

Abstract
The mGluR-dependent endocytosis of AMPA receptors (AMPARs) in the CA1 region is protein synthesis dependent. However, why this form of trafficking, and not that mediated by NMDA receptor activation, is dependent on protein translation is unclear. Here we have studied the contribution of the cytoskeletal microtubule-associated protein 1B (MAP1B) to the pathway-specific internalization of AMPARs. Treatments of cultured neurons with 3,4-dihydroxyphenylglycol (DHPG) or NMDA, both of which drive AMPAR endocytosis, caused a translation-dependent increase in the dendritic levels of MAP1B protein. Although interfering with protein synthesis using short interfering RNA (siRNA) to eEF2 kinase (eukaryotic elongation factor 2 kinase) blocked the dendritic MAP1B increase by both pathways, it selectively blocked the DHPG- and not the NMDA-induced AMPAR endocytosis. In support of MAP1B synthesis contributing to metabotropic glutamate receptor (mGluR)-mediated AMPAR endocytosis, siRNA against MAP1B in CA1 cultured neurons specifically blocked the DHPG-induced AMPAR internalization. Previous studies suggest a direct interaction between MAP1B and the AMPAR-binding protein GRIP1. Biochemical studies establish that MAP1B associates with GRIP1 and forms a complex with GluR2 in vivo in rat hippocampus. Furthermore, the interaction between MAP1B and GRIP1 increased significantly in acute slices after treatment with DHPG and not NMDA. Together, these findings suggest that MAP1B plays a selective role in the DHPG-induced endocytosis of AMPARs, perhaps through its interaction with GRIP1.
Keywords: microtubule-associated protein 1B (MAP1B), GRIP, AMPA receptor endocytosis, metabotropic glutamate receptor-dependent LTD, NMDA receptor-dependent LTD, eukaryotic elongation factor 2 kinase (eEF2k), dendritic protein synthesis
Introduction
New dendritic protein synthesis is necessary for metabotropic glutamate receptor-dependent long-term depression (mGluR-LTD) in the CA1 region (Huber et al., 2000; Schuman et al., 2006), and its role in this process and the relevant translation targets are the subject of intense investigation. The fragile X mental retardation protein (FMRP) plays an important role in modulating protein synthesis during mGluR-LTD. Studies with Fmr1 knock-out (KO) mice demonstrate that they have enhanced CA1 hippocampal mGluR-LTD (Huber et al., 2002), which together with AMPA receptor (AMPAR) endocytosis persists in the absence of new protein synthesis (Nosyreva and Huber, 2006). The translation of a number of proteins that may participate in mGluR-LTD is normally repressed by FMRP (Weiler et al., 2004). One of these proteins is microtubule-associated protein 1B (MAP1B), whose mRNA colocalizes with FMRP at synapses in hippocampal pyramidal neurons (Lu et al., 2004; Antar et al., 2005). Several characteristics of MAP1B make it an interesting candidate to play a role in mGluR plasticity and AMPAR trafficking. Whereas the levels of MAP1B protein increase during 3,4-dihydroxyphenylglycol (DHPG)-LTD in the CA1 region in wild-type mice, the basal levels of MAP1B are increased in the hippocampus of Fmr1 KO mice (Lu et al., 2004) and do not increase further in Fmr1 KO mice (Hou et al., 2006). mGluR-LTD, similarly to NMDA receptor (NMDAR)-LTD in the CA1 region, is thought to be expressed through postsynaptic AMPAR endocytosis (Carroll et al., 1999; Snyder et al., 2001; Xiao et al., 2001). MAP1B is a component of the neuronal cytoskeleton and binds to actin (Togel et al., 1998; Cueille et al., 2007), which has been implicated in AMPAR internalization (Zhou et al., 2001; Tatsukawa et al., 2006). Additionally, the regulated endocytosis of AMPARs during LTD is dependent on interactions of the GluR2 C terminus with PDZ proteins, including GRIP1 (Matsuda et al., 1999; Braithwaite et al., 2002; Lu and Ziff, 2005), whose second PDZ domain is reported to interact with the light chain of MAP1B (Seog, 2004).
We investigated the role of MAP1B in mGluR-dependent AMPAR endocytosis in hippocampus. We have characterized the expression of MAP1B and its interaction with GRIP1 after treatments with DHPG or NMDA, which induce AMPAR endocytosis and LTD. Both treatments increased the synthesis of MAP1B protein, but the increased dendritic MAP1B levels were necessary only for DHPG-induced AMPAR endocytosis. Reducing the dendritic levels of MAP1B protein using short interfering RNA (siRNA) decreased the basal surface levels of AMPARs and interfered with the DHPG-induced endocytosis of AMPARs. The interaction between MAP1B and GRIP1 increased significantly after treatment with DHPG but not NMDA. These finding support a specific role of MAP1B in mGluR-dependent AMPAR endocytosis involving its interaction with GRIP1.
Materials and Methods
Acute hippocampal slices.
Hippocampal slices (400 μm) were prepared from 21-d-old Sprague Dawley rats. Slices were kept in artificial CSF (ACSF) containing (in mm) 119 NaCl, 26 NaHCO3, 10 glucose, 2.5 KCl, 1 NaH2PO4, 1.3 MgSO4, and 2.5 CaCl2 saturated with 95% O2/5% CO2 and were equilibrated to room temperature (RT) for the drug treatments.
CA1 neuronal cultures.
Hippocampal CA1 regions from postnatal day 0 rat pups were dissociated by papain (Worthington Biochemical, Lakewood, NJ) digestion and plated onto poly-l-lysine-coated coverslips in minimum essential medium with fetal bovine serum (both from Invitrogen, Grand Island, NY) for 24–36 h. The neurons were grown on coverslips in 24-well plates in Neurobasal medium supplemented with Glutamax (both from Invitrogen) and were used for transfections, drug treatments, and immunocytochemical assays at 12–21 d in vitro.
Antibodies.
The following primary antibodies were used: mouse monoclonal MAP1B and MAP1 light chain (Abcam, Cambridge, MA); rabbit polyclonal MAP1B (750 and NR; kind gift from Dr. Itzhak Fischer, Drexel University, Philadelphia, PA); mouse monoclonal GRIP (BD Biosciences, San Jose, CA); rabbit polyclonal GRIP1 (C-terminal epitope-specific; kind gift from Dr. Richard Huganir, Johns Hopkins University, Baltimore, MD); mouse monoclonal GluR2 and rabbit polyclonal GluR1 (Millipore, Billerica, MA); rabbit polyclonal eukaryotic elongation factor 2 kinase (eEF2k; Cell Signaling Technology, Danvers, MA); and mouse monoclonal PSD95 and β-actin (Sigma, St. Louis, MO). FITC-, Cy3-, and horseradish peroxidase (HRP)-conjugated secondary antibodies were from Jackson ImmunoResearch (West Grove, PA).
Drug treatments.
Hippocampal slices were incubated at 37°C in ACSF containing 10 μm CNQX and 50 μm APV (control); 20 μm NMDA with CNQX, 1 μm MPEP, and 1 μm LY367385 for 3 min (NMDA); or 50 μm DHPG with CNQX and APV for 15 min (DHPG). Where indicated, slices were pretreated with rapamycin (200 nm in ACSF) or with vehicle (0.01% DMSO in ACSF) for 30 min at 37°C. CA1 cultures were treated with conditioned culture medium containing 10 μm CNQX and 50 μm APV (control), 20 μm NMDA, CNQX, 1 μm MPEP, and 1 μm LY367385 for 3 min (NMDA) or 20 μm DHPG, CNQX, and APV for 10–15 min (DHPG). Where indicated, anisomycin (20 μm final concentration; Sigma) or rapamycin (200 nm final concentration) was added to the cells or slices for 30 min. All drugs unless specified otherwise were from Tocris (Ellisville, MO).
Transfection of cultures with siRNAs.
Predesigned and annealed 21-mer siRNAs to rat eEF2 kinase (exon 2, region of mRNA sequence, NM_012947), rat MAP1B (three siRNAs targeted to different regions of the MAP1B mRNA sequence, XM_215469) and control scramble siRNA (#1 and #2) were purchased from Ambion (Austin, TX). Transfections of neurons grown onto coverslips were performed using Lipofectamine 2000 (Invitrogen). Lipofectamine and siRNAs were incubated in 100 μl of conditioned medium for 20 min at 37°C, and the complexes were added to the cultures grown in 500 μl of medium. After 24 h, the complexes were removed, and the neurons were washed and cultured for 48 h before being subjected to various treatments and assays (72 h after transfection).
Biotinylation of proteins and surface GluR2 assay.
After drug treatments, hippocampal slices were rinsed in ACSF supplemented with HEPES buffer (ACSF-HEPES) and were incubated in ACSF-HEPES containing 1 mg/ml EZ-link sulfo-NHS-LC Biotin (Pierce, Rockford, IL) at 4°C for 40 min with shaking. Slices were washed with ACSF and homogenized in lysis buffer [Tris-HCl, pH 7.6, containing 0.5% Triton X-100 and EDTA-free protease inhibitor mixture (Roche Diagnostics, Mannheim, Germany)]. Lysates were centrifuged (2000 rpm, 2 min) and the supernatants were subjected to second homogenization followed by centrifugation (12,000 rpm, 10 min). Protein concentrations of the supernatants were measured using BCA protein assay kit (Pierce). Biotinylated proteins (200–400 μg) were incubated with UltraLink Immobilized Streptavidin (Pierce) for 2 h. Streptavidin–protein complexes were washed with PBS and pellets were resuspended in 20 μl of PBS. Surface protein samples and 1/10 (20–40 μg) of the total protein extracts were denatured by boiling in 5× SDS buffer and separated on 10% SDS-PAGE gels. Proteins were transferred onto ECL Hybond nitrocellulose membranes (GE Healthcare, Piscataway, NJ). Blots were probed with rabbit polyclonal GluR2 antibody (1:1000 dilution in TBS containing 5% milk) for 1 h at RT, washed in TBS containing 0.01% Triton X-100 (TBS/Triton X-100), and incubated with secondary HRP-conjugated donkey anti-rabbit antibody (1:1000 dilution in TBS containing 5% milk) for 1 h at RT. Blots were washed in TBS/Triton X-100, incubated with ECL detection reagent (GE Healthcare) and exposed to Biomax film (Kodak, Rochester, NY). The films were scanned and analyzed using MetaMorph (Universal Imaging, Downingtown, PA).
Coimmunoprecipitation and Western blotting.
Protein extracts were prepared from hippocampal slices as described above. Proteins (500–700 μg) were incubated with 5 μg of GRIP, MAP1B, or GluR2 mouse monoclonal antibodies overnight at 4°C with shaking. Protein G-agarose beads (Sigma) were incubated with the coimmunoprecipitates for 2 h at 4°C with shaking. The protein–bead complexes were washed, resuspended in sample buffer, denatured, and separated on 10% SDS-PAGE gels together with the corresponding protein inputs (1/10). The blotted proteins were incubated with primary GRIP (1:1000), MAP1B (1:500), or GluR2 (1:1000) mouse monoclonal antibodies, followed by secondary HRP-conjugated anti-mouse antibody (1:1000). Chemiluminescent detection, exposure to film, and imaging were as described above.
Immunocytochemistry.
Cultured neurons were fixed for 10 min in PBS containing 4% paraformaldehyde, and nonspecific binding was blocked with TBS containing 2.5% BSA for 30 min (for cell surface antigens). For assaying intracellular antigens, neurons were permeabilized with TBS containing 2.5% BSA and 0.1% Triton X-100 for 15 min before the primary antibody. Cells were incubated with primary antibodies for 1–2 h at RT, followed by washing in TBS and incubation with secondary antibodies for 1 h at RT. Samples were washed in TBS and mounted with Vectashield (Vector Laboratories, Burlingame, CA).
Imaging.
Immunostained neurons were photographed with a Hamamatsu Orca ER camera attached to inverted Nikon fluorescent microscope with 60× Plan Apo lens. Exposures were adjusted to ensure signals throughout the neuron within the linear range. Images analyzed using MetaMorph were background subtracted and thresholded, and the fluorescence integrated signal intensity was normalized to area. For each condition, 7–14 cells were analyzed per experiment.
Statistics.
Statistical comparisons were performed using unpaired or paired Student's t test, as appropriate.
Results
Both DHPG and NMDA treatments increase the dendritic synthesis of MAP1B protein
Although both mGluR- and NMDAR-dependent LTD are associated with the internalization of AMPARs, generally the initial phase of only mGluR-LTD has been reported to be protein synthesis dependent (Huber et al., 2000). The mechanism by which the process of AMPAR trafficking requires protein translation specifically after mGluR activation has not been established. We investigated whether MAP1B, whose translation is stimulated by mGluR activation, may contribute to mGluR-mediated AMPAR endocytosis. Immunocytochemistry shows that MAP1B is abundantly expressed in dendrites of CA1 pyramidal neurons (Fig. 1A). Diffuse and punctate MAP1B immunoreactivity was found primarily in the dendritic shafts with a low level of staining in some dendritic spine-like structures. To establish whether MAP1B protein is expressed in the PSD of CA1 pyramidal neurons, we performed coimmunostaining with MAP1B and PSD95 antibodies. As shown in Figure 1A, MAP1B infrequently colocalizes with PSD95 in spine-like structures of these hippocampal neurons.
Figure 1.
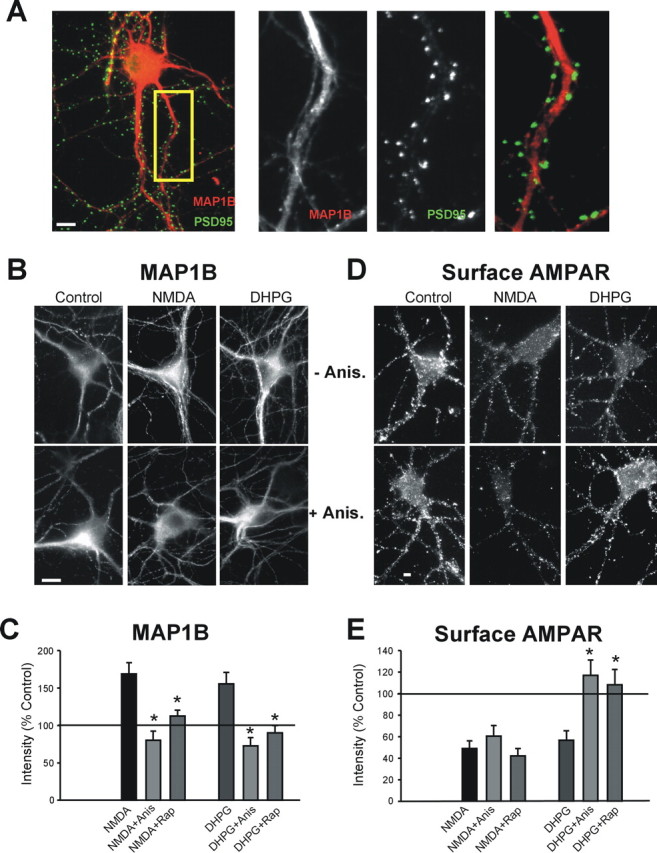
Immunocytochemical expression of MAP1B in dendrites of cultured CA1 pyramidal neurons. A, Colocalization (yellow) between MAP1B (red) and PSD95 (green) in hippocampal neurons. MAP1B, PSD-95, and merged labeling in the inset is enlarged on the right. B–E, NMDA or DHPG treatments, which induce AMPAR endocytosis, increase the dendritic levels and synthesis of MAP1B protein. B, MAP1B immunostaining without and with anisomycin (Anis.) pretreatment in control and DHPG- or NMDA-treated cells. C, Quantitation of MAP1B immunostaining after DHPG in the presence and absence of anisomycin and rapamycin (Rap) (Con, DHPG n = 8; anisomycin n = 5; rapamycin n = 5; *p ≤ 0.05). D, Surface GluR1 immunostaining in neurons labeled live with N-terminal-specific GluR1 antibody in NMDA- or DHPG-treated neurons in the presence or absence of anisomycin. E, Quantitation of surface GluR1 after DHPG treatment in the presence and absence of anisomycin or rapamycin (Con, DHPG n = 8, anisomycin, rapamycin n = 4; *p ≤ 0.05). Scale bars, 10 μm.
Surprisingly, we found that both DHPG and NMDA treatments significantly increased the dendritic levels of MAP1B protein in dendrites of CA1 neurons measured immunocytochemically (Fig. 1B,C). The increased MAP1B levels were attributable to de novo protein synthesis, because both the DHPG- and NMDA-mediated effects were blocked by pretreatment with anisomycin (Fig. 1B,C). Our results are in agreement with Hou et al. (2006), who reported that the MAP1B protein levels increase in CA1 after treatment with DHPG. Although anisomycin blocked both the DHPG- and NMDA-induced increase in dendritic MAP1B protein, it prevented only the DHPG-induced GluR1 AMPAR endocytosis after DHPG treatment (Fig. 1B–E). We also pretreated primary hippocampal cultures with rapamycin to block mammalian target of rapamycin (mTOR) activity, which has been linked to mGluR-LTD (Hou and Klann, 2004; Banko et al., 2006) and which regulates translation by modulating the activity of several translation factors and enzymes, including p70S6K and eEF2. Similarly to anisomycin, rapamycin pretreatment (200 nm, 30 min) blocked the DHPG- and NMDA-induced increase in dendritic MAP1B protein levels but prevented only the endocytosis of GluR1 AMPARs after treatment with DHPG but not NMDA (Fig. 1C,E).
eEF2 kinase siRNA inhibits DHPG-induced AMPAR endocytosis
To further study the activity-induced MAP1B protein increase by NMDAR and mGluR activation, we investigated the role of eukaryotic elongation factor 2 kinase. eEF2 kinase phosphorylates and inactivates eEF2 and thereby stops translational elongation (Ryazanov et al., 1988). eEF2 kinase is implicated in protein synthesis in response NMDAR activation (Scheetz et al., 2000) and lies downstream of mTOR. Unlike translational initiation, the role of translational elongation during synaptic plasticity is not extensively studied. Reducing the dendritic levels of eEF2 kinase with siRNA (75.6 ± 8.4% decrease in comparison with scramble siRNA, n = 50 cells from two transfections) significantly reduced the DHPG- or NMDA-induced increases in the dendritic levels of MAP1B protein 72 h after the transfection (Fig. 2A,B). This suggests that eEF2 kinase is involved the activity-dependent translation of MAP1B in hippocampal neurons. Similarly to anisomycin, eEF2 kinase siRNA interfered with the endocytosis of GluR2 AMPARs after treatment with DHPG but not NMDA (Fig. 2C,D).
Figure 2.

Acute knockdown of eEF2 kinase blocks DHPG- and NMDA-induced increases in dendritic MAP1B protein and DHPG-induced AMPAR endocytosis. A, eEF2 kinase and MAP1B immunostaining in neurons transfected with scrambled (Scr.) control or eEF2 kinase siRNA. B, Quantitation of dendritic MAP1B immunostaining. n = 4 transfections; *p < 0.01. C, Surface GluR2 immunostaining in transfected neurons labeled live with N-terminal-specific GluR2 antibody. D, Quantitation of surface GluR2 immunostaining. n = 5 (NMDA) and 4 (DHPG) transfections; *p < 0.05. Scale bars, 10 μm. si, siRNA; NS, not significantly different.
MAP1B siRNA blocks the DHPG-induced AMPAR endocytosis in CA1 neurons
If MAP1B plays any specific role in the protein translation-dependent AMPAR endocytosis after mGluR, but not NMDAR activation, our data suggest that it cannot be accounted for by the ability of these receptors to differentially modulate synthesis of this protein. We therefore investigated whether MAP1B expression itself could be linked to NMDAR- or mGluR-mediated AMPAR endocytosis. Transfection of CA1 cultures with a combination of three MAP1B-targeted siRNAs resulted in ∼50% reduction in the levels of dendritic MAP1B protein measured immunocytochemically 72 h after transfection in comparison with the control scrambled siRNA. MAP1B siRNA did not significantly affect basal levels of surface GluR2 in control neurons (Fig. 3A). Additionally, we did not find any changes in the neuronal dendritic arbors induced by transfection with MAP1B siRNA (Fig. 3C). However, MAP1B siRNA blocked the DHPG-dependent internalization of GluR2 induced by treatment with DHPG (Fig. 3A,B). In contrast, NMDAR-mediated AMPAR endocytosis was not inhibited by the acute reduction in MAP1B expression (Fig. 3A,B).
Figure 3.

MAP1B siRNA blocks DHPG-induced AMPAR endocytosis. A, Surface GluR2 immunostaining in neurons transfected with scrambled or MAP1B siRNAs (si). After labeling of GluR2 receptors in live neurons, cells were treated with NMDA or DHPG. Scale bar, 10 μm. B, Graph depicts effects of MAP1B siRNA on surface GluR2 as depicted in A. *p ≤ 0.05. C, MAP1B siRNA does not have effects on number of primary dendrites (# Primary Dend.) and total dendritic length (Dend. Length).
DHPG treatment increases the interaction between MAP1B and GRIP1 in vivo
A recent report suggests that MAP1B can interact with the AMPAR-binding protein GRIP in whole mouse brain lysate (Seog, 2004) Because GRIP has been implicated in the endocytosis of AMPARs (Braithwaite et al., 2002; Seidenman et al., 2003), we investigated the binding of MAP1B and GRIP in hippocampal neurons. Double staining of GRIP and MAP1B demonstrates that these two proteins are found colocalized in the dendritic shafts of neurons, with little costaining apparent in spiny structures (Fig. 4A). Using GRIP coimmunoprecipitation, we found that MAP1B interacts with GRIP1 in rat hippocampus (Fig. 4B). We further found that MAP1 light chain also interacts with GRIP (supplemental Figs. 1, 2, available at www.jneurosci.org as supplemental material). Using GluR2 immunoprecipitation, we found that MAP1B and GRIP associate in a complex with GluR2 but not PSD95 (Fig. 4B).
Figure 4.
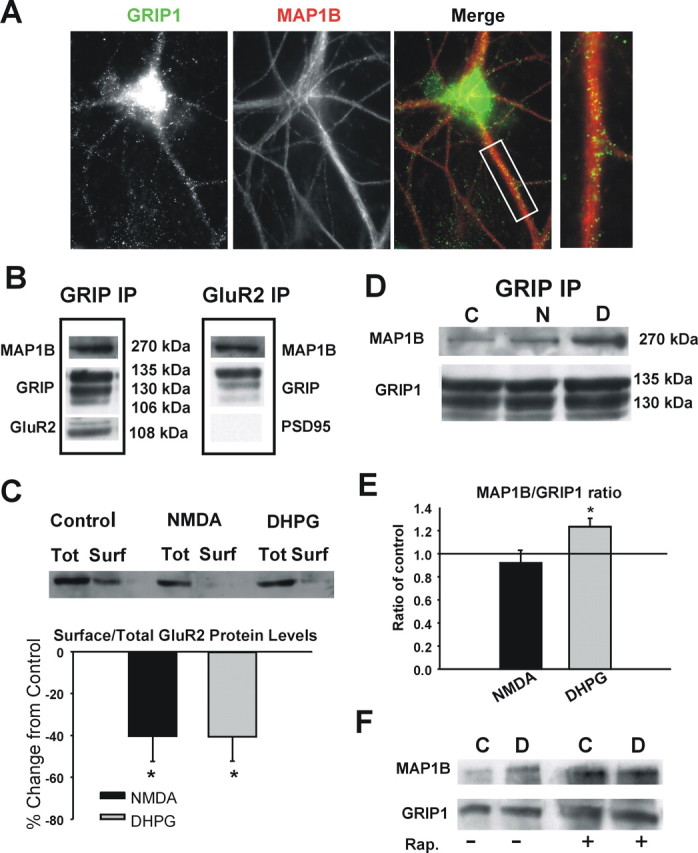
MAP1B-GRIP1 interaction in vivo in hippocampus. A, MAP1B colocalizes with GRIP1 in dendritic shafts of hippocampal pyramidal neurons. Scale bar, 10 μm. B, GRIP or GluR2 coimmunoprecipitation demonstrates a complex between MAP1B, GRIP, and GluR2 but not PSD95. C, Western blot of biotinylated proteins showing that NMDA or DHPG treatments induce AMPAR endocytosis. n = 3 experiments; *p < 0.05. Total AMPAR (Tot) and isolated biotin-labeled surface GluR2 protein (Surf) from NMDA- or DHPG-treated hippocampal lysates were analyzed by Western blot. D, GRIP coimmunoprecipitation showing that DHPG treatment is associated with increased binding of MAP1B to GRIP1. E, Quantitation of the MAP1B/GRIP1 ratio; NMDA n = 4, DHPG n = 6; *p < 0.05. F, Coimmunoprecipitation experiment demonstrates that rapamycin (Rap.) pretreatment blocks the DHPG-dependent increase in GRIP–MAP1B association. C, Control; N, NMDA; D, DHPG.
We characterized the interaction between MAP1B and GRIP1 in slices after treatment with DHPG or NMDA using GRIP coimmunoprecipitation. NMDA and DHPG both caused reductions in surface GluR2 levels as a result of endocytosis as measured by surface protein biotinylation assays (Fig. 4C). DHPG treatment was associated with significant increase in the interaction between MAP1B and GRIP1 in comparison with the control, whereas comparable NMDA treatment was not (Fig. 4D,E) (control vs DHPG, p = 0.002; n = 6 experiments). Consistent with this association being dependent on mTOR-dependent synthesis, the increased association of MAP1B and GRIP was blocked when slices were preincubated with rapamycin (Fig. 1F).
Discussion
Here we present evidence for a specific role of MAP1B in mGluR-dependent endocytosis of AMPARs. NMDAR and mGluR activation associated with AMPAR endocytosis in the CA1 region both increased dendritic levels and synthesis of MAP1B protein in an eEF2 kinase-dependent manner. However, blocking protein synthesis and MAP1B protein increases prevent only DHPG-induced AMPAR endocytosis. We find that MAP1B forms a complex with GRIP1 and GluR2 in vivo in hippocampus, which is increased after treatment with DHPG but not NMDA, which induces AMPAR endocytosis. These results suggest that MAP1B plays a role in DHPG-induced AMPAR endocytosis in hippocampus through interaction with GRIP1.
Our study provides evidence that both NMDAR and mGluR activation can modulate dendritic protein synthesis through the regulation of eEF2 kinase. eEF2 kinase phosphorylates and inactivates eEF2, resulting in inhibition of peptide chain elongation (Ryazanov and Davydova, 1989; Redpath et al., 1993). eEF2 kinase itself is regulated by phosphorylation by variety of kinases, including mTOR and p70 S6 kinase, which inhibit eEF2 kinase activity, resulting in enhanced eEF2 activity and increased protein synthesis (Wang et al., 2001; Browne and Proud, 2004). The block of NMDAR- and mGluR-mediated increases in dendritic MAP1B by acute knockdown of eEF2 kinase suggests that each of these receptors can couple to the phosphorylation of eEF2, leading to enhanced MAP1B translation. It appears that the DHPG- and NMDA-induced increases in dendritic MAP1B protein do not have identical significance for AMPAR endocytosis at the time points studied, because blocking MAP1B protein levels interferes only with DHPG-induced AMPAR internalization. Importantly, the mTOR pathway has been found to play a role in mGluR-LTD by the regulation of translation initiation factors (Hou and Klann, 2004; Banko et al., 2006). The ability of eEF2 kinase knock-down to block mGluR-mediated AMPAR endocytosis, a mechanism of LTD, suggests that both translation initiation and elongation are critical targets of regulation by the mTOR pathway during mGluR-mediated plasticity in the hippocampus.
Our data suggest that endogenous levels of MAP1B are necessary for mGluR-mediated AMPAR endocytosis. The blocking effect of MAP1B siRNA on AMPAR endocytosis does not appear to be attributable to some general disruption of AMPARs or the machinery that controls their trafficking, because NMDAR-dependent AMPAR internalization remained intact. The protein synthesis dependence of mGluR-mediated AMPAR endocytosis suggests that increases in MAP1B that accompany mGluR activation, and not basal levels, are specifically necessary for the process of endocytosis. However, because NMDAR-dependent increases in MAP1B levels are not necessary for endocytosis, a role for MAP1B specifically activated by the mGluR pathway must be involved. The observation that the MAP1B-GRIP1 interaction is enhanced by mGluR, but not NMDAR activation, provides insight that this interaction may play important role in AMPAR endocytosis.
The MAP1B–GRIP1 interaction could contribute to DHPG-induced AMPAR endocytosis in a number of ways. Our observation that MAP1B is expressed predominantly at nonsynaptic sites in dendrites of pyramidal neurons suggests that MAP1B and its interaction with GRIP1 function away from the synapse. A disruption of the spine actin cytostructure and a consequent effect directly on AMPAR endocytosis seems unlikely, because this would be expected to also disrupt NMDAR-mediated endocytosis (Zhou et al., 2001). The proposed role of GRIP1 in stabilizing surface AMPARs (Osten et al., 2000; Kim et al., 2001; Seidenman et al., 2003; Lu and Ziff, 2005) suggests another possibility. The DHPG-dependent increases in MAP1B–GRIP1 binding may serve to retain GRIP1 away from synaptic sites after AMPAR endocytosis. This could in turn prevent the stable return of AMPARs to the membrane surface. Certainly, MAP1B may have additional roles in AMPAR trafficking not dependent on interaction with GRIP1.
In conclusion, this study identifies a role of MAP1B, possibly through interaction with GRIP1, in DHPG-induced AMPAR endocytosis in the CA1 region. These results may have significance for understanding the mechanisms of AMPAR trafficking in fragile X syndrome, in which there is enhanced mGluR-LTD and AMPAR endocytosis.
Footnotes
This work was supported by National Institutes of Health/National Institute of Neurological Disorders and Stroke Grant NS 049661(R.C.C.). G.D. was supported by an Albert Einstein College of Medicine Neuroscience Fellowship.
References
- Antar LN, Dictenberg JB, Plociniak M, Afroz R, Bassell GJ. Localization of FMRP-associated mRNA granules and requirement of microtubules for activity-dependent trafficking in hippocampal neurons. Genes Brain Behav. 2005;4:350–359. doi: 10.1111/j.1601-183X.2005.00128.x. [DOI] [PubMed] [Google Scholar]
- Banko JL, Hou L, Poulin F, Sonenberg N, Klann E. Regulation of eukaryotic initiation factor 4E by converging signaling pathways during metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2006;26:2167–2173. doi: 10.1523/JNEUROSCI.5196-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braithwaite SP, Xia H, Malenka RC. Differential roles for NSF and GRIP/ABP in AMPA receptor cycling. Proc Natl Acad Sci USA. 2002;99:7096–7101. doi: 10.1073/pnas.102156099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne GJ, Proud CG. A novel mTOR-regulated phosphorylation site in elongation factor 2 kinase modulates the activity of the kinase and its binding to calmodulin. Mol Cell Biol. 2004;24:2986–2997. doi: 10.1128/MCB.24.7.2986-2997.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll RC, Lissin DV, von Zastrow M, Nicoll RA, Malenka RC. Rapid redistribution of glutamate receptors contributes to long-term depression in hippocampal cultures. Nat Neurosci. 1999;2:454–460. doi: 10.1038/8123. [DOI] [PubMed] [Google Scholar]
- Cueille N, Tallichet Blanc C, Popa-Nita S, Catsicas S, Dietler G, Riederer BM. Characterization of MAP1B heavy chain interaction with actin. Brain Res Bull. 2007;71:610–618. doi: 10.1016/j.brainresbull.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Hou L, Klann E. Activation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2004;24:6352–6361. doi: 10.1523/JNEUROSCI.0995-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou L, Nation MD, Hu D, Spencer CM, Paylor R, Klann E. Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron. 2006;51:441–454. doi: 10.1016/j.neuron.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Huber KM, Kayser MS, Bear MF. Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science. 2000;288:1254–1256. doi: 10.1126/science.288.5469.1254. [DOI] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci USA. 2002;99:7746–7750. doi: 10.1073/pnas.122205699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CH, Chung HJ, Lee HK, Huganir RL. Interaction of the AMPA receptor subunit GluR2/3 with PDZ domains regulates hippocampal long-term depression. Proc Natl Acad Sci USA. 2001;98:11725–11730. doi: 10.1073/pnas.211132798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R, Wang H, Liang Z, Ku L, O'Donnell WT, Li W, Warren ST, Feng Y. The fragile X protein controls microtubule-associated protein 1B translation and microtubule stability in brain neuron development. Proc Natl Acad Sci USA. 2004;101:15201–15206. doi: 10.1073/pnas.0404995101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Ziff EB. PICK1 interacts with ABP/GRIP to regulate AMPA receptor trafficking. Neuron. 2005;47:407–421. doi: 10.1016/j.neuron.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Matsuda S, Mikawa S, Hirai H. Phosphorylation of serine-880 in GluR2 by protein kinase C prevents its C terminus from binding with glutamate receptor-interacting protein. J Neurochem. 1999;73:1765–1768. doi: 10.1046/j.1471-4159.1999.731765.x. [DOI] [PubMed] [Google Scholar]
- Nosyreva ED, Huber KM. Metabotropic receptor-dependent long-term depression persists in the absence of protein synthesis in the mouse model of fragile X syndrome. J Neurophysiol. 2006;95:3291–3295. doi: 10.1152/jn.01316.2005. [DOI] [PubMed] [Google Scholar]
- Osten P, Khatri L, Perez JL, Köhr G, Giese G, Daly C, Schultz TW, Wensky A, Lee LM, Ziff EB. Mutagenesis reveals a role for ABP/GRIP binding to GluR2 in synaptic surface accumulation of the AMPA receptor. Neuron. 2000;27:313–325. doi: 10.1016/s0896-6273(00)00039-8. [DOI] [PubMed] [Google Scholar]
- Redpath NT, Price NT, Severinov KV, Proud CG. Regulation of elongation factor-2 by multisite phosphorylation. Eur J Biochem. 1993;213:689–699. doi: 10.1111/j.1432-1033.1993.tb17809.x. [DOI] [PubMed] [Google Scholar]
- Ryazanov AG, Shestakova EA, Natapov PG. Phosphorylation of elongation factor 2 by EF-2 kinase affects rate of translation. Nature. 1988;334:170–173. doi: 10.1038/334170a0. [DOI] [PubMed] [Google Scholar]
- Ryazanov AG, Davydova EK. Mechanism of elongation factor 2 (EF-2) inactivation upon phosphorylation. Phosphorylated EF-2 is unable to catalyze translocation. FEBS Lett. 1989;251:187–190. doi: 10.1016/0014-5793(89)81452-8. [DOI] [PubMed] [Google Scholar]
- Scheetz AJ, Nairn AC, Constantine-Paton M. NMDA receptor-mediated control of protein synthesis at developing synapses. Nat Neurosci. 2000;3:211–216. doi: 10.1038/72915. [DOI] [PubMed] [Google Scholar]
- Schuman EM, Dynes JL, Steward O. Synaptic regulation of translation of dendritic mRNAs. J Neurosci. 2006;26:7143–7146. doi: 10.1523/JNEUROSCI.1796-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidenman KJ, Steinberg JP, Huganir R, Malinow R. Glutamate receptor subunit 2 serine 880 phosphorylation modulates synaptic transmission and mediates plasticity in CA1 pyramidal cells. J Neurosci. 2003;23:9220–9228. doi: 10.1523/JNEUROSCI.23-27-09220.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seog D-H. Glutamate receptor interacting protein binds to the microtubule-associated protein. Biosci Biotechnol Biochem. 2004;68:1808–1810. doi: 10.1271/bbb.68.1808. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Philipot BD, Huber KM, Dong X, Fallon JR, Bear MF. Internalization of ionotropic glutamate receptors in response to mGluR activation. Nat Neurosci. 2001;4:1079–1085. doi: 10.1038/nn746. [DOI] [PubMed] [Google Scholar]
- Tatsukawa T, Chimura T, Miwakawa H, Yamaguchi K. Involvement of basal protein kinase C and extracellular signal-regulated kinase 1 /2 activities in constitutive internalization of AMPA receptors in cerebellar Purkinje cells. J Neurosci. 2006;26:4820–4825. doi: 10.1523/JNEUROSCI.0535-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togel M, Wiche G, Propst F. Novel features of the light chain of microtubule-associated protein 1B: microtubule stabilization, self interaction, actin filament binding, and regulation by the heavy chain. J Cell Biol. 1998;143:695–707. doi: 10.1083/jcb.143.3.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Li W, Williams M, Terada N, Alessi DR, Proud CG. Regulation of elongation factor 2 kinase by p90RSK1 and p70 S6 kinase. EMBO J. 2001;20:4370–4379. doi: 10.1093/emboj/20.16.4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiler IJ, Spangler CC, Klintsova AY, Grossman AW, Kim SH, Bertaina-Anglade V, Khaliq H, de Vries FE, Lambers FA, Hatia F, Base CK, Greenough WT. Fragile X mental retardation protein is necessary for neurotransmitter-activated protein translation at synapses. Proc Natl Acad Sci USA. 2004;101:17504–17509. doi: 10.1073/pnas.0407533101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao M-Y, Zhou Q, Nicoll RA. Metabotropic glutamate receptor activation causes a rapid redistribution of AMPA receptors. Neuropharmacology. 2001;41:664–671. doi: 10.1016/s0028-3908(01)00134-4. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Xiao M, Nicoll RA. Contribution of the cytoskeleton to the internalization of AMPA receptors. Proc Natl Acad Sci USA. 2001;98:1261–1266. doi: 10.1073/pnas.031573798. [DOI] [PMC free article] [PubMed] [Google Scholar]