

Abstract
Regulator of calcineurin 1 (RCAN1/MCIP1/DSCR1) regulates the calmodulin-dependent phosphatase calcineurin. Because it is located on human chromosome 21, RCAN1 has been postulated to contribute to mental retardation in Down syndrome and has been reported to be associated with neuronal degeneration in Alzheimer's disease. The studies herein are the first to assess the role of RCAN1 in memory and synaptic plasticity by examining the behavioral and electrophysiological properties of RCAN1 knock-out mice. These mice exhibit deficits in spatial learning and memory, reduced associative cued memory, and impaired late-phase long-term potentiation (L-LTP), phenotypes similar to those of transgenic mice with increased calcineurin activity. Consistent with this, the RCAN1 knock-out mice display increased enzymatic calcineurin activity, increased abundance of a cleaved calcineurin fragment, and decreased phosphorylation of the calcineurin substrate dopamine and cAMP-regulated phosphoprotein-32. We propose a model in which RCAN1 plays a positive role in L-LTP and memory by constraining phosphatase signaling.
Keywords: calcineurin, protein phosphatase, mental retardation, hippocampus, learning and memory, synaptic plasticity
Introduction
The regulator of calcineurin proteins (RCANs) are a family of small, highly conserved proteins that can bind and inhibit the calcium-regulated protein phosphatase calcineurin (Fuentes et al., 2000; Kingsbury and Cunningham, 2000; Rothermel et al., 2003). The RCAN1 gene, also known as DSCR1, MCIP1, or calcipressin 1, encodes several protein isoforms through differential promoter usage (Fuentes et al., 1997; Rothermel et al., 2003). Exon 1 (RCAN1.1) and exon 4 (RCAN1.4) are the predominant isoforms. RCAN1 is highly expressed in brain, heart, and skeletal muscle and is elevated in the brains Alzheimer's disease (AD) patients (Ermak et al., 2001). Importantly, the RCAN1 gene is located on the long arm of human chromosome 21, trisomy of which causes Down syndrome.
Calcineurin controls expression of numerous genes, including RCAN1.4 (Yang et al., 2000; Rothermel et al., 2003) via the transcription factor nuclear activator of T-cells (NFAT). Newly synthesized RCAN1.4 protein binds calcineurin, thereby forming a negative regulatory feedback loop (Yang et al., 2000; Rothermel et al., 2003). Although numerous in vitro studies demonstrate that RCAN proteins are potent inhibitors of calcineurin activity, there is substantial controversy regarding their actions in vivo. Genetic studies from fungi and mice suggest that low levels of RCAN proteins may actually be required to facilitate some calcineurin-dependent signals (Kingsbury and Cunningham, 2000; Vega et al., 2003; Hilioti et al., 2004; Fox and Heitman, 2005; Sanna et al., 2006). Therefore, RCANs may both inhibit and facilitate calcineurin signaling depending on cellular and developmental context.
Calcineurin is intimately involved in many facets of synaptic plasticity and memory formation because of its ability to directly dephosphorylate critical targets in both the presynaptic and postsynaptic compartments of neurons (Mansuy, 2003). Calcineurin also can indirectly promote dephosphorylation of many other proteins by increasing protein phosphatase 1 (PP1) activity. Calcineurin accomplishes this through dephosphorylation of the PP1 inhibitors inhibitor-1 (I-1) and dopamine and cAMP-regulated phosphoprotein-32 (DARPP-32). Behavioral and electrophysiological studies using both genetic and pharmacological methods to alter calcineurin activity indicate that calcineurin is likely a negative regulator of learning, memory, and synaptic plasticity (Yakel, 1997; Winder and Sweatt, 2001; Mansuy, 2003). Finally, dysregulation of calcineurin activity is associated with many forms of brain disease and injury, including Alzheimer's disease and excitotoxic ischemia (Klee et al., 1998; Mansuy, 2003).
Because of the importance of calcineurin in regulating diverse neuronal processes, there is growing interest in the potential for trisomy of RCAN1 to contribute to the plethora of phenotypes associated with Down syndrome through its interaction with calcineurin. Thus, understanding how RCAN1 contributes normally to mechanisms of learning and memory formation is critical to understanding its role in the deficits observed in Down syndrome and Alzheimer's patients. We hypothesized that RCAN1 is critical for synaptic plasticity and memory because it serves to maintain a balance between neuronal kinase and phosphatase activities. To test this hypothesis, we compared mice with a disruption of the RCAN1 gene with their wild-type littermates. These experiments provide the first evidence that RCAN1 serves to positively regulate hippocampus-dependent memory and hippocampal late-phase long-term potentiation (L-LTP) via suppression of calcineurin.
Materials and Methods
RCAN1 knock-out mice.
Generation of RCAN1 knock-out mice has been described previously (Vega et al., 2003). Mouse genotyping was performed by PCR using mutant and wild-type specific primer sets (Vega et al., 2003). PCR products were visualized by agarose gel electrophoresis.
In situ hybridization.
A clone specific for RCAN1.4 transcripts was generated from a 160 bp fragment spanning the 3′ untranslated region and coding regions of exon 4 of the mouse RCAN1 gene (Yang et al., 2000). The plasmid was linearized, gel purified, and used as template for [35S]UTP-labeled sense and antisense riboprobe synthesis (Maxiscript T7/T3 kit; Ambion, Austin, TX). Specific activity of both probes was >1000 Ci/mmol. Two-month-old male C57BL/6 mice were killed and perfused with freshly prepared, chilled 4% paraformaldehyde/DEPC–PBS. Brains were removed and fixed further by immersion in 4% paraformaldehyde/DEPC–PBS overnight at 4°C with gentle agitation. Tissues were stored in DEPC–PBS at 4°C until paraffin embedment and sectioning.
In situ hybridizations were performed according to procedures described previously (Shelton et al., 2000). Briefly, 4 μm paraffin sections mounted on microscope slides were dewaxed, permeabilized, and acetylated before hybridization at 70°C. For hybridization, riboprobes were diluted in a mixture containing 50% formamide, 0.75 m NaCl, 20 mm Tris-HCl, pH 8.0, 5 mm EDTA, pH 8.0, 10 mm NaPO4, pH 8.0, 10% dextran sulfate, 1× Denhardt's solution, and 0.5 mg/ml tRNA. After hybridization, the sections were rinsed with increasing stringency washes, subjected to RNase A (2 μg/ml, 30 min at 37°C), and dehydrated before dipping in K5 nuclear emulsion gel (Ilford, Poole, UK). Autoradiographic exposure was performed at 3 weeks.
Behavioral studies.
For the Morris water maze (MWM), male RCAN1 knock-out mice and wild-type littermates were used. The pool had dimensions of 48 inch diameter × 36 inch height. Water in the pool was kept at a constant 21°C. The training for the hidden platform version of the MWM consisted of four trials [60 s maximum; intertrial interval (ITI), 30 min] each day for 8 consecutive days. The probe trial was administered 1 h after the completion of training on day 8. The visible platform task consisted of four trials (ITI, 20 min) each day for 2 consecutive days with the escape platform marked by a visible cue and moved randomly between two locations. The trajectories of the mice were recorded with a video tracking system (HVS Image Analyzing VP-200). Data represent mean ± SEM. ANOVA with repeated measures, genotype as between-group measure, day of testing as repeated measure, were used for statistical analysis with p < 0.05 as significance criteria.
For fear conditioning, RCAN1 knock-out and their wild-type littermates (females and males, 2–4 months old) were used. Data obtained from male and female mice were analyzed separately to determine whether there were gender-specific differences in behavior; none were found and the data were pooled. The training sessions for contextual and cued fear conditioning consisted of a 150 s exploration period followed by two conditioned stimulus–unconditioned stimulus (CS–US) pairings separated by 1 min (footshock intensity, 0.5 mA, 2 s duration; tone, 85 db white noise, 30 s duration). Context tests were performed in the training chamber after 1 and 24 h. Cue tests were performed in an environmentally altered testing chamber either 2 or 24 h after training. Baseline freezing was monitored (3 min) before presentation of the tone (85 db white noise, 3 min duration). For drug injection experiments, mice were injected intraperitoneally with vehicle [30% DMSO, 70% artificial CSF (ACSF)] or vehicle containing drug (5 mg/kg FK506) or (6 μg/kg Calyculin A). Mice were allowed to recover for 60 min before fear conditioning. Effectiveness of the drug injection was determined by phosphatase activity analysis (supplemental Fig. 2a, available at www.jneurosci.org as supplemental material). Data represent mean ± SEM. One-way ANOVA was used for statistical analysis with p < 0.05 as significance criteria.
Electrophysiology.
Transverse hippocampal slices (400 μm) were prepared from age-matched mice (12–24 weeks of age) using conventional techniques. Slices were maintained at 30°C in an interface chamber perfused with oxygenated ACSF containing the following (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 25 d-glucose, 2 CaCl2, and 1 MgCl2 (2 ml/min). When indicated, ACSF was supplemented with FK506 (A.G. Scientific, San Diego, CA) and Calyculin A (Axxora, San Diego, CA). Extracellular field EPSPs (fEPSPs) were evoked by stimulation of Schaeffer collateral pathway afferents and were measured by recording in stratum radiatum of area CA1. Stable baseline synaptic transmission was established for 20–30 min with a stimulus intensity of 40–50% of the maximum fEPSP before LTP-inducing high-frequency stimulation (HFS). Stimulus intensity of the HFS was matched to the intensity used in the baseline recordings. LTP was induced by either one or four trains (5 min intertrain interval) of 100 Hz HFS for 1 s. Data were collected and presented as the average slope of the fEPSP from six individual traces collected over 2 min and then normalized to baseline recordings of fEPSPs. Hippocampal slices from RCAN1 knock-out and wild-type mice were prepared simultaneously and placed in a chamber outfitted with dual-recording equipment, thereby minimizing day-to-day variability in slice preparations and recordings. Wild-type control LTP and RCAN1 knock-out control LTP data were recapitulated in Figure 4a–d. When specified, ACSF was supplemented with either FK506 (10 μm) or Calyculin A (750 nm). One-way ANOVA and post hoc tests were used for electrophysiological data analysis with p < 0.05 as significance criteria.
Figure 4.
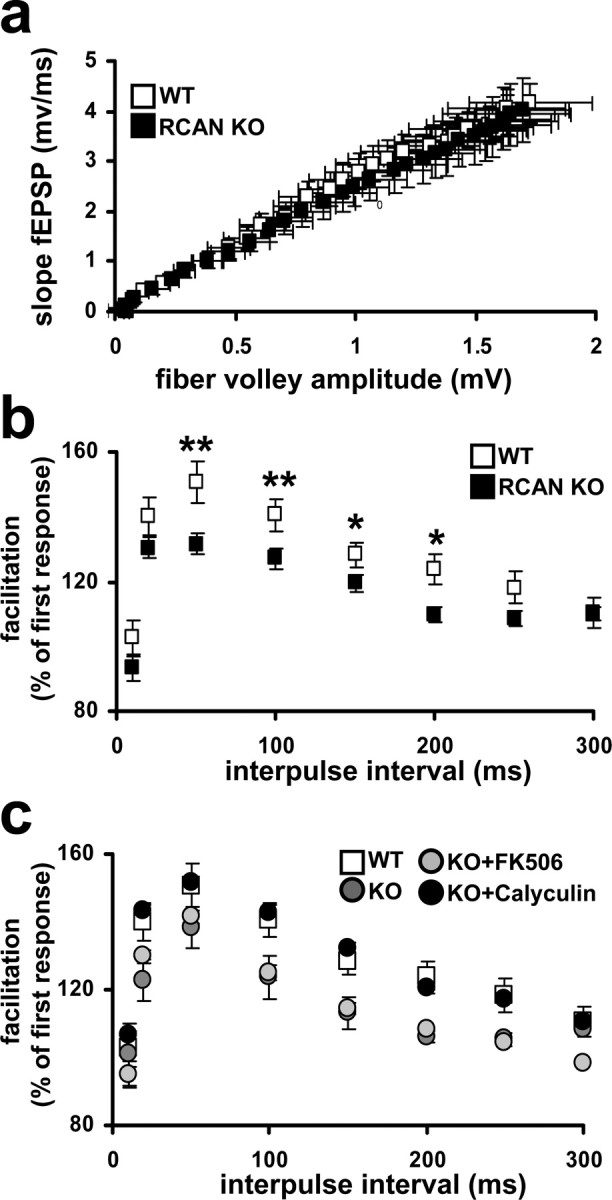
RCAN1 knock-out mice have normal basal synaptic transmission but have deficits in PPF. a, Input versus output plot shows that wild-type and RCAN1 knock-out mice had comparable fEPSP slopes evoked by increasing stimulation. Wild-type (WT) mice, RCAN1 knock-out mice (RCAN KO), n = 24 slices, 16 mice per genotype (p > 0.05, Student's t test). b, RCAN1 knock-out mice had reduced PPF compared with their wild-type littermates. The percentage facilitation, determined by the ratio of the second fEPSP to the first fEPSP, is shown at interpulse intervals from 10 to 300 ms. Wild-type (WT) mice, RCAN1 knock-out mice (RCAN KO) n = 22 slices, 14 mice per genotype (*p < 0.05, **p < 0.01, ANOVA). c, PPF deficits in RCAN1 knock-out mice are rescued by pretreatment with the PP1 inhibitor Calyculin A but not the calcineurin inhibitor FK506. Wild-type (WT) mice, RCAN1 knock-out mice (KO), RCAN1 knock-out mice pretreated with 10 μm FK506 (KO+FK506), RCAN1 knock-out mice pretreated with 750 nm Calyculin A (KO+Calyculin), n = 16 slices, 8 mice per genotype or treatment (KO+Calyculin compared with WT at all time points, p > 0.05, ANOVA).
Immunoblotting.
Soluble protein extracts were prepared by homogenizing the tissue samples in ice-cold buffer [in mm: 50 Tris-HCl, pH 7.5, 150 KCl, 1 DTT, 1 EDTA, 1× complete protease inhibitor cocktail III (Sigma-Aldrich, St. Louis, MO), 1× phosphatase inhibitor cocktail I (Sigma-Aldrich); 1× phosphatase inhibitor cocktail II (Sigma-Aldrich)]. Soluble cytoplasmic and pellet fractions were prepared by ultracentrifugation (100,000 × g for 45 min at 4°C) using a Beckman Coulter (Fullerton, CA) Optima TL ultracentrifuge. Proteins were resolved on SDS-polyacrylamide gels, and immunoblotting was performed using standard techniques. A rabbit polyclonal antiserum raised against RCAN1 that detects both isoforms 1.1. and 1.4 was diluted 1:1000 in 10% milk–TTBS (Tween 20 Tris-buffered saline) and used to detect RCAN isoforms 1.1 and 1.4 (a generous gift from Myogen, Denver, CO) (Bush et al., 2004). Phospho- (Thr-34) DARPP-32 and total DARPP-32 antibodies (Cell Signaling Technologies, Beverly, MA) were diluted 1:1000. Calcineurin A (CnA) and Calcineurin B (CnB) antibodies (Abcam, Cambridge, MA) were diluted 1:1000. β-Actin antibody was diluted 1:5000. Laminin AC (Cell Signaling Technology) and voltage-dependent anion channel (VDAC) antibodies (Novus Biologicals, Littleton, CO) were diluted 1:1000. Anti-rabbit and anti-goat HRP tagged antibodies (Promega, Madison, WI) were diluted 1:5000. All antibodies used were diluted in 0.2% I-Block (Tropix, Foster City, CA). All blots were developed using enhanced chemiluminescence detection (GE Healthcare, Fairfield, CT). The bands of each Western blot were quantified from film exposures (Eastman Kodak, Rochester, NY) in the linear range (determined by multiple exposures) for each antibody and normalized initially by total protein and subsequently via densitometry using a desktop scanner and NIH ImageJ software. Data represent mean ± SEM. A Student's t test was used for Western blot analysis with p < 0.05 as significance criteria.
For fractionation of organelles, hippocampi from three animals were pooled, weighed, and homogenized on ice in 5–10 vol of extraction buffer (in mm: 4 HEPES, pH 7.4, 320 sucrose, 1 DTT, 1 orthovanadate, and 1× complete protease inhibitor cocktail) using a 2 ml Kontes Dounce tissue grinder (A and B pestle, 20 strokes each). The homogenate was centrifuged 3 min at 800 × g, 4°C. The nuclear-enriched pellet (P1) was washed twice with extraction buffer. The supernatant then was centrifuged for 55 min at 17,000 × g, 4°C. The mitochondrial-enriched pellet (P2) was washed twice. The supernatant from P2 contained the soluble cytoplasmic fraction.
Calcineurin activity assay.
Hippocampal tissue was harvested from wild-type and RCAN1 knock-out mice (pooled samples contain at least three whole hippocampi) collected from mice aged (12–24 weeks) at identical times during the day to reduce variability because of possible circadian regulation of calcineurin activity. Soluble protein extracts were prepared by homogenizing the tissue samples in ice-cold lysis buffer containing protein inhibitor cocktail III (Sigma-Aldrich). Soluble cytoplasmic and pellet protein fractions were prepared by ultracentrifugation (100,000 × g for 45 min at 4°C). The pelleted fraction was resuspended in assay buffer. Excess sample phosphate was removed using freshly prepared columns containing Desalting Resin (Bio-Rad, Hercules, CA). Protein extracts were quantified and equal protein amounts were incubated for 30 min after general instructional guidelines for the Biomol Green QuantiZyme Assay System (Biomol, Plymouth, PA). Liberated phosphate was colorimetrically measured using a KC Junior plate reader at λ620. The resulting calcineurin activity was plotted as arbitrary units, and data represented as means ± SEM. Student's t test was used for Western blot analysis with p < 0.05 as significance criteria.
Protein phosphatase-1 activity assay.
Hippocampal lysates were prepared in the same way as for calcineurin assays. Protein extracts were quantified, and equal protein amounts were incubated for 30 min following general instructional guidelines for the EnzCheck Phosphatase Assay kit, using DiFUMP as a substrate (Invitrogen, Carlsbad, CA). Liberated fluorescent phosphatase substrate was measured using a Synergy2 microplate reader using an excitation 360λ and emission 460λ. PP1 activity was determined by subtracting total phosphatase activity from phosphatase activity after 20 min preincubation of lysate with 100 nm nuclear inhibitor of protein phosphatase 1 (EMD Biochemicals, San Diego, CA). The resulting PP1 activity was plotted as fold of wild-type, and data represented as means ± SEM. Single-factor ANOVA analysis was used for analysis with p < 0.05 as significance criteria.
Results
RCAN1.4 is most abundant in areas of the brain in which calcineurin is most abundant
Although it is well established that RCAN1 is highly expressed in the brain, no studies to date have distinguished between the various RCAN1 isoforms. We were specifically interested in determining the pattern of expression for RCAN1.4 because this transcript is under the control of calcineurin via a cluster of NFAT sites in the promoter directly upstream of exon 4 (Yang et al., 2000). By monitoring RCAN1.4 protein levels, we sought to determine in which regions of the brain the RCAN1/calcineurin feedback loop is active. In situ hybridization using an oligonucleotide antisense probe specific for RCAN1.4 revealed that RCAN1.4 mRNA is highly expressed in the mouse brain and is particularly enriched in hippocampal pyramidal neurons (Fig. 1aii,av). A sense probe for the same sequence produced no signal (Fig. 1aiii,avi). Brain sections from wild-type and RCAN1 knock-out mice were examined at different ages during postnatal development. No gross anatomical difference between wild-type and RCAN1 knock-out mice was detected (supplemental Fig. 1a, available at www.jneurosci.org as supplemental material). RCAN1 protein levels were assessed by Western blots using an antibody that detects both the RCAN1.1 and RCAN1.4 isoforms. RCAN1.1 protein was abundant throughout the brain, with highest levels in the cortex and hippocampus (Fig. 1b). RCAN1.4 was less abundant than RCAN1.1 and more variable region to region. RCAN1.4 protein levels were enriched in the hippocampus striatum, cortex, and prefrontal cortex (supplemental Fig. 1b,d, available at www.jneurosci.org as supplemental material). Notably, the levels of RCAN1.4 protein correlated with the level of the catalytic subunit of calcineurin, CnA. Regions of the brain with the highest levels of CnA also contained the highest levels of RCAN1.4 (Fig. 1b). Brain extracts from RCAN1 knock-out mice lacked both RCAN1 isoforms but contained normal levels of CnA, suggesting that, although RCAN1.4 protein levels are dependent on calcineurin, calcineurin levels are independent of RCAN1 (supplemental Fig. 1c, available at www.jneurosci.org as supplemental material).
Figure 1.
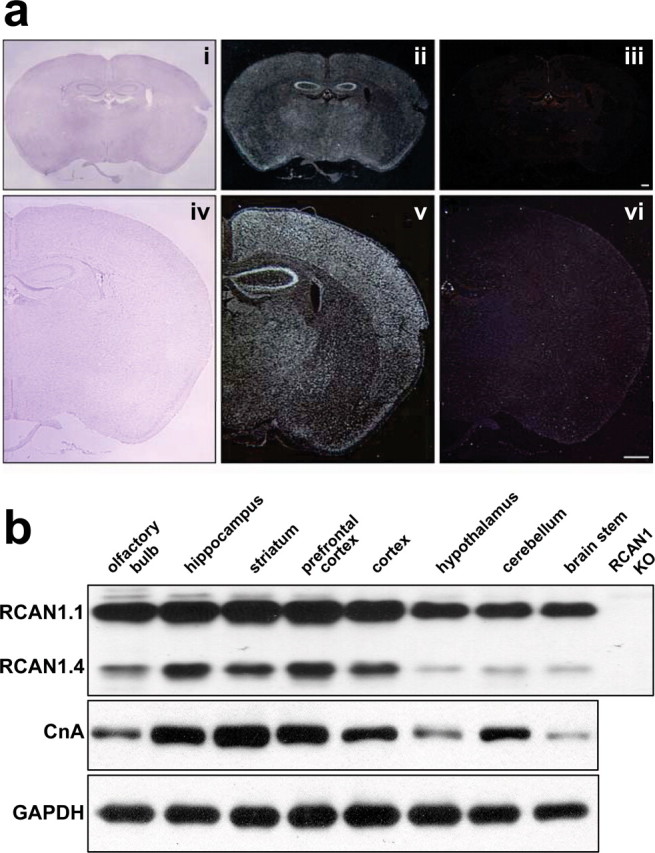
Analysis of RCAN1.4 mRNA and protein levels in the mouse brain. a, In situ hybridization using an antisense and sense RCAN1.4 probes and Nissl staining of sectioned mouse brains. Nissl staining from coronal sections of a wild-type RCAN1 mouse for reference (i, iv). In wild-type mice, an antisense RCAN1.4 riboprobe shows that RCAN1.4 is expressed throughout the cortex and is highly enriched in the hippocampus and cortex (ii, v). A sense probe failed to generate any detectable signal (iii, vi). Hybridization using an antisense RCAN1.4 probe did not generate signal in RCAN1 knock-out mice (data not shown) Scale bar, 300 μm. b, Western blot analysis using antibodies to RCAN1 and calcineurin-A (CnA)-specific antibodies (60 kDa). Both RCAN1.1 (38 kDa, top band) and 1.4 (26 kDa, bottom band) are recognized by the RCAN1 antibody. RCAN1.1 and RCAN1.4 were present in all regions of the mouse brain examined; RCAN1.4 is enriched in tissue in which calcineurin expression is the highest. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 37 kDa) is provided as a loading control. In RCAN1 knock-out (KO) mice, RCAN1.1 and 1.4 are completely absent.
RCAN1 knock-out mice exhibit impaired spatial learning and memory
To test whether RCAN1 is involved in learning and memory, RCAN1 knock-out mice and their wild-type littermates were trained in the hidden platform version of the MWM, which measures the ability to learn the relationship between distal environmental cues and the location of an escape platform obscured by opaque water (Morris, 1984). During the acquisition phase of the task, both RCAN1 knock-out mice and their wild-type littermates exhibited decreasing escape latencies throughout the MWM training, indicating that both groups of mice learned the location of the escape platform. However, escape latencies of the RCAN1 knock-out mice were significantly longer than their wild-type littermates (Fig. 2a). This impairment was not attributable to differences in swim speed or impaired vision, as assessed by visual platform on days 9 and 10 (Fig. 2a). Moreover, RCAN1 knock-out mice performed similar to wild-type littermates in motor performance and learning as determined by the rotating rod test (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). These results indicate that the RCAN1 knock-out mice have a spatial learning impairment rather than a deficit in gross sensorimotor ability.
Figure 2.
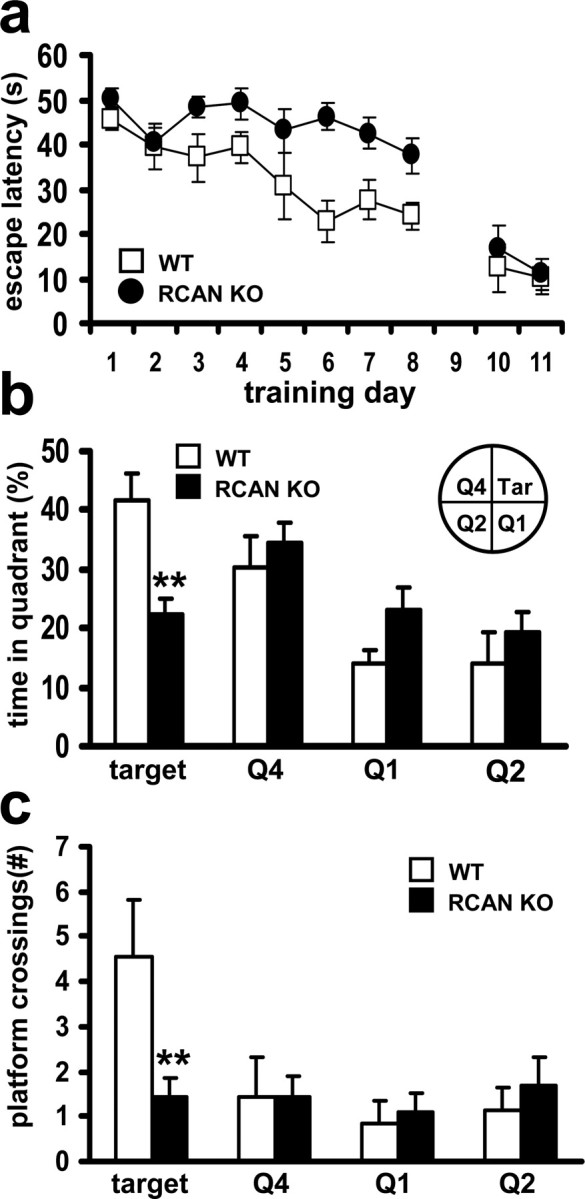
The RCAN1 knock-out mice display impaired spatial learning and memory. a, Escape latency times in hidden platform Morris water maze training. For acquisition (days 1–8), p < 0.01, ANOVA; for visible platform performance (days 10–11), p > 0.05, ANOVA. b, The mean time spent in each maze quadrant (Q) during the probe test (**p < 0.0001, ANOVA). c, The mean number of platform crossings (equivalent location) for each quadrant during probe test. Wild-type (WT) mice, n = 9; RCAN1 knock-out mice (RCAN KO), n = 11 (**p < 0.0001, ANOVA).
To assess spatial memory, the RCAN1 mutant mice were tested in a probe trial in which the escape platform was removed from the maze. In the probe test, RCAN1 knock-out mice did not demonstrate a specific preference for the target quadrant (Fig. 2b). In addition, the RCAN1-deficient mice exhibited a significant reduction in the number of platform crossings when compared with their wild-type littermates (Fig. 2c). The combined results from MWM training and probe testing indicate compromised spatial learning and memory in the mice lacking RCAN1.
RCAN1 knock-out mice display normal contextual fear memory but reduced cued fear memory that is rescued by inhibition of calcineurin
To determine how the loss of RCAN1 affects associative learning and memory, we tested the mice using a conditioned fear paradigm. In this paradigm, mice display memory for environmental context or an emotionally neutral CS (white noise tone) resulting from its temporal association with an aversive US (footshock) through a stereotypical behavioral response known as “freezing” (Phillips and LeDoux, 1992).
For contextual and cued conditioning, RCAN1 knock-out mice and their wild-type littermates were trained with a pair of CS–US stimulations during exploration of a new environment (context). RCAN1-deficient mice exhibited comparable performance in both short-term memory (STM) and long-term memory (LTM) for the training context (Fig. 3a), which suggests that RCAN1-deficient mice have normal associative learning and unaltered contextual fear memory. In contrast to contextual conditioning, the RCAN1 knock-out mice demonstrated a deficit in both short- and long-term cued memory (Fig. 3b). The memory impairment was more pronounced during STM testing but was still evident 24 h later during LTM testing (Fig. 3b). Because RCAN1 is a potent inhibitor of calcineurin activity, we examined fear memory after pretraining with the calcineurin inhibitor FK506. Injection of FK506 60 min before fear conditioning rescued the deficits in both STM and LTM observed in RCAN1 mutant mice, returning the levels of freezing to those observed in wild-type mice (Fig. 3c). Because calcineurin can regulate PP1 via phosphorylation of either DARPP or I-1, we also examined whether pretraining PP1 blockade would also rescue the memory deficits observed in RCAN1 knock-out mice. In contrast, injection of the PP1/protein phosphatase 2A (PP2A) inhibitor Calyculin A did not rescue either the STM or LTM deficits displayed by the RCAN1 mutant mice (Fig. 3c). Interestingly, injection of Calyculin A significantly reduced LTM for cued conditioning in wild-type mice (Fig. 3c). The effectiveness of the drugs to inhibit phosphatase activity in vivo was confirmed by phosphatase activity assays from hippocampal extracts (supplemental Fig. 2a, available at www.jneurosci.org as supplemental material). These results indicate that RCAN1 plays a role in associative fear memory. Moreover, the impairments in fear memory caused by the deletion of RCAN1 can be ameliorated by inhibiting calcineurin activity, which suggests that the memory deficits observed in RCAN1 mutant mice are not exclusively developmental in nature. Altogether, these findings indicate that RCAN1 is required for the formation and consolidation of some forms of memory but is dispensable for others.
Figure 3.
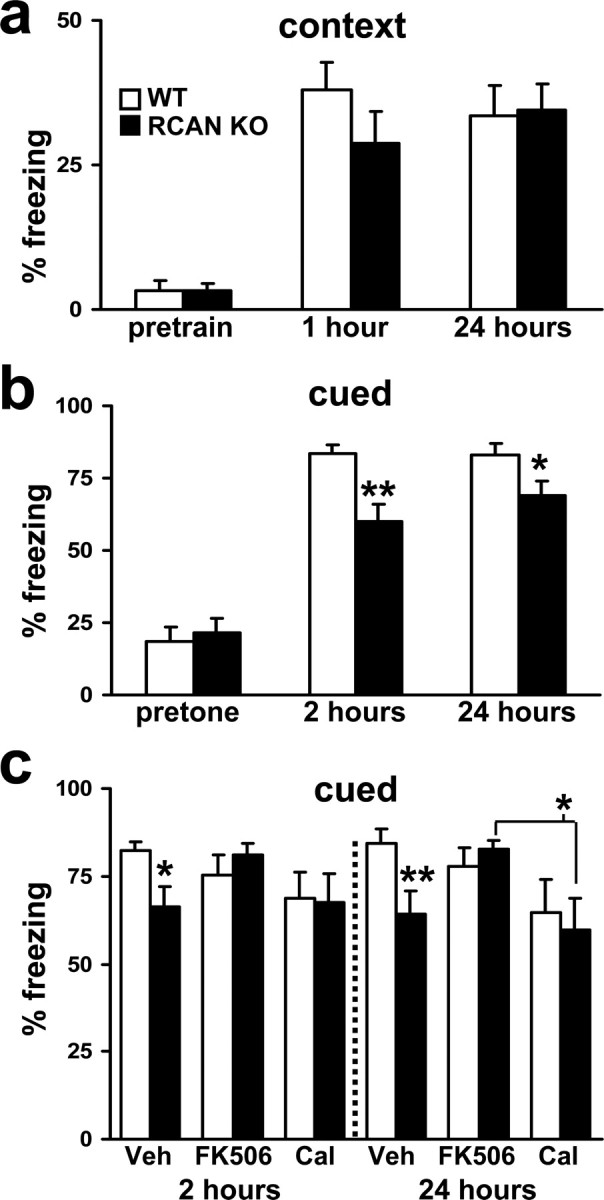
RCAN1 knock-out mice display cued associative fear memory deficits. a, Mean freezing behavior of wild-type and RCAN1 knock-out mice. Both genotypes performed similarly during training and had similar freezing behavior in both short-term (1 h) and long-term (24 h) contextual memory tests (p > 0.05, ANOVA). b, RCAN1 knock-out mice displayed impaired and short-term (2 h) and long-term (24 h) cued fear memory but had similar levels of pretone freezing (p > 0.05, ANOVA). Wild-type (WT) mice, n = 22; RCAN1 knock-out mice (RCAN KO), n = 16 (*p < 0.05, **p < 0.01, ANOVA). c, Inhibition of calcineurin rescues deficits in both STM and LTM for cued conditioning in the RCAN1 knock-out mice. Vehicle-injected (Veh): wild-type (WT) mice, n = 10; RCAN1 knock-out mice (RCAN KO), n = 10. FK506-injected (FK506): WT, n = 9; RCAN KO n = 8. Calyculin A-injected (Cal): WT, n = 8; RCAN KO, n = 8 (*p < 0.05, **p < 0.01, ANOVA).
Deletion of RCAN1 does not alter basal synaptic transmission but blunts paired-pulse facilitation in hippocampal area CA1
We next investigated synaptic plasticity at the Schaffer collateral–CA1 synapse in hippocampal slices from RCAN1 mutant mice. Because calcineurin can influence presynaptic plasticity via regulation of synaptic vesicle release and uptake (Wang and Kelly, 1997), it is possible that the loss of RCAN1 affects basal synaptic function. We found that basal synaptic transmission, measured as synaptic output in response to a stimulatory input, was normal in RCAN1 knock-out mice (Fig. 4a). We next examined whether RCAN1 was required for normal paired-pulse facilitation (PPF), a form of presynaptic plasticity evoked by two temporally linked stimuli (Katz and Miledi, 1968). PPF was reduced at several time intervals in the RCAN1-deficient mice compared with wild-type littermates (Fig. 4b). This reduction in PPF was similar to that observed in a previous study examining the role of calcineurin in hippocampal synaptic plasticity (Winder et al., 1998). In this report, overexpression of a constitutively active form of the calcineurin catalytic subunit resulted in alterations in synaptic plasticity, some of which could be rescued via pharmacological blockade of phosphatase activity with Calyculin A. To assess whether the alterations in PPF observed in the RCAN1 knock-out mice occur through similar mechanisms, we attempted to rescue the reduction in PPF with either Calyculin A or FK506. We found that Calyculin A reversed the reduced PPF in RCAN1-deficient mice (Fig. 4c). Direct inhibition of calcineurin activity with FK506 failed to rescue the PPF phenotype in the RCAN1 mutant mice (Fig. 4c). The effectiveness of the drugs to inhibit phosphatase activity in slices was confirmed by phosphatase activity assays from extracts obtained from hippocampal slices (supplemental Fig. 2b, available at www.jneurosci.org as supplemental material). The short-term plasticity deficits exhibited by the RCAN1 knock-out mice appear to be insensitive to pharmacological inhibition of calcineurin activity, but they may be acutely rescued by an inhibitor of downstream phosphatases regulated by calcineurin through DARPP-32 dephosphorylation.
RCAN1 knock-out mice exhibit impaired L-LTP in hippocampal area CA1
LTP can be divided into at least two components: early-phase (E-LTP) and L-LTP. A single 100 Hz train of HFS elicits E-LTP, an enhancement of synaptic transmission that requires neither protein kinase A (PKA) activity nor new RNA and protein synthesis. The more robust and stable L-LTP that can be elicited by four trains of HFS requires PKA activity and both RNA and protein synthesis (Huang et al., 1996). Previous studies using pharmacological or genetic manipulation of calcineurin activity have shown variable effects on E-LTP (Winder et al., 1998; Zhuo et al., 1999; Malleret et al., 2001; Zeng et al., 2001). However, it is generally accepted that calcineurin acts to gate the transition of E-LTP to more enduring forms such as L-LTP that are dependent on PKA (Wang and Kelly, 1996; Winder et al., 1998; Malleret et al., 2001).
First, we examined the effects RCAN1 deletion on E-LTP induced with one train of HFS. This pattern of stimulation elicited E-LTP in RCAN1 knock-out mice indistinguishable from that in wild-type littermates (Fig. 5a). To test whether the loss of RCAN1 altered L-LTP, we stimulated hippocampal slices with four spaced trains of HFS. L-LTP in the RCAN1-deficient mice was significantly impaired, in both the initial amplitude and the overall duration of potentiation, when compared with their wild-type littermates (Fig. 5b). This result also is consistent with findings from studies of L-LTP in mice with increased expression of constitutively active calcineurin (Winder et al., 1998). We next investigated whether the observed LTP deficit could be rescued by pharmacological blockade of protein phosphatases. Hippocampal slices were preincubated with either FK506 or Calyculin A, and then L-LTP was elicited using four trains of HFS. As with PPF, we found that the calcineurin-specific inhibitor FK506 failed to rescue the L-LTP impairments in the RCAN1 knock-out mice (Fig. 5c). Furthermore, FK506 also impaired L-LTP in wild-type mice (Fig. 5c). Conversely, Calyculin A pretreatment restored L-LTP in the RCAN1 knock-out mice to levels of potentiation observed in wild-type mice. In contrast to its effect on LTP in slices from RCAN1 knock-out mice, Calyculin A produced L-LTP deficits in wild-type slices similar to those observed in FK506-treated slices (Fig. 5d). Reduced L-LTP in wild-type hippocampal slices by FK506 and Calyculin A was not entirely unexpected, because a reduction in L-LTP has been observed in previous studies using calcineurin and PP1 inhibitors (Lu et al., 1996; Winder et al., 1998). Together, our findings closely resemble the results obtained from transgenic mice with increased calcineurin activity. This implies that RCAN1 facilitates long-lasting synaptic plasticity through inhibition of calcineurin.
Figure 5.

RCAN1 knock-out mice express normal E-LTP but have deficits in L-LTP. a, A single train of HFS (100 Hz for 1 s) evoked similar levels of E-LTP in wild-type and RCAN1 knock-out mice that decayed to baseline levels after 80 min. Wild-type (WT) mice, RCAN1 knock-out mice (RCAN KO), n = 14 slices, 9 mice per genotype (p > 0.05, ANOVA). b, Four trains of HFS (100 Hz trains for 1 s, intertrain interval of 5 min) evoked L-LTP in wild-type mice that persisted at least 3 h but did not evoke L-LTP in the RCAN1 knock-out mice. Wild-type (WT) mice, RCAN1 knock-out mice (RCAN KO), n = 18 slices, 11 animals per genotype (p < 0.001, ANOVA). c, The L-LTP deficit in RCAN1 knock-out mice was not rescued by pretreatment with the calcineurin inhibitor FK506. Pretreatment with FK506 also impaired L-LTP in wild-type slices. Wild-type (WT) mice, RCAN1 knock-out mice (RCAN KO), wild-type mice pretreated with 10 μm FK506 (WT+FK506), RCAN1 knock-out mice pretreated with 10 μm FK506 (RCAN KO+FK506), n = 12 slices, 7 mice per genotype or treatment (p < 0.01, WT vs WT + FK506; p > 0.05, WT+FK506 vs RCAN KO; ANOVA). d, The L-LTP deficit in RCAN1 knock-out mice was rescued by pretreatment with the protein phosphatase-1 inhibitor Calyculin A. Pretreatment with Calyculin A also resulted in impaired L-LTP in wild-type mice. Wild-type (WT) mice, RCAN1 knock-out mice (RCAN KO), wild-type mice pretreated with 750 nm Calyculin A (WT+Cal), RCAN1 knock-out mice pretreated with 750 nm Calyculin A (RCAN KO+Cal), n = 16 slices, 10 animals per genotype or treatment (p < 0.001, WT vs WT+Cal; p > 0.05, WT vs RCAN KO+Cal; ANOVA). Veh, Vehicle.
Specific pools of calcineurin are more active in the hippocampus of RCAN1 knock-out mice
The preceding behavioral and electrophysiological studies suggest that calcineurin activity is increased in the hippocampus of RCAN1 knock-out mice. To assess whether the capacity for calcineurin signaling is altered in RCAN1 mutant mice, we used enzymatic assays to measure calcineurin activity in extracts made from hippocampal slices. This assay was performed at saturating levels of calcium and calmodulin to fully activate all of the calcineurin in the extract. Thus, this assay is a measure of maximal potential activity in the tissue rather than the actual in vivo activity. We found an increase in hippocampal calcineurin activity in total cell lysates from RCAN1 knock-out animals compared with wild-type mice (Fig. 6a). Because calcineurin can be localized to specific subcellular domains, we performed fractionation studies in which organelles and membranes were separated from cytoplasm by centrifugation. We found that soluble cytoplasmic calcineurin activity was not significantly different in the RCAN1-deficient mice compared with wild-type mice (Fig. 6a). In contrast, calcineurin activity was elevated in the pellet fraction of lysates from RCAN1 knock-outs. This finding supports a model in which RCAN1 primarily functions as an inhibitor of calcineurin activity in the hippocampus. Moreover, it suggests that the action of RCAN1 may be selective for specific subcellular pools of calcineurin.
Figure 6.

RCAN1 knock-out mice exhibit elevated calcineurin activity. a, Calcineurin activity is upregulated in hippocampal extracts from RCAN1 knock-out mice compared with wild-type mice. Total lysate, n = 3 both genotypes (**p < 0.01, Student's t test); crude pellet fraction (nuclear/mitochondrial enriched), n = 9 both genotypes (**p < 0.01, Student's t test); soluble fraction (cytoplasmic), n = 11 both genotypes (p > 0.05, Student's t test). b, Western blot analysis of calcineurin isoforms in hippocampal extracts. There was no difference in calcineurin levels between wild-type and RCAN1 knock-out mice with respect to the 60 kDa isoform. A 48 kDa calcineurin isoform was present in greater levels in crude pellet (nuclear/mitochondrial enriched) extracts (represented as percentage of wild-type soluble cytoplasmic levels for each isoform). n = 9, each genotype, wild-type (WT) mice, RCAN1 knock-out mice (RCAN KO) (*p < 0.05, ANOVA). c, RCAN1.1 and 1.4 are absent from the pellet and soluble fractions of RCAN1 knock-out mice after crude lysate fractionation. Mitochondrial (VDAC) and nuclear [Laminin AC (Lamin A/C)] proteins are both present in the crude pellet fraction. d, Organelle fractionation of hippocampi from wild-type mice: soluble cytoplasmic fraction (S), mitochondrial-enriched fraction (M), and nuclear-enriched fraction (N). CnA, RCAN1.1, and RCAN1.4 were found in both the soluble and mitochondrial-enriched fractions. The nuclear protein Laminin AC and the mitochondrial protein VDAC were used to confirm the purity of the fractionation. Extracts in C were isolated without phosphatase inhibitors so that calcineurin activity could be measured. Extracts in D were isolated with phosphatase inhibitors, so phosphorylation of RCAN1.4 was preserved, resulting in the observed doublet.
We proceeded to determine whether the increase in calcineurin activity in the RCAN1 knock-out mice was a result of changes in either the abundance or subcellular location of calcineurin protein with Western blot analysis. The full-length, 60 kDa CnA subunit was present in both the soluble cytosolic and pellet (containing both mitochondrial and nuclear compartment) fractions at comparable levels in wild-type and RCAN1 knock-out mice (supplemental Fig. 1c, available at www.jneurosci.org as supplemental material) (Fig. 6b). Therefore, the elevation in calcineurin activity in the hippocampus from RCAN1 mutant mice is not because of a corresponding change in the abundance of the catalytic subunit of calcineurin but more likely attributable to a loss of inhibition by RCAN1. We also noticed an increase in a 48 kDa constitutively active fragment of CnA that can be generated by calpain cleavage (Wu et al., 2004; Shioda et al., 2006) was present in the hippocampus of RCAN1 mutant mice. No difference in the abundance of the 48 kDa fragment of CnA was observed in the soluble supernatant fraction between RCAN1 mutant mice and wild-type mice. However, in the pellet fraction, we observed increased levels of the 48 kDa isoform in the RCAN1-deficient mice (Fig. 6b). The increased abundance of the 48 kDa isoform in RCAN1 knock-out mice does suggest that, in addition to inhibiting calcineurin activity, RCAN1 may also help protect full-length CnA from cleavage by calpain. Although RCAN1 protein was present in both the supernatant and pellet fractions from wild-type mice (Fig. 6c), calcineurin activity and CnA cleavage was increased only in the pelleted fraction, which is enriched for nuclei and mitochondria (Fig. 6b). We further fractionated these subcellular compartments using differential centrifugation of whole hippocampus pooled from several animals to assess the location of RCAN1 and calcineurin proteins. The purity of the fractionation was verified using antibodies for nuclear-specific Laminin AC and mitochondrial-specific VDAC (Fig. 6d). CnA was present in both the soluble and mitochondrial-enriched fractions but was absent from nuclei. RCAN1.1 and RCAN1.4 shared the same pattern of distribution. These results demonstrate that RCAN1 and calcineurin proteins are available to interact in either the cytoplasm or mitochondria of the hippocampus. The lack of either calcineurin or RCAN1 in the nuclear-enriched fraction in Figure 6d suggests that the elevated calcineurin activity measured in Figure 6a is primarily from calcineurin localized in mitochondria. Previous studies have shown that both calcineurin (Shibasaki et al., 1997) and RCAN (Chang and Min, 2005) proteins can localize to mitochondria. Furthermore, substantial experimental data indicate that mitochondrial activities can influence synaptic plasticity (Ben-Shachar and Laifenfeld, 2004; Jonas, 2006; Mattson, 2006). Whether RCAN1 and calcineurin localized to mitochondria are involved in this process remains to be determined.
Phosphorylation of DARPP-32 is decreased in the hippocampus of RCAN1-deficient mice
Calcineurin activity promotes dephosphorylation of a number of proteins important for synaptic plasticity either directly or indirectly by releasing repression of PP1 activity (Yakel, 1997; Klee et al., 1998; Snyder et al., 1998). We examined the phosphorylation state of a direct target of calcineurin, DARPP-32. PKA-phosphorylated forms of DARPP-32 and the closely related I-1 inhibit PP1 activity. Calcineurin dephosphorylates both DARPP-32 and I-1, releasing repression of PP1 and promoting dephosphorylation of PP1 substrates (Hemmings et al., 1984; Snyder et al., 1998; Bibb et al., 1999). Because we detected elevated CnA activity in hippocampal lysates from RCAN1 knock-out mice, we posited that PP1 activity would also be elevated in RCAN1 knock-out mice. Indeed, PP1 activity was elevated in hippocampal lysates from the RCAN1 mutant mice (Fig. 7a). In preparations in which cytoplasmic, mitochondrial, and nuclear compartments were isolated, elevated PP1 activity was detected primarily in the mitochondrial fraction (Fig. 7a). This finding is consistent with the levels of calcineurin activity in the RCAN1 knock-out mice after crude fractionation (Fig. 6a). We then separated hippocampal lysates into crude supernatant and pellet (mixture of cytoplasmic/nuclear) fractions and then quantified total and phosphorylated DARPP-32. Total DARPP-32 protein levels were similar in RCAN1 knock-out and wild-type lysates. In contrast, there was a significant reduction in the level of DARPP-32 phosphorylated at threonine-34, the calcineurin dephosphorylation site, in both the soluble and crude pellet fractions of hippocampal lysates from RCAN1 mutant mice (Fig. 7). The decreased DARPP-32 phosphorylation is consistent with an increase in calcineurin activity and a derepression of PP1 activity in the hippocampus of the RCAN1-deficient mice. Repression of this downstream elevation in PP1 activity may be the mechanism through which Calyculin A rescues the L-LTP impairment in the RCAN1 knock-out mice (Fig. 5d).
Figure 7.
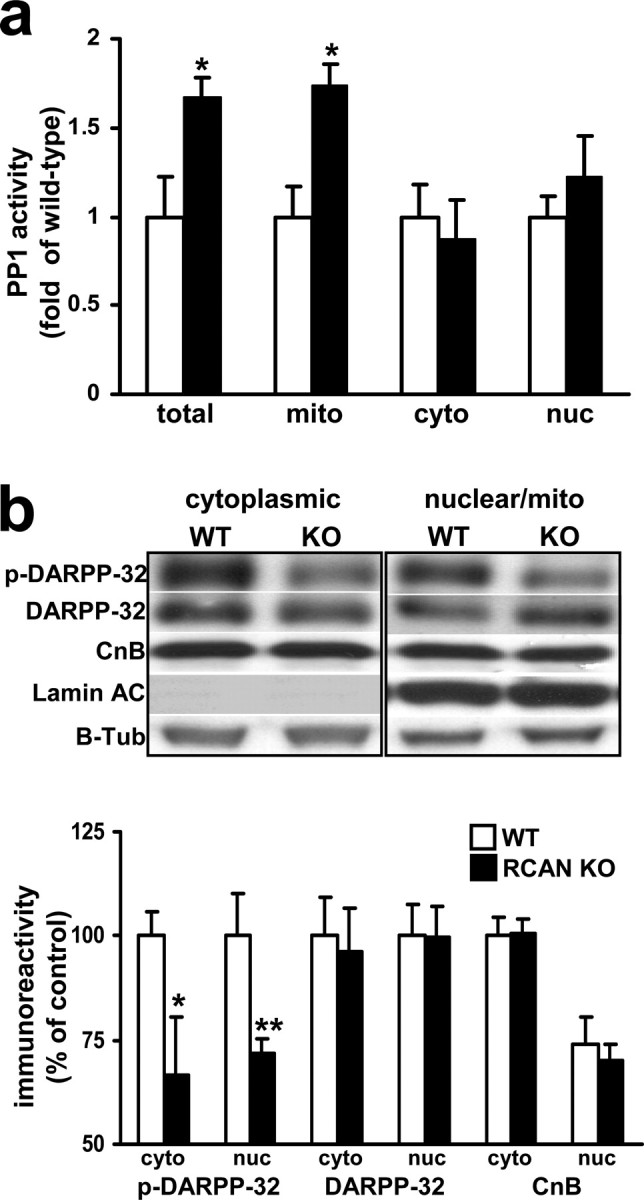
RCAN1 knock-out mice exhibit elevated PP1 activity and decreased DARPP-32 phosphorylation. a, PP1 activity is upregulated in hippocampal extracts from RCAN1 knock-out mice compared with their wild-type littermates. Total lysate (tot), n = 5 for both genotypes (*p < 0.05); mitochondrial fraction (mito), n = 5 for both genotypes (*p < 0.05); cytoplasmic soluble fraction (cyto), n = 4 both genotypes [p > 0.05, nuclear (nuc), n = 4 both genotypes, Student's t test]. b, DARPP-32 phosphorylation is decreased in RCAN1 knock-out mice. Western blot analyses from crude hippocampal pellet extracts (enriched for the nuclear and cytoplasmic compartments). Proteins examined are involved in calcineurin regulation or are downstream targets of calcineurin activity. p-DARPP-32, DARPP phosphorylated at Thr-34; DARPP, total DARPP; CnB, calcineurin regulatory subunit; Lamin AC, nuclear Laminin protein AC; B-Tub, β-tubulin. n = 9, each genotype, wild-type (WT), RCAN1 knock-out mice (RCAN KO) (*p < 0.05, **p < 0.01, cytoplasm; p < 0.01, nuclear; ANOVA).
Discussion
In this study, we used mice with a genetic disruption of the RCAN1 locus to assess the role of RCAN1 in learning and memory. RCAN1 knock-out mice lack expression of all known RCAN1 transcripts and their encoded proteins, including the RCAN1.1 and RCAN1.4 isoforms that are abundant in the hippocampus. We found that the RCAN1 knock-out mice displayed deficits in learning and memory for several tasks, as well as hippocampal L-LTP. Biochemical analysis of hippocampal lysates from the RCAN1 knock-out mice revealed that they have increased enzymatic activity of both calcineurin and PP1 and decreased phosphorylation of the calcineurin substrate DARPP-32, consistent with an elevation in calcineurin activity in the hippocampus of RCAN1 knock-out mice. The findings presented here represent, to the best of our knowledge, the first studies demonstrating a critical role for RCAN1 in the proper manifestation of memory and L-LTP.
We found that the RCAN1 knock-out mice have pronounced spatial learning and memory deficits in the MWM task (Fig. 2) and STM and LTM impairments after associative cued fear conditioning (Fig. 3). These deficits were similar to those found in mice with inducible, hippocampal-restricted overexpression of constitutively active calcineurin (Mansuy et al., 1998a,b) and the direct opposite of learning behaviors of animals in which calcineurin was inhibited by either transgenic expression of a calcineurin inhibitory domain or application of antisense oligonucleotides (Ikegami and Inokuchi, 2000; Malleret et al., 2001). This suggests that RCAN1 provides a constraint on calcineurin activity during learning and memory and that this constraint is absent in the RCAN1 knock-out mice. Importantly, the cued fear memory deficit displayed by RCAN1 knock-out mice was rescued by acutely inhibiting calcineurin. This finding argues against a developmental defect as the primary cause of the learning and memory deficits displayed by the RCAN1 knock-out mice. Interestingly, the RCAN1-deficient mice did not exhibit alterations in contextual fear memory (Fig. 3a). Inducible expression of constitutively active calcineurin produced similar results (Mansuy et al., 1998a,b), arguing that calcineurin activation does not influence contextual fear memory. Together, these findings suggest that proper calcineurin/RCAN1 interactions are required for multiple types of learning and memory.
Synaptic plasticity is thought to underlie the acquisition and storage of information (Martin et al., 2000) and therefore is often used as a cellular model of learning and memory. The RCAN1 knock-out mice exhibited deficits in specific forms of short- and long-term synaptic plasticity. PPF, a form of short-term plasticity dependent on calcineurin activity (Wang and Kelly, 1997), was reduced in RCAN1 knock-out mice (Fig. 4c). However, RCAN1 apparently is not required for the proper manifestation of all forms of short-term plasticity. E-LTP, a form of LTP that does not depend on either new PKA activity or protein synthesis (Huang et al., 1996), was normal in the RCAN1 knock-out mice (Fig. 5a) similar to transgenic mice that overexpress a constitutively active calcineurin (Winder et al., 1998). These findings further support the concept that RCAN1 and calcineurin are involved in some, but not all, functional aspects of synaptic plasticity.
The RCAN1 knock-out mice also displayed significant deficits in L-LTP, a form of synaptic plasticity dependent on PKA, de novo gene expression, and protein translation (Huang et al., 1996; Abel et al., 1997) (Fig. 5b), corroborating studies demonstrating L-LTP deficits in mice that overexpress a constitutively active calcineurin (Winder et al., 1998) and enhancement of LTP acquisition when calcineurin is inhibited (Ikegami and Inokuchi, 2000; Malleret et al., 2001). Our findings are consistent with a model in which RCAN1 constrains calcineurin activity to promote L-LTP. Whether newly synthesized RCAN1 proteins or existing pools of RCAN1 perform these functions will require additional investigation, as will the relative contributions of RCAN1.1 and RCAN1.4 to L-LTP.
It is generally accepted that kinase activities promote synaptic plasticity, whereas phosphatases inhibit synaptic plasticity. The ability of Calyculin A, a PP1/PP2A inhibitor, to rescue both the PPF and L-LTP deficits is consistent with the idea that the RCAN1 knock-out mice have elevated phosphatase activities and argues against a purely developmental defect as the cause of the deficits we observe in the RCAN1-deficient mouse. Decreased phosphorylation of the PP1 inhibitor DARPP-32, a calcineurin substrate (Fig. 7), suggests that loss of RCAN1 can lead to both a direct elevation in calcineurin activity and an indirect derepression of PP1 activity.
In contrast to our cued memory experiments, FK506, a direct calcineurin inhibitor, failed to rescue the PPF and L-LTP deficits. Furthermore, FK506 treatment actually blocked L-LTP in wild-type mice (Fig. 5c). This may simply be attributable to differences in phosphatase signaling in specific brain regions. Cued fear memory is primarily an amygdala-dependent task, whereas our synaptic plasticity studies were performed in area CA1 of the hippocampus. Studies using pharmacological inhibitors of calcineurin (FK506 and cyclosporine A) have produced a spectrum of effects on both short- and long-term plasticity (Wang and Stelzer, 1994; Lu et al., 1996; Wang and Kelly, 1996; Ikegami and Inokuchi, 2000).
The failure of FK506 to rescue the synaptic plasticity deficits in RCAN1 mutant mice may be because the drug concentration was insufficient or had secondary effects unrelated to calcineurin activity. However, we propose two alternative explanations that are more intriguing. One is that RCAN1 functions are not limited to calcineurin inhibition. Another possibility is that RCAN1 does not have the same effect on all cellular pools of calcineurin. This is supported by our findings that the enzymatic activity of calcineurin and the 48 kDa isoform were elevated only in the pellet fraction in hippocampal extracts from RCAN1 knock-out mice (Fig. 6). Calcineurin has numerous intracellular targets in addition to DARPP-32 (Fig. 8). Which of these targets are influenced by the presence of RCAN1 is still unknown. Inhibition by FK506 presumably would act on the entire cellular pool of calcineurin. Mice with a forebrain-specific knock-out of the CnB regulatory subunit have impaired LTP, working memory deficits, and a generalized perturbation of brain functions (Zeng et al., 2001; Miyakawa et al., 2003), suggesting that, although calcineurin negatively modulates learning, memory, and synaptic plasticity, complete loss of calcineurin activity is not beneficial for cognitive function. It seems likely that there are mechanisms in place to maintain the proper balance in kinase and phosphatase activities at the synapse. Our studies suggest that RCAN1 is an important component of this regulatory network and acts by placing a constraint on calcineurin activity.
Figure 8.

Schematic model of presynaptic and postsynaptic calcineurin (CnA) signaling. Direct substrates of CnA include dynamin I, synapsin, NFAT, DARPP-32, and I-1. In addition, CnA can promote PP1 activity through dephosphorylation of the PP1 inhibitors I-1 and DARPP-32. Through this mechanism, CnA activity could increase the rate of inactivation of critical effectors such as the translation initiation factor eIF2α and the transcription factor cAMP response element-binding protein (CREB). RCAN1 helps maintain the phosphorylation of these targets by constraining CnA activity.
Similar conclusions that calcineurin activity is elevated in RCAN1 knock-out mice have been drawn from studies of T-cell activation (Ryeom et al., 2003). Conversely, studies of cardiac hypertrophy in RCAN1 knock-out mice by our group and others (Vega et al., 2003; Sanna et al., 2006) suggest that calcineurin activity is lower in the hearts of these animals. A model in which the RCAN1 gene product can be conditionally knocked out or restored in adult mice will be required to fully delineate the positive and negative effects of RCAN1 on calcineurin/PP1 signaling cascades and their contribution to LTP and memory.
The RCAN1/DSCR1 gene is located on the long arm of human chromosome 21. There has been substantial speculation that trisomy of this locus contributes to learning and memory deficits in individuals with Down syndrome as well as accelerating the onset of an Alzheimer's-like dementia. Based on the studies we present here, one might predict that trisomy of RCAN1/DSCR1 would increase inhibition of calcineurin signaling and improve learning and memory, which is clearly not the case in Down syndrome. Thus, perturbation of calcineurin activity, regardless of direction, may upset a critical balance in kinase and phosphatase activities sufficient to disrupt optimal neuronal function. Drosophila display learning deficits with either overexpression or deletion of the RCAN1 homolog (Chang et al., 2003), demonstrating that any alteration in RCAN1 likely perturbs essential learning processes. It is unlikely that trisomy of RCAN1 alone causes Down syndrome. Rather, it is postulated that several genes, including intersectin-1 and the kinase DYRK1A, cooperate with RCAN1/DSCR1 to amplify gene–dosage imbalances (de la Luna and Estivill, 2006; Gwack et al., 2006; Keating et al., 2006). Indeed, our results using FK506 blockade of elevated calcineurin activity in RCAN1 mutants strongly suggest the critical need for dynamically balancing neuronal kinase and phosphatase pathways for proper cognitive function.
Overexpression of RCAN1 also has been observed in AD patients (Ermak et al., 2001; Harris et al., 2007), but the impact of this on calcineurin activity is not clear. Calcineurin activity has been reported to increase (Liu et al., 2005) and decrease (Gong et al., 1994; Lian et al., 2001) in AD patients. Decreased calcineurin activity has been proposed to result in increased tau phosphorylation, leading to the formation of neurofibrillary tangles (Ermak et al., 2001; Harris et al., 2007). Evidence for increased calcineurin activity in AD was provided by direct enzymatic assays of human AD tissue (Liu et al., 2005) and of tissue from mouse models of AD (Norris et al., 2005). Moreover, recent findings indicate that acute blockade of calcineurin activity improves memory and cognitive function in AD model mice (Dineley et al., 2007). Cleavage of calcineurin by calpain increases levels of the constitutively active 48 kDa CnA isoform in response to ischemic injury and in AD (Wu et al., 2004; Liu et al., 2005; Shioda et al., 2006). An increase in this cleaved form of CnA in the hippocampus of RCAN1 knock-out mice (Fig. 6b) suggests that RCAN1 may be involved in proteolytic processing of calcineurin.
Calcineurin activity also has been implicated in the pathologies of ischemic brain injury and neurotoxicity (Liu et al., 2005; Norris et al., 2005; Shioda et al., 2006). These processes involve elevated levels of oxidative stress, calcium, and calcineurin activity (Norenberg and Rao, 2007). Oxidative stress is associated with both Down syndrome and AD (Perry et al., 2000; Lott et al., 2006), and it was demonstrated recently that overexpression of RCAN1 renders neurons more susceptible to oxidative stress (Porta et al., 2007). Mitochondria play an important role in oxidative stress (Norenberg and Rao, 2007), and RCAN1, in addition to regulating calcineurin, may be important for normal mitochondrial function. Perturbation of the Drosophila homolog of RCAN1, sarah, resulted in mitochondrial abnormalities and elevated levels of reactive oxygen species (Chang and Min, 2005). Our finding of increased levels of the 48 kDa isoform of CnA from mitochondrial-associated subcellular fractions may provide for a mechanistic link between oxidative stress and calcineurin function.
In summary, our findings strongly suggest that RCAN1 facilitates synaptic plasticity and memory by constraining phosphatase signaling via inhibition of calcineurin and its downstream target PP1. Thus, RCAN1 represents an important potential therapeutic target for the treatment of numerous neurological disorders whose pathologies involve the dysregulation of calcineurin.
Footnotes
This work was supported by National Institutes of Health Grants NS047384 and NS034007 (E.K.), T32 HL07676 (C.A.H.), and HL072016 (B.A.R.). We thank Drs. Jonathan Levenson, Lingfei Hou, Caterina Hernandez, Corrine Spencer, Richard Paylor, and Erik Bush for providing reagents and expert technical assistance. We also thank Laura Villasana for technical assistance.
References
- Abel T, Nguyen PV, Barad M, Deuel TA, Kandel ER, Bourtchouladze R. Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell. 1997;88:615–626. doi: 10.1016/s0092-8674(00)81904-2. [DOI] [PubMed] [Google Scholar]
- Ben-Shachar D, Laifenfeld D. Mitochondria, synaptic plasticity, and schizophrenia. Int Rev Neurobiol. 2004;59:273–296. doi: 10.1016/S0074-7742(04)59011-6. [DOI] [PubMed] [Google Scholar]
- Bibb JA, Snyder GL, Nishi A, Yan Z, Meijer L, Fienberg AA, Tsai LH, Kwon YT, Girault JA, Czernik AJ, Huganir RL, Hemmings HC, Jr, Nairn AC, Greengard P. Phosphorylation of DARPP-32 by Cdk5 modulates dopamine signalling in neurons. Nature. 1999;402:669–671. doi: 10.1038/45251. [DOI] [PubMed] [Google Scholar]
- Bush E, Fielitz J, Melvin L, Martinez-Arnold M, McKinsey TA, Plichta R, Olson EN. A small molecular activator of cardiac hypertrophy uncovered in a chemical screen for modifiers of the calcineurin signaling pathway. Proc Natl Acad Sci USA. 2004;101:2870–2875. doi: 10.1073/pnas.0308723101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KT, Min KT. Drosophila melanogaster homolog of Down syndrome critical region 1 is critical for mitochondrial function. Nat Neurosci. 2005;8:1577–1585. doi: 10.1038/nn1564. [DOI] [PubMed] [Google Scholar]
- Chang KT, Shi YJ, Min KT. The Drosophila homolog of Down's syndrome critical region 1 gene regulates learning: implications for mental retardation. Proc Natl Acad Sci USA. 2003;100:15794–15799. doi: 10.1073/pnas.2536696100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Luna S, Estivill X. Cooperation to amplify gene-dosage-imbalance effects. Trends Mol Med. 2006;12:451–454. doi: 10.1016/j.molmed.2006.08.001. [DOI] [PubMed] [Google Scholar]
- Dineley KT, Hogan D, Zhang WR, Taglialatela G. Acute inhibition of calcineurin restores associative learning and memory in Tg2576 APP transgenic mice. Neurobiol Learn Mem. 2007;88:217–224. doi: 10.1016/j.nlm.2007.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ermak G, Morgan TE, Davies KJ. Chronic overexpression of the calcineurin inhibitory gene DSCR1 (Adapt78) is associated with Alzheimer's disease. J Biol Chem. 2001;276:38787–38794. doi: 10.1074/jbc.M102829200. [DOI] [PubMed] [Google Scholar]
- Fox DS, Heitman J. Calcineurin-binding protein Cbp1 directs the specificity of calcineurin-dependent hyphal elongation during mating in Cryptococcus neoformans. Eukaryot Cell. 2005;4:1526–1538. doi: 10.1128/EC.4.9.1526-1538.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentes JJ, Pritchard MA, Estivill X. Genomic organization, alternative splicing, and expression patterns of the DSCR1 (Down syndrome candidate region 1) gene. Genomics. 1997;44:358–361. doi: 10.1006/geno.1997.4866. [DOI] [PubMed] [Google Scholar]
- Fuentes JJ, Genesca L, Kingsbury TJ, Cunningham KW, Perez-Riba M, Estivill X, de la Luna S. DSCR1, overexpressed in Down syndrome, is an inhibitor of calcineurin-mediated signaling pathways. Hum Mol Genet. 2000;9:1681–1690. doi: 10.1093/hmg/9.11.1681. [DOI] [PubMed] [Google Scholar]
- Gong CX, Singh TJ, Grundke-Iqbal I, Iqbal K. Alzheimer's disease abnormally phosphorylated tau is dephosphorylated by protein phosphatase-2B (calcineurin) J Neurochem. 1994;62:803–806. doi: 10.1046/j.1471-4159.1994.62020803.x. [DOI] [PubMed] [Google Scholar]
- Gwack Y, Sharma S, Nardone J, Tanasa B, Iuga A, Srikanth S, Okamura H, Bolton D, Feske S, Hogan PG, Rao A. A genome-wide Drosophila RNAi screen identifies DYRK-family kinases as regulators of NFAT. Nature. 2006;441:646–650. doi: 10.1038/nature04631. [DOI] [PubMed] [Google Scholar]
- Harris CD, Ermak G, Davies KJ. RCAN1–1L is overexpressed in neurons of Alzheimer's disease patients. FEBS J. 2007;274:1715–1724. doi: 10.1111/j.1742-4658.2007.05717.x. [DOI] [PubMed] [Google Scholar]
- Hemmings HC, Jr, Greengard P, Tung HY, Cohen P. DARPP-32, a dopamine-regulated neuronal phosphoprotein, is a potent inhibitor of protein phosphatase-1. Nature. 1984;310:503–505. doi: 10.1038/310503a0. [DOI] [PubMed] [Google Scholar]
- Hilioti Z, Gallagher DA, Low-Nam ST, Ramaswamy P, Gajer P, Kingsbury TJ, Birchwood CJ, Levchenko A, Cunningham KW. GSK-3 kinases enhance calcineurin signaling by phosphorylation of RCNs. Genes Dev. 2004;18:35–47. doi: 10.1101/gad.1159204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YY, Nguyen PV, Abel T, Kandel ER. Long-lasting forms of synaptic potentiation in the mammalian hippocampus. Learn Mem. 1996;3:74–85. doi: 10.1101/lm.3.2-3.74. [DOI] [PubMed] [Google Scholar]
- Ikegami S, Inokuchi K. Antisense DNA against calcineurin facilitates memory in contextual fear conditioning by lowering the threshold for hippocampal long-term potentiation induction. Neuroscience. 2000;98:637–646. doi: 10.1016/s0306-4522(00)00161-5. [DOI] [PubMed] [Google Scholar]
- Jonas E. BCL-xL regulates synaptic plasticity. Mol Interv. 2006;6:208–222. doi: 10.1124/mi.6.4.7. [DOI] [PubMed] [Google Scholar]
- Katz B, Miledi R. The role of calcium in neuromuscular facilitation. J Physiol (Lond) 1968;195:481–492. doi: 10.1113/jphysiol.1968.sp008469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keating DJ, Chen C, Pritchard MA. Alzheimer's disease and endocytic dysfunction: clues from the Down syndrome-related proteins, DSCR1 and ITSN1. Ageing Res Rev. 2006;5:388–401. doi: 10.1016/j.arr.2005.11.001. [DOI] [PubMed] [Google Scholar]
- Kingsbury TJ, Cunningham KW. A conserved family of calcineurin regulators. Genes Dev. 2000;14:1595–1604. [PMC free article] [PubMed] [Google Scholar]
- Klee CB, Ren H, Wang X. Regulation of the calmodulin-stimulated protein phosphatase, calcineurin. J Biol Chem. 1998;273:13367–13370. doi: 10.1074/jbc.273.22.13367. [DOI] [PubMed] [Google Scholar]
- Lian Q, Ladner CJ, Magnuson D, Lee JM. Selective changes of calcineurin (protein phosphatase 2B) activity in Alzheimer's disease cerebral cortex. Exp Neurol. 2001;167:158–165. doi: 10.1006/exnr.2000.7534. [DOI] [PubMed] [Google Scholar]
- Liu F, Grundke-Iqbal I, Iqbal K, Oda Y, Tomizawa K, Gong CX. Truncation and activation of calcineurin A by calpain I in Alzheimer disease brain. J Biol Chem. 2005;280:37755–37762. doi: 10.1074/jbc.M507475200. [DOI] [PubMed] [Google Scholar]
- Lott IT, Head E, Doran E, Busciglio J. Beta-amyloid, oxidative stress and down syndrome. Curr Alzheimer Res. 2006;3:521–528. doi: 10.2174/156720506779025305. [DOI] [PubMed] [Google Scholar]
- Lu YF, Hayashi Y, Moriwaki A, Tomizawa K, Matsui H. FK506, a Ca2+/calmodulin-dependent phosphatase inhibitor, inhibits the induction of long-term potentiation in the rat hippocampus. Neurosci Lett. 1996;205:103–106. doi: 10.1016/0304-3940(96)12384-3. [DOI] [PubMed] [Google Scholar]
- Malleret G, Haditsch U, Genoux D, Jones MW, Bliss TV, Vanhoose AM, Weitlauf C, Kandel ER, Winder DG, Mansuy IM. Inducible and reversible enhancement of learning, memory, and long-term potentiation by genetic inhibition of calcineurin. Cell. 2001;104:675–686. doi: 10.1016/s0092-8674(01)00264-1. [DOI] [PubMed] [Google Scholar]
- Mansuy IM. Calcineurin in memory and bidirectional plasticity. Biochem Biophys Res Commun. 2003;311:1195–1208. doi: 10.1016/j.bbrc.2003.10.046. [DOI] [PubMed] [Google Scholar]
- Mansuy IM, Mayford M, Jacob B, Kandel ER, Bach ME. Restricted and regulated overexpression reveals calcineurin as a key component in the transition from short-term to long-term memory. Cell. 1998a;92:39–49. doi: 10.1016/s0092-8674(00)80897-1. [DOI] [PubMed] [Google Scholar]
- Mansuy IM, Winder DG, Moallem TM, Osman M, Mayford M, Hawkins RD, Kandel ER. Inducible and reversible gene expression with the rtTA system for the study of memory. Neuron. 1998b;21:257–265. doi: 10.1016/s0896-6273(00)80533-4. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Grimwood PD, Morris RG. Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci. 2000;23:649–711. doi: 10.1146/annurev.neuro.23.1.649. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Mitochondrial regulation of neuronal plasticity. Neurochem Res. 2006;32:707–715. doi: 10.1007/s11064-006-9170-3. [DOI] [PubMed] [Google Scholar]
- Miyakawa T, Leiter LM, Gerber DJ, Gainetdinov RR, Sotnikova TD, Zeng H, Caron MG, Tonegawa S. Conditional calcineurin knock-out mice exhibit multiple abnormal behaviors related to schizophrenia. Proc Natl Acad Sci USA. 2003;100:8987–8992. doi: 10.1073/pnas.1432926100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- Norenberg MD, Rao KV. The mitochondrial permeability transition in neurologic disease. Neurochem Int. 2007;50:983–997. doi: 10.1016/j.neuint.2007.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris CM, Kadish I, Blalock EM, Chen KC, Thibault V, Porter NM, Landfield PW, Kraner SD. Calcineurin triggers reactive/inflammatory processes in astrocytes and is upregulated in aging and Alzheimer's models. J Neurosci. 2005;25:4649–4658. doi: 10.1523/JNEUROSCI.0365-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry G, Nunomura A, Hirai K, Takeda A, Aliev G, Smith MA. Oxidative damage in Alzheimer's disease: the metabolic dimension. Int J Dev Neurosci. 2000;18:417–421. doi: 10.1016/s0736-5748(00)00006-x. [DOI] [PubMed] [Google Scholar]
- Phillips RG, LeDoux JE. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci. 1992;106:274–285. doi: 10.1037//0735-7044.106.2.274. [DOI] [PubMed] [Google Scholar]
- Porta S, Marti E, de la Luna S, Arbones ML. Differential expression of members of the RCAN family of calcineurin regulators suggests selective functions for these proteins in the brain. Eur J Neurosci. 2007;26:1213–1226. doi: 10.1111/j.1460-9568.2007.05749.x. [DOI] [PubMed] [Google Scholar]
- Rothermel BA, Vega RB, Williams RS. The role of modulatory calcineurin-interacting proteins in calcineurin signaling. Trends Cardiovasc Med. 2003;13:15–21. doi: 10.1016/s1050-1738(02)00188-3. [DOI] [PubMed] [Google Scholar]
- Ryeom S, Greenwald RJ, Sharpe AH, McKeon F. The threshold pattern of calcineurin-dependent gene expression is altered by loss of the endogenous inhibitor calcipressin. Nat Immunol. 2003;4:874–881. doi: 10.1038/ni966. [DOI] [PubMed] [Google Scholar]
- Sanna B, Brandt EB, Kaiser RA, Pfluger P, Witt SA, Kimball TR, van Rooij E, De Windt LJ, Rothenberg ME, Tschop MH, Benoit SC, Molkentin JD. Modulatory calcineurin-interacting proteins 1 and 2 function as calcineurin facilitators in vivo. Proc Natl Acad Sci USA. 2006;103:7327–7332. doi: 10.1073/pnas.0509340103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton JM, Lee MH, Richardson JA, Patel SB. Microsomal triglyceride transfer protein expression during mouse development. J Lipid Res. 2000;41:532–537. [PubMed] [Google Scholar]
- Shibasaki F, Kondo E, Akagi T, McKeon F. Suppression of signalling through transcription factor NF-AT by interactions between calcineurin and Bcl-2. Nature. 1997;386:728–731. doi: 10.1038/386728a0. [DOI] [PubMed] [Google Scholar]
- Shioda N, Moriguchi S, Shirasaki Y, Fukunaga K. Generation of constitutively active calcineurin by calpain contributes to delayed neuronal death following mouse brain ischemia. J Neurochem. 2006;98:310–320. doi: 10.1111/j.1471-4159.2006.03874.x. [DOI] [PubMed] [Google Scholar]
- Snyder GL, Fienberg AA, Huganir RL, Greengard P. A dopamine/D1 receptor/protein kinase A/dopamine- and cAMP-regulated phosphoprotein (Mr 32 kDa)/protein phosphatase-1 pathway regulates dephosphorylation of the NMDA receptor. J Neurosci. 1998;18:10297–10303. doi: 10.1523/JNEUROSCI.18-24-10297.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega RB, Rothermel BA, Weinheimer CJ, Kovacs A, Naseem RH, Bassel-Duby R, Williams RS, Olson EN. Dual roles of modulatory calcineurin-interacting protein 1 in cardiac hypertrophy. Proc Natl Acad Sci USA. 2003;100:669–674. doi: 10.1073/pnas.0237225100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JH, Kelly PT. The balance between postsynaptic Ca2+-dependent protein kinase and phosphatase activities controlling synaptic strength. Learn Mem. 1996;3:170–181. doi: 10.1101/lm.3.2-3.170. [DOI] [PubMed] [Google Scholar]
- Wang JH, Kelly PT. Attenuation of paired-pulse facilitation associated with synaptic potentiation mediated by postsynaptic mechanisms. J Neurophysiol. 1997;78:2707–2716. doi: 10.1152/jn.1997.78.5.2707. [DOI] [PubMed] [Google Scholar]
- Wang JH, Stelzer A. Inhibition of phosphatase 2B prevents expression of hippocampal long-term potentiation. NeuroReport. 1994;5:2377–2380. doi: 10.1097/00001756-199411000-00041. [DOI] [PubMed] [Google Scholar]
- Winder DG, Sweatt JD. Roles of serine/threonine phosphatases in hippocampal synaptic plasticity. Nat Rev Neurosci. 2001;2:461–474. doi: 10.1038/35081514. [DOI] [PubMed] [Google Scholar]
- Winder DG, Mansuy IM, Osman M, Moallem TM, Kandel ER. Genetic and pharmacological evidence for a novel, intermediate phase of long-term potentiation suppressed by calcineurin. Cell. 1998;92:25–37. doi: 10.1016/s0092-8674(00)80896-x. [DOI] [PubMed] [Google Scholar]
- Wu HY, Tomizawa K, Oda Y, Wei FY, Lu YF, Matsushita M, Li ST, Moriwaki A, Matsui H. Critical role of calpain-mediated cleavage of calcineurin in excitotoxic neurodegeneration. J Biol Chem. 2004;279:4929–4940. doi: 10.1074/jbc.M309767200. [DOI] [PubMed] [Google Scholar]
- Yakel JL. Calcineurin regulation of synaptic function: from ion channels to transmitter release and gene transcription. Trends Pharmacol Sci. 1997;18:124–134. doi: 10.1016/s0165-6147(97)01046-8. [DOI] [PubMed] [Google Scholar]
- Yang J, Rothermel B, Vega RB, Frey N, McKinsey TA, Olson EN, Bassel-Duby R, Williams RS. Independent signals control expression of the calcineurin inhibitory proteins MCIP1 and MCIP2 in striated muscles. Circ Res. 2000;87:E61–E68. doi: 10.1161/01.res.87.12.e61. [DOI] [PubMed] [Google Scholar]
- Zeng H, Chattarji S, Barbarosie M, Rondi-Reig L, Philpot BD, Miyakawa T, Bear MF, Tonegawa S. Forebrain-specific calcineurin knock-out selectively impairs bidirectional synaptic plasticity and working/episodic-like memory. Cell. 2001;107:617–629. doi: 10.1016/s0092-8674(01)00585-2. [DOI] [PubMed] [Google Scholar]
- Zhuo M, Zhang W, Son H, Mansuy I, Sobel RA, Seidman J, Kandel ER. A selective role of calcineurin aalpha in synaptic depotentiation in hippocampus. Proc Natl Acad Sci USA. 1999;96:4650–4655. doi: 10.1073/pnas.96.8.4650. [DOI] [PMC free article] [PubMed] [Google Scholar]