

Abstract
Acid-sensing ion channels (ASICs) are a class of ion channels activated by extracellular protons and are believed to mediate the pain caused by tissue acidosis. Although ASICs have been widely studied, little is known about their regulation by inflammatory mediators. Here, we provide evidence that nitric oxide (NO) potentiates the activity of ASICs. Whole-cell patch-clamp recordings were performed on neonatal rat cultured dorsal root ganglion neurons and on ASIC isoforms expressed in CHO cells. The NO donor S-nitroso-N-acetylpenicillamine (SNAP) potentiates proton-gated currents in DRG neurons and proton-gated currents in CHO cells expressing each of the acid-sensitive ASIC subunits. Modulators of the cGMP/PKG pathway had no effect on the potentiation, but in excised patches from CHO cells expressing ASIC2a, the potentiation could be reversed by externally applied reducing agents. NO therefore has a direct external effect on the ASIC ion channel, probably through oxidization of cysteine residues. Complementary psychophysiological studies were performed using iontophoresis of acidic solutions through the skin of human volunteers. Topical application of the NO donor glyceryl trinitrate significantly increased acid-evoked pain but did not affect heat or mechanical pain thresholds. ASICs may therefore play an important role in the pain associated with metabolic stress and inflammation, where both tissue acidosis and a high level of NO are present.
Keywords: pain, inflammation, ASIC, proton-gated currents, nitric oxide, acid
Introduction
Acid-sensitive ion currents were first reported in sensory neurons (Krishtal and Pidoplichko, 1980; Krishtal, 2003). The ion channels thought to be responsible are the acid-sensing ion channels (ASICs), and four genes, two of which are alternatively spliced, generate the six known members of the ASIC family: ASIC1a, ASIC1b, ASIC2a, ASIC2b, ASIC3, and ASIC4 (Price et al., 1996; Lingueglia et al., 1997; Waldmann et al., 1997a,b; Chen et al., 1998; Akopian et al., 2000). A major function of ASICs is thought to be the initiation of acid-mediated pain, and the properties of acid perception in humans correlate well with those of the ASICs (Jones et al., 2004).
In contrast with the heat-gated ion channel TRPV1, which is modulated by a number of proinflammatory mediators (Huang et al., 2006), ASICs seem to be unresponsive to many of the same mediators. Among the known interactions, peptides such as FMRFamide can enhance ASIC1 and 3 activity (Xie et al., 2003), and arachidonic acid can potentiate ASIC activation (Allen and Attwell, 2002; Smith et al., 2007).
Nitric oxide (NO) is a short-lived mediator whose release is strongly potentiated by inflammation (Rawlingson, 2003; Mander et al., 2005) mainly because an inducible form of the enzyme responsible for synthesizing NO from l-arginine [inducible nitric oxide synthase (iNOS)] is upregulated by inflammation (Grabowski et al., 1997). NO modifies the function of a wide range of proteins, including ion channels, by two main pathways (Ahern et al., 2002). An indirect mechanism involves the production of cGMP and the activation of protein kinase G (PKG). NO can also directly modify the tertiary structure of proteins by S-nitrosylating the thiol side-chains of cysteine residues, leading to the formation of disulfide bonds between neighboring cysteine residues (Stamler, 1994; Stamler et al., 1997; Davis et al., 2001; Jaffrey et al., 2001). The direct mechanism has been shown to be important in modulating the activity of a number of ion channels (Lei et al., 1992; Manzoni and Bockaert, 1993; Bolotina et al., 1994; Broillet and Firestein, 1996; Castel and Vaudry, 2001; Yoshida et al., 2006). ASICs are a potential target for a similar direct mechanism of action in view of the large number of cysteine residues on their external surface (Waldmann et al., 1999). Three recent studies have shown that oxidizing agents can inhibit acid-evoked currents in neurons and heterologously expressed ASICs (Andrey et al., 2005; Chu et al., 2006; Cho and Askwith, 2007), but possible effects of NO on members of the ASIC family have not so far been investigated. In the present study we find that proton-gated currents of both sensory neurons and ASIC isoforms are potentiated, rather than being inhibited, by the oxidizing agents NO and H2O2. Intracellular pathways modulated by NO are not involved in the enhancement of acid-gated currents in neurons, and the mechanism of the effect is instead via a direct action on the extracellular surface of the ion channel. We also provide evidence that acid-mediated pain in humans is potentiated by agents releasing NO.
Materials and Methods
Dorsal root ganglion neuronal culture
Experiments were performed on cultured DRG neurons taken from neonatal Wistar rats (3–10 d old). Rat pups were killed by cervical dislocation followed by decapitation. Culture methods were as described by Smith et al. (2007). Briefly, individual DRG were removed and enzymatically isolated using collagenase. Cells were then plated on 35 mm poly-l-lysine- (1 mg/ml; Sigma, Poole, UK) coated plastic Petri dishes and maintained for 3 d in culture at 37°C and 5% CO2/air in DMEM (Invitrogen, Carlsbad, CA) containing 10% FBS, 1% penicillin/streptomycin, and 50 ng/ml NGF (nerve growth factor; Promega, Madison, WI).
Cell line
Chinese hamster ovary (CHO) cells were obtained from the American Type Culture Collection and were cultured in DMEM (Invitrogen) supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin at 37°C, 5% CO2/air. Cells were plated onto plastic Petri dishes coated with poly-l-lysine and transfected 24 h after plating using calcium phosphate (Calphos; Clontech, Mountain View, CA) according to the manufacturer's instructions. For single-channel experiments, cells were plated onto glass coverslips at a density at 3000 cells/ml. ASIC1a, 1b, 2a, and 3 contained in pCI vector were kindly donated by M. Lazdunski (Institut de Pharmacologie Moléculaire et Cellulaire, Valbonne Sophia-Antipolis, France) and were cotransfected with GFP (Clontech) at a ratio of 4:1. The quantities of DNA per dish were 2 μg for ASIC1a and 4 μg for ASIC1b, 2a, and 3.
Electrophysiology
Whole-cell experiments.
All recordings were made from the somata of medium/small diameter DRG neurons (13–30 μm) and from CHO cells using the whole-cell configuration of the patch-clamp technique. Recordings were performed at a holding potential of −80 mV for DRG neurons and −60 mV for CHO cells, respectively. Experiments were performed at room temperature (20–24°C). Currents were amplified by an Axopatch 200B amplifier (Axon Instruments, Foster City, CA) controlled by pClamp software (versions 6 and 8). Patch pipettes of resistance 3–5.5 MΩ were made from borosilicate glass using a Sutter Instruments P-80 (Novato, CA) horizontal micropipette puller. In both DRG and CHO cells, 60–80% of series resistance was compensated. A rapid extracellular solution change (<50 ms) was performed using a home-made multibarrel perfusion system, moved in front of the recorded cell by a computer-controlled stepping motor. Only one recording was performed on each culture dish, to ensure that data were not obtained from cells that had been inadvertently exposed to anything other than the standard extracellular solution. In the experiments presented here and for both cell types, the extracellular solution contained the following (in mm): 140 NaCl, 1.8 CaCl2, 1 MgCl2, 4 KCl, 10 HEPES (N-2-[hydroxyethyl]piperazine-N′-[2-ethanesulfonic acid]), and 4 glucose, adjusted to pH 7.4 or other pH (from 6.0 to 7.2) using NaOH and to 300–310 mOsm osmolarity. To rule out the possibility that HEPES was inadequately buffering the pH at the bottom of this pH range, some control experiments using HEPES (5 mm) and 2-morpholinoethanesulfonic acid (5 mm) were also conducted at pH 6.3 with no significant difference in the amplitude of acid-evoked currents. The pipette solution contained the following (in mm): 140 KCl, 1.6 MgCl2, 2 EGTA, 2.5 MgATP, 0.5 Li2GTP, and 10 HEPES, adjusted to pH 7.3 using KOH.
To investigate the potentiation of proton-gated currents by agents such as the NO donor S-nitroso-N-acetylpenicillamine (SNAP; Sigma), we used the following standard protocol. Eight pulses of 5 s each of low-pH solution at 30 s intervals were given, with the response to the final pulse used for analysis. Five pulses of low-pH solution were given in the presence of an NO donor (in both acid and pH 7.4 solutions), with the response to the third pulse used for analysis. After return to control solution, the response to the fourth pulse was used for analysis. SNAP (100 μm) was in general used as the NO donor, although sodium nitroprusside (SNP; 10 μm) was used for some experiments as noted in the text. SNAP was freshly prepared every 3 h and was kept protected from light. The possibilities that SNAP may have an effect on ASIC function other than by acting as an NO donor or that DMSO may have an effect were investigated by leaving the solution for 4–6 h in the syringe, which completely abolished its potentiating effect (see Results).
For the dose–response experiments 5 s pulses of solution of progressively decreasing pH where given at 30 s intervals and the same protocol was then repeated with SNAP present in all the solutions. Maximum response amplitudes were fitted with a smooth curve using the Hill equation: I/Imax = [H+]b/([H+]0.5b + [H+]b), where [H+]0.5 denotes the proton concentration producing half-maximal response, and b is the Hill coefficient.
Single-channel recordings.
Single-channel currents were recorded using an Axopatch 200B amplifier and pClamp9 software (Axon Instruments). Pipettes (10–15 MΩ) were pulled from thick-walled borosilicate glass (150F-10; Clark Instruments, Salisbury, UK) and fire-polished before use. Coverslips with transfected cells were placed in a fast exchange perfusion chamber (Warner Instruments, Hamden, CT), and the effect of drugs was studied by exchanging the bath solution. For inside-out recordings, the pipette solution contained an extracellular solution (in mm): 144 NaCl, 2 CaCl2, 1 MgCl2, 10 HEPES; the pH was adjusted to pH 6.0 using NaOH. An isotonic KCl solution was used as extracellular solution and consisted of the following (in mm): 144 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES; the pH was adjusted to 7.4 using KOH. For outside-out recordings, the pipette solution was isotonic KCl as detailed above, and the extracellular solution was as detailed above except that the pH was adjusted to pH 7.4 (or pH 6.0 for acid pulses) using NaOH. SNAP (100 μm), H2O2 0.05%, and 2-mercaptoethanol (2-ME; 2 mm) were prepared in the extracellular solution appropriate for each type of experiment. The data were collected with a filtering frequency of 2 kHz and sampled at 10 kHz. For presentation, data were filtered with a digital Gaussian filter at 500 Hz. Data analysis was performed on stretches longer than 30 s from patches that contained a single channel, as judged by the existence of a single open state. Single channel recordings were analyzed with Clampfit. Channel openings were detected using the half-amplitude threshold crossing method, and open probability (po) was calculated as the ratio of time the channel spent in the open state to the total recording time.
Both whole-cell and single-channel traces were imported into SigmaPlot software for presentation. All data are presented as mean ± SEM (n = number of experiments). The t test (paired or unpaired, as appropriate) was used for all statistical tests, which were performed on the raw data. For presentational purposes, the data in Figures 1 and 3 are normalized to control levels.
Figure 1.
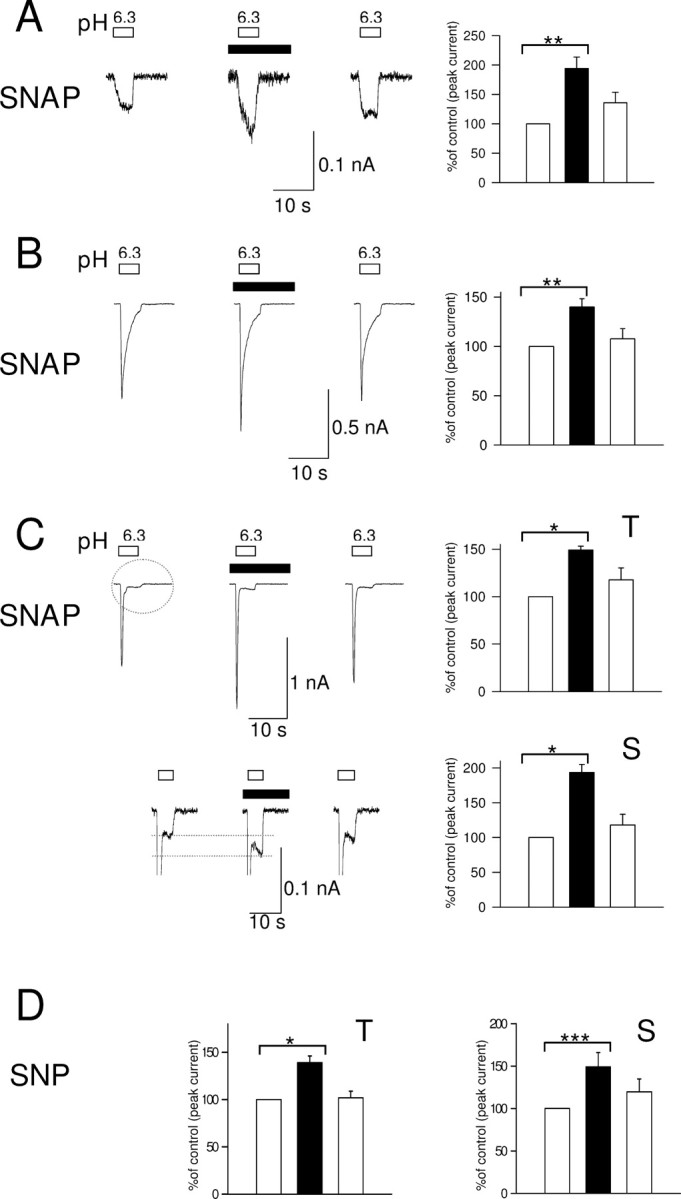
A–C, Potentiation of the three types of proton-gated currents in rat neonatal DRG neurons by the NO donor SNAP (100 μm). Whole-cell patch clamp, holding potential −80 mV. Left panels, Current evoked by eighth application of pH 6.3 solution (white bar), third application of pH 6.3 in the presence of SNAP (100 μm, black box), and fourth application of pH 6.3 after wash. Right panel shows current normalized to control (100%, left column). A, Potentiation of sustained current by SNAP (n = 6). B, Potentiation of slowly inactivating current by SNAP (n = 8). C, Potentiation of rapidly inactivating current by SNAP (n = 8). Top shows potentiation of peak of rapidly inactivating current (T), and bottom shows potentiation of sustained component (S, circled). D, Potentiation of transient (T) and sustained (S) phases of rapidly inactivating current by SNP (10 μm). Data in all bar graphs are shown relative to control. Error bars show ±SEM. Statistical tests in this and other figures were performed on raw data using paired t test, and significance is shown as follows: *p < 0.05; **p < 0.01; ***p < 0.001.
Figure 3.

Potentiation of proton-gated current in CHO cells expressing ASICs. Whole-cell patch clamp, holding potential −60 mV. A, ASIC1a: application of pH 6.9 solution (open bar) before (left trace), during (middle trace), and after (right trace) the application of 100 μm SNAP (black bar). B, Corresponding bar graph in which the current is normalized to the control (100%, left column; n = 9; **p < 0.01). C, Current in ASIC1a transfected CHO cells in response to low pH in the absence (top) and in the presence (black box) of 100 μm SNAP. D, Dependence of peak current on pH in absence (open squares) and presence (black squares) of SNAP (n = 7). E–G, Enhancement of activity of ASIC1b, ASIC2a, and ASIC3 by SNAP (5 s pulses of pH 6.0, 6.0, and 6.9 respectively). *p < 0.05; **p < 0.01.
Human experiments
Subjects.
Experiments were performed on healthy volunteers (male and female, n = 13) aged between 21 and 31. Written consent in accordance with the Declaration of Helsinki was given by subjects before participation in the study. Exclusions included pregnancy, asthma, chronic skin condition, known sensitivity to glyceryl trinitrate (GTN), or known circulatory problems. The subjects were blind to all treatments and test solutions applied. Subjects underwent an initial training session to familiarize them with the testing and rating procedures and then attended one or more sessions, each lasting ∼1 h. All procedures and protocols were approved by St Thomas' Hospital Ethics Committee.
Iontophoresis of test solutions.
Skin on the volar forearm of healthy adult volunteers was used in this study. The experimental skin was cleaned with an alcohol swab, an iontophoresis chamber was attached with an adhesive disc, and an indifferent electrode placed at a distant site on the hand. The chamber, 9 mm in diameter, was filled with acid (hydrochloric acid, pH 2), and a constant current of 0.3 mA was applied for 4 min to the cathode, located in the iontophoresis chamber, with a commercial iontophoresis unit (Phoresor II PM 700; IOMED, Salt Lake City, UT) to drive protons into the skin. This constituted one trial. Different experimental areas of skin were used for each trial. Iontophoresis of saline (0.9% NaCl w/v) was used as control.
Magnitude of perceived pain rating.
The magnitude of the perceived pain was assessed continually throughout the 4 min trial using a visual analog scale (VAS). Briefly, a scale was viewed on the monitor of a computer with the ends labeled “0” and “100.” Subjects were told that 0 represented no pain and 100 represented the worst pain imaginable. The volunteers were then prompted every 20 s by the computer to rate the pain they were experiencing at the experimental site, and they indicated the level of perceived pain by moving a pointer on the VAS scale with a computer mouse.
Topical application of NO donor.
Commercially available topically applied GTN (15 mm; Percutol, 2% w/w; Dominion Pharma, Haslemere, UK) was applied to a 6 × 4 cm area of skin and covered by occlusion dressing for ∼15 min. The occlusion dressing was then removed before iontophoresis of acid and saline. Both GTN and placebo (KY jelly, an inert lubricating jelly; Johnson and Johnson, Maidenhead, UK) were left on the skin during iontophoresis of test solutions.
Determination of heat pain threshold.
Skin pretreated with GTN or placebo was subjected to measured heating to determine the effects of GTN on the heat pain threshold. A commercial thermal sensory analyzer (Medoc Advanced Medical Systems, Ramat Yishai, Israel) was used to heat the experimental skin area from a holding temperature of 32°C at 0.5°C/s. Subjects were asked to indicate when the stimulus was clearly painful. This was repeated at least twice for each site, and the thresholds were averaged.
Determination of mechanical pain threshold.
To determine the effects of GTN on the mechanical pain threshold, skin pretreated with GTN and placebo cream was subjected to measured mechanical stimuli using a commercially available electronic von Frey System (Somedic, Hörby, Sweden). Briefly, the von Frey monofilament (1 mm in diameter) was applied to the experimental skin, and the force applied by the experimenter (monitored on a computer screen) was gradually increased (4 g/s) until the subject indicated that the stimulus was painful by pressing a button.
Solutions.
Concentrated hydrochloric acid (1 ml; Sigma) was diluted in 1 L of distilled water to make a 12 mm solution with a pH of approximately pH 2. Sodium chloride (BDH Laboratory Supplies, Poole, UK) was dissolved in distilled water to make a 0.9% w/v saline solution.
Statistical analysis.
Statistical analysis was performed using SigmaStat software. Tests for normality were performed on the data, and subsequent parametric tests, one-way repeated-measure ANOVA (followed by Tukey post hoc test) and paired t tests, were performed as appropriate. All data are expressed as mean ± SEM, and the significance level is taken as 5%.
Results
Effects of NO on proton-gated currents in DRG neurons
Figure 1 shows the three main types of acid-evoked membrane current observed in small to medium diameter cultured rat dorsal root ganglion neurons (15–30 μm diameter). We used a modest pH excursion, to 6.3, for the majority of experiments in this study in view of the evidence that pH excursions are not large even in extreme inflammation and anoxia (Cobbe et al., 1982; Issberner et al., 1996). In most DRG neurons (95%), an inward current was evoked by application of a pH 6.3 solution (Smith et al., 2007). A non-inactivating current of small amplitude (−43.7 ± 8.3 pA, n = 6) (Fig. 1A) was present in 15% of cells. A slowly inactivating current of large amplitude (−897.7 ± 306.3 pA, n = 8) (Fig. 1B) was present in 47% of cells. Finally, a large rapidly inactivating current (−1148.3 ± 335.4 pA, n = 8) (Fig. 1C) followed by a small sustained component (−53.7 ± 17.9 pA, n = 8) (Fig. 1D) was present in 32% of neurons. The slowly and rapidly inactivating acid-evoked currents form two distinct populations (Smith et al., 2007).
The transient phases of the acid-evoked inward currents in Figure 1A–C are consistent with activation of ASICs (see Fig. 3 below), but the sustained phases could be caused by activation of TRPV1, which is known to be pH sensitive (Tominaga et al., 1998). The sustained phases may be of particular interest in acid-evoked pain sensation, in view of the need to signal pain continuously. We investigated the possibility that TRPV1 may contribute to the sustained phases by the use of the potent and specific TRPV1 blocker N-(4-tertiarybutylphenyl)-4-(3-chloropyridin-2-yl)tetrahydropyrazine-1(2H)-carbox-amide (BCTC) (Valenzano et al., 2003). BCTC had little effect on the transient current evoked by a large pH excursion to pH 5, but largely blocked the sustained inward current (Fig. 2A), as expected if the inward current activated by a large pH excursion consists of a transient phase attributable to activation of one or more ASICs, followed by a sustained phase attributable mainly to activation of TRPV1. However, the sustained inward current activated by a modest pH excursion to pH 6.3 was little affected by BCTC. The effect on both the pure sustained inward current (Fig. 2B, left) and on the sustained component of the rapidly inactivating current (Fig. 2B, right) was similar: in the majority of cases, BCTC had no effect, showing that TRPV1 did not contribute, whereas in some cases the current was partially blocked, showing that TRPV1 makes a partial contribution. In no case was the sustained current completely abolished by BCTC. These data show, in agreement with other studies (Liu et al., 2004), that the sustained phase of inward current activated by modest pH excursions cannot be attributed solely to activation of TRPV1. The observation that the sustained current is enhanced by NO in all cells (see below), whereas a component attributable to TRPV1 was present in only a minority, also shows that the effect of NO on the sustained component cannot be attributable to an action of NO on TRPV1 alone.
Figure 2.

Effect of the TRPV1 blocker BCTC on acid-evoked currents. A, BCTC (100 nm) blocks most of the sustained phase of inward current evoked by a large acidification to pH 5.0 but has little effect on the transient phase. B, The sustained inward current (left, S of S; compare Fig. 1A) and the sustained phase of the rapidly inactivating current (right, S of T/S; compare Fig. 1C) evoked by mild acidification (to pH 6.3) are partially inhibited in some cells by BCTC but unaffected in the majority.
Application of the NO donor SNAP elicited an increase in all three types of proton-gated currents. The amplitude of the sustained current increased to 194.3 ± 19.4% of that before application of SNAP (n = 6, p < 0.01), the slowly inactivating current to 140.0 ± 8.3% (n = 8, p < 0.01), and the transient component of the rapidly inactivating current to 149.2 ± 4.0% (n = 8, p < 0.05). The sustained component of the rapidly inactivating current showed a similar augmentation to that observed for the sustained component when present alone (193.4 ± 11.7%, n = 8).
SNAP also modulated the kinetics of both transient currents. For the slowly inactivating current the time to peak shortened from 529.2 ± 54.2 ms to 313.5 ± 23.4 ms (n = 8, p < 0.01) and the inactivation time constant from 1239.4 ± 220.0 ms to 808.7 ± 167.6 ms (n = 8, p < 0.05). The activation time of the rapidly inactivating current was shortened from 451.5 ± 82.7 ms (n = 8) to 268.5 ± 82.9 ms (n = 8, p < 0.001), and the inactivation time constant from 268.2 ± 44.8 ms to 202.8 ± 33.7 ms (n = 8, p < 0.01). The shortening of the time to peak of the transient phases of acid-evoked current is a consequence of the speeding of the inactivation rate. This more rapid inactivation also has the effect of reducing the enhancement of the transient phases caused by SNAP. The apparently smaller enhancement of the transient currents when compared with the sustained currents (see above) therefore probably does not represent a real difference in the actions of SNAP on these distinct phases.
All the increases in acid-evoked currents were dependent on release of NO by SNAP, because no increase was observed when the release of NO was allowed to decay by leaving the SNAP solution for 4–6 h (sustained current 97.5 ± 7.28% of control, n = 3; slowly inactivating current 91.6 ± 0.8%, n = 2; transient component of rapidly inactivating current 96.8 ± 8.6%, n = 2; sustained component of rapidly inactivating current 117.2 ± 17.4%, n = 2). We also studied the effect of a second NO donor, SNP (10 μm), on the rapidly inactivating current (Fig. 1D), and found that both the sustained component and the transient component also increased significantly (to 149 ± 17% and 139 ± 7% of control, respectively, n = 10).
Nitric oxide can potentiate ion channel function by activation of soluble guanylate cyclase, which catalyzes an increase in cGMP and a consequent activation of protein kinase G (Ahern et al., 2002). However, application of the cell-permeable cGMP analog dibutyryl-cGMP (db-cGMP, 500 μm) had no effect on the amplitude of any of the three acid-evoked current types identified in DRG neurons. In these experiments, the sustained current was −118 ± 44 pA before and −130 ± 34 pA after application of db-cGMP (not significant, p = 0.559, n = 3); the slowly inactivating current was −2003 ± 637 pA before and −2081 ± 709 pA after application of db-cGMP (not significant, p = 0.476, n = 12); the transient phase of the rapidly inactivating current was −1878 ± 434 pA before and −1798 ± 40 pA after db-cGMP (not significant, p = 0.678, n = 4); and the sustained phase was −88 ± 24 pA before and −89 ± 22 pA after db-cGMP (not significant, p = 0.881, n = 4). To check that db-cGMP was able to cross the membrane and activate PKG, we observed its effect on GABAA channels, which are known to be inhibited by elevations in cGMP (Bie and Zhao, 2001). Perfusing the DRG neurons with db-cGMP (500 μm) decreased the amplitude of the current evoked by GABA (100 μm) to 39.82 ± 11.54%, showing that PKG is activated in DRG neurons by application of db-cGMP. In a second series of experiments, we observed the effect of the soluble PKG inhibitor 1H-[1,2,4] oxadiazole [4,3-a]quinoxalin-1-one (ODQ; 10 μm), which has been demonstrated to inhibit guanylate cyclase in rat DRG neurons (Yoshimura et al., 2001). In the presence of ODQ, application of SNAP caused an increase to 144.3 ± 12.4% (n = 5) in the transient phase and 172.0 ± 21.7% in the sustained phase of the rapidly inactivating current, values that are not significantly different from those without ODQ (p = 0.36 and p = 0.66, respectively). The failure of db-cGMP to enhance acid-modulated ion currents, and the failure of ODQ to inhibit the effect of SNAP, shows that these currents are not modulated by the intracellular cGMP-PKG pathway, and reinforces other evidence (see below) that the action of NO is external.
Effects of NO on ASICs
The effects of NO on the rat ASIC1a isoform were investigated by expression in CHO cells. ASIC1a was chosen for detailed study because it requires less extreme pH excursions than ASIC2a to activate it fully, and CHO cells were preferred for expression because they do not show any major change ion currents in response to low-pH solution application (Garcia-Anoveros et al., 2001; Yermolaieva et al., 2004; Smith et al., 2007). We confirmed that untransfected CHO cells showed little proton-activated current at pH 6.0 (−15.2 ± 3.8 pA, n = 8), and SNAP had no significant potentiating effect (change to 105.2 ± 4.0% of control, n = 5, not significant).
The NO donor SNAP caused an increase to 184.4 ± 16.2% of control (n = 9, p < 0.01) in the amplitude of the current elicited by a 5 s pulse of pH 6.9 applied to CHO cells expressing ASIC1a (Fig. 3A,B). The NO donor also had an effect on the kinetics of activation and inactivation. The time to peak current shortened from 546.6 ± 30.9 ms to 386.6 ± 36.5 (n = 9, p < 0.001), and the inactivation time constant decreased from 756.1 ± 89.8 ms to 605.9 ± 75.6 ms (n = 9, p < 0.05).
The effect of NO on the dose–response relation between H+ concentration and peak inward current carried by ASIC1a was next investigated, as shown in Figure 3, C and D. As found in other studies, inward current was steeply activated between pH 6.9 and 6.0 (Waldmann et al., 1997b), and the Hill coefficient of 2.76 ± 0.38 (n = 9) implies that three or more protons must bind before activation of the ASIC1a ion channel. The increase in current caused by exposure to SNAP was most pronounced at pH 6.9 and 6.6, whereas at pH 6.0 there was no difference between currents recorded in the presence and absence of the NO donor. This is illustrated in Figure 3D, which shows the dose–response curves in the presence and absence of SNAP. The pH50 changed from 6.40 ± 0.08 to 6.80 ± 0.03 (n = 8, p < 0.05) in the presence of the NO donor. The most striking change, however, was in the steepness of the curve. The Hill coefficient changed from 2.76 ± 0.38 in control conditions to 9.31 ± 2.77 in the presence of SNAP (n = 8, p < 0.05). The likely explanation is that NO causes a conformational change in ASIC1a which unmasks further active protonation sites.
The effect of SNAP on the pH-dependent currents in other proton-gated ASIC isoforms was tested at a single pH value, as detailed below (Fig. 3E–G).
ASIC1b
A pH of 6.0 evoked a transient inward current (−109.3 ± 22.6 pA, n = 6) which was enhanced to 157.5 ± 17.9% of control by SNAP (Fig. 3E) (p < 0.05). The inactivation time constant was shortened by NO from 1054.0 ± 81.6 ms to 852.6 ± 66.9 ms (n = 6, p < 0.05). The enhancement of the ASIC1b current by the NO donor and the change in the inactivation time constant were irreversible even after long washing, perhaps because disulfide bonds formed after exposure to NO in ASIC1b (see below) are buried at a relatively inaccessible site and so are slowly reduced on return to normal conditions.
ASIC2a
The current generated by ASIC2a is relatively sustained, in contrast with the highly transient currents generated by ASIC1a and ASIC3 (Adams et al., 1998; Zhang and Canessa, 2002). The peak current generated at pH 6.0 by ASIC2a (63.7 ± 8.6 pA, n = 5) was enhanced to 257.5 ± 17.7% of control by SNAP, an effect that was largely reversible (Fig. 3F) (p < 0.01).
ASIC3
The transient current generated by ASIC3 (Waldmann et al., 1997a; Zhang and Canessa, 2002) had peak amplitude 271 ± 92 pA at pH 6.9 and was enhanced to 225 ± 27% of control by SNAP (Fig. 3G) (n = 7, p < 0.05). The inactivation time constant was shortened by NO from 2184 ± 219 ms to 1699 ± 143 ms (n = 7, p < 0.05). The effect was largely reversible. The effect depended on NO as the current was 97.3 ± 3.4% of control (n = 5) when SNAP was allowed to decay for 4–6 h (see also similar experiments on DRG neurons described above).
In summary, all H+-gated ASIC isoforms are potentiated by nitric oxide in a manner similar to the native proton-gated currents in DRG neurons. We examined ASIC1a in greatest detail and found that the steepness of the gating by protons is enhanced by NO, consistent with the creation of new protonation site(s) by cross-linking of cysteine residues.
Site of action of NO
To investigate further the site of action of NO and other free radicals on proton-gated channels, we performed inside-out and outside-out recordings of single ASIC channels in cell-free membrane patches. We chose ASIC2a for these studies because it is the only isoform that generates a substantial sustained current at low pH (Adams et al., 1998; Zhang and Canessa, 2002), and it is therefore the only isoform suitable for single-channel experiments in which a sustained and stable period of activation is required to measure the effect of NO on channel open/closed state occupancy. In inside-out patches from nontransfected CHO cells, pH 6.0 applied to the external membrane surface did not induce single-channel activity in any patch (Fig. 4A, top trace) (n = 13), but in cells transfected with ASIC2a, single-channel activity was observed in the majority of inside-out patches (Fig. 4A, bottom traces). At −40 mV, single-channel events had an amplitude of −1.01 ± 0.04 pA (n = 11), and the open probability was low (po = 0.034 ± 0.009, n = 11). The single-channel conductance measured from the single-channel amplitude as a function of membrane potential was 11.5 ± 0.5 pS, n = 3 (data not shown).
Figure 4.
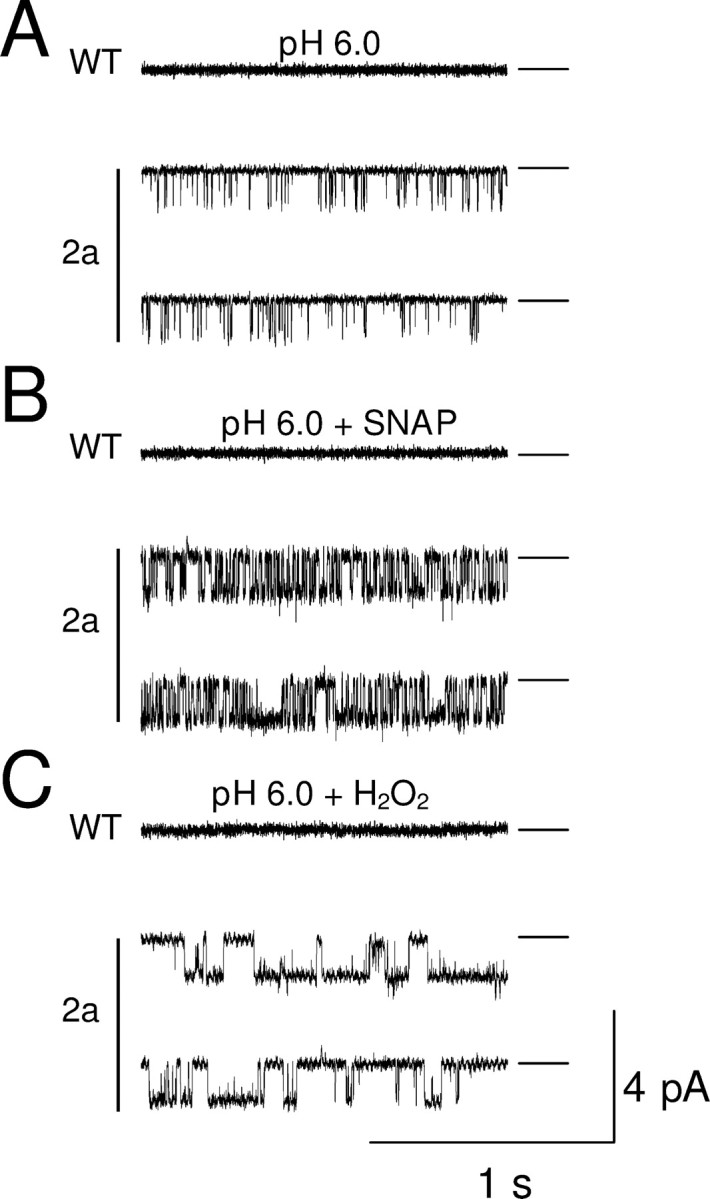
SNAP and H2O2 applied to cytoplasmic surface increase ASIC2a single-channel activity in inside-out patches from CHO cells. Applied potential −40 mV. Zero current (closed state) shown by horizontal line. A, Single-channel activity during application of pH 6.0 to external surface of patches from transfected cell (2a, bottom traces) and control cell (WT, top trace). B, Similar traces in presence of SNAP (100 μm). C, Effect of H2O2 (0.05%).
Perfusing the cytoplasmic face of inside-out patches with 100 μm SNAP dramatically enhanced ASIC2a single-channel activity (Fig. 4B; top trace shows that no activity was observed in the absence of ASIC2a). Maximal activity was seen after 1–2 min of perfusion and remained elevated as long as SNAP was present. The open probability increased to 0.564 ± 0.023 (p < 0.001, n = 7) (Fig. 5B). Openings became more frequent, had a longer duration, and occurred in clusters, whereas long closures disappeared. The amplitude of the single-channel events (−1.10 ± 0.04 pA) did not differ significantly from that in the absence of SNAP (p = 0.110). Interestingly, the increase in single-channel open probability, of 8- to 16-fold, is much larger than the increase in ASIC2a whole-cell current recorded from intact cells (2.5×) (Fig. 3F). The difference may be caused by a reduction of NO concentrations near the membrane in the whole-cell configuration by the high concentrations of the intracellular reducing agent glutathione.
Figure 5.

2-ME abolishes potentiation by SNAP of the ASIC2a proton-gated current in outside-out but not in inside-out patches. Applied potential −40 mV (inside − outside) in each case. Horizontal lines show closed state. A, pH 6.0 (top trace), pH 6.0 plus 100 μm SNAP (middle trace), and pH 6.0 plus SNAP (100 μm) and 2-ME (2 mm) in inside-out (left) and outside-out (right) configurations. B, Bar graph shows the open probabilities in inside-out (left) and outside-out (right). ***p < 0.001.
We also studied the effect of hydrogen peroxide, H2O2, another oxidizing agent that is able to oxidize thiol groups of cysteine residues in proteins (Lee et al., 2002) but is not able to activate guanylate cyclase directly. H2O2 also increased po (0.582 ± 0.029, n = 3, p < 0.001) with no change in unitary current amplitude (−1.04 ± 0.08 pA, n = 3). There was no statistical difference in amplitude or open probability between currents potentiated by SNAP and H2O2 (Student's unpaired t test: p = 0.47 and p = 0.67 respectively), but in the presence of H2O2, the transitions between the closed and the open state were longer and more stable (Fig. 4C).
NO and H2O2 are both membrane permeable and can therefore interact with cysteines on both sides of the membrane. To investigate whether extracellular or intracellular residues are involved in the potentiation of ASIC2a by NO, the effect of the thiol reducing agent 2-ME (2 mm) on SNAP-potentiated ASIC2a single-channel activity was examined in inside- and outside-out patches. 2-ME is not membrane permeable and so will act on one side of the patch of membrane only. As noted in Figure 4A, in inside-out patches from ASIC2a-transfected cells, single-channel events were seen at a low frequency when the external membrane surface (i.e., in the pipette) was exposed to pH 6.0, and perfusing the intracellular membrane surface with SNAP greatly enhances the open probability of the single-channel events (Fig. 5A, left, top and middle trace). Addition of 2-ME to the intracellular side of the membrane had no effect on the enhanced single-channel open probability caused by NO (po = 0.539 ± 0.075, n = 3) (Fig. 5A, left, bottom trace, B). This experiment suggests that NO acts at an external site. We investigated this further using outside-out patches. In the outside-out configuration, no single-channel activity was observed at pH 7.4 (n = 4) (data not shown), and exposing the external membrane surface to pH 6.0 solution evoked single-channel events indistinguishable from those seen in inside-out patches (Fig. 5A, right panel, top trace) (po = 0.076 ± 0.047 and single-channel amplitude = −1.03 ± 0.08 pA, n = 4). Addition of SNAP increased the open probability, similar to its effect on channels in the inside-out configuration (po = 0.587 ± 0.035, single-channel amplitude = −1.09 ± 0.07 pA, n = 4) (Fig. 5B). In outside-out patches, however, addition of 2-ME reduced po to 0.131 ± 0.062 (n = 4), significantly lower than that without 2-ME (Student's unpaired t test, p < 0.001), and not significantly different from the activity at pH 6.0 alone (p = 0.5) (Fig. 5B, right). The reversal of the action of NO by 2-ME present at the external membrane surface shows that the site of action of NO on ASIC2a is extracellular.
NO enhances acid-evoked pain in humans
The physiological relevance of NO for acid-evoked pain in humans was investigated by studying the action of an NO donor on the pain evoked by iontophoresis of an acid solution through the skin. Volunteers were asked to rate the intensity of pain on a visual analog scale as described previously (Jones et al., 2004). Iontophoresis of acid evoked a painful sensation which declined in intensity over the 4 min test period (Fig. 6B). Prior topical application of the NO donor glyceryl trinitrate potentiated the pain perceived during iontophoresis of acid. Potentiation of the acid-evoked pain was most marked during the first 2 min of the iontophoretic trial, with the perceived pain reaching a peak within the first minute of iontophoresis (Fig. 6B). The potentiation over this period was statistically significant when mean VAS scores were compared between GTN-treated skin and vehicle-treated skin (p < 0.05, one-way repeated measure ANOVA followed by Tukey test; n = 13) and between GTN-treated skin and untreated (naive) skin (p < 0.05, one-way repeated measure ANOVA followed by Tukey test; n = 13) (Fig. 6A). Prior treatment of the skin with GTN did not significantly potentiate the perceived pain resulting from iontophoresis of saline (Fig. 6A). Furthermore, prior treatment of the skin with GTN did not significantly affect the heat pain threshold or the mechanical pain threshold (p > 0.05, paired t test; n = 12) (Fig. 6C,D) when GTN and placebo treated skin were compared. Thus the potentiating effects of nitric oxide are specific for acid-mediated pain.
Figure 6.

Perceived pain during iontophoresis of acid before and after topical application of glyceryl trinitrate. Recordings were made by iontophoresis of acid solution, pH 2.0, or saline at a current of 0.3 mA. A, Topical application of GTN significantly increased the perceived acid mediated-pain compared with application of vehicle or to untreated skin (naive). B, Time course of the mean VAS with application of GTN, vehicle or no treatment (naive). The effect of GTN was most prominent in the first 2 min after beginning iontophoresis. C, D, Treatment of the skin with GTN did not affect heat pain threshold (C) or mechanical pain threshold (D).
Discussion
We found in the present study that the physiological proinflammatory mediator nitric oxide enhances the activation of ASICs by protons. The action is at the external membrane surface and is likely to involve the formation of new cysteine-cysteine bonds. The dependence of channel activation on pH is steepened, consistent with an unmasking of additional residues whose protonation leads to channel activation.
Proton-gated currents in DRG neurons
Application of a pH 6.3 solution to DRG neurons was found to evoke in different neurons a sustained, slowly inactivating, or rapidly inactivating inward current, in agreement with other studies (Petruska et al., 2002; Liu et al., 2004; Smith et al., 2007). The slowly and rapidly inactivating currents had similar amplitudes, but the inactivation time constants fell into two distinct groups (1239.4 ± 220.0 ms and 268.2 ± 44.8 ms, significantly different, p < 0.01) (see also Smith et al., 2007). The ASIC subunits responsible for generating these currents have not been unequivocally determined, although heteromultimers involving ASIC3 have been proposed to underlie some of these currents (Benson et al., 2002; Xie et al., 2002; Jiang et al., 2006). The sustained current component evoked at pH 6.3 may involve ASIC2a, which generates a sustained inward current in response to acidification, or ASIC3, which some studies have found to generate a sustained inward current (Waldmann et al., 1997a), although other studies have not (Zhang and Canessa, 2002). In some neurons TRPV1 contributed to the sustained phase (5/12) (Fig. 2). The sensitivity of TRPV1 to NO, which has been established in other studies (Yoshida et al., 2006), cannot, however, explain the effect of NO on the sustained phase because this phase was enhanced by NO in all neurons, whereas TRPV1 contributed to the sustained current in only a minority of neurons.
Little is known about the regulation of ASICs by inflammatory mediators. In the present study, we show that the amplitude of all three distinct acid-evoked currents in DRG neurons, and of each of the four acid-sensitive homomeric ASIC subunits, is approximately doubled by nitric oxide. A similar effect was observed when ASIC2a was exposed to the oxidizing agent H2O2.
NO acts on ASIC subunits via a direct mechanism
NO activates guanylate cyclase, increasing [cGMP] and activating PKG, but this mechanism does not contribute to the modulation of ASICs by NO. First, the potentiation was not mimicked by the cell-permeable cGMP analog db-cGMP. Second, ODQ, a guanylate cyclase inhibitor, did not reduce potentiation. Third, H2O2 does not activate guanylate cyclase, but it potentiates ASIC2a in a similar manner to NO. Finally, NO increased the open probability of the ASIC2a subunit in isolated membrane patches where the intracellular content, including the ATP necessary to sustain kinase activity, is washed away.
NO acts on ASICs through S-nitrosylation of cysteine residues
NO is present in solution as three forms, nitrosomium (NO+), nitric oxide (NO·), and nitroxyl (NO−) (Stamler et al., 1992, 1997). NO+ is the most likely of these to react with cysteine to form nitrosothiol groups, which are unstable and can combine with other adjacent nitrosothiol groups to form stable disulfide bounds. This ability to react with thiol groups is also shared with oxidizing agents such as H2O2 (Forman et al., 2004), which we found was able to mimic the effects of NO. A possible alternative mechanism of action of NO is to induce irreversible peroxidation of membrane fatty acids (Resh, 1996), but this mechanism is unlikely to account for the modulation of ASIC subunits by NO because the enhancement was reversible (except in the case of ASIC1b).
Site of action
The sequence of hASIC1a contains 15 cysteine residues in the extracellular loop. Our single-channel experiments confirm that the site of action of NO is the extracellular loop, because the membrane impermeant reducing agent 2-mercaptoethanol was able to negate the effect of NO only when applied to the extracellular domain of ASIC2a. A recent structural study shows that, in the case of ASIC1a at low pH, 14 of the extracellular cysteine residues form disulfide bonds (Jasti et al., 2007), but our observation that oxidation by NO can dramatically alter activation of ASICs implies that fewer cysteines form disulfide bonds in physiological conditions. Thiol groups of cysteines have a high reactivity because of their redox properties and can alter ion channel function by forming additional disulfide bonds after oxidation. Nitrosylation of only one cysteine residue is responsible for the activation of the olfactory cyclic nucleotide-gated channel by nitric oxide (Broillet and Firestein, 1996). In our study, in contrast with the cyclic nucleotide-gated channel, NO was unable to activate ASIC subunits directly but instead potentiated activation by protons. The formation of new disulfide bonds apparently led to the unmasking or creation of new protonation sites, because the Hill coefficient for activation by protons increased from 2.76 to 9.31.
Regulation of ASICs by oxidizing agents
Three recent studies have shown that acid-evoked currents in neurons and in heterologously expressed ASICs are modulated by a range of redox reagents (Andrey et al., 2005; Chu et al., 2006; Cho and Askwith, 2007), but in contrast to the results of the present study, these studies found that acid-evoked currents were decreased by oxidizing agents such as 5,5′-dithio-bis(2-nitrobenzoic acid (DTNB). Chu et al. (2006) found that only the ASIC1a subunit was sensitive to redox reagents, a result in contrast to the results of present study, in which we find that the activity of all ASIC isoforms is potentiated more or less equally by NO. A further difference is that we find that NO increases the ASIC-mediated current by enhancing the affinity for protons with no change in the maximum current, whereas both Andrey et al. (2005) and Chu et al. (2006) found an increase in the maximal current.
The action of the physiological oxidizing agent NO is therefore completely different from that of the nonphysiological oxidizing agents used in previous studies. The possibility that different oxidizing agents may have different effects on ion channels is supported by a recent study by Aracena-Parks et al. (2006), who studied the ryanodine receptor 1 (RyR1). Only 12 of the 100 cysteine residues can be redox-modified (the “hyper-reactive” cysteines), presumably because the majority of cysteines are buried deep in the protein and so are inaccessible to oxidizing agents. Of the 12 hyper-reactive cysteines, two are able to be S-nitrosylated but not S-glutathionylated, whereas for a further two, the reverse applies. Thus the opposite effects of NO and DTNB on members of the ASIC family can probably be attributed to oxidation of different cysteine residues by these two different oxidizing agents. Our observation that NO enhances the activation of acid-evoked currents in sensory neurons and heterologously expressed ASICs, and moreover that a similar enhancement of acid sensation is observed in human subjects after application of an NO donor, supports the idea that NO may play a role in physiological enhancement of ASIC function in vivo.
NO potentiates acid-mediated pain in humans
Iontophoresis of protons into human skin causes pain (Jones et al., 2004), and the results presented here show that topical application of an NO donor enhances acid-evoked pain but does not affect the threshold for heat or mechanical pain. In previous work, we showed that the acid-evoked pain is blocked by the ASIC blocker amiloride but is not affected by changes in temperature, and that it is therefore probably mediated by activation of members of the ASIC family rather than TRPV1 (Jones et al., 2004). The observation that acid-evoked pain is modulated by NO (Fig. 6A,B), whereas the threshold for heat-evoked pain is unaffected (Fig. 6C), provides additional evidence in support of the proposal that physiological acidification is sensed by members of the ASIC family rather than TRPV1, and in addition suggests that modulation of TRPV1 by NO (Yoshida et al., 2006) is less physiologically significant than modulation of ASICs. We note, however, that all our experiments were performed on normal skin, and the picture may be different after inflammation.
Possible pathophysiological implications
The potentiation of ASICs activity by nitric oxide is likely to be of physiological relevance in pathologies in which both tissue acidosis and a high level of NO and/or other free radicals may occur together. For example, in rheumatoid arthritis, a disease characterized by a progressive destruction of articular bone and cartilage, the pH can drop dramatically at the site of the inflammation (Grinstein et al., 1991). In inflammatory conditions, iNOS in chondrocytes is stimulated by interleukin 17, leading to the production of NO (Miljkovic and Trajkovic, 2004). In contrast to the two other isoforms of the enzyme, iNOS can produce nitric oxide in the nanomolar range, concentrations that are cytotoxic. Pathophysiological conditions such as heart failure see a pronounced enhancement in NO-based signaling (for review, see Hare and Stamler, 2005). ASIC3 has been proposed to mediate cardiac pain, because the proton-gated current in sensory neurons innervating the heart resembles that observed from ASIC3 homomers in an expression system (Immke and McCleskey, 2001; Sutherland et al., 2001). An enhancement of ASIC3 function by NO, as we show in the present study, may therefore play an important role in cardiac pain.
Footnotes
This work was supported by a Biotechnology and Biological Sciences Research Council grant to P. McNaughton and S. B. McMahon, by a Medical Research Council PhD studentship to E. St. J. Smith, and by a PhD studentship funded by Merck, Sharpe, and Dohme to M. Studer. We thank Prof. Michel Lazdunski for providing us with ASIC clones, Dr. Robin Hiley for useful discussions about the physiology and chemistry of NO, and Dr. Felix Viana for providing us with BCTC.
References
- Adams CM, Snyder PM, Price MP, Welsh MJ. Protons activate brain Na+ channel 1 by inducing a conformational change that exposes a residue associated with neurodegeneration. J Biol Chem. 1998;273:30204–30207. doi: 10.1074/jbc.273.46.30204. [DOI] [PubMed] [Google Scholar]
- Ahern GP, Klyachko VA, Jackson MB. cGMP and S-nitrosylation: two routes for modulation of neuronal excitability by NO. Trends Neurosci. 2002;25:510–517. doi: 10.1016/s0166-2236(02)02254-3. [DOI] [PubMed] [Google Scholar]
- Akopian AN, Chen CC, Ding Y, Cesare P, Wood JN. A new member of the acid-sensing ion channel family. NeuroReport. 2000;11:2217–2222. doi: 10.1097/00001756-200007140-00031. [DOI] [PubMed] [Google Scholar]
- Allen NJ, Attwell D. Modulation of ASIC channels in rat cerebellar Purkinje neurons by ischaemia-related signals. J Physiol (Lond) 2002;543:521–529. doi: 10.1113/jphysiol.2002.020297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrey F, Tsintsadze T, Volkova T, Lozovaya N, Krishtal O. Acid sensing ionic channels: modulation by redox reagents. Biochim Biophys Acta. 2005;1745:1–6. doi: 10.1016/j.bbamcr.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Aracena-Parks P, Goonasekera SA, Gilman CP, Dirksen RT, Hidalgo C, Hamilton SL. Identification of cysteines involved in S-nitrosylation, S-glutathionylation, and oxidation to disulfides in ryanodine receptor type 1. J Biol Chem. 2006;281:40354–40368. doi: 10.1074/jbc.M600876200. [DOI] [PubMed] [Google Scholar]
- Benson CJ, Xie J, Wemmie JA, Price MP, Henss JM, Welsh MJ, Snyder PM. Heteromultimers of DEG/ENaC subunits form H+-gated channels in mouse sensory neurons. Proc Natl Acad Sci USA. 2002;99:2338–2343. doi: 10.1073/pnas.032678399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bie BH, Zhao ZQ. Nitric oxide inhibits GABA-evoked current in dorsal root ganglion neuron via PKG-dependent pathway. Brain Res Bull. 2001;55:335–339. doi: 10.1016/s0361-9230(01)00500-7. [DOI] [PubMed] [Google Scholar]
- Bolotina VM, Najibi S, Palacino JJ, Pagano PJ, Cohen RA. Nitric oxide directly activates calcium-dependent potassium channels in vascular smooth muscle. Nature. 1994;368:850–853. doi: 10.1038/368850a0. [DOI] [PubMed] [Google Scholar]
- Broillet MC, Firestein S. Direct activation of the olfactory cyclic nucleotide-gated channel through modification of sulfhydryl groups by NO compounds. Neuron. 1996;16:377–385. doi: 10.1016/s0896-6273(00)80055-0. [DOI] [PubMed] [Google Scholar]
- Castel H, Vaudry H. Nitric oxide directly activates GABA(A) receptor function through a cGMP/protein kinase-independent pathway in frog pituitary melanotrophs. J Neuroendocrinol. 2001;13:695–705. doi: 10.1046/j.1365-2826.2001.00683.x. [DOI] [PubMed] [Google Scholar]
- Chen CC, England S, Akopian AN, Wood JN. A sensory neuron-specific, proton-gated ion channel. Proc Natl Acad Sci USA. 1998;95:10240–10245. doi: 10.1073/pnas.95.17.10240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho JH, Askwith C. Potentiation of acid-sensing ion channels by sulfhydryl compounds. Am J Physiol Cell Physiol. 2007:C2161–C2174. doi: 10.1152/ajpcell.00598.2006. [DOI] [PubMed] [Google Scholar]
- Chu XP, Close N, Saugstad JA, Xiong ZG. ASIC1a-specific modulation of acid-sensing ion channels in mouse cortical neurons by redox reagents. J Neurosci. 2006;26:5329–5339. doi: 10.1523/JNEUROSCI.0938-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobbe SM, Parker DJ, Poole-Wilson PA. Tissue and coronary venous pH in ischemic canine myocardium. Clin Cardiol. 1982;5:153–156. doi: 10.1002/clc.4960050206. [DOI] [PubMed] [Google Scholar]
- Davis KL, Martin E, Turko IV, Murad F. Novel effects of nitric oxide. Annu Rev Pharmacol Toxicol. 2001;41:203–236. doi: 10.1146/annurev.pharmtox.41.1.203. [DOI] [PubMed] [Google Scholar]
- Forman HJ, Fukuto JM, Torres M. Redox signaling: thiol chemistry defines which reactive oxygen and nitrogen species can act as second messengers. Am J Physiol Cell Physiol. 2004;287:C246–C256. doi: 10.1152/ajpcell.00516.2003. [DOI] [PubMed] [Google Scholar]
- Garcia-Anoveros J, Samad TA, Zuvela-Jelaska L, Woolf CJ, Corey DP. Transport and localization of the DEG/ENaC ion channel BNaC1alpha to peripheral mechanosensory terminals of dorsal root ganglia neurons. J Neurosci. 2001;21:2678–2686. doi: 10.1523/JNEUROSCI.21-08-02678.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabowski PS, Wright PK, Van 't Hof RJ, Helfrich MH, Ohshima H, Ralston SH. Immunolocalization of inducible nitric oxide synthase in synovium and cartilage in rheumatoid arthritis and osteoarthritis. Br J Rheumatol. 1997;36:651–655. doi: 10.1093/rheumatology/36.6.651. [DOI] [PubMed] [Google Scholar]
- Grinstein S, Swallow CJ, Rotstein OD. Regulation of cytoplasmic pH in phagocytic cell function and dysfunction. Clin Biochem. 1991;24:241–247. doi: 10.1016/0009-9120(91)80014-t. [DOI] [PubMed] [Google Scholar]
- Hare JM, Stamler JS. NO/redox disequilibrium in the failing heart and cardiovascular system. J Clin Invest. 2005;115:509–517. doi: 10.1172/JCI200524459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Zhang X, McNaughton PA. Inflammatory pain: the cellular basis of heat hyperalgesia. Current Neuropharmacology. 2006;4:197–206. doi: 10.2174/157015906778019554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Immke DC, McCleskey EW. ASIC3: a lactic acid sensor for cardiac pain. ScientificWorldJournal. 2001;1:510–512. doi: 10.1100/tsw.2001.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issberner U, Reeh PW, Steen KH. Pain due to tissue acidosis: a mechanism for inflammatory and ischemic myalgia? Neurosci Lett. 1996;208:191–194. doi: 10.1016/0304-3940(96)12576-3. [DOI] [PubMed] [Google Scholar]
- Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol. 2001;3:193–197. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- Jasti J, Furukawa H, Gonzales EB, Gouaux E. Structure of acid-sensing ion channel 1 at 1.9 A resolution and low pH. Nature. 2007;449:316–323. doi: 10.1038/nature06163. [DOI] [PubMed] [Google Scholar]
- Jiang N, Rau KK, Johnson RD, Cooper BY. Proton sensitivity Ca2+ permeability and molecular basis of acid-sensing ion channels expressed in glabrous and hairy skin afferents. J Neurophysiol. 2006;95:2466–2478. doi: 10.1152/jn.00861.2005. [DOI] [PubMed] [Google Scholar]
- Jones NG, Slater R, Cadiou H, McNaughton P, McMahon SB. Acid-induced pain and its modulation in humans. J Neurosci. 2004;24:10974–10979. doi: 10.1523/JNEUROSCI.2619-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishtal O. The ASICs: signaling molecules? Modulators? Trends Neurosci. 2003;26:477–483. doi: 10.1016/S0166-2236(03)00210-8. [DOI] [PubMed] [Google Scholar]
- Krishtal OA, Pidoplichko VI. A receptor for protons in the nerve cell membrane. Neuroscience. 1980;5:2325–2327. doi: 10.1016/0306-4522(80)90149-9. [DOI] [PubMed] [Google Scholar]
- Lee CS, Jang YY, Song JS, Song JH, Han ES. Ambroxol inhibits peroxynitrite-induced damage of alpha1-antiproteinase and free radical production in activated phagocytic cells. Pharmacol Toxicol. 2002;91:140–149. doi: 10.1034/j.1600-0773.2002.910309.x. [DOI] [PubMed] [Google Scholar]
- Lei SZ, Pan ZH, Aggarwal SK, Chen HS, Hartman J, Sucher NJ, Lipton SA. Effect of nitric oxide production on the redox modulatory site of the NMDA receptor-channel complex. Neuron. 1992;8:1087–1099. doi: 10.1016/0896-6273(92)90130-6. [DOI] [PubMed] [Google Scholar]
- Lingueglia E, De Weille JR, Bassilana F, Heurteaux C, Sakai H, Waldmann R, Lazdunski M. A modulatory subunit of acid sensing ion channels in brain and dorsal root ganglion cells. J Biol Chem. 1997;272:29778–29783. doi: 10.1074/jbc.272.47.29778. [DOI] [PubMed] [Google Scholar]
- Liu M, Willmott NJ, Michael GJ, Priestley JV. Differential pH and capsaicin responses of Griffonia simplicifolia IB4 (IB4)-positive and IB4-negative small sensory neurons. Neuroscience. 2004;127:659–672. doi: 10.1016/j.neuroscience.2004.05.041. [DOI] [PubMed] [Google Scholar]
- Mander P, Borutaite V, Moncada S, Brown GC. Nitric oxide from inflammatory-activated glia synergizes with hypoxia to induce neuronal death. J Neurosci Res. 2005;79:208–215. doi: 10.1002/jnr.20285. [DOI] [PubMed] [Google Scholar]
- Manzoni O, Bockaert J. Nitric oxide synthase activity endogenously modulates NMDA receptors. J Neurochem. 1993;61:368–370. doi: 10.1111/j.1471-4159.1993.tb03580.x. [DOI] [PubMed] [Google Scholar]
- Miljkovic D, Trajkovic V. Inducible nitric oxide synthase activation by interleukin-17. Cytokine Growth Factor Rev. 2004;15:21–32. doi: 10.1016/j.cytogfr.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Petruska JC, Napaporn J, Johnson RD, Cooper BY. Chemical responsiveness and histochemical phenotype of electrophysiologically classified cells of the adult rat dorsal root ganglion. Neuroscience. 2002;115:15–30. doi: 10.1016/s0306-4522(02)00409-8. [DOI] [PubMed] [Google Scholar]
- Price MP, Snyder PM, Welsh MJ. Cloning and expression of a novel human brain Na+ channel. J Biol Chem. 1996;271:7879–7882. doi: 10.1074/jbc.271.14.7879. [DOI] [PubMed] [Google Scholar]
- Rawlingson A. Nitric oxide, inflammation and acute burn injury. Burns. 2003;29:631–640. doi: 10.1016/s0305-4179(03)00079-2. [DOI] [PubMed] [Google Scholar]
- Resh MD. Regulation of cellular signalling by fatty acid acylation and prenylation of signal transduction proteins. Cell Signal. 1996;8:403–412. doi: 10.1016/s0898-6568(96)00088-5. [DOI] [PubMed] [Google Scholar]
- Smith ES, Cadiou H, McNaughton PA. Arachidonic acid potentiates acid-sensing ion channels in rat sensory neurons by a direct action. Neuroscience. 2007;145:686–698. doi: 10.1016/j.neuroscience.2006.12.024. [DOI] [PubMed] [Google Scholar]
- Stamler JS. Redox signaling: nitrosylation and related target interactions of nitric oxide. Cell. 1994;78:931–936. doi: 10.1016/0092-8674(94)90269-0. [DOI] [PubMed] [Google Scholar]
- Stamler JS, Singel DJ, Loscalzo J. Biochemistry of nitric oxide and its redox-activated forms. Science. 1992;258:1898–1902. doi: 10.1126/science.1281928. [DOI] [PubMed] [Google Scholar]
- Stamler JS, Toone EJ, Lipton SA, Sucher NJ. (S)NO signals: translocation, regulation, and a consensus motif. Neuron. 1997;18:691–696. doi: 10.1016/s0896-6273(00)80310-4. [DOI] [PubMed] [Google Scholar]
- Sutherland SP, Benson CJ, Adelman JP, McCleskey EW. Acid-sensing ion channel 3 matches the acid-gated current in cardiac ischemia-sensing neurons. Proc Natl Acad Sci USA. 2001;98:711–716. doi: 10.1073/pnas.011404498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, Skinner K, Raumann BE, Basbaum AI, Julius D. The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron. 1998;21:531–543. doi: 10.1016/s0896-6273(00)80564-4. [DOI] [PubMed] [Google Scholar]
- Valenzano KJ, Grant ER, Wu G, Hachicha M, Schmid L, Tafesse L, Sun Q, Rotshteyn Y, Francis J, Limberis J, Malik S, Whittemore ER, Hodges D. N-(4-tertiarybutylphenyl)-4-(3-chloropyridin-2-yl)tetrahydropyrazine-1(2H)-carbox-amide (BCTC), a novel, orally effective vanilloid receptor 1 antagonist with analgesic properties: I. In vitro characterization and pharmacokinetic properties. J Pharmacol Exp Ther. 2003;306:377–386. doi: 10.1124/jpet.102.045674. [DOI] [PubMed] [Google Scholar]
- Waldmann R, Bassilana F, de Weille J, Champigny G, Heurteaux C, Lazdunski M. Molecular cloning of a non-inactivating proton-gated Na+ channel specific for sensory neurons. J Biol Chem. 1997a;272:20975–20978. doi: 10.1074/jbc.272.34.20975. [DOI] [PubMed] [Google Scholar]
- Waldmann R, Champigny G, Bassilana F, Heurteaux C, Lazdunski M. A proton-gated cation channel involved in acid-sensing. Nature. 1997b;386:173–177. doi: 10.1038/386173a0. [DOI] [PubMed] [Google Scholar]
- Waldmann R, Champigny G, Lingueglia E, De Weille JR, Heurteaux C, Lazdunski M. H(+)-gated cation channels. Ann NY Acad Sci. 1999;868:67–76. doi: 10.1111/j.1749-6632.1999.tb11274.x. [DOI] [PubMed] [Google Scholar]
- Xie J, Price MP, Berger AL, Welsh MJ. DRASIC contributes to pH-gated currents in large dorsal root ganglion sensory neurons by forming heteromultimeric channels. J Neurophysiol. 2002;87:2835–2843. doi: 10.1152/jn.2002.87.6.2835. [DOI] [PubMed] [Google Scholar]
- Xie J, Price MP, Wemmie JA, Askwith CC, Welsh MJ. ASIC3 and ASIC1 mediate FMRFamide-related peptide enhancement of H+-gated currents in cultured dorsal root ganglion neurons. J Neurophysiol. 2003;89:2459–2465. doi: 10.1152/jn.00707.2002. [DOI] [PubMed] [Google Scholar]
- Yermolaieva O, Leonard AS, Schnizler MK, Abboud FM, Welsh MJ. Extracellular acidosis increases neuronal cell calcium by activating acid-sensing ion channel 1a. Proc Natl Acad Sci USA. 2004;101:6752–6757. doi: 10.1073/pnas.0308636100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Inoue R, Morii T, Takahashi N, Yamamoto S, Hara Y, Tominaga M, Shimizu S, Sato Y, Mori Y. Nitric oxide activates TRP channels by cysteine S-nitrosylation. Nat Chem Biol. 2006;2:596–607. doi: 10.1038/nchembio821. [DOI] [PubMed] [Google Scholar]
- Yoshimura N, Seki S, de Groat WC. Nitric oxide modulates Ca(2+) channels in dorsal root ganglion neurons innervating rat urinary bladder. J Neurophysiol. 2001;86:304–311. doi: 10.1152/jn.2001.86.1.304. [DOI] [PubMed] [Google Scholar]
- Zhang P, Canessa CM. Single channel properties of rat acid-sensitive ion channel-1alpha, -2a, and -3 expressed in Xenopus oocytes. J Gen Physiol. 2002;120:553–566. doi: 10.1085/jgp.20028574. [DOI] [PMC free article] [PubMed] [Google Scholar]