estrogen-based hormone therapy may reduce this risk. However, the effects of progesterone both alone and in combination with estrogen on AD neuropathology remain unknown. In this study, we used the triple transgenic mouse model of AD (3xTg-AD) to investigate the individual and combined effects of estrogen and progesterone on β-amyloid (Aβ) accumulation, tau hyperphosphorylation, and hippocampal-dependent behavioral impairments. In gonadally intact female 3xTg-AD mice, AD-like neuropathology was apparent by 3 months of age and progressively increased through age 12 months, a time course that was paralleled by behavioral impairment. Ovariectomy-induced depletion of sex steroid hormones in adult female 3xTg-AD mice significantly increased Aβ accumulation and worsened memory performance. Treatment of ovariectomized 3xTg-AD mice with estrogen, but not progesterone, prevented these effects. When estrogen and progesterone were administered in combination, progesterone blocked the beneficial effect of estrogen on Aβ accumulation but not on behavioral performance. Interestingly, progesterone significantly reduced tau hyperphosphorylation when administered both alone and in combination with estrogen. These results demonstrate that estrogen and progesterone independently and interactively regulate AD-like neuropathology and suggest that an optimized hormone therapy may be useful in reducing the risk of AD in postmenopausal women.
Keywords: estrogen, progesterone, Alzheimer's disease, β-amyloid, tau, Y-maze
Introduction
Alzheimer's disease (AD) is an age-related neurodegenerative disorder characterized by accumulation of β-amyloid (Aβ) and neurofibrillary tangles, progressive neuron loss, and cognitive deficits (Hardy and Selkoe, 2002). Abundant evidence suggests that the depletion of the sex steroid hormones estrogen and progesterone (P4) at menopause is a significant risk factor for the development of AD in women (Paganini-Hill and Henderson, 1994, 1996; Tang et al., 1996; Kawas et al., 1997). Furthermore, prospective and case-control studies have demonstrated that hormone therapy (HT) can reduce the risk of AD in women (Paganini-Hill and Henderson, 1996; Tang et al., 1996; Kawas et al., 1997). Given this background, findings from the Women's Health Initiative Memory Study (WHIMS) demonstrating a higher incidence of dementia in subjects receiving estrogen-based HT in the absence (Espeland et al., 2004) and presence (Rapp et al., 2003; Shumaker et al., 2003) of progestin were unexpected. The WHIMS findings raised many important issues, highlighting the needs to better understand the role of estrogen and progesterone in AD pathogenesis and to optimize HT.
One of the important issues raised by the WHIMS trial is the role of progestogens in HT (Brinton, 2004; Craig et al., 2005). Although the many beneficial actions of estrogen in the brain are well established, the roles of progesterone, both alone and in combination with estrogen, remain incompletely defined. Some evidence suggests that progesterone improves hippocampal-dependent spatial memory (Roof et al., 1994) and protects neurons in some (Roof et al., 1994; Alkayed et al., 2000; Hoffman et al., 2003; De Nicola et al., 2006) but not all (Azcoitia et al., 1999; Toung et al., 2004) rodent models of neuronal injury. Similarly, coadministration of estrogen and progesterone rescues neuronal loss in some rodent paradigms of neural injury (Azcoitia et al., 1999; Toung et al., 2004) but not others (Rosario et al., 2006a). The effects of progesterone, both alone and in combination with estrogen, on neuropathology in animal models of AD are not known.
To begin investigating the individual and combined effects of estrogen and progesterone on AD-like neuropathology, we used the triple transgenic mouse model of AD (3xTg-AD), which exhibits many features of AD neuropathology (Oddo et al., 2003b). In this study, we evaluated the effects of experimental manipulation of estrogen and progesterone levels in adult female 3xTg-AD mice on the development of Aβ accumulation, tau hyperphosphorylation, and hippocampal-dependent memory performance.
Materials and Methods
Animals
Colonies of 3xTg-AD (Oddo et al., 2003b) and background strain, wild-type (WT) (C57BL/6/129S; The Jackson Laboratory, Bar Harbor, ME) mice were bred and maintained at the University of Southern California in accordance with National Institutes of Health guidelines on use of laboratory animals and an approved protocol by the University of Southern California (Los Angeles, CA) Institutional Animal Care and Use Committee. Female mice used in these studies were housed individually on 12 h light/dark cycles and provided ad libitum access to food and water.
Experimental design
This study consists of three experiments that were conducted using separate groups of animals, except when indicated.
Treatment groups for experiment 1.
To investigate the age-related development of AD-like neuropathology in female 3xTg-AD mice, gonadally intact females were randomly assigned to one of the following four groups (n = 7 per group) representing age at death: 3, 6, 9, and 12 months. WT mice age 6 months were used as a comparison group.
Treatment groups for experiment 2.
To investigate the development of AD-like neuropathology after estrogen manipulation, 3-month-old female 3xTg-AD and WT mice were assigned to one of the following three treatment groups (n = 7 per group): sham ovariectomized (OVX), OVX, and OVX plus 17β-estradiol (E2). Mice were bilaterally OVX and immediately implanted with a subcutaneous, continuous-release 90 d pellet (Innovative Research of America, Sarasota FL) containing either 0.25 mg of E2 (OVX+E2 group) or placebo (sham OVX and OVX groups). Animals in the sham OVX group were the same mice that comprised the 6 month group in experiment 1. All animals in experiment 2 were killed at age 6 months, 3 months after the initiation of hormone manipulations.
Treatment groups for experiment 3.
To investigate the development of AD-like neuropathology after progesterone manipulation, 3-month-old female 3xTg-AD and WT mice were divided into the following treatment groups (n = 7 per group): OVX, OVX+P4, and OVX+E2+P4. Mice were bilaterally OVX, immediately implanted with a subcutaneous, continuous-release 90 d pellet containing 25.0 mg of P4, both 0.25 mg of E2 and 25 mg of P4, or vehicle (Innovative Research of America), and killed at 6 months of age.
The hormone pellet doses used in experiments 2 and 3 have been shown to produce serum levels of estrogen and progesterone in the physiological range (Pomp et al., 1995; Kadish and Van Groen, 2002; Shultz et al., 2004). All hormone-treated mice were killed at 6 months of age, 3 months after initiation of hormone treatment. For all experiments, animals were behaviorally tested on the morning they were killed. Afterward, they were deeply anesthetized (100 mg/kg sodium pentobarbital), transcardially perfused with cold PBS, and decapitated. To confirm the efficacy of hormone treatments, (1) uteri were dissected, blotted, and weighed, and (2) blood was collected for analysis of serum hormone levels. Hemibrains were immersion fixed in 4% paraformaldehyde for 48 h and then stored in 4°C in PBS/1% sodium azide until use.
Serum hormone levels
E2 and P4 serum levels were measured using radioimmunoassay (RIA) as described previously (Slater et al., 2001).
Immunohistochemistry
Fixed hemibrains were blocked, sectioned (40 μm) exhaustively in the horizontal plane using a vibratome, and then processed for immunohistochemistry using a standard protocol (Pike, 1999; Rosario et al., 2006b). Briefly, every eighth section (∼12 per brain) was immunostained using antibodies directed against (1) Aβ (71-5800 Aβ, 1:300 dilution; Zymed Laboratories, San Francisco CA), (2) Aβ precursor protein C-terminal fragments (CTFs) (CT20, 1:16,000 dilution; Calbiochem, San Diego, CA), or (3) hyperphosphorylated tau (AT8, 1:1000 dilution; Pierce, Rockford, IL) using ABC Vector Elite and diaminobenzidine kits (Vector Laboratories, Burlingame, CA). Antigen unmasking treatment, consisting of 5 min rinse in 99% formic acid, was performed to enhance Aβ immunoreactivity (IR) (Cummings et al., 2002).
Quantification of immunohistochemistry
Immunohistochemistry was quantified using two different methods by a researcher blinded to experimental condition. First, levels of positive immunoreactivity for Aβ and CTF antibodies were determined by immunohistochemistry load technique (Cummings et al., 2002; Rosario et al., 2006b). Load values were determined from selected 420 × 330 μm fields of immunolabeled sections that were captured and digitized using a video capture system [black and white CCD camera coupled to an Olympus Optical (Tokyo, Japan) BX40 upright microscope]. Using NIH Image software 1.61, digital grayscale images were converted into binary positive/negative data using a constant threshold limit. The percentage of positive pixels (i.e., IR area) was quantified for each image and then averaged across images for each brain region in each animal to generate immunoreactive load values. Quantified fields were selected systematically using a predetermined pattern to maximize analysis of immunoreactivity in each brain region. For hippocampus CA1 and subiculum, every eighth horizontal section was analyzed across the entire hippocampus (a total of ∼12 sections per brain), beginning with a randomly selected section from the first eight sections of dorsal hippocampus. In hippocampus CA1, the capture frame was centered over the pyramidal layer with the first captured field corresponding to the narrow zone that marks the division between the end of the regio inferior (CA2/3) and the start of regio superior (CA1) as defined by West et al. (1991). Progressing across CA1 toward the subiculum, the first three adjacent but nonoverlapping fields were captured for load analysis. In the subiculum, we similarly captured two adjacent nonoverlapping fields per section beginning at the sharp transition from the large pyramidal cells of the CA1 to the smaller cells of the presubiculum (West et al., 1991). In the frontal cortex, fields were captured from every sixth coronal section beginning rostrally with the appearance of somatosensory cortex (Franklin and Paxinos, 1997) for a total of six sections per brain. For each section, the frame was centered over layers 4–5, and five adjacent nonoverlapping fields were captured beginning in cingulate cortex area 1 and progressing laterally to somatosensory cortex (Franklin and Paxinos, 1997).
Second, the numbers of Aβ deposits and AT8-immunoreactive neurons were counted, as described previously (Rosario et al., 2006b). Because of the relatively low numbers of both Aβ deposits and AT8-immunoreactive cells, we counted all immunoreactive objects meeting criteria within the combined CA1 hippocampus and subiculum regions, as defined above, for every 11th section in each animal. AT8-immunoreactive neurons were defined as cells showing strong AT8 immunolabeling over most of the cell surface. Aβ deposits were defined as diffuse or dense extracellular accumulations of Aβ immunoreactivity with approximate diameter at least twice the size of a neuron (to prevent misidentification of intraneuronal Aβ immunoreactivity).
Spontaneous alternation behavior
Mice were tested for working memory using spontaneous alternation behavior (SAB) in a Y-maze, as described previously (Rosario et al., 2006b). The maze was constructed from black-colored Plexiglas (short arms A and B, 6 × 5 × 3 inches; long arm C, 8 × 5 × 3 inches). Each animal was placed in arm C of the maze and allowed to explore freely for 8 min. The total numbers of arm choices and alternations were recorded. An arm choice was defined as both forepaws and hindpaws fully entering the arm. The maze was cleaned with 70% ethanol between animals to minimize odor cues. SAB score was calculated as the proportion of alternations (an arm choice differing from the previous two choices) to the total number of alternation opportunities (total arm entries, two) (King and Arendash, 2002).
Statistical analyses
To statistically evaluate treatment effects, raw data were first analyzed by ANOVA and then subjected to between-group comparisons using the Fisher's least significant difference test. Effects achieving 95% probability (i.e., p < 0.05) were interpreted as statistically significant.
Results
Experiment 1: development of neuropathology in female 3xTg-AD mice
Age-related increase in Aβ accumulation
As a first step in determining the effects of E2 and P4 on AD-like neuropathology in female 3xTg-AD mice, we assessed the age-related development of Aβ accumulation in gonadally intact female 3xTg-AD mice at 3, 6, 9, and 12 months of age. Aβ accumulation was evaluated in three hormone-responsive brain regions affected in AD: CA1 of hippocampus, subiculum, and frontal cortex. We observed intraneuronal Aβ-IR in all three brain regions that was apparent at low levels by age 3 months and increased through age 12 months (Fig. 1). In frontal cortex, Aβ-IR was primarily restricted to layers IV and V and in hippocampus was most prominent in CA1 pyramidal layer. Relatively lower levels of Aβ-IR were observed in basolateral amygdala and entorhinal cortex (data not shown). Only light, nonspecific immunolabeling was observed in WT female mice in all brain regions at age 6 months (Fig. 1 A,F,K) and later ages (data not shown). Load quantification of Aβ-IR showed that Aβ levels were greatest in subiculum, followed by frontal cortex, then hippocampus CA1 (Fig. 2). Aβ accumulated most quickly in frontal cortex and exhibited the strongest trend for continued accumulation beyond age 12 months in subiculum (Fig. 2). In addition to accumulating intraneuronally, Aβ-IR was also observed in the form of extracellular, plaque-like deposits beginning at age 9 months and strongly increasing at age 12 months (Fig. 2 D,E). Extracellular Aβ deposits were observed almost exclusively in subiculum through age 12 months, although they were also occasionally observed in hippocampus CA1 and entorhinal cortex.
Figure 1.

Age-related increase in Aβ immunoreactivity. Representative images show Aβ-IR in hippocampus CA1 (A–E), subiculum (Sub.; F–J), and layers IV and V of frontal cortex (F.Ctx.; K–O) from female 3xTg-AD mice at ages 3 months (B, G, L), 6 months (C, H, M), 9 months (D, I, N), and 12 months (E, J, O) and from WT female mice at age 6 months (A, F, K). Specific Aβ-IR is not observed in WT mice but in 3xTg-AD is apparent intraneuronally by age 3 months and extracellularly (arrows) beginning at age 9 months. Scale bar, 100 μm.
Figure 2.

Quantification of age-related increase in Aβ accumulation. Aβ load values were quantified in hippocampus CA1 (A), subiculum (Sub.; B), and layers IV and V of frontal cortex (F.Ctx.; C) from WT mice at 6 months of age and from female 3xTg-AD mice at 3, 6, 9, and 12 months of age. Data represent mean ± SEM Aβ load values from two to three nonoverlapping fields per section, 6–12 sections per brain (n = 7 per condition). ANOVA showed significant between group differences in Aβ load in hippocampus CA1 (F = 5.98, p = 0.008), subiculum (F = 31.85, p < 0.001), and frontal cortex (F = 7.6, p = 0.008). D, Aβ-IR also included extracellular Aβ deposits that were observed primarily in subiculum. Scale bar, 50 μm. E, Data show mean ± SEM counts of extracellular Aβ deposits in subiculum and hippocampus CA1 from 12 sections per animal at ages 3, 6, 9, and 12 months in 3xTg-AD mice and age 6 months in WT mice. A significant age-related increase in Aβ deposits was observed in 3xTg-AD mice (F = 9.70, p < 0.001). *p < 0.05 compared with 3 months; # p < 0.05 compared with 6 months.
To confirm that our evaluation of Aβ pathology reflects amyloidogenic species of Aβ rather than non-amyloidogenic CTFs of amyloid precursor protein, we examined CTF-IR and quantified CTF load in the subiculum. We observed that CTF-IR exhibited markedly different staining pattern compared with Aβ-IR; CTF-IR was localized to the periphery of the cell body (Fig. 3 H), whereas Aβ-IR was localized throughout the cell body, often with a punctate distribution (Fig. 3 I). High levels of CTF load were observed at similar levels in female 3xTg-AD mice across all ages examined (Fig. 3 B–F). Comparably low levels of CTF load were seen in 6-month-old female WT mice (Fig. 3 A,G).
Figure 3.
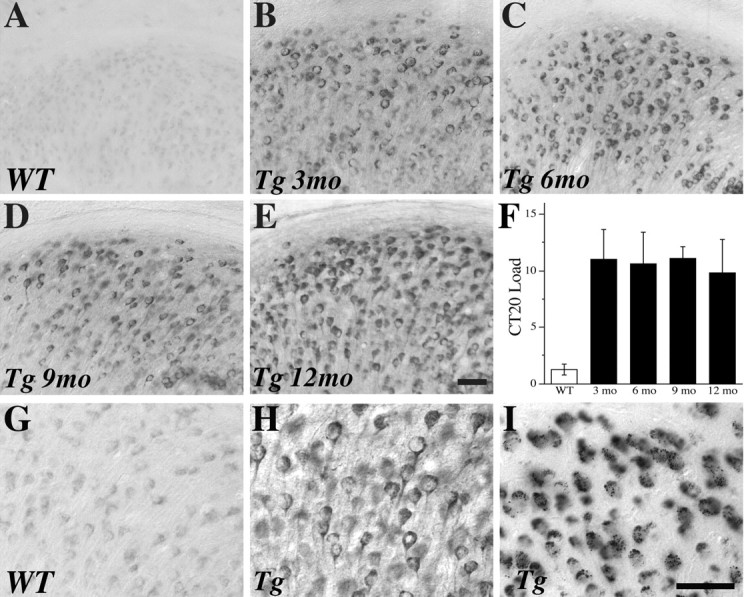
CTF levels do not change across conditions in 3xTg-AD mice. Representative images show CTF-IR in the subiculum of 6-month-old WT female mice (A) and female 3xTg-AD mice at 3 months (B), 6 months (C), 9 months (D), and 12 months (E) of age. F, Quantification of CTF load across conditions shows high CTF levels in all 3xTg-AD groups that does not significantly vary by age (F = 0.14, p = 0.94). A similar, extranuclear distribution of CTF-IR is observed at high magnification (400×) in WT mice (G) and 3xTg-AD mice (H) that qualitatively differs from the more uniform and often punctate appearance of Aβ-IR in 3xTg-AD mice (I). Scale bar, 50 μm.
Age-related increase in tau hyperphosphorylation
To further investigate the development of AD-like neuropathology in female 3xTg-AD mice, we assessed the age-related progression of tau hyperphosphorylation. We quantified the number of cells strongly immunoreactive with the AT8 antibody, which recognizes pathological phosphorylation of tau at Ser 202 and 305 that is associated with AD (Goedert et al., 1995). Intensely stained AT8-immunoreactive neurons were reliably detected, albeit in low numbers, at 9 months of age and increased in abundance through 12 months of age (Fig. 4). AT8-immunoreactive neurons were most common in the subiculum and observed occasionally in hippocampus CA1 but were not found in frontal cortex by age 12 months.
Figure 4.

Age-related increase in tau hyperphosphorylation. Tau hyperphosphorylation is absent in 6-month-old WT mice (A) but exhibits an age-related increase in female 3xTg-AD mice at 3 months (B), 6 months (C), 9 months (D), and 12 months (E) of age. Representative images show sections immunostained with AT8 antibody (dark brown label), which recognizes abnormally phosphorylated tau, and counterstained with cresyl violet. Scale bar, 50 μm. F, Quantification of numbers of intensely AT8-immunoreactive neurons in hippocampus CA1 and subiculum demonstrates the magnitude of this effect (F = 5.59, p = 0.007). *p < 0.05 relative to 3 months; # p < 0.05 relative to 6 month 3xTg-AD groups.
Age-related decline in spontaneous alternation behavior
Finally, we evaluated working memory in female 3xTg-AD mice by assessing performance of hippocampal-dependent SAB in the Y-maze. SAB performance of female 3xTg-AD mice at ages 3 and 6 months was not significantly impaired compared with WT female mice. However, SAB performance was significantly and increasingly impaired at 9 and 12 months of age (Fig. 5 A). In contrast, female WT mice did not show a significant age-related decline in SAB performance through age 12 months (data not shown). To ensure that observed changes in SAB in 3xTg-AD mice were not the result of changes in activity level, we measured the number of arm entries in the Y-maze. SAB performance was not associated with the number of arm entries (Fig. 5 B).
Figure 5.

Age-related decline in spontaneous alternation behavior. Hippocampal-dependent working memory performance in female 3xTg-AD mice was investigated by assessing SAB in a Y-maze. A, Compared with WT mice (open bars), SAB performance in female 3xTg-AD mice (filled bars) showed impairment that began at age 6 months and became statistically significant at ages 9 and 12 months (F = 16.2, p < 0.001). *p < 0.05 compared with WT. B, The number of arm entries, measured as a negative control for activity level, did not significantly vary across conditions (F = 1.14, p = 0.37).
Experiment 2: effect of estrogen on neuropathology in female 3xTg-AD mice
Confirmation of estrogen treatment
To investigate the effects of hormone status on the development of AD-like pathology, female 3xTg-AD mice were depleted of sex steroid hormones by OVX at age 3 months and then treated immediately with continuous E2 or placebo for a period of 3 months. Efficacy of these hormone manipulations was confirmed directly by measuring serum E2 levels using radioimmunoassay and indirectly using a bioassay of uterine weight. Serum E2 levels were found to be within physiological range (sham OVX, 98 ± 7 pg/ml; OVX, 28 ± 1 pg/ml; OVX+E2, 104 ± 28 pg/ml; F = 4.28, p = 0.04). The OVX group had significantly lower E2 levels than the sham OVX group (p = 0.016), whereas the OVX+E2 group had serum E2 levels significantly higher than the OVX group (p = 0.012) but not significantly greater than the sham OVX group (p = 0.81). Similar results were observed with uterine weights (sham OVX, 86.6 ± 9.6 mg; OVX, 15.5 ± 1.9 mg; OVX+E2, 162.2 ± 31.1 mg; F = 12.86, p < 0.001). OVX-induced hormone depletion caused a significant decrease in uterine weight compared with the sham OVX condition (p = 0.027), and E2 treatment caused a significant increase compared with both the OVX (p < 0.001) and sham OVX (p = 0.01) groups.
Estrogen regulates Aβ accumulation
If E2 beneficially regulates Aβ accumulation, then OVX-induced hormone depletion should increase Aβ accumulation, whereas E2 treatment of OVX animals should prevent this increase. To investigate this hypothesis, female 3xTg-AD mice were assessed for Aβ accumulation at age 6 months, after the 3 month period of hormone manipulation. Consistent with our prediction, we observed that, compared with the sham OVX group, the OVX group exhibited a robust increase in Aβ load in hippocampus CA1, subiculum, and frontal cortex (Fig. 6 B,F,J). E2 treatment in OVX animals partially attenuated the increased Aβ load in subiculum and entirely prevented it in hippocampus CA1 and frontal cortex (Fig. 6 C,G,K). The second measure of Aβ pathology, Aβ plaque number, was not counted in E2-manipulated mice because of the small number of plaques present in 6-month-old animals. To confirm that estrogen regulation of Aβ-IR reflects amyloidogenic species of Aβ rather than CTFs, we quantified and compared CTF-IR in E2-manipulated mice. As expected, we observed that CTF load was not significantly different across the sham OVX, OVX, and OVX+E2 groups (F = 0.36, p = 0.70).
Figure 6.

E2 regulates Aβ accumulation. Aβ-IR was visualized in female 3xTg-AD mice at age 6 months in the sham OVX (A, E, I), OVX (B, F, J), and OVX+E2 (C, G, K) groups in hippocampus CA1 (A–C), subiculum (Sub.; E–G), and frontal cortex (F.Ctx.; I–K), as shown in representative images. Scale bar, 100 μm. Data demonstrate that mean ± SEM Aβ load differed significantly across treatment groups in hippocampus CA1 (F = 4.40, p = 0.028) (D), subiculum (F = 6.77, p = 0.007) (H), and frontal cortex (F = 4.30, p = 0.031) (L). *p < 0.05 relative to OVX group; **p < 0.05 relative to sham OVX group.
Estrogen regulation of tau hyperphosphorylation
To further investigate estrogen regulation of AD-like neuropathology, we also determined the effect of estrogen status on levels of tau hyperphosphorylation by counting the combined number of AT8-immunoreactive neurons in subiculum and hippocampus CA1 of E2-manipulated female 3xTg-AD mice. We found that the number of AT8-immunoreactive neurons was not significantly altered in the OVX group or the OVX+E2 group (F = 2.29, p = 0.13) (Fig. 7).
Figure 7.

E2 regulation of tau hyperphosphorylation. Representative images of AT8-IR (counterstained with cresyl violet) show neurons with abnormal tau phosphorylation in female 3xTg-AD mice at age 6 months in sham OVX (A), OVX (B), and OVX+E2 (C) conditions. Scale bar, 50 μm. D, Quantification of numbers of intensely AT8-immunoreactive cells in hippocampus CA1 and subiculum show that neither OVX nor E2 treatment significantly altered tau phosphorylation (F = 2.29, p = 0.13).
Estrogen effects on spontaneous alternation behavior
Next, we investigated the possibility that E2 regulation of AD-like neuropathology in female 3xTg-AD mice may also affect memory function as measured by SAB performance in the Y-maze test. We found that OVX resulted in a significant decline in SAB performance and that E2 treatment of OVX animals reversed this effect, restoring SAB performance to the level of sham OVX animals (Fig. 8 A). These effects were unrelated to general activity levels because the numbers of arm entries were similar across treatment groups (Fig. 8 B). To confirm that the effects of hormone manipulations on SAB reflected underlying neuropathology rather than direct hormone effects on SAB, we also evaluated the effects of OVX with and without E2 treatment on SAB in WT female mice at age 6 months. There were no significant differences across the sham OVX, OVX, and OVX+E2 groups of WT female mice in SAB performance (Fig. 8 C).
Figure 8.

E2 regulates spontaneous alternation behavior. Estrogen status significantly affected SAB performance in 6-month-old female 3xTg-AD mice but not in age-matched WT female mice. A, Compared with the sham OVX group (Sham) of 3xTg-AD mice, the OVX group exhibited significantly impaired SAB that was prevented in the OVX+E2 group (F = 14.48, p < 0.001). *p < 0.05 from relative to OVX; **p < 0.05 relative to sham. B, The number of arm entries in the task did not significantly differ across 3xTg-AD treatment groups (F = 1.87, p = 0.19). C, In contrast to results in 3xTg-AD mice, there was no significant difference in SAB performance across sham, OVX, and OVX+E2 groups in WT mice (F = 0.94, p = 0.41).
Experiment 3: effect of progesterone on neuropathology in female 3xTg-AD mice
Confirmation of progesterone treatment
To investigate the effects of progesterone on the development of AD-like neuropathology, female 3xTg-AD mice were depleted of endogenous sex steroid hormones by OVX at age 3 months and then treated immediately with either placebo (OVX group) or continuous P4 in the absence (OVX+P4 group) or presence of continuous E2 (OVX+E2+P4 group) for a period of 3 months. Efficacy of P4 treatment was confirmed directly by measuring serum P4 levels using RIA. Effects on uterine weight were also determined. Serum levels of P4 resulting from hormone treatment were within the expected physiological range and significantly greater than observed in gonadally intact female mice (sham OVX, 1.6 ± 1.1 ng/ml; OVX+P4, 7.3 ± 0.5 ng/ml; OVX+E2+P4, 3.9 ± 0.7 ng/ml; F = 4.59, p = 0.003). Evaluation of uterine weights (OVX, 17.1 ± 1.4 mg; OVX+P4, 40.3 ± 5.1 mg; OVX+E2+P4, 108.1 ± 20.4 mg; F = 12.84, p < 0.001) showed that P4 treatment had a mild uterotrophic effect that did not reach statistical significance (p = 0.19), although the E2+P4 treatment had a significant proliferative effect (p < 0.001) compared with OVX mice.
Progesterone attenuates the effect of estrogen on Aβ accumulation
If P4 shares with E2 the ability to reduce Aβ accumulation, then P4 treatment would be predicted to prevent the increase in Aβ accumulation resulting from OVX-induced hormone depletion. However, we observed no significant difference in Aβ load between the OVX and OVX+P4 conditions in hippocampus CA1, subiculum, and frontal cortex (Fig. 9). We also investigated the clinically relevant issue of whether P4 affects the actions of E2 to reduce the OVX-induced increase in Aβ accumulation. We observed that, unlike the Aβ-lowering effect of continuous E2 alone (Fig. 6), combined continuous treatment with E2 and P4 treatment for 3 months did not significantly affect Aβ accumulation in hippocampus CA1, subiculum, or frontal cortex compared with the OVX condition (Fig. 9). There were also no significant differences in CTF load across treatment groups (F = 0.11, p = 0.90).
Figure 9.

P4 attenuates the effects of E2 on Aβ accumulation. Aβ-IR was visualized in female 3xTg-AD mice at age 6 months in the OVX (A, E, I), OVX+P4 (B, F, J), and OVX+E2+P4 (C, G, K) groups in hippocampus CA1 (A–C), subiculum (Sub.; E–G), and frontal cortex (F.Ctx.; I–K), as shown in representative images. Scale bar, 100 μm. Data demonstrate that mean ± SEM Aβ load did not differ significantly across treatment groups in hippocampus CA1 (F = 0.85, p = 0.45) (D), subiculum (F = 2.10, p = 0.15) (H), and frontal cortex (F = 0.58, p = 0.57) (L).
Progesterone regulates tau hyperphosphorylation
We also investigated the effect of P4 treatments on tau hyperphosphorylation by counting the number of AT8-immunoreactive neurons in hippocampus CA1 and subiculum of female 3xTg-AD mice. Interestingly, P4 treatment robustly decreased the number of AT8-immunoreactive neurons compared with the OVX group, an effect that persisted when P4 was delivered in combination with E2 (Fig. 10).
Figure 10.

P4 regulates tau hyperphosphorylation. Representative images of AT8-IR (counterstained with cresyl violet) show neurons with abnormal tau phosphorylation in female 3xTg-AD mice at age 6 months in the OVX (A), OVX+P4 (B), and OVX+E2+P4 (C) conditions. Scale bar, 50 μm. D, Quantification of numbers of intensely AT8-immunoreactive cells in hippocampus CA1 and subiculum show that P4 treatments significantly decreased tau hyperphosphorylation compared with the OVX group (F = 9.71, p = 0.002). *p < 0.05 relative to OVX group.
Progesterone effects on spontaneous alternation behavior
Finally, we investigated whether the P4 treatments affected SAB performance. Compared with the poor SAB performance of OVX 3xTg-AD mice (Fig. 8), we observed that the OVX+E2+P4 group but not the OVX+P4 group showed improved performance (Fig. 11 A). As expected, SAB did not reflect changes in activity level, because the number of arm entries was not significantly different across the 3xTg-AD treatment groups (Fig. 11 B). To ensure that SAB performance was not significantly affected by direct effects of female sex steroid hormones, we evaluated SAB in WT mice in the OVX, OVX+P4, and OVX+E2+P4 conditions and found no significant between group differences (Fig. 11 C).
Figure 11.

P4 regulation of spontaneous alternation behavior. Progesterone treatment did not significantly affect SAB performance in either 6-month-old female 3xTg-AD mice or in age-matched WT female mice. A, Compared with the OVX group of 3xTg-AD mice, P4 significantly improved the impaired SAB performance only when cotreated with E2 (F = 4.36, p = 0.03). *p < 0.05 relative to OVX. B, The number of arm entries in the task did not significantly differ across 3xTg-AD treatment groups (F = 0.37, p = 0.70). C, In WT mice, there were no significant differences in SAB performance across OVX, OVX+P4, and OVX+E2+P4 groups (F = 1.68, p = 0.22).
Discussion
In this study, we investigated the relationship between the sex steroid hormones estrogen and progesterone and the development of AD-like neuropathology in the 3xTg-AD mouse model of AD. Consistent with previous findings (Oddo et al., 2003a), we observed that AD-like pathologies in female 3xTg-AD mice, including Aβ accumulation, tau hyperphosphorylation, and deficits in working memory, appear within a few months of age and progressively worsen. Specifically, intraneuronal Aβ accumulation occurred first, followed by tau hyperphosphorylation, and then impaired SAB performance. Significantly, we found that experimental depletion of endogenous estrogen and progesterone by OVX exacerbated these pathologies, suggesting independent and or combined actions of these hormones in regulating AD-like pathology. Consistent with a protective role of estrogen, we found that E2 treatment in OVX mice prevented the worsening in Aβ accumulation and SAB impairment. However, estrogen status was not associated with significant changes in tau phosphorylation. In contrast, P4 treatment in OVX mice did not affect either Aβ accumulation or SAB, although it did strongly reduce tau hyperphosphorylation. When P4 treatment was combined with E2, the protective effect of E2 on Aβ accumulation was attenuated, but both reduced tau hyperphosphorylation and improved SAB were observed compared with OVX mice. These data, which represent the first report of progesterone effects in an animal model of AD, implicate both separate and combined actions of estrogen and progesterone in regulation of AD-like pathologies.
Our results support the hypothesis that estrogen protects against development of AD-like neuropathology. Similar to our findings, other studies have reported increased Aβ levels after OVX in WT guinea pigs (Petanceska et al., 2000) and in transgenic mouse models of AD (Zheng et al., 2002). Furthermore, E2 treatment has been associated with decreased brain levels of Aβ in guinea pigs (Petanceska et al., 2000) as well as in the APPSWE (mice expressing the Swedish form of amyloid precursor protein) (Levin-Allerhand et al., 2002), Tg2576 (Zheng et al., 2002), and Tg2576×PS1 (transgenic 2576 mice expressing presenilin 1) (Zheng et al., 2002) AD mouse models. In contrast, other studies have reported that neither OVX nor E2 treatment significantly altered Aβ levels in the PDAPP (mice expressing the Parkinson's disease form of amyloid precursor protein) (Green et al., 2005), APPSWE×PS1 (Heikkinen et al., 2004), and APP23 (Yue et al., 2005) transgenic mouse models of AD. It is unclear why estrogen status is associated with Aβ levels in some models but not others. There are several factors that vary between the studies and may contribute to the inconsistent findings. One potentially important difference across studies has been varying methods of Aβ quantification, each of which preferentially detects different pools of Aβ ranging from soluble monomeric Aβ to oligomeric and deposited forms. The effect of estrogen on the various Aβ forms has yet to be determined. In this study, we found that estrogen decreases Aβ as determined by immunoreactive load method, which measures Aβ that accumulates both intracellularly and extracellularly and presumably reflects relatively insoluble Aβ forms. Because oligomeric Aβ is increasingly recognized as pathologically significant (Walsh et al., 2005), we have begun to consider the potential hormone regulation of this Aβ pool. In preliminary slot blot studies using an oligomeric-specific anti-Aβ antibody, we observed elevated levels of oligomeric Aβ in aged female 3xTg-AD mice (our unpublished observations), consistent with a previous report (Oddo et al., 2006a). However, we did not consistently observe oligomeric Aβ in female 3xTg-AD mice at age 6 months and thus were unable to evaluate whether hormonal status affects levels of oligomeric Aβ. Future studies will need to address not only how sex steroid hormones regulate Aβ but also which Aβ pool(s) they regulate.
Beyond investigating estrogen effects, this study is the first to directly examine the effects of progesterone on AD-like neuropathology. The most significant findings from this study are the observations that, although continuous P4 treatment by itself had no effect on Aβ accumulation, P4 attenuated the beneficial effect of E2 treatment on lowering Aβ accumulation in female 3xTg-AD mice. Our finding that P4 lacks independent action but attenuates E2 action is consistent with recent observations in other paradigms. For example, we recently reported that, although P4 treatment in female OVX rats did not affect the extent of kainate-induced neuron loss, it completely blocked E2 neuroprotection (Rosario et al., 2006a). Similarly, continuous P4 treatment has been reported to inhibit E2-mediated increases in neurotrophin expression (Bimonte-Nelson et al., 2004) and spatial memory performance (Bimonte-Nelson et al., 2006). One particularly interesting component of this relationship with hypothesized clinical relevance is the effect of continuous versus cyclic P4 treatment. Previous work by Gibbs (2000) demonstrated that hippocampal cholinergic activity in OVX female rats was increased by cyclic treatment with E2 and P4 but decreased by continuous treatment with E2 and P4. Potential differences between continuous and cyclic P4 treatment in 3xTg-AD mice are currently under study.
In addition to the hormone-mediated effects on Aβ accumulation, we also observed that progesterone significantly regulated tau hyperphosphorylation in female 3xTg-AD mice. Although we observed a statistically nonsignificant trend of reduced tau hyperphosphorylation in the presence of estrogen, we found that P4 treatment significantly decreased tau hyperphosphorylation, reducing AT8-IR to levels even lower than those observed in gonadally intact 3xTg-AD mice. These observations are generally consistent with previous studies demonstrating that E2 and P4 can modulate activities of kinases involved in regulating tau phosphorylation, including glycogen synthase kinase-3β (GSK-3β) (Alvarez-de-la-Rosa et al., 2005; Goodenough et al., 2005). Specifically, E2 has been reported to reduce GSK-3β activity (Goodenough et al., 2005), a kinase that phosphorylates tau sites associated with neuropathology (Ishizawa et al., 2003). Recent data suggest that acute P4 treatment can decrease expression of both tau and GSK-3β in rat cerebellum, but net increases in both GSK-3β activity and tau phosphorylation were also observed (Guerra-Araiza et al., 2007). It is unclear how continuous P4 affects GSK-3β, although our data predict that extended P4 exposure alters the balance of tau-relevant kinase and phosphatase activities to yield a net decrease in pathological tau phosphorylation. Interestingly, previous studies have demonstrated that GSK-3β-dependent regulation of tau hyperphosphorylation is related to behavioral performance in 3xTg-AD mice (Billings et al., 2007), which suggests a potentially important connection between the effects of progesterone on tau hyperphosphorylation and cognition in AD patients.
The results of this study also provide interesting insight into the relationships between Aβ accumulation, tau hyperphosphorylation, and behavioral impairment. In gonadally intact 3xTg-AD females, we found a strong negative association between the progression of Aβ accumulation in hippocampus CA1 and subiculum and performance in the hippocampal-dependent SAB task. In addition, we observed a correlation between E2-mediated changes in Aβ accumulation and behavioral impairment, additional evidence of a cause-and-effect relationship between these two pathologies. However, combined E2+P4 treatment revealed that behavioral impairments in the 3xTg-AD mouse are not simply a function of Aβ load. Combined E2+P4 treatment did not decrease Aβ load yet reduced tau hyperphosphorylation and significantly improved SAB performance, suggesting that the relationship between tau hyperphosphorylation and behavioral impairment is presumably dependent on interactions with Aβ accumulation. For example, despite strongly reducing AT8 immunoreactivity, independent P4 treatment did not significantly affect either Aβ load or SAB performance. These results are consistent with recent observations by Oddo et al. (2006b) that improving behavioral performance in the 3xTg-AD mouse model by Aβ immunotherapy strategies required not only decreasing soluble Aβ levels but also lowering soluble tau levels. Together, these results suggest that behavioral impairment in 3xTg-AD mice involves both Aβ accumulation and tau hyperphosphorylation, pathologies that are differentially affected by E2 and P4 treatments.
In summary, the results of this study provide novel insights into the roles of estrogen and progesterone in regulating AD neuropathology. Although HT use in postmenopausal women can significantly reduce the risk of developing AD (Paganini-Hill and Henderson, 1996; Tang et al., 1996; Kawas et al., 1997; Hogervorst et al., 2000; Zandi et al., 2002; Maki, 2005), the recent WHIMS study has raised several important issues that may significantly affect the efficacy of HT, including the estrogen and progestogen composition of treatment, the timing of treatment initiation, and cyclic versus continuous application of treatment hormones (Brinton, 2004; Craig et al., 2005). Our findings in an animal model of AD support the hypothesis that the loss of sex steroid hormones promotes development of AD neuropathology and that hormone treatments can effectively reduce this effect. Our results are also consistent with the complex, seemingly conflicting clinical data, which indicate both apparent benefits and risks with HT use in women. In the 3xTg-AD mouse, our results indicate that continuous E2 treatment combats Aβ accumulation and behavioral impairments. However, continuous P4 treatment had both a positive effect, reduction of tau hyperphosphorylation, and a negative outcome, attenuation of Aβ lowering by E2. Additional studies are needed to not only elucidate the relationships between estrogen and progesterone interactions but also to optimize the ability of E2 and P4 treatments to reduce AD-like neuropathology. We believe that the continued evaluation of hormone treatments in animal models of AD will yield important insights into the design of future HT regimens that will safely and effectively reduce the development of AD in postmenopausal women.
Footnotes
This work was funded by National Institutes of Health Grants AG026572, AG00093 (J.C.C.), and NS52143 (E.R.R.).
References
- Alkayed NJ, Murphy SJ, Traystman RJ, Hurn PD, Miller VM. Neuroprotective effects of female gonadal steroids in reproductively senescent female rat. Stroke. 2000;31:3041–3046. doi: 10.1161/01.str.31.1.161. [DOI] [PubMed] [Google Scholar]
- Alvarez-de-la-Rosa M, Silva I, Nilsen J, Perez MM, Garcia-Segura LM, Avila J, Naftolin F. Estradiol prevents neural tau hyperphosphorylation characteristic of Alzheimer's disease. Ann NY Acad Sci. 2005;1052:210–224. doi: 10.1196/annals.1347.016. [DOI] [PubMed] [Google Scholar]
- Azcoitia I, Fernandez-Galaz C, Sierra A, Garcia-Segura LM. Gonadal hormones affect neuronal vulnerability to excitotoxin-induced degeneration. J Neurocytol. 1999;28:699–710. doi: 10.1023/a:1007025219044. [DOI] [PubMed] [Google Scholar]
- Billings LM, Green KN, McGaugh JL, LaFerla FM. Learning decreases Aβ*56 and tau pathology and ameliorates behavioral decline in 3xTg-AD mice. J Neurosci. 2007;27:751–761. doi: 10.1523/JNEUROSCI.4800-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bimonte-Nelson HA, Nelson ME, Granholm AC. Progesterone counteracts estrogen-induced increases in neurotrophins in the aged female rat brain. NeuroReport. 2004;15:2659–2663. doi: 10.1097/00001756-200412030-00021. [DOI] [PubMed] [Google Scholar]
- Bimonte-Nelson HA, Francis KR, Umphlet CD, Granholm AC. Progesterone reverses the spatial memory enhancements initiated by tonic and cyclic oestrogen therapy in middle-aged ovariectomized female rats. Eur J Neurosci. 2006;24:229–242. doi: 10.1111/j.1460-9568.2006.04867.x. [DOI] [PubMed] [Google Scholar]
- Brinton RD. Impact of estrogen therapy on Alzheimer's disease: a fork in the road? CNS Drugs. 2004;18:405–422. doi: 10.2165/00023210-200418070-00001. [DOI] [PubMed] [Google Scholar]
- Craig MC, Maki PM, Murphy DG. The Women's Health Initiative Memory Study: findings and implications for treatment. Lancet Neurol. 2005;4:190–194. doi: 10.1016/S1474-4422(05)01016-1. [DOI] [PubMed] [Google Scholar]
- Cummings BJ, Mason AJ, Kim RC, Sheu PC, Anderson AJ. Optimization of techniques for the maximal detection and quantification of Alzheimer's-related neuropathology with digital imaging. Neurobiol Aging. 2002;23:161–170. doi: 10.1016/s0197-4580(01)00316-5. [DOI] [PubMed] [Google Scholar]
- De Nicola AF, Gonzalez SL, Labombarda F, Deniselle MC, Garay L, Guennoun R, Schumacher M. Progesterone treatment of spinal cord injury: effects on receptors, neurotrophins, and myelination. J Mol Neurosci. 2006;28:3–15. doi: 10.1385/jmn:28:1:3. [DOI] [PubMed] [Google Scholar]
- Espeland MA, Rapp SR, Shumaker SA, Brunner R, Manson JE, Sherwin BB, Hsia J, Margolis KL, Hogan PE, Wallace R, Dailey M, Freeman R, Hays J. Conjugated equine estrogens and global cognitive function in postmenopausal women: Women's Health Initiative Memory Study. JAMA. 2004;291:3005–3007. doi: 10.1001/jama.291.24.2959. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. San Diego: Academic; 1997. The mouse brain in stereotaxic coordinates. [Google Scholar]
- Goedert M, Jakes R, Vanmechelen E. Monoclonal antibody AT8 recognizes tau protein phosphorylated at both serine 202 and threonine 205. Neurosci Lett. 1995;189:167–169. doi: 10.1016/0304-3940(95)11484-e. [DOI] [PubMed] [Google Scholar]
- Goodenough S, Schleusner D, Pietrzik C, Skutella T, Behl C. Glycogen synthase kinase 3beta links neuroprotection by 17beta-estradiol to key Alzheimer processes. Neuroscience. 2005;132:581–589. doi: 10.1016/j.neuroscience.2004.12.029. [DOI] [PubMed] [Google Scholar]
- Gibbs RB. Effects of gonadal hormone replacement on measures of basal forebrain cholinergic function. Neuroscience. 2000;101:931–938. doi: 10.1016/s0306-4522(00)00433-4. [DOI] [PubMed] [Google Scholar]
- Green PS, Bales K, Paul S, Bu G. Estrogen therapy fails to alter amyloid deposition in the PDAPP model of Alzheimer's disease. Endocrinology. 2005;146:2774–2781. doi: 10.1210/en.2004-1433. [DOI] [PubMed] [Google Scholar]
- Guerra-Araiza C, Amorim MA, Camacho-Arroyo I, Garcia-Segura LM. Effects of progesterone and its reduced metabolites, dihydroprogesterone and tetrahydroprogesterone, on the expression and phosphorylation of glycogen synthase kinase-3 and the microtubule-associated protein tau in the rat cerebellum. Dev Neurobiol. 2007;67:510–520. doi: 10.1002/dneu.20383. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Heikkinen T, Kalesnykas G, Rissanen A, Tapiola T, Iivonen S, Wang J, Chaudhuri J, Tanila H, Miettinen R, Puolivali J. Estrogen treatment improves spatial learning in APP + PS1 mice but does not affect beta amyloid accumulation and plaque formation. Exp Neurol. 2004;187:105–117. doi: 10.1016/j.expneurol.2004.01.015. [DOI] [PubMed] [Google Scholar]
- Hoffman GE, Moore N, Fiskum G, Murphy AZ. Ovarian steroid modulation of seizure severity and hippocampal cell death after kainic acid treatment. Exp Neurol. 2003;182:124–134. doi: 10.1016/s0014-4886(03)00104-3. [DOI] [PubMed] [Google Scholar]
- Hogervorst E, Williams J, Budge M, Riedel W, Jolles J. The nature of the effect of female gonadal hormone replacement therapy on cognitive function in post-menopausal women: a meta-analysis. Neuroscience. 2000;101:485–512. doi: 10.1016/s0306-4522(00)00410-3. [DOI] [PubMed] [Google Scholar]
- Ishizawa T, Sahara N, Ishiguro K, Kersh J, McGowan E, Lewis J, Hutton M, Dickson DW, Yen SH. Co-localization of glycogen synthase kinase-3 with neurofibrillary tangles and granulovacuolar degeneration in transgenic mice. Am J Pathol. 2003;163:1057–1067. doi: 10.1016/S0002-9440(10)63465-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadish I, Van Groen T. Low levels of estrogen significantly diminish axonal sprouting after entorhinal cortex lesions in the mouse. J Neurosci. 2002;22:4095–4102. doi: 10.1523/JNEUROSCI.22-10-04095.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawas C, Resnick S, Morrison A, Brookmeyer R, Corrada M, Zonderman A, Bacal C, Lingle DD, Metter E. A prospective study of estrogen replacement therapy and the risk of developing Alzheimer's disease: the Baltimore Longitudinal Study of Aging. Neurology. 1997;48:1517–1521. doi: 10.1212/wnl.48.6.1517. [DOI] [PubMed] [Google Scholar]
- King DL, Arendash GW. Behavioral characterization of the Tg2576 transgenic model of Alzheimer's disease through 19 months. Physiol Behav. 2002;75:627–642. doi: 10.1016/s0031-9384(02)00639-x. [DOI] [PubMed] [Google Scholar]
- Levin-Allerhand JA, Lominska CE, Wang J, Smith JD. 17Alpha-estradiol and 17beta-estradiol treatments are effective in lowering cerebral amyloid-beta levels in AbetaPPSWE transgenic mice. J Alzheimers Dis. 2002;4:449–457. doi: 10.3233/jad-2002-4601. [DOI] [PubMed] [Google Scholar]
- Maki PM. A systematic review of clinical trials of hormone therapy on cognitive function: effects of age at initiation and progestin use. Ann NY Acad Sci. 2005;1052:182–197. doi: 10.1196/annals.1347.012. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Kitazawa M, Tseng BP, LaFerla FM. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer's disease. Neurobiol Aging. 2003a;24:1063–1070. doi: 10.1016/j.neurobiolaging.2003.08.012. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003b;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Tran L, Lambert MP, Glabe CG, Klein WL, LaFerla FM. Temporal profile of amyloid-β (Aβ) oligomerization in an in vivo model of Alzheimer disease. J Biol Chem. 2006a;281:1599–1604. doi: 10.1074/jbc.M507892200. [DOI] [PubMed] [Google Scholar]
- Oddo S, Vasilevko V, Caccamo A, Kitazawa M, Cribbs DH, LaFerla FM. Reduction of soluble Abeta and tau, but not soluble Abeta alone, ameliorates cognitive decline in transgenic mice with plaques and tangles. J Biol Chem. 2006b;281:39413–39423. doi: 10.1074/jbc.M608485200. [DOI] [PubMed] [Google Scholar]
- Paganini-Hill A, Henderson VW. Estrogen deficiency and risk of Alzheimer's disease in women. Am J Epidemiol. 1994;140:256–261. doi: 10.1093/oxfordjournals.aje.a117244. [DOI] [PubMed] [Google Scholar]
- Paganini-Hill A, Henderson VW. Estrogen replacement therapy and risk of Alzheimer disease. Arch Intern Med. 1996;156:2213–2217. [PubMed] [Google Scholar]
- Petanceska SS, Nagy V, Frail D, Gandy S. Ovariectomy and 17beta-estradiol modulate the levels of Alzheimer's amyloid beta peptides in brain. Exp Gerontol. 2000;35:1317–1325. doi: 10.1016/s0531-5565(00)00157-1. [DOI] [PubMed] [Google Scholar]
- Pike CJ. Estrogen modulates neuronal Bcl-xL expression and beta-amyloid-induced apoptosis: relevance to Alzheimer's disease. J Neurochem. 1999;72:1552–1563. doi: 10.1046/j.1471-4159.1999.721552.x. [DOI] [PubMed] [Google Scholar]
- Pomp D, Geisert RD, Durham CM, Murray JD. Rescue of pregnancy and maintenance of corpora lutea in infertile transgenic mice expressing an ovine metallothionein 1a-ovine growth hormone fusion gene. Biol Reprod. 1995;52:170–178. doi: 10.1095/biolreprod52.1.170. [DOI] [PubMed] [Google Scholar]
- Rapp SR, Espeland MA, Shumaker SA, Henderson VW, Brunner RL, Manson JE, Gass ML, Stefanick ML, Lane DS, Hays J, Johnson KC, Coker LH, Dailey M, Bowen D. Effect of estrogen plus progestin on global cognitive function in postmenopausal women: the Women's Health Initiative Memory Study: a randomized controlled trial. JAMA. 2003;289:2663–2672. doi: 10.1001/jama.289.20.2663. [DOI] [PubMed] [Google Scholar]
- Roof RL, Duvdevani R, Braswell L, Stein DG. Progesterone facilitates cognitive recovery and reduces secondary neuronal loss caused by cortical contusion injury in male rats. Exp Neurol. 1994;129:64–69. doi: 10.1006/exnr.1994.1147. [DOI] [PubMed] [Google Scholar]
- Rosario ER, Ramsden M, Pike CJ. Progestins inhibit the neuroprotective effects of estrogen in rat hippocampus. Brain Res. 2006a;1099:206–210. doi: 10.1016/j.brainres.2006.03.127. [DOI] [PubMed] [Google Scholar]
- Rosario ER, Carroll JC, Oddo S, LaFerla FM, Pike CJ. Androgens regulate the development of neuropathology in a triple transgenic mouse model of Alzheimer's disease. J Neurosci. 2006b;26:13384–13389. doi: 10.1523/JNEUROSCI.2514-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shultz JM, Zhu XD, Knopp RH, Leboeuf RC, Rosenfeld ME. Norgestimate and medroxyprogesterone acetate do not attenuate the atheroprotective effects of 17beta-estradiol in ovariectomized, apolipoprotein E-deficient mice. Fertil Steril. 2004;82(Suppl 3):1133–1139. doi: 10.1016/j.fertnstert.2004.05.069. [DOI] [PubMed] [Google Scholar]
- Shumaker SA, Legault C, Rapp SR, Thal L, Wallace RB, Ockene JK, Hendrix SL, Jones BN, Assaf AR, Jackson RD, Kotchen JM, Wassertheil-Smoller S, Wactawski-Wende J. Estrogen plus progestin and the incidence of dementia and mild cognitive impairment in postmenopausal women: the Women's Health Initiative Memory Study: a randomized controlled trial. JAMA. 2003;289:2651–2662. doi: 10.1001/jama.289.20.2651. [DOI] [PubMed] [Google Scholar]
- Slater CC, Zhang C, Hodis HN, Mack WJ, Boostanfar R, Shoupe D, Paulson RJ, Stanczyk FZ. Comparison of estrogen and androgen levels after oral estrogen replacement therapy. J Reprod Med. 2001;46:1052–1056. [PubMed] [Google Scholar]
- Tang MX, Jacobs D, Stern Y, Marder K, Schofield P, Gurland B, Andrews H, Mayeux R. Effect of oestrogen during menopause on risk and age at onset of Alzheimer's disease. Lancet. 1996;348:429–432. doi: 10.1016/S0140-6736(96)03356-9. [DOI] [PubMed] [Google Scholar]
- Toung TJ, Chen TY, Littleton-Kearney MT, Hurn PD, Murphy SJ. Effects of combined estrogen and progesterone on brain infarction in reproductively senescent female rats. J Cereb Blood Flow Metab. 2004;24:1160–1166. doi: 10.1097/01.WCB.0000135594.13576.D2. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Shankar GM, Townsend M, Fadeeva JV, Betts V, Podlisny MB, Cleary JP, Ashe KH, Rowan MJ, Selkoe DJ. The role of cell-derived oligomers of Abeta in Alzheimer's disease and avenues for therapeutic intervention. Biochem Soc Trans. 2005;33:1087–1090. doi: 10.1042/BST20051087. [DOI] [PubMed] [Google Scholar]
- West MJ, Slomianka L, Gundersen HJG. Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using optical fractionator. Anat Rec. 1991;231:482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- Yue X, Lu M, Lancaster T, Cao P, Honda S, Staufenbiel M, Harada N, Zhong Z, Shen Y, Li R. Brain estrogen deficiency accelerates Abeta plaque formation in an Alzheimer's disease animal model. Proc Natl Acad Sci USA. 2005;102:19198–19203. doi: 10.1073/pnas.0505203102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandi PP, Carlson MC, Plassman BL, Welsh-Bohmer KA, Mayer LS, Steffens DC, Breitner JC. Hormone replacement therapy and incidence of Alzheimer disease in older women: the Cache County Study. JAMA. 2002;288:2123–2129. doi: 10.1001/jama.288.17.2123. [DOI] [PubMed] [Google Scholar]
- Zheng H, Xu H, Uljon SN, Gross R, Hardy K, Gaynor J, Lafrancois J, Simpkins J, Refolo LM, Petanceska S, Wang R, Duff K. Modulation of A(beta) peptides by estrogen in mouse models. J Neurochem. 2002;80:191–196. doi: 10.1046/j.0022-3042.2001.00690.x. [DOI] [PubMed] [Google Scholar]