

Abstract
Mice heterozygous for the radiation-induced Sprawling (Swl) mutation display an early-onset sensory neuropathy with muscle spindle deficiency. The lack of an H reflex despite normal motor nerve function in the hindlimbs of these mutants strongly suggests defective proprioception. Immunohistochemical analyses reveal that proprioceptive sensory neurons are severely compromised in the lumbar dorsal root ganglia of newborn Swl/+ mice, whereas motor neuron numbers remain unaltered even in aged animals. We have used positional cloning to identify a nine base-pair deletion in the cytoplasmic dynein heavy chain 1 gene (Dync1h1) in this mutant. Furthermore, we demonstrate that Loa/+ mice, which have previously been shown to carry a missense point mutation in Dync1h1 that results in late-onset motor neuron loss, also present with a severe, early-onset proprioceptive sensory neuropathy. Interestingly, in contrast to the Loa mutation, the Swl mutation does not delay disease progression in a motor neuron disease mouse model overexpressing a human mutant superoxide dismutase (SOD1G93A) transgene. Together, we provide in vivo evidence that distinct mutations in cytoplasmic dynein can either result in a pure sensory neuropathy or in a sensory neuropathy with motor neuron involvement.
Keywords: Charcot-Marie-Tooth disease, dynein, sensory neuron degeneration, dorsal root ganglia, gene deletion, mouse mutant
Introduction
Occurring with an estimated prevalence of 1 in 2500, Charcot-Marie-Tooth disease (CMT) encompasses a group of clinically and genetically heterogeneous disorders of the peripheral nervous system that affect both motor and sensory neurons (Skre, 1974). In contrast, in hereditary sensory neuropathy (HSN) sensory neuron involvement predominates (Verpoorten et al., 2006). The clinical hallmarks of HSN include sensory loss with a varying degree of motor involvement, decreased or absent reflexes, foot deformities and various autonomic features.
Previous reports have indicated that axonal trafficking may play an important role in sensory neuron degeneration underlying HSN. Mutations in the neurotrophic tyrosine kinase receptor type 1 (TrkA) gene and the nerve growth factor (NGF) β gene have been found in patients with the loss of nociception (Indo et al., 1996; Einarsdottir et al., 2004). The activated NGF/TrkA complex, essential for sensory neuron survival, is thought to be retrogradely transported by a minus-end-directed motor protein, dynein, from nerve terminals to neuronal cell bodies (Heerssen et al., 2004). Moreover, null mutations in the bullous pemphigoid antigen 1 (BPAG1) gene have been shown to result in progressive sensory neuron degeneration in Dystonia Musculorum (dt) mice, an animal model for HSN in humans (Guo et al., 1995). In vitro evidence has suggested that a neuronal isoform of BPAG1, interacting with retrograde motor proteins, is essential for sensory neuronal survival (Liu et al., 2003).
Although less well understood, defects in axonal transport attributable to mutated motor proteins have recently been implicated in neuronal degeneration. KIF1B is a plus-end-directed kinesin superfamily motor protein that is thought to transport mitochondria (Nangaku et al., 1994). Mice that are heterozygous for a Kif1b null allele display a neuropathy-like phenotype (Zhao et al., 2001). Two independent N-ethyl-N-nitrosourea (ENU)-induced missense point mutations in the cytoplasmic dynein heavy chain 1 (DYNC1H1) gene, Loa and Cra1, demonstrate late-onset motor neuron degeneration in heterozygous mice (Hafezparast et al., 2003). Similarly, mutations in a dynein-associated protein, dynactin, have been identified in families with slowly progressive forms of motor neuron disease (Puls et al., 2005). Although recent studies have underscored the importance of dynein-dependent axonal transport in the survival of motor neurons, the role of the dynein motor proteins in sensory neuron survival has not been described.
Mice heterozygous for the radiation-induced mutation Sprawling (Swl) display an early-onset HSN with muscle spindle deficiency (Duchen, 1974, 1975; Duchen and Scaravilli, 1977a,b; Scaravilli and Duchen, 1980a,b; Brook and Duchen, 1990). In this report, we use a positional cloning approach to identify a nine base-pair deletion in Dync1h1 in the Swl mutants. We demonstrate that the mutation markedly disrupts the viability of proprioceptive sensory neurons. Additionally, we show that the Swl Dync1h1 mutation, in contrast to the Loa mutation, does not delay disease progression in a motor neuron disease mouse model expressing a human mutant SODG93A transgene. We thus provide evidence that mutations in cytoplasmic dynein lead to sensory neuron degeneration in mice.
Materials and Methods
Mice.
Swl/+ mutant mice were derived from frozen embryos provided by the Mammalian Genetic Unit/Harwell (Oxfordshire, UK). The mutant line is maintained on a C3H/101 hybrid genetic background. The presence of hindlimb flexion while suspended by the tail after postnatal day 7 (P7) and the typical unsteady gait in adulthood were used to determine the mutant genotype in the linkage mapping experiment. The polymorphic markers closely associated with the Swl locus were used to genotype the mice until the Swl mutation was identified. Genotyping was performed by PCR of tail DNA. The amplification of exon 12 of Dync1h1 by the forward primer 5′-GGTGGGAGTGCATTACGAGT-3′, and the reverse primer 5′-TCTCTCGCTCTCTTGGTGGT-3′ yields a 269-bp PCR product. The mutant allele is 260 bp in size. The Loa congenic line, which originated in Dr. Elizabeth Fisher's laboratory (Hafezparast et al., 2003), was maintained on the C57BL/6J background. Genotyping of Loa mice was performed by PCR and RsaI restriction enzyme digestion as described previously (Hafezparast et al., 2003). Hemizygote male SOD1G93A mice (Stock# 002726; The Jackson Laboratory, Bar Harbor, ME) were maintained on the B6/SJL background and crossed with female Swl/+ mice or Loa/+ mice. All progeny were genotype for the human SOD1 transgene as reported (Gurney et al., 1994). The disease end-stage in the mice was defined as a loss of the righting reflex and 20% body weight loss (Kieran et al., 2005). For developmental studies, the day on which a plug was detected in the morning was considered as embryonic day 0.5 (E0.5). We monitored mice daily, and all procedures were approved by the University of Chicago Laboratory Animal Care and Use Committee.
Positional cloning of Swl mice.
In light of the fully penetrant, autosomal dominant inheritance of the Swl mutation (Duchen, 1974), one male rederived Swl/+ mouse was crossed with 8–10 female C57BL/6J mice (The Jackson Laboratory, Bar Harbor, ME), and F1 progeny were then backcrossed to the C57BL/6J mice. Only markers showing polymorphism between C57BL/6J strain (The Jackson Laboratory) and C3H/HeH strain or 101/H strain (Mammalian Genetic Unit/Harwell) were used for PCR-based typing. Some polymorphic markers, such as D12mgh9, were developed by selecting a number of noncoding sequences evenly distributed within the interval for PCR amplification from both C3H/HeH and C57BL/6J genomic DNA. All genotyping data were analyzed using MapManger QTX software (Manly and Olson, 1999). Direct sequencing of candidate genes was performed using standard dye-terminator chemistry at the Cancer Research Center DNA Sequencing Facility of the University of Chicago. Chromatograms were generated using the Sequencher software (Gene Codes, Ann Arbor, MI).
Grip strength test and tail flick test.
Grip strength test was measured using a computerized grip strength meter from Columbus Instruments (Columbus, OH) as described (Levedakou et al., 2005). Ten consecutive measurements were averaged as tension force (Newton, N) for each animal. Thermal nociception was measured using an analgesia tail flick apparatus from Columbus Instruments (Columbus, OH). A beam of intense light was applied to the mouse's tail, resulting in discomfort that induces the animal to flick its tail out of the beam. The elapsed time between beam activation and animal reaction was recorded at low intensity (level 4) and high intensity (level 10). Two trials of three measurements per trial were averaged for each mouse.
Electrophysiology.
Nerve conduction velocity (NCV) and H reflex tests were performed with a Viking Quest System from Nicolet Biomedical (Madison, WI). Motor NCV was performed in the sciatic nerves as described previously (Levedakou et al., 2005). Sensory NCV was assessed in the sural nerves stimulated in the distal nerve terminal behind the knee and recorded proximally. Mice were anesthetized by intraperitoneal injection with Avertin (0.5 mg/g) and the body temperature was maintained at 30°C-32°C under a heating lamp. The H reflex tests were performed on awakening animals immobilized by an acrylic restrainer. Single stimuli produced by the constant-current stimulator were delivered to the tibial nerves in the popliteal fossa or at the medial malleolus. Recording needle electrodes were placed subcutaneously in the hind footpad. Submaximal stimulation was used to obtain the maximal H wave, either before M wave was seen or after a small M wave was observed. Each side of hindlimbs was examined before a negative result of the H reflex was reported.
Antibodies.
Rabbit polyclonal anti-collagen type IV antibody (dilution, 1:40) and mouse monoclonal anti-neuronal nuclei (NeuN) antibody (dilution, 1:200) were purchased from Chemicon (Temecula, CA). Rabbit polyclonal antibody raised against amino acids 4320–4644 of rat dynein heavy chain (dilution, 1:50) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit polyclonal anti-parvalbumin (PV) antibody (dilution, 1:5000) was purchased from Swant (Bellinzona, Switzerland). Rabbit polyclonal anti-calcitonin gene related peptide (CGRP) antibody (dilution, 1:100) was obtained from Biogenesis (Oxford, UK). Mouse monoclonal antibody S46 (dilution, 1:50) was a generous gift from Dr. Frank Stockdale (Stanford University). Cy3-conjugated and FITC-conjugated secondary antibodies were obtained from Jackson ImmunoResearch (West Grove, PA).
Histology, Epon section, and immunofluorescence staining.
Histology, Epon section and immunofluorescence stainings were performed as described (Levedakou et al., 2005). Briefly, animals, once deeply anesthetized by intraperitoneal injection with Avertin (0.5 mg/g), were subjected to transcardial perfusion with either 4% paraformaldehyde in 0.1 m Millonig's buffer, pH 7.4, for paraffin and frozen block processing, or a mixture of 4% paraformaldehyde and 2.5% glutaraldehyde in 0.1 m Millonig's buffer, pH 7.4, for Epon section processing. After 30 min to overnight fixation in the same solution, harvested tissues were either cryoprotected by 30% sucrose in 1 × PBS for cryosection or processed for paraffin and Epon embedding. For dorsal root ganglia (DRG) neuron analysis, the cervical and lumbar spinal segments were subsequently decalcified by immersion in 14% EDTA solution, pH 7.0, for 2–3 weeks. Embryos were immersion fixed by 4% paraformaldehyde in 0.1 m Millonig's buffer, pH 7.4, for 2 h at room temperature.
Reverse transcriptase-PCR.
Reverse transcriptase (RT)-PCR was performed as described (Levedakou et al., 2005) and was analyzed by 12% PAGE.
Neuron counts.
Neuron counts were performed on 7 μm serial paraffin-embedded sections of mouse spinal cords and DRGs. For motor neuron count, every ninth section in the series from lumbar (L)2–L4 spinal cord segment was immunostained with NeuN antibody. A total of 49 sections per animal were analyzed using NIH Image J at a calibration of 1.5 pixels/μm. A circular area of 0.2 mm2 within the anterior horn of the spinal cord was selected. Only cells that had a visible nucleolus, characteristic morphological features of α-motor neuron, and a minimum diameter of 13.0 μm were counted (Hafezparast et al., 2003). For DRG neuron counts, every sixth (P91) or every fourth (E15.5 and P0.5) section in the series from the cervical (C)4 or L4 DRGs was stained with hematoxylin and eosin (H&E). The second set and the third set of sections from the P91 L4 DRGs were immunostained with PV antibody and CGRP antibody, respectively, for the DRG subpopulation analysis. Neurons were counted at a magnification of ×20 using an Axiovert-25 microscope with digital imaging from Carl Zeiss MicroImaging (Göttingen, Germany).
Statistical analysis.
All data are expressed as the mean ± SEM. Data from body weight, grip strength test, tail flick test, electrophysiologic tests, muscle spindle and neuron counts were analyzed by a two-tailed distribution, two-sample equal, or unequal variance Student's t test. The variance equality was examined by F test. If the data were nonparametric, the Mann–Whitney rank-sum test was used. For survival time, the Kaplan-Meier survival analyses by log-rank test for curve equality were performed using Stata software (StataCorp LP, College Station, TX). Differences were considered to be statistically significant if p < 0.05.
Results
Reduced hindlimb grip strength in Swl/+ mice does not result from an impaired motor pathway
Swl/+ mice are distinguishable ∼1 week after birth by the presence of hindlimb flexion during tail suspension, which is more evident in adulthood (Fig. 1A). At 3–4 weeks of age, they develop a typical unsteady gait characterized by jerky and wobbly locomotion (Fig. 1B) (supplemental video 1, available at www.jneurosci.org as supplemental material). At rest, the hindlimbs of Swl/+ mice are splayed and flexed forward. Additionally, their hindpaws are incapable of gripping structures (Fig. 1C) (supplemental video 2, available at www.jneurosci.org as supplemental material). The forelimb's locomotion is not noticeably impaired. Mutant mice have a normal gestation period, lactation ability, litter size, and life span. No obvious progression of the phenotype with age is noted in Swl/+ mice. There is no overt atrophy observed in limb or truncal muscles.
Figure 1.

Swl/+ mice display abnormal hindlimb function. Abnormal postures of Swl/+ mice while being lifted by the tail (A), during locomotion (B), and being placed on a wired structure (C). D, Motor function examined by a grip strength test shows no difference in forelimb between male Swl/+ mice and WT mice, whereas hindlimb grip strength is reduced by 50% in Swl/+ mice (*p < 0.01, Student's t test). E, Nociceptive function measured by a tail flick test is comparable between Swl/+ mice and WT mice. F, H reflex, which represents the integrity of peripheral proprioception and spinal monosynaptic pathway, is absent in Swl/+ mice, whereas it was presented in WT mice.
A grip strength test and motor NCV were performed to discern whether motor function is impaired in Swl/+ mice. Forelimb and hindlimb grip strength was measured in 13-week-old male Swl/+ mutants and male wild-type (WT) littermate mice. The forelimb grip strength of Swl/+ mutants was comparable with that of WT mice, whereas a 50% reduction of grip strength was detected in the hindlimbs of Swl/+ mice (n = 5) (Fig. 1D). Supramaximal stimuli were delivered to the sciatic nerves of 13-week-old Swl/+ and WT littermate mice, predominantly activating the large-caliber motor nerve fibers. Neither a slowing of velocity nor a reduction in amplitude of the compound muscle action potential (M wave) was detected in Swl/+ mice (n = 13), indicating that the peripheral motor nerve function was normal (Table 1).
Table 1.
Electrophysiologic analysis of motor nerves and sensory nerves in Swl/+ and wild-type mice
| Parameters | Units | WT (n = 12) | Swl/+ (n = 13) |
|---|---|---|---|
| MDL | ms | 1.4 ± 0.09 | 1.4 ± 0.08 |
| MPL | ms | 1.9 ± 0.10 | 1.9 ± 0.09 |
| MCV | m/s | 30.1 ± 1.32 | 27.7 ± 1.25 |
| MAmp (D) | mV | 27.0 ± 3.95 | 28.2 ± 3.54 |
| MAmp (P) | mV | 24.8 ± 3.75 | 26.5 ± 3.64 |
| SL | ms | 0.5 ± 0.02 | 0.6 ± 0.03 |
| SAmp | μV | 21.3 ± 1.13 | 14.5 ± 1.30* |
| SCV | m/s | 22.9 ± 0.85 | 20.9 ± 0.97 |
In sciatic nerves, distal latencies (MDL), proximal latencies (MPL), motor nerve conduction velocities (MCV), and distal (D) and proximal (P) compound muscle action potential amplitudes (MAmp) are measured. In sural nerves latencies (SL), sensory nerve action potential amplitudes (SAmp) and sensory nerve conduction velocity (SCV) are measured. Tests are performed in 13-week-old Swl/+ mice and age-matched WT mice. No significant difference is detected between two groups, except that the sensory potential amplitude is reduced by 32% in Swl/+ mice.
*p < 0.01, Mann–Whitney rank-sum test.
To determine whether the reduced hindlimb grip strength in Swl/+ mice was attributable to anterior horn α-motor neuron defects in the lumbar spinal cord, we examined the morphology and number of α-motor neurons in animals as old as two years. Neither overt morphological alterations nor a significant reduction in total number of α-motor neurons were detected in the lumbar spinal cords in Swl/+ mice (1355 ± 139 neurons in L2-L4 segment of Swl/+ mice vs 1447 ± 79 neurons in L2-L4 segment of WT mice, p > 0.05, Student's t test, n = 3 per genotype).
Swl/+ mice demonstrate a specific proprioceptive sensory defect
Swl/+ mice were previously characterized as having thin peripheral nerves and sensory roots, a reduced number of DRG neurons, small dorsal columns of the spinal cord, and a marked reduction in the number of sensory receptors (Duchen, 1975). We asked whether a specific subpopulation of sensory ganglion neurons is affected in Swl/+ mice. First, nociceptive sensory function was assessed using a tail flick test in which thermal stimuli at low and high intensities were applied to the tails of mice. These tests demonstrated that nociception is unaffected in Swl/± mice (n = 9) (Fig. 1E), which is corroborated by an absence of limb amputation in these animals.
Next, sensory NCV was measured in the sural nerves to evaluate sensory nerve function in 13-week-old Swl/+ mice and WT littermate mice. Although sensory NCV of Swl/+ mice was comparable with WT mice, the sensory nerve action potential amplitude was reduced by 32% (n = 12 for WT and n = 13 for Swl/+) (Table 1). Both NCV and tail flick tests suggest that Swl/+ mice have a moderate sensory neuropathy that does not affect the nociceptive modality or motor function.
The distinctive unsteady gait and inability to grip despite normal motor function prompted us to investigate the possibility of a proprioceptive sensory defect in Swl/+ mice. We tested the H reflex in the tibial nerves of 13-week-old Swl/+ mutants and WT littermate mice. The H reflex, which measures the latency over the monosynaptic reflex arc through the afferent Ia fibers and efferent α-motor fibers, primarily represents proprioceptive function (Shibasaki et al., 2004). Strikingly, the H reflex was not detectable in the hindlimbs of any of the Swl/+ mice examined (n = 8), but it was present in all WT mice (n = 6) (Fig. 1F). The absence of the H reflex despite normal motor nerve function indicates that the proprioceptive modality of the sensory function is compromised in Swl/+ mice. Consistent with a proprioceptive defect in sensory nerves, Swl/+ mice display a marked decrease in the number of proprioceptive sensory receptors. We examined the number of mature muscle spindles in Swl/+ hindlimbs by using the S46 antibody, a marker recognizing the slow-tonic myosin heavy chain within muscle spindles (Crow and Stockdale, 1986), and the collagen IV antibody, which labels the spindle capsule. In agreement with previous morphological characterizations (Duchen, 1975), the Swl/+ hindlimbs displayed 88% fewer muscle spindles than the WT hindlimbs (n = 10) (Figs. 2A,B, 3B).
Figure 2.

Proprioceptive sensory neurons and muscle spindles are marked reduced in adult Swl/+ mice. A, Mature muscle spindles, stained by S46 antibody (green) and collagen IV antibody (ColgIV, red), are primarily absent in the Swl/+ hindlimbs. B, Quantitative analysis of muscle spindles demonstrates that Swl/+ mice have 88% fewer muscle spindles than those of WT mice. *p < 0.01 (Student's t test). C, Representative immunostained sections show that proprioceptive neurons (PV+) are more severely compromised than nociceptive neurons (CGRP+) in the lumbar Swl/+ DRGs. Scale bars (in A, C), 100 μm. D, Quantitative analysis indicates that the total number of DRG neurons is significantly reduced in the lumbar (L4) DRGs but is not significantly reduced in the cervical (C4) DRGs of Swl/+ mice. *p < 0.01 (Student's t test). E, Quantitative analysis of subpopulation in the L4 DRGs demonstrates that the number of proprioceptive neurons (PV+) is reduced by 69% in Swl/+ mice (*p < 0.01, Student's t test), whereas only a 31% reduction of the number of nociceptive neurons (CGRP+) is detected in Swl/+ mice (p > 0.05, Student's t test).
Figure 3.

Swl/+ mice exhibit lumbar proprioceptive DRG neuron and muscle spindle degeneration during development. A, Immunofluorescence stainings of developing (Egr3+) and mature (S46+) muscle spindles show that the gross morphology of muscle spindle during different stages is comparable between Swl/+ mice and WT littermate mice. Scale bar, 50 μm. B, Quantitative analysis of muscle spindle indicates that the spindle count in the Swl/+ hindlimbs is comparable with those in WT hindlimbs at E15.5 (p > 0.05, Student's t test). However, at P0.5 and P91, the number of muscle spindles is reduced by 87 and 88%, respectively, in Swl/+ mice compared with those in WT mice. *p < 0.01 (Student's t test). C, Quantitative analysis of L4 PV+ DRG neurons reveals no significant change in the number of the lumbar proprioceptive DRG neurons in Swl/+ mice at E15.5 (p > 0.05, Student's t test) but a 69% reduction in the number of Swl/+ lumbar proprioceptive DRG neurons at both P0.5 and P91 in Swl/+ mice compared with those of WT mice. *p < 0.05 (Student's t test).
We further evaluated the extent of neuron loss in the DRGs of 13-week-old Swl/+ mice by using H&E staining. The reduction in total neuron number was found to be greater in lumbar segments than cervical segments in Swl/+ mice (n = 5) (Fig. 2D), consistent with the observation that the forelimbs are much less affected in Swl/+ mice. To identify the subpopulation of sensory neuron lost in lumbar DRGs of Swl/+ mice, we used the anti-PV antibody to label neurotrophin (NT) 3-dependent proprioceptive sensory neurons and their processes (Celio, 1990), and the anti-CGRP antibody to label nociceptive sensory neurons (Grunditz et al., 1986). We found that the number of proprioceptive sensory neurons in the Swl/+ lumbar DRGs was reduced by 69% compared with that in the WT DRGs (n = 4) (Figs. 2C,E, 3C), whereas, the number of nociceptive sensory neurons was reduced by 31% in the Swl/+ lumbar DRGs compared with that in the WT DRGs (n = 4) (Fig. 2C,E). These data indicate that the proprioceptive sensory neuron subpopulation was more severely compromised than the nociceptive sensory neuron subpopulation in the lumbar DRGs of Swl/+ mice.
Swl/+ mice exhibit lumbar proprioceptive DRG neuron and muscle spindle degeneration during development
To determine the development stage at which lumbar proprioceptive DRG neurons and hindlimb muscle spindles are lost, we analyzed Swl/+ animals of different ages. The muscle spindles in the hindlimbs of Swl/+ and WT littermate mice at E15.5 and P0.5 were labeled by the Egr3 antibody, a marker for developing muscle spindles (Tourtellotte and Milbrandt, 1998), and the S46 antibody, a marker for mature muscle spindles, respectively. Egr3-positive muscle spindles were found to form properly in Swl/+ mice at E15.5 (Fig. 3A), the time at which proprioceptive innervation signals type I myotube differentiation to muscle spindle (Kucera et al., 1995). Additionally, no reduction of the spindle count was detected in the Swl/+ hindlimbs at this stage (7.7 ± 0.7 counts/section, n = 3) compared with WT mice (8.3 ± 0.5 counts/section, n = 4) (Fig. 3B). Nevertheless, at P0.5, the number of muscle spindles was reduced by 87% in Swl/+ mice (3.5 ± 1.0 counts/section, n = 4) compared with that in WT mice (27.0 ± 1.0 counts/section, n = 5) (Fig. 3B), although existent Swl/+ muscle spindles, visualized at P0.5 and P91, appeared unaltered compared with the WT muscle spindles (Fig. 3A). Similarly, we did not detect a change in the number of the lumbar proprioceptive DRG neurons in Swl/+ mice at E15.5 (n = 3) (Fig. 3C), whereas there was a 69% reduction in the number of Swl/+ lumbar DRG neurons compared with that of WT mice at P0.5 (n = 3) (Fig. 3C). These findings suggest that the lumbar proprioceptive DRG neuron loss is concomitant with muscle spindle degeneration during late embryonic development in Swl/+ mice.
Positional cloning of the Swl mutation identifies a 9-bp deletion in Dync1h1
The Swl mutation arose in descendents of X-irradiated C3H/HeH × 101/H hybrid mice (Duchen, 1975). We examined these animals for gross chromosomal rearrangements, such as translocations, inversions, and deletions. No cytogenetic abnormalities were observed in heterozygous Swl mutant mice by means of the Giemsa banding assay for well spread metaphase chromosomes (data not shown).
To identify the gene responsible for the Swl/+ phenotype, we performed a two-generation backcross with the C57BL/6J strain to generate N2 progeny mice. Forty-two N2 mice and a set of 35 polymorphic markers with a spacing of 10–20 cM across the mouse genome (MapPairs Mouse Markers, Invitrogen, Carlsbad, CA) were used for whole-genome localization. The D12Nds2 locus, which is 59 cM from the centromere of chromosome 12, had a recombination frequency of 0.0 and an LOD score of 12.6, strongly indicating that the Swl locus is located in a region on distal chromosome 12. Subsequently, 816 N2 backcross mice were generated and additional genetic markers on distal chromosome 12 were used for high-resolution linkage analysis to localize the mutation to a 4.6-Mb critical interval between markers D12Mit123 and D12Mit20 (Fig. 4A,B). For informative markers that are polymorphic between the C3HeH and 101H alleles, only the C3HeH alleles were detected in haplotypes around the Swl locus, indicating that the Swl mutation most likely arose on the ancestral C3HeH chromosome.
Figure 4.
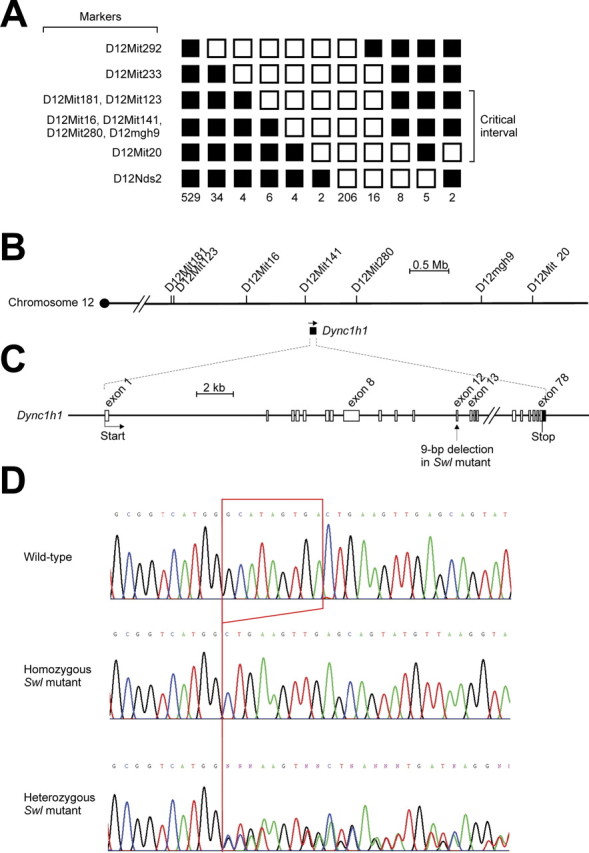
Positional cloning of the Swl mutation identifies a 9 bp deletion in Dync1h1. A, Genetic linkage mapping defined D12Mit123 as the proximal flanking polymorphic marker by a recombinant frequency of 0.86% (7 of 816) and D12Mit20 as the distal flanking polymorphic marker by a recombinant frequency of 1.84% (15 of 816) based on the analysis of 816 backcross progeny. Filled and open boxes indicate the presence and absence, respectively, of the C57BL/6J allele. B, Region of mouse chromosome 12 surrounding the critical interval. Distance between markers is drawn to scale of physical distance. Among 82 known genes and expressed sequence tags within the 4.6 Mb critical interval, Dync1h1 was selected as a candidate gene for the Swl mutation. C, Genomic structure of Dync1h1. Position of 9 bp deletion in Swl mutants is indicated. D, Chromatogram shows the 9 bp deletion in exon 12 of Dync1h1 in homozygous and heterozygous Swl mice.
This critical region contained 82 known genes and expressed sequence tags. From them, Dync1h1 was chosen as a candidate gene primarily on the basis of the relevance of axonal transport to neuronal survival (Harada et al., 1998; Hafezparast et al., 2003). We sequenced all 78 exons of the gene and identified a 9-bp deletion in exon 12 of Dync1h1 in Swl mice (Fig. 4C,D). The mutation occurs at residues 1040–1043, which results in the change of four amino acids [glycine (GGC), isoleucine (ATA), valine (GTG), and threonine (ACT)] to an alanine (GCT) in Swl. The mutation lies in the cargo-binding domain of the DYNC1H1 protein (Fig. 5A), which is also thought to be a potential site of homodimerization (Hafezparast et al., 2003). Although Swl/Swl mutants were identified at the expected frequency at E7.5, no viable Swl/Swl mutants were detected after E8.5 (data not shown), indicating that Swl/Swl mutants have a late implantation or early gastrulation lethal phenotype. The age by which Swl/Swl mutants die is the same as that of Dync1h1 homozygous knock-out mutant mice (Harada et al., 1998). To genetically prove that the Dync1h1 deletion is responsible for the Swl/+ phenotype, we crossed Swl/+ mice with heterozygotes of the Dync1h1 point mutation-carrying strain, Loa/±. None of the 78 surviving offspring from 19 litters were of the Swl/Loa genotype, suggesting that the Swl/Loa phenotype, like that of Swl/Swl and Loa/Loa mice, is embryonic lethal. This lack of complementation indicates that Swl and Loa are two different alleles of the same gene, Dync1h1.
Figure 5.

Expression of Dync1h1 is not altered in Swl mice. A, Graphical illustration of the domain structure of the DYNC1H1 protein, adapted from the publication of Hafezparast et al. (2006), shows the relative position of the Swl, Loa, and Cra1 mutations. The cargo-binding domain and motor domain of DYNC1H1 are shown by the black box and the gray box, respectively. HC, Heavy chain binding site; IC, intermediate chain binding site; LIC, light intermediate chain binding site. B, RT-PCR analysis indicates that Dync1h1 mRNA expression is not altered in Swl/Swl mutants (E7.5) or Swl/+ mutants (E7.5 and P91) compared with WT (+/+) animals. Cyclophilin is amplified separately as a positive control. C, No differences in the level or localization of DYNC1H1 is detected by using immunofluorescence staining of dynein heavy chain antibody (R-325) in E7.5 Swl/Swl embryos compared with E7.5 WT (+/+) embryos. Scale bar, 100 μm.
RT-PCR analysis demonstrated that Dync1h1 mRNA expression was not altered in Swl/+ and Swl/Swl mice (Fig. 5B). Immunofluorescence staining of E7.5 embryos using a DYNC1H1 antibody detected no gross changes in the level or localization of DYNC1H1 in Swl mice (Fig. 5C).
The proprioceptive sensory defect is a common feature in both Loa/+ and Swl/+ mice
Because Swl and Loa are different alleles of Dync1h1, we asked whether Swl/+ mice and Loa/+ mice have similar nervous system defects. Like Swl/+ mice, Loa/+ mice display hindlimb flexion by tail suspension, as well as a mild uncoordinated gait (Hafezparast et al., 2003). In addition, Loa/+ mice demonstrated a 52% reduction in hindlimb grip strength with unaffected forelimb grip strength (n = 7 for Loa/+ and n = 6 for WT) (Fig. 6A). The number of muscle spindles was reduced by 86% in the hindlimbs of 13-week-old Loa/+ mice (n = 4 for Loa/+ and n = 3 for WT) (Fig. 6B). Also, the number of cervical DRG neurons was not reduced in Loa/+ mice, whereas the number of lumbar DRG neurons was reduced by 42% in these mice (n = 3) (Fig. 6C). The dorsal roots in the lumbar segment were significantly thinner than the ventral roots in Loa/+ mice, to an extent similar to those in Swl/+ mice (Fig. 6D). The H reflex was absent in 13-week-old Loa/+ mice examined (n = 4) but present in 3 of 4 WT littermates examined (Fig. 6E). The phenotypic similarities between the two mutant strains indicate that the early-onset proprioceptive sensory defect represents a common feature in both Dync1h1 mutations.
Figure 6.

Proprioceptive sensory deficit is a common feature in Loa and Swl Dync1h1 mutations. A, Hindlimb grip strength is reduced by 52% in 13-week-old male Loa/+ mice, whereas no difference is detected in their forelimbs. *p < 0.01 (Student's t test). B, Quantitative analysis of muscle spindles demonstrates that Loa/+ mice have 86% fewer muscle spindles than WT mice. *p < 0.01 (Student's t test). C, Quantitative analysis of DRG neurons indicates that the number of DRG neurons is significantly reduced in L4 segment but not in C4 segment of Loa/+ mice. *p < 0.01 (Student's t test). D, In contrast to the ventral root (VR), the dorsal root (DR) and the sciatic nerve (SN) are markedly thinner in Swl/+ and Loa/+ mice compared with the WT littermate mice. Scale bar, 100 μm. E, The H reflex is absent in Loa/+ mice (n = 4) but present in three of four WT littermates.
The Swl Dync1h1 mutation does not attenuate disease progression in SOD1G93A mice
Loa/+ and Cra1/+ mice, when crossed with a motor neuron disease mouse model resulting from the over-expression of a human mutant SOD1G93A transgene, have been shown to delay motor neuron degeneration and prolong the life span of these mice (Kieran et al., 2005; Teuchert et al., 2006). We investigated whether the Swl allele can similarly delay disease progression of the SOD1G93A mice. Crossing male SOD1G93A/+ mice with female Swl/+ mice or Loa/+ mice produced four distinct genotypes respectively. Each progeny was examined for survival time, body weight and forelimb grip strength from the 10th week after birth. In agreement with the earlier reports (Kieran et al., 2005; Teuchert et al., 2006), the survival time of Loa/SOD1G93A mice (156 d ± 2.4 SEM, n = 12) increased by 21% compared with that of SOD1G93A/+ mice (129 d ± 2.9 SEM, n = 16) (Fig. 7B). Changes in the body weight and forelimb grip strength of Loa/SOD1G93A mice were significantly delayed relative to SODG93A mice during the observation period (Fig. 7D,F). Intriguingly, Swl/SOD1G93A mice showed no difference in the average survival time (124 d ± 1.5 SEM, n = 15, in Swl/SOD1G93A mice vs 122 d ± 1.4 SEM, n = 15, in SOD1G93A/+ mice) (Fig. 7A). Similarly, Swl/SOD1G93A mice have a comparable body weight and forelimb grip strength to those of SOD1G93A/+ mice during the observation period (Fig. 7C,D). This data indicate that the Swl Dync1h1 mutation does not attenuate disease progression in SOD1G93A mice.
Figure 7.

The Swl Dync1h1 mutation does not delay disease progression in SOD1G93A mice. A, C, E, Swl/+ × SOD1G93A hemizygote; B, D, F, Loa/+ × SOD1G93A hemizygote. The Kaplan–Meier survival analysis demonstrates that Swl/SOD1G93A mice have a comparable average survival time with that of SOD1G93A/+ mice (A), whereas the average survival time of Loa/SOD1G93A mice is significantly delayed compared with that of SOD1G93A/+ mice (B; p < 0.01, log-rank test for equality of survival curves). In contrast to Swl/SOD1G93A mice (C), the body weight loss of Loa/SOD1G93A mice is significantly delayed compared with that of SOD1G93A/+ mice during the observation period (D; p < 0.01, Student's t test). Similarly, the motor function determined by forelimb grip strength test shows no significant difference between Swl/SOD1G93A mice and SOD1G93A/+ mice (E), whereas Loa/SOD1G93A mice have a significant delay in motor dysfunction compared with that of SOD1G93A/+ mice during the observation period (F; p < 0.01, Student's t test).
Discussion
In this study, we describe the identification of a novel mutation in Dync1h1 in Swl/+ mice, which display an early-onset proprioceptive sensory neuropathy. Detailed comparison of Swl/+ mice with Loa/+ mice, another Dync1h1 mutant, demonstrate that both strains are characterized by a common proprioceptive sensory deficit. Thus, we provide in vivo evidence that dysfunction of cytoplasmic dynein can lead to an early-onset proprioceptive sensory neuropathy in mice.
Cytoplasmic dynein is the major microtubule minus end-directed motor protein, which is composed of two heavy chains (DYNC1H1), two intermediate chains, four light intermediate chains, and several light chains (Karki and Holzbaur, 1999; Hafezparast et al., 2003; Vallee et al., 2004). DYNC1H1 has a C-terminal motor domain head and an N-terminal cargo-binding domain stalk, and homodimerizes at the N-terminal. Most of the accessory subunits of cytoplasmic dynein associate with the N-terminal portion of DYNC1H1. Mediated by dynactin, a multi-subunit complex essential for most dynein functions, cytoplasmic dynein interacts with cargo. The largest subunit of dynactin, p150glued, binds to both microtubules and the intermediate chain of dynein (Karki and Holzbaur, 1999; Vallee et al., 2004).
Cytoplasmic dynein is expressed ubiquitously and participates in many cellular processes in higher eukaryotes, including cell division (Karki and Holzbaur, 1999), membranous organelle trafficking (Karki and Holzbaur, 1999) and nuclear envelop breakdown (Salina et al., 2002). Homozygosity for null mutations of the gene cause early embryonic lethality in both Drosophila and mice (Gepner et al., 1996; Harada et al., 1998). Similarly, the abrogation of these essential functions by the Swl mutation most likely accounts for the late implantation or early gastrulation lethal phenotype in Swl/Swl embryos. Studies have also shown that this motor protein is involved in multiple neuron-specific processes, such as neuronal migration (Sasaki et al., 2000), the growth and development of neuritis (Barakat-Walter and Riederer, 1996), synapse formation (Cheng et al., 2006), as well as axonal transport of microtubules, neurofilaments (He et al., 2005) and organelles (Schnapp et al., 1989). Among them, the axonal transport role of cytoplasmic dynein has recently been associated with motor neuron survival (Hafezparast et al., 2003; Puls et al., 2005).
The lack of an H reflex in the Swl hindlimbs, despite normal motor function and a moderate decrease of sensory potential amplitude, strongly suggests that these mice possess a proprioceptive deficit with pronounced dorsal root involvement. Detailed immunohistochemical analyses reveal that both lumbar proprioceptive sensory neurons and hindlimb muscle spindles are markedly lost in Swl/+ mice. In contrast to the sensory defects, the number of α-motor neurons in the lumbar segments is not significantly reduced in Swl/+ mice, even in aged animals.
Proprioceptive sensory neurons are a subpopulation of DRG neurons characterized by medium and large cell soma areas, and TrkC-expressing proprioceptive neurons represent ∼20% of adult DRG neurons (Mu et al., 1993). Like α-motor neurons, this subpopulation of sensory neurons possesses a high energy demand, and requires significant trafficking capacity, and hence may be particularly vulnerable to vesicular and axonal transport defects. Previous studies have shown that large motor and sensory neurons are more vulnerable to diseased conditions than small-sized neurons (McIlwain, 1991). Because cytoplasmic dynein moves membranous vesicles and organelles along microtubules from the nerve terminals to neuron cell bodies, the neurons with longer axons are most likely affected by the Swl Dync1h1 mutation, thus explaining why the hindlimbs involvement is much more severe than forelimbs in these mutant mice.
The pronounced loss of proprioceptive DRG neurons and muscle spindles displayed by Swl/+ mice is reminiscent of those in TrkC null and NT-3 null mutants (Ernfors et al., 1994; Klein et al., 1994). Unlike TrkC null mutants, other DRG neuron subgroups are also lost in NT-3 null mutants (Ernfors et al., 1994) as well as in Swl/+ mice. Moreover, in vitro studies have suggested that NT-3 functions to support sensory neuron survival at early developmental stages whereas NGF is required at a later stage, and sensory neurons change from dependence on brain-derived neurotrophic factor (BDNF) or NT-3 to dependence on NGF at approximately E13 (Buchman et al., 1993). We have observed an excess of apoptosis between E11.5 to E15.5 (data not shown) and a marked neuron loss between E15.5 to P0.5 in the Swl/+ lumbar DRGs. This time frame of DRG cell loss in Swl/+ mice is consistent with the actions of neurotrophins on DRG neurons during development, suggesting that disrupted neurotrophin/Trk signaling may be responsible for sensory neuron loss in Swl/+ mice. Because cytoplasmic dynein has the ability to bind Trk receptors directly (Yano et al., 2001), and activated target-derived neurotrophic factor/Trk complexes essential for the survival of DRG neurons are transported in a retrograde manner (Heerssen et al., 2004), it is possible that the degeneration of proprioceptive sensory neurons in Swl/+ mice is a consequence of defective dynein-based axonal transport. Nevertheless, the possibility that other neuron-specific dynein functions contribute to sensory neuron loss in these animals cannot be ruled out.
Our genetic analysis indicates that Swl and Loa are allelic to Dync1h1. Nevertheless, the Swl mutation appears to exclusively affect sensory neuron viability, whereas the Loa mutation also leads to a more subtle, late-onset loss of motor neurons. In addition, two independent studies have shown that interactions between mutant SOD1G93A and the dynein mutations with a significant involvement of motor neurons can markedly attenuate motor neuron degeneration in double-heterozygote mice (Kieran et al., 2005; Teuchert et al., 2006). In contrast, we did not find a significant delay of average survival time, body weight loss or motor dysfunction in Swl/SOD1G93A mice. This finding further suggests that the Swl mutation affects sensory neurons rather than motor neurons.
The separate DYNC1H1 regions disrupted by the Swl versus Loa mutations, perhaps resulting in the disruption of distinct protein binding domains, might be responsible for the differences in neuron susceptibility of these alleles. The Loa mutation is a missense point mutation at residue 580 of DYNC1H1 (Hafezparast et al., 2003), which resides within the highly conserved DYNC1H1 domain responsible for binding the dynein intermediate chains, as well as for homodimerization (Tynan et al., 2000). In contrast, the Swl mutation is a nine base-pair deletion at residues 1040–1043 of DYNC1H1, which lies outside the dynein intermediate chain binding region but within the C-terminal end of the putative homodimerization region (Tynan et al., 2000). Given that the targeted null Dync1h1 mutation leads to early embryonic lethality in homozygous mice and a normal phenotype in heterozygous mice (Harada et al., 1998), the semidominant Swl and Loa mutations in Dync1h1 likely results in a gain of function, either increasing the activity of DYNC1H1, introducing a novel toxic property to DYNC1H1 such as protein misfolding, or reducing the activity of endogenous DYNC1H1 by a dominant-negative effect. Together, these dynein mutant animals are important resources for dissecting the molecular mechanism of dynein-dependent neuronal degeneration.
The gene encoding DYNC1H1 is particularly large, which may make it susceptible to a high frequency of mutation. The recent evidence that suggests that this protein plays a role in motor neuron survival (Hafezparast et al., 2003; Puls et al., 2005) has led to the hypothesis that cytoplasmic dynein may be a causative gene or susceptibility factor in human motor neuron disease (Ahmad-Annuar et al., 2003). Nonetheless, two human studies have failed to reveal an association of DYNC1H1 with motor neuron disease (Ahmad-Annuar et al., 2003; Shah et al., 2006). Here, we provide strong evidence that mutations in cytoplasmic dynein compromise the function of sensory neurons to a much greater extent, and at an earlier onset, than that of motor neurons. It is therefore possible that DYNC1H1 mutations are linked to HSN or some axonal degenerative forms of CMT with sensory involvement.
In summary, we have identified a novel mutation in Dync1h1 in Swl mice and provided in vivo evidence that mutations in this gene lead to early-onset sensory neuron degeneration with marked proprioceptive deficits in mice. We also show that distinct mutations in Dync1h1 can lead to either a pure sensory neuropathy or to a disorder that includes both sensory and motor abnormalities.
Footnotes
This work was supported in part by a grant from the Miriam and Sheldon Adelson Program in Neural Repair and Rehabilitation (B.P.). We thank Frank Stockdale for the S46 antibody; Elizabeth Fisher and Don Cleveland for the Loa/+ mice; Peter Glenister for the frozen Swl/+ embryos; Steven Scherer and Scott Brady for insightful suggestions; Nicole Campbell for initiating the histology, electrophysiology, and behavioral experiments; Victoria Yan and Erdong Liu for their contributions to electron microscopy; Hanson Ho for performing the electrophysiology and behavioral experiments; Gloria Wright for help with figure preparation; Eliot Popko for videotaping the mice; and Darlene Douglas for critically reading this manuscript.
References
- Ahmad-Annuar A, Shah P, Hafezparast M, Hummerich H, Witherden AS, Morrison KE, Shaw PJ, Kirby J, Warner TT, Crosby A, Proukakis C, Wilkinson P, Orrell RW, Bradley L, Martin JE, Fisher EM. No association with common Caucasian genotypes in exons 8, 13 and 14 of the human cytoplasmic dynein heavy chain gene (DNCHC1) and familial motor neuron disorders. Amyotroph Lateral Scler Other Motor Neuron Disord. 2003;4:150–157. doi: 10.1080/14660820310011737. [DOI] [PubMed] [Google Scholar]
- Barakat-Walter I, Riederer BM. Triiodothyronine and nerve growth factor are required to induce cytoplasmic dynein expression in rat dorsal root ganglion cultures. Brain Res Dev Brain Res. 1996;96:109–119. [PubMed] [Google Scholar]
- Brook GA, Duchen LW. End-plates, transmission and contractile characteristics of muscles without spindles in the hereditary sensory neuropathy of the Sprawling mouse. Brain. 1990;113:867–891. doi: 10.1093/brain/113.4.867. [DOI] [PubMed] [Google Scholar]
- Buchman VL, Davies AM. Different neurotrophins are expressed and act in a developmental sequence to promote the survival of embryonic sensory neurons. Development. 1993;118:989–1001. doi: 10.1242/dev.118.3.989. [DOI] [PubMed] [Google Scholar]
- Celio MR. Calbindin D-28k and parvalbumin in the rat nervous system. Neuroscience. 1990;35:375–475. doi: 10.1016/0306-4522(90)90091-h. [DOI] [PubMed] [Google Scholar]
- Cheng HH, Liu SH, Lee HC, Lin YS, Huang ZH, Hsu CI, Chen YC, Chang YC. Heavy chain of cytoplasmic dynein is a major component of the postsynaptic density fraction. J Neurosci Res. 2006;84:244–254. doi: 10.1002/jnr.20898. [DOI] [PubMed] [Google Scholar]
- Crow MT, Stockdale FE. Myosin expression and specialization among the earliest muscle fibers of the developing avian limb. Dev Biol. 1986;113:238–254. doi: 10.1016/0012-1606(86)90126-0. [DOI] [PubMed] [Google Scholar]
- Duchen LW. A dominant hereditary sensory disorder in the mouse with deficiency of muscle spindles: the mutant Sprawling. J Physiol (Lond) 1974;237:10P–11P. [PubMed] [Google Scholar]
- Duchen LW. “Sprawling”: a new mutant mouse with failure of myelination of sensory axons and a deficiency of muscle spindles. Neuropath Applied Neurobiol. 1975;1:89–101. [Google Scholar]
- Duchen LW, Scaravilli F. Quantitative and electron microscopic studies of sensory ganglion cells of the Sprawling mouse. J Neurocytol. 1977a;6:465–481. doi: 10.1007/BF01178229. [DOI] [PubMed] [Google Scholar]
- Duchen LW, Scaravilli F. The structure and composition of peripheral nerves and nerve roots in the Sprawling mouse. J Anat. 1977b;123:763–775. [PMC free article] [PubMed] [Google Scholar]
- Einarsdottir E, Carlsson A, Minde J, Toolanen G, Svensson O, Solders G, Holmgren G, Holmberg D, Holmberg M. A mutation in the nerve growth factor beta gene (NGFB) causes loss of pain perception. Hum Mol Genet. 2004;13:799–805. doi: 10.1093/hmg/ddh096. [DOI] [PubMed] [Google Scholar]
- Ernfors P, Lee KF, Kucera J, Jaenisch R. Lack of neurotrophin-3 leads to deficiencies in the peripheral nervous system and loss of limb proprioceptive afferents. Cell. 1994;77:503–512. doi: 10.1016/0092-8674(94)90213-5. [DOI] [PubMed] [Google Scholar]
- Gepner J, Li M, Ludmann S, Kortas C, Boylan K, Iyadurai SJ, McGrail M, Hays TS. Cytoplasmic dynein function is essential in Drosophila melanogaster. Genetics. 1996;142:865–878. doi: 10.1093/genetics/142.3.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunditz T, Ekman R, Hakanson R, Rerup C, Sundler F, Uddman R. Calcitonin gene-related peptide in thyroid nerve fibers and C cells: effects on thyroid hormone secretion and response to hypercalcemia. Endocrinology. 1986;119:2313–2324. doi: 10.1210/endo-119-5-2313. [DOI] [PubMed] [Google Scholar]
- Guo L, Degenstein L, Dowling J, Yu QC, Wollmann R, Perman B, Fuchs E. Gene targeting of BPAG1: abnormalities in mechanical strength and cell migration in stratified epithelia and neurologic degeneration. Cell. 1995;81:233–243. doi: 10.1016/0092-8674(95)90333-x. [DOI] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, Chen W, Zhai P, Sufit RL, Siddique T. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Hafezparast M, Klocke R, Ruhrberg C, Marquardt A, Ahmad-Annuar A, Bowen S, Lalli G, Witherden AS, Hummerich H, Nicholson S, Morgan PJ, Oozageer R, Priestley JV, Averill S, King VR, Ball S, Peters J, Toda T, Yamamoto A, Hiraoka Y. Mutations in dynein link motor neuron degeneration to defects in retrograde transport. Science. 2003;300:808–812. doi: 10.1126/science.1083129. [DOI] [PubMed] [Google Scholar]
- Harada A, Takei Y, Kanai Y, Tanaka Y, Nonaka S, Hirokawa N. Golgi vesiculation and lysosome dispersion in cells lacking cytoplasmic dynein. J Cell Biol. 1998;141:51–59. doi: 10.1083/jcb.141.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Francis F, Myers KA, Yu W, Black MM, Baas PW. Role of cytoplasmic dynein in the axonal transport of microtubules and neurofilaments. J Cell Biol. 2005;168:697–703. doi: 10.1083/jcb.200407191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heerssen HM, Pazyra MF, Segal RA. Dynein motors transport activated Trks to promote survival of target-dependent neurons. Nat Neurosci. 2004;7:596–604. doi: 10.1038/nn1242. [DOI] [PubMed] [Google Scholar]
- Indo Y, Tsuruta M, Hayashida Y, Karim MA, Ohta K, Kawano T, Mitsubuchi H, Tonoki H, Awaya Y, Matsuda I. Mutations in the TRKA/NGF receptor gene in patients with congenital insensitivity to pain with anhidrosis. Nat Genet. 1996;13:485–488. doi: 10.1038/ng0896-485. [DOI] [PubMed] [Google Scholar]
- Karki S, Holzbaur EL. Cytoplasmic dynein and dynactin in cell division and intracellular transport. Curr Opin Cell Biol. 1999;11:45–53. doi: 10.1016/s0955-0674(99)80006-4. [DOI] [PubMed] [Google Scholar]
- Kieran D, Hafezparast M, Bohnert S, Dick JR, Martin J, Schiavo G, Fisher EM, Greensmith L. A mutation in dynein rescues axonal transport defects and extends the life span of ALS mice. J Cell Biol. 2005;169:561–567. doi: 10.1083/jcb.200501085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein R, Silos-Santiago I, Smeyne RJ, Lira SA, Brambilla R, Bryant S, Zhang L, Snider WD, Barbacid M. Disruption of the neurotrophin-3 receptor gene trkC eliminates Ia muscle afferents and results in abnormal movements. Nature. 1994;368:249–251. doi: 10.1038/368249a0. [DOI] [PubMed] [Google Scholar]
- Kucera J, Fan G, Jaenisch R, Linnarsson S, Ernfors P. Dependence of developing group Ia afferents on neurotrophin-3. J Comp Neurol. 1995;363:307–320. doi: 10.1002/cne.903630211. [DOI] [PubMed] [Google Scholar]
- Levedakou EN, Chen XJ, Soliven B, Popko B. Disruption of the mouse Large gene in the enr and myd mutants results in nerve, muscle, and neuromuscular junction defects. Mol Cell Neurosci. 2005;28:757–769. doi: 10.1016/j.mcn.2004.12.007. [DOI] [PubMed] [Google Scholar]
- Liu JJ, Ding J, Kowal AS, Nardine T, Allen E, Delcroix JD, Wu C, Mobley W, Fuchs E, Yang Y. BPAG1n4 is essential for retrograde axonal transport in sensory neurons. J Cell Biol. 2003;163:223–229. doi: 10.1083/jcb.200306075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manly KF, Olson JM. Overview of QTL mapping software and introduction to map manager QT. Mamm Genome. 1999;10:327–334. doi: 10.1007/s003359900997. [DOI] [PubMed] [Google Scholar]
- McIlwain DL. Nuclear and cell body size in spinal motor neurons. Adv Neurol. 1991;56:67–74. [PubMed] [Google Scholar]
- Mu X, Silos-Santiago I, Carroll SL, Snider WD. Neurotrophin receptor genes are expressed in distinct patterns in developing dorsal root ganglia. J Neurosci. 1993;13:4029–4041. doi: 10.1523/JNEUROSCI.13-09-04029.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nangaku M, Sato-Yoshitake R, Okada Y, Noda Y, Takemura R, Yamazaki H, Hirokawa N. KIF1B, a novel microtubule plus end-directed monomeric motor protein for transport of mitochondria. Cell. 1994;79:1209–1220. doi: 10.1016/0092-8674(94)90012-4. [DOI] [PubMed] [Google Scholar]
- Puls I, Oh SJ, Sumner CJ, Wallace KE, Floeter MK, Mann EA, Kennedy WR, Wendelschafer-Crabb G, Vortmeyer A, Powers R, Finnegan K, Holzbaur EL, Fischbeck KH, Ludlow CL. Distal spinal and bulbar muscular atrophy caused by dynactin mutation. Ann Neurol. 2005;57:687–694. doi: 10.1002/ana.20468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salina D, Bodoor K, Eckley DM, Schroer TA, Rattner JB, Burke B. Cytoplasmic dynein as a facilitator of nuclear envelope breakdown. Cell. 2002;108:97–107. doi: 10.1016/s0092-8674(01)00628-6. [DOI] [PubMed] [Google Scholar]
- Sasaki S, Shionoya A, Ishida M, Gambello MJ, Yingling J, Wynshaw-Boris A, Hirotsune S. A LIS1/NUDEL/cytoplasmic dynein heavy chain complex in the developing and adult nervous system. Neuron. 2000;28:681–696. doi: 10.1016/s0896-6273(00)00146-x. [DOI] [PubMed] [Google Scholar]
- Scaravilli F, Duchen LW. The development of sensory ganglion cells in normal and Sprawling mutant mice. J Neurocytol. 1980a;9:363–371. doi: 10.1007/BF01181542. [DOI] [PubMed] [Google Scholar]
- Scaravilli F, Duchen LW. Electron microscopic and quantitative studies of cell necrosis in developing sensory ganglia in normal and Sprawling mutant mice. J Neurocytol. 1980b;9:373–380. doi: 10.1007/BF01181543. [DOI] [PubMed] [Google Scholar]
- Schnapp BJ, Reese TS. Dynein is the motor for retrograde axonal transport of organelles. Proc Natl Acad Sci USA. 1989;86:1548–1552. doi: 10.1073/pnas.86.5.1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah PR, Ahmad-Annuar A, Ahmadi KR, Russ C, Sapp PC, Horvitz HR, Brown RH, Jr, Goldstein DB, Fisher EM. No association of DYNC1H1 with sporadic ALS in a case-control study of a northern Eur derived population: a tagging SNP approach. Amyotroph Lateral Scler. 2006;7:46–56. doi: 10.1080/14660820500397057. [DOI] [PubMed] [Google Scholar]
- Shibasaki H, Hitomi T, Mezaki T, Kihara T, Tomimoto H, Ikeda A, Shimohama S, Ito M, Oka N. A new form of congenital proprioceptive sensory neuropathy associated with arthrogryposis multiplex. J Neurol. 2004;251:1340–1344. doi: 10.1007/s00415-004-0539-4. [DOI] [PubMed] [Google Scholar]
- Skre H. Genetic and clinical aspects of Charcot-Marie-Tooth's disease. Clin Genet. 1974;6:98–118. doi: 10.1111/j.1399-0004.1974.tb00638.x. [DOI] [PubMed] [Google Scholar]
- Teuchert M, Fischer D, Schwalenstoecker B, Habisch HJ, Böckers TM, Ludolph AC. A dynein mutation attenuates motor neuron degeneration in SOD1(G93A) mice. Exp Neurol. 2006;198:271–274. doi: 10.1016/j.expneurol.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Tourtellotte WG, Milbrandt J. Sensory ataxia and muscle spindle agenesis in mice lacking the transcription factor Egr3. Nat Genet. 1998;20:87–91. doi: 10.1038/1757. [DOI] [PubMed] [Google Scholar]
- Tynan SH, Gee MA, Vallee RB. Distinct but overlapping sites within the cytoplasmic dynein heavy chain for dimerization and for intermediate chain and light intermediate chain binding. J Biol Chem. 2000;275:32769–32774. doi: 10.1074/jbc.M001537200. [DOI] [PubMed] [Google Scholar]
- Vallee RB, Williams JC, Varma D, Barnhart LE. Dynein: an ancient motor protein involved in multiple modes of transport. J Neurobiol. 2004;58:189–200. doi: 10.1002/neu.10314. [DOI] [PubMed] [Google Scholar]
- Verpoorten N, De Jonghe P, Timmerman V. Disease mechanisms in hereditary sensory and autonomic neuropathies. Neurobiol Dis. 2006;21:247–255. doi: 10.1016/j.nbd.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Yano H, Lee FS, Kong H, Chuang J, Arevalo J, Perez P, Sung C, Chao MV. Association of Trk neurotrophin receptors with components of the cytoplasmic dynein motor. J Neurosci. 2001;21(RC125):1–7. doi: 10.1523/JNEUROSCI.21-03-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Takita J, Tanaka Y, Setou M, Nakagawa T, Takeda S, Yang HW, Terada S, Nakata T, Takei Y, Saito M, Tsuji S, Hayashi Y, Hirokawa N. Charcot-Marie-Tooth disease type 2A caused by mutation in a microtubule motor KIF1Bbeta. Cell. 2001;105:587–597. doi: 10.1016/s0092-8674(01)00363-4. [DOI] [PubMed] [Google Scholar]