

Abstract
Physiological processing of the β-amyloid precursor protein (APP) generates amyloid β-protein, which can assemble into oligomers that mediate synaptic failure in Alzheimer's disease. Two decades of research have led to human trials of compounds that chronically target this processing, and yet the normal function of APP in vivo remains unclear. We used the method of in utero electroporation of shRNA constructs into the developing cortex to acutely knock down APP in rodents. This approach revealed that neuronal precursor cells in embryonic cortex require APP to migrate correctly into the nascent cortical plate. cDNAs encoding human APP or its homologues, amyloid precursor-like protein 1 (APLP1) or APLP2, fully rescued the shRNA-mediated migration defect. Analysis of an array of mutations and deletions in APP revealed that both the extracellular and cytoplasmic domains of APP are required for efficient rescue. Whereas knock-down of APP inhibited cortical plate entry, overexpression of APP caused accelerated migration of cells past the cortical plate boundary, confirming that normal APP levels are required for correct neuronal migration. In addition, we found that Disabled-1 (Dab1), an adaptor protein with a well established role in cortical cell migration, acts downstream of APP for this function in cortical plate entry. We conclude that full-length APP functions as an important factor for proper migration of neuronal precursors into the cortical plate during the development of the mammalian brain.
Keywords: brain development, Alzheimer's disease, amyloid β-protein, neuronal migration, neural precursor, adhesion, integrins
Introduction
β-amyloid precursor protein (APP) has become the subject of intense study because of its apparently causative role in the pathogenesis of Alzheimer's disease. APP is a type I transmembrane glycoprotein that is cleaved by either α- or β-secretase to release its large extracellular domain (APPs). Subsequently, the membrane-retained C-terminal fragments are subjected to an intramembranous scission by the γ-secretase complex (Wolfe et al., 1999) to generate the APP intracellular domain and simultaneously release p3 (after α-secretase cleavage) or Aβ (after β-secretase cleavage). Over 20 mutations in APP have been linked to familial AD, all of which enhance the generation and/or aggregation of Aβ42 (Hardy and Selkoe, 2002). Because several therapeutic strategies chronically target the enzymes that process APP, it is essential to understand the physiological function of APP and its proteolytic derivatives in vivo.
In vitro studies have provided insights into the functional activities of APP. Cell culture studies have shown that APP can mediate cell-cell and cell-matrix adhesion (for review, see Mattson, 1997). APP is expressed in part at the cell surface, and its large extracellular domain is capable of binding to several extracellular matrix proteins. In neurons, APP is present in growth cones of neurites and at both presynaptic and postsynaptic sites (Ferreira et al., 1993; Yamazaki et al., 1997; Sabo et al., 2003) and studies in cultured neurons have shown that APP plays a role in neurite outgrowth (Small et al., 1994; Perez et al., 1997) and synaptogenesis (Priller et al., 2006).
Studies in mice lacking APP have suggested roles in aspects of neuronal development and function, but the defects observed have been subtle relative to the functional activities observed for APP in vitro (for review, see Anliker and Muller, 2006). Lack of a robust phenotype is believed to result in part from compensation by its family members, amyloid precursor-like protein 1 (APLP1) and APLP2 (Zheng et al., 1995; von Koch et al., 1997; Heber et al., 2000). Mice with a triple deletion of all three family members exhibit early postnatal lethality, with two thirds of the embryos displaying focal regions of neuronal overmigration in the cortex (Herms et al., 2004). However, in these mice only small subsets of cortical cells are affected, despite the fact that APP, APLP1 and APLP2 are expressed in the developing and mature cerebral cortex (Wasco et al., 1993; Slunt et al., 1994; Lorent et al., 1995). These findings suggest that additional factors either play redundant roles with APP family members or compensate for their chronic loss.
To circumvent compensatory mechanisms that may be masking in vivo functions for APP, we sought to acutely eliminate APP expression in neuronal precursor cells of the living cortex, using the method of in utero electroporation of shRNAs. Using this approach, we identify a role for APP in neuronal migration into the cortical plate and show that both the extra- and intracellular domains of APP are required for this function. This experimental paradigm allows for the efficient introduction of multiple DNA constructs into subsets of cortical cells, providing direct insights into how APP mediates this process in vivo.
Materials and Methods
Plasmid generation.
ShRNA constructs were generated in the pENTR-U6 vector (Invitrogen, Eugene, OR). APP target sequences were as follows: shRNA-1 (partial), gcacatgaatgtgcagaatgg; shRNA-2 (active), gcactaacttgcacgactatg; shRNA-3 (inactive), gctgacaagaaggccgttatc; Dab shRNA, gcatcaatgcgagctcatggag. For testing shRNA constructs, full-length mouse APP (695-residue variant) was C-terminally tagged with the FLAG epitope (Asp-Tyr-Lys-Asp-Asp-Asp-Asp-Lys) in pCDNA 3.1. All of the other constructs used here were generated in the pCAG vector, which has the chick β-actin promoter and cytomegalovirus immediate-early gene enhancer. All APP constructs were generated in human APP 695 unless otherwise stated, and amino acid numbering listed below is for hAPP 695. APP Δ C-term encodes amino acids 1–651 followed by a stop codon. APP AenAtA was generated through mutation of tyrosine 682, proline 685, and tyrosine 687 to alanine. C99 encodes the C-terminal 99 amino acids of APP (amino acids 597–695), from the β-secretase cleavage site. APP-sα (amino acids 1–612) and APP-sβ (amino acids 1–596) are the N-terminal amino acids from the start methionine to the α- and β-secretase cleavage sites. The APP construct harboring two familial Alzheimer's disease (FAD) mutations [Swedish (Mullan et al., 1992) and Indiana (Murrell et al., 1991)] had the following mutations: K676N; M677L; and V642F.
In utero electroporation.
Sprague Dawley rats (Charles River Laboratories, Wilmington, MA) were housed and cared for under the guidelines established by University of Connecticut and Harvard University's Institutional Animal Care and Use Committees in compliance with federal standards. Timed pregnant rats [embryonic day 13 (E13)–E17] were anesthetized with ketamine/xylazine (100/10 mixture, 0.1 mg/g body weight, i.p.). The uterine horns were exposed, and a lateral ventricle of each embryo injected with DNA constructs and Fast Green (2 mg/ml; Sigma, St. Louis, MO) via a microinjector (Picospritzer III; General Valve, Fairfield, NJ) and pulled glass capillaries. For characterization of APP and Dab1 shRNA phenotypes, 1.0–1.5 μg/μl of shRNA was coelectroporated with 0.5 μg/μl pCAG-green fluorescent protein (GFP). For rescue experiments, 0.5 μg/μl shRNA was coelectroporated with 0.5 μg/μl pCAG-GFP and 3.0 μg/μl rescue constructs, unless otherwise stated. For C60 and C60NATA electroporations, 3 μg/μl was coelectroporated with 0.5 μg/μl pCAG-GFP. For 3 d harvests, APP and APPAenAtA was electroporated at 2 μg/μl with 0.5 μg/μl pCAG-GFP. Electroporation was accomplished by discharging a 500 μF capacitor charged to 50–100 V with a sequencing power supply or with a BTX square wave electroporator, at 50–75 V, for 50 ms on followed my 950 ms off for 5 pulses. The voltage was discharged across copper alloy oval plates placed on the uterine wall across the head of the embryo. Brains from rat embryos or postnatal pups were harvested in 4% paraformaldehyde by immersion or cardiac perfusion, respectively.
Immunofluorescent staining and confocal microscopy.
Paraformaldehyde-fixed brains were washed in PBS, embedded in 2% agarose, and vibratome sectioned (50–100 μm). Sections were incubated in blocking buffer (2% donkey or goat serum; 0.001%–0.1% Triton X-100 in PBS) for >1 h. Sections were then incubated in primary antibody [anti-APP C9, Selkoe Lab, 1:500; anti-APP 22C11, 1:200 (Millipore, Temecula, CA); anti-MAP2, 1:10,000 (Abcam, Cambridge, UK); anti-NeuN, 1:800 (Millipore); anti-APLP1 1Cta, 1:500, gift from D. Walsh (University College Dublin, Dublin, Ireland); anti-APLP2 W2CT, 1:500, gift from D. Walsh; anti-GFAP, 1:1000 (Abcam); anti-nestin, 1:500 (BD Biosciences, San Jose, CA); anti-Tbr1, 1:2500, gift from R. Hevner (University of Washington, Seattle, WA); anti-Oct6, 1:200 (Santa Cruz Biotechnology, Santa Cruz, CA); anti-CTIP2, 1:500 (Abcam)] between 6 h and overnight at 4°C, followed by three washes in PBS. Sections were then incubated with Cy3-, Cy2- and Cy5-conjugated secondary antibodies (1:500–1:1000; Jackson Immunoresearch, West Grove, PA) for >2 h followed by four PBS washes. Sections were mounted on glass slides using GelMount (Biomeda, Foster City, CA). Images were acquired using a Zeiss (Oberkochen, Germany) LSM 510 confocal microscope with Axiovert 100M system.
Quantitative analyses of cortical plate entry.
For quantitative analyses, all electroporations were performed targeting the same region of the developing cortex. This resulted in a reliable electroporation of the dorsal-lateral region of the neocortex adjacent to the lateral ventricle (see Fig. 2 A,B). After harvest, brains were vibratome sectioned (100 μm) in the coronal plane and immunostained for microtubule-associated protein 2 (MAP2) to delineate the cortical plate. For each electroporation condition (i.e., each set of electroporated DNAs), greater than a total of 1500 cells were counted and assessed for their location in either the MAP2+ cortical plate or the intermediate zone. To determine significant changes relative to control electroporations, at least three independent brains were electroporated and analyzed for each DNA condition. For each independent brain, the percentage of cells in the intermediate zone (IZ) and cortical plate (CP) were calculated. These values were then compared between electroporation conditions using GraphPad (San Diego, CA) InStat. Using this program, one-way ANOVA tests were performed with the Bonferroni multiple comparisons test. This analysis was used to determine that the percentage of cells in the CP in the noted electroporation conditions was significantly different from the percentage cells in the CP of the control electroporations (similarly, these same conditions were statistically significant when comparing the percentage cells in the IZ for each condition).
Figure 2.
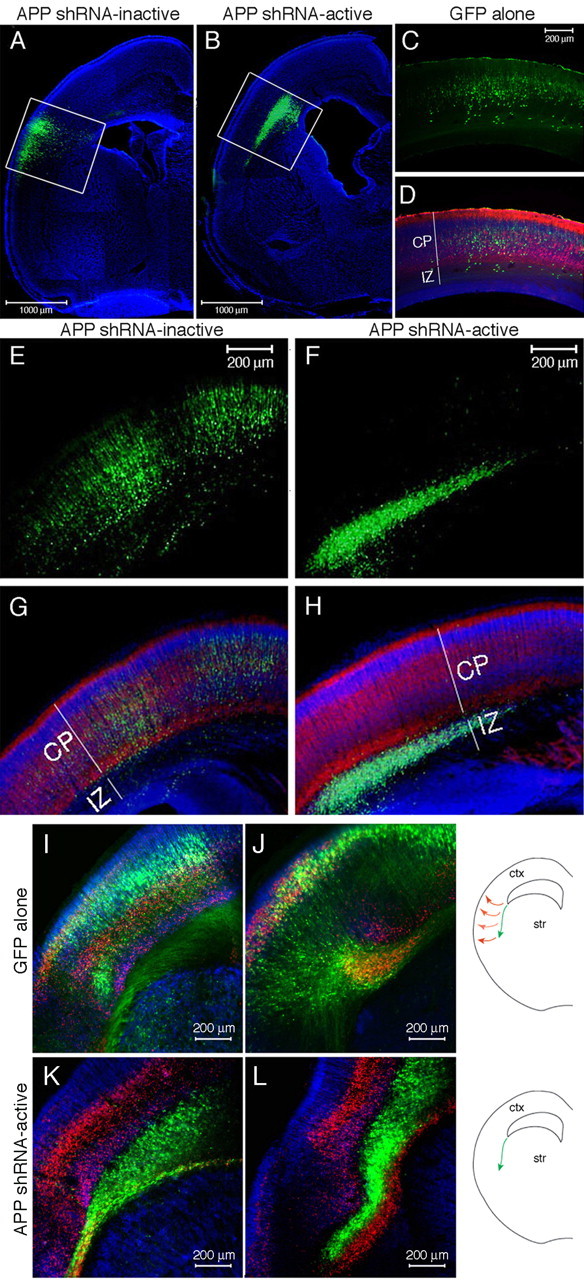
Electroporation of APP shRNA in embryonic rat cortex. DNA encoding GFP and APP shRNA was coelectroporated into one lateral ventricle of an E13 rat and harvested 6 d later. Cells of the ventricular zone lining the ventricle are electroporated. A–H, Location of the electroporated cells (green) is shown 6 d after electroporation of GFP alone (C, D), GFP with an inactive shRNA (A, E, G), or GFP with an active shRNA targeting APP (B, F, H). White boxes delineate electroporated regions (A, B) that are magnified (E–H). I–L, Electroporation of the corticostriatal boundary with GFP alone (I, J) or APP shRNA-active (K, L). A–H, Immunostaining for MAP2 (red) denotes the differentiated neurons of the cortical plate. Nuclei are stained with DAPI (blue). I–L, Immunostaining for MAP2 is shown in blue, and Tbr-1 is shown in red. Str, Striatum; ctx, cortex.
Cell lines and Western blot analysis.
To test shRNA constructs, COS cells (monkey kidney cell line) were transiently transfected using Lipofectamine 2000 (Invitrogen) in 6-well dishes in duplicate with pCDNA-mAPP-FLAG (500 ng), pCAG-GFP (500 ng), and each shRNA construct (1000 ng). Cells were lysed after 48 h in 1% Nonidet P-40 (NP-40) STEN buffer [150 mm sodium chloride, 50 mm Tris, 2 mm EDTA, and 1.0% (v/v) NP-40]. Lysates were electrophoresed on 10–20% Tricine gels (Invitrogen) and transferred to nitrocellulose. Western blotting was performed with anti-M2 FLAG (1:1000; Sigma), anti-GFP (1:1000; Invitrogen), and IRDye680- and IRDye800-conjugated secondary antibodies (1:10,000; Rockland Immunochemicals, Gilbertsville, PA), and detected using the LiCor detection system.
To test for expression of various APP constructs, COS cells were transiently transfected using Lipofectamine 2000 in 6-well dishes in duplicate with each DNA construct (2 μg). After 48 h, media were collected and cells lysed in 1% NP-40 STEN buffer. Conditioned media or lysates were electrophoresed on 10–20% Tricine gels (Invitrogen). Western blotting was performed with anti-APP antibodies: C9 (1:1000, Selkoe laboratory), 22C11 (1:500, Millipore), and 1736 (1:2000, Selkoe laboratory) and detected as above.
Results
Expression of APP in the developing cortex
Previous studies have used in situ hybridization to describe APP mRNA expression in the cortical plate and intermediate zone of the embryonic mouse cortex (Lorent et al., 1995). To confirm the localization of APP protein in the developing cortex, E15 rat cortices were sectioned coronally and immunostained with an antibody to the APP C terminus (C9) (Kimberly et al., 2005). APP immunoreactivity was widespread throughout the cortical plate, and also was observed in a subset of cells in the intermediate zone and ventricular zone (Fig. 1 A). These APP immunoreactive cells were frequently aligned with nestin-positive radial glial fibers (Fig. 1 A,B), a result that indicates the expression of APP in migrating neuronal precursors.
Figure 1.
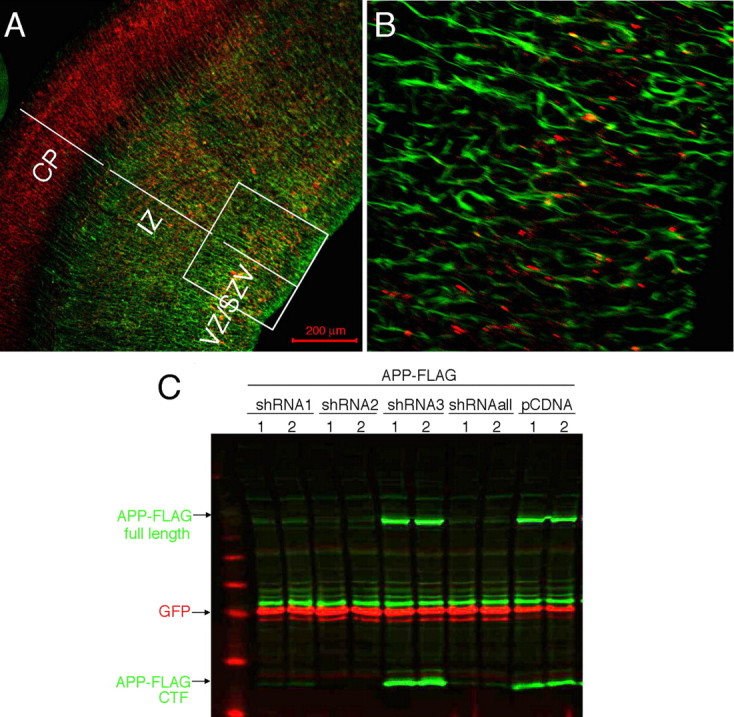
Generation of shRNA constructs targeting endogenous rodent APP in embryonic cortex. A, B, Coronal sections of E15 mouse cortex immunostained for APP (red) and nestin (green). Magnified view of IZ/SVZ/VZ (box in A) is shown in B. SVZ, Subventricular zone; VZ, ventricular zone. C, FLAG-tagged murine APP construct was cotransfected into CHO cells with an empty vector or with 3 different shRNA constructs targeting rodent APP (shRNA1, shRNA2, shRNA3), along with a GFP construct to control for transfection efficiency. Transfections in duplicate wells are shown. 48 h post-transfection, cells were lysed and Western blotted for FLAG (green) and GFP (red). shRNAall, Mix of all three shRNAs.
Generation of shRNA constructs to knock-down APP
To investigate the function of APP in the developing cortex, we sought to decrease the expression of APP in neural precursor cells of the cortex using shRNA. Three shRNA constructs were designed to target both mouse and rat APP. BLAST (NCBI) searches of the shRNA sequences revealed no significant homology to genes other than APP in the mouse or rat genomes. The shRNAs were driven by the U6 promoter in the pENTR vector (Invitrogen). Each construct was tested for its ability to knock-down APP levels in a transient transfection assay, in which C-terminally FLAG-tagged murine APP was cotransfected with GFP and the shRNA construct into Chinese hamster ovary (CHO) cells. Cells were lysed 48 h post-transfection, and Western blots were performed with anti-FLAG and anti-GFP antibodies. Cotransfection with APP shRNA 3 (subsequently called shRNA-inactive) did not reduce APP expression, such that APP-FLAG levels were similar to transfection with an empty shRNA vector (Fig. 1 C). In contrast, transfection with APP shRNA 2 (shRNA-active) fully eliminated expression of APP-FLAG (Fig. 1 C). A third construct, shRNA 1, had an intermediate effect on APP levels (APP shRNA-partial). These shRNA constructs did not affect expression of APLP1 or APLP2 using a transient transfection assay (data not shown).
In utero electroporation allows for acute introduction of DNA constructs to knock-down or misexpress genes in the embryonic cortex. Coelectroporation of a DNA encoding a fluorescent protein (such as GFP or red fluorescent protein) allows the electroporated cells to be readily identified, as >90% of electroporated cells have been shown to express both plasmids using this technique (Bai et al., 2003). Active or inactive APP shRNA constructs were coinjected with GFP plasmid into a lateral ventricle of E13 rat embryos. Application of a small voltage across the head of each embryo resulted in coelectroporation of the DNAs into a subset of cells of the ventricular zone lining the ventricle, where neural precursor cells reside (Tabata and Nakajima, 2001; Bai et al., 2003). The embryonic brains were harvested 6 d later (E19). Coronal sectioning and APP immunostaining of these brains revealed that cells coelectroporated with GFP and APP shRNA-active no longer expressed detectable APP (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Migration of neuronal precursors after APP knock-down
Electroporation of GFP alone or of GFP plus an inactive shRNA did not affect the normal migration of the electroporated cells from the ventricular zone into the cortical plate over 6 d. In contrast, electroporation of APP shRNA-active prevented migration of these cells; Figure 2, A and B, shows the extent of GFP/shRNA-electroporated cells in the cortex. MAP2 immunostaining was used to mark the differentiated neurons of the cortical plate (Fig. 2 D,G,H). Cells electroporated at E13 with GFP alone or GFP plus APP shRNA-inactive were primarily distributed throughout the cortical plate by E19, with a limited number of cells still in the intermediate zone (Fig. 2 A,C–E,G). In contrast, the majority of cells electroporated with APP shRNA-active were tightly packed directly below the cortical plate in the intermediate zone, with a small minority of cells in the cortical plate (Fig. 2 B,F,H). This phenotype was observed in every brain electroporated with APP shRNA-active, in >30 rat brains from more than 20 independent experiments. This phenotype was never observed with shRNA-inactive electroporation, which was indistinguishable from electroporation of GFP alone.
Examination of coronal sections of these electroporated brains revealed that electroporation of APP shRNA-active induced ectopic MAP2 expression in the retarded cells in the intermediate zone (supplemental Fig. 2C,F, available at www.jneurosci.org as supplemental material). Electroporation of APP shRNA-partial resulted in the same migration phenotype but with a smaller subset of cells retained in the intermediate zone, and these cells also expressed MAP2 (supplemental Fig. 2B,E, available at www.jneurosci.org as supplemental material).
Altering the position of the electrodes for voltage application can target different subsets of cells lining the lateral ventricle. Electroporation of slightly more lateral cells at the cortical–striatal boundary targets cells known to migrate along the lateral cortical stream (LCS). These cells migrate along radial glial fibers to the LCS and then migrate radially into the cortical plate of the ventral-most forebrain (Bayer and Altman, 1991; Bayer et al., 1991). Targeting of APP shRNA-active to the cortical-striatal boundary resulted in initially normal migration along the LCS, but an impaired ability to then migrate radially into the cortical plate (Fig. 2 I–L).
Initial effects of altered APP levels in neuronal precursor cells
To examine the more immediate effects of APP knock-down, embryos electroporated at E13 were harvested 3 d after electroporation (E16). In performing the in utero electroporations, cells lining the ventricular zone are transfected. These include both newly postmitotic neuronal precursor cells and radial glial cells. However, 72 h after electroporation, radial glial cells lose expression of GFP, perhaps because of their rapid division. To examine the morphology of radial glial fibers, coronal sections from electroporated brains were immunostained with an anti-brain lipid-binding protein (BLBP) antibody. Cortical sections from brains electroporated with GFP alone (Fig. 3 A,C,E,G) or coelectroporated with GFP and inactive shRNA (data not shown) displayed radially aligned BLBP-positive glial fibers, and GFP-positive electroporated cells extended radial processes along these BLBP-positive fibers. BLBP-positive radial glial fibers looked unchanged in sections from brains electroporated with GFP plus APP shRNA-active versus GFP alone (Fig. 3 B). However, the cells receiving APP shRNA did not extend radial processes along the BLBP-positive glial fibers 3 d postelectroporation (Fig. 3 D) and arrested just below the MAP2+, cell-dense cortical plate (Fig. 3 D,H). Although there does not appear to be an effect of APP shRNA-active on radial glial morphology, there may be a cell nonautonomous effect of APP shRNA-active to retard in the intermediate zone a subset of neighboring cells that had not received the APP shRNA and GFP constructs (supplemental Fig. 2D–F, available at www.jneurosci.org as supplemental material).
Figure 3.
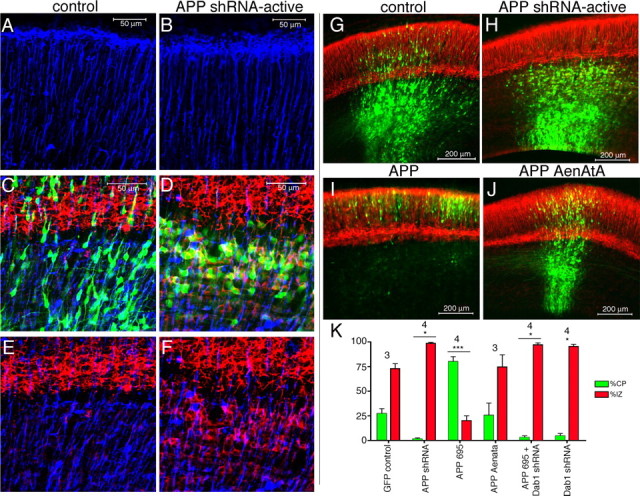
Early effects of acutely altering APP levels in embryonic cortex. A–J, Electroporation of GFP alone (A, C, E, G) or coelectroporation of GFP plus active APP shRNA (B, D, F, H), GFP plus APP (I), or GFP plus mutant APP AenAtA (J) into E13 cortex and harvesting 3 d later. Electroporated cells are green (GFP). Immunostaining for BLBP (A–F) is blue and MAP2 is red (C–J). A, B, BLBP immunostaining in upper cortical plate in electroporated region. C–F, MAP2 with BLBP double staining at the cortical plate/intermediate zone boundary. K, Quantification of cells in the IZ and CP for each condition shown in G–J. Error bars indicate SD; *p < 0.05; ***p < 0.001, percentage of cells in IZ and CP relative to the corresponding values in GFP control. Numbers above bars show the number of independent brains analyzed for each condition.
Coronal sections from these electroporations also were immunostained for MAP2. At this time point (E16), cells transfected with GFP alone had begun to enter the cortical plate, but many cells were in the process of migrating through the intermediate zone (Fig. 3 C,G). In the case of cells electroporated with APP shRNA-active, few had entered the cortical plate by E16, as expected, so that the majority of these cells remained in the intermediate zone (Fig. 3 H). Interestingly, these APP shRNA receiving cells retained in the intermediate zone already expressed the neuronal marker MAP2 (Fig. 3 D,F), whereas the cells in the intermediate zone that had been electroporated with GFP alone did not express MAP2 at E16 (Fig. 3 C,E).
To examine whether the altered positioning of neuronal precursor cells having decreased APP levels is directly related to their migration into the cortical plate, we performed experiments to examine the effects of APP overexpression. Overexpression of wild-type APP (the 695-residue splice variant) at E13 did not appear grossly different from control electroporations when harvested 6 d later at E19 (data not shown). However, harvest of APP-electroporated brains 3 d after electroporation revealed that overexpression of APP caused an acceleration of migration into the cortical plate (Fig. 3 I,K). Over 75% of cells electroporated with APP at E13 had already entered the cortical plate after 3 d, in contrast to just 25% in control (GFP only) electroporations (Fig. 3 G,K). Electroporation of APP with mutations in its YENPTY domain (APP AENATA) at E13 eliminated the acceleration phenotype of APP overexpression at E16 (Fig. 3 J,K).
Late effects of embryonic APP knock-down in the mature brain
To determine the ultimate fate of APP shRNA electroporated cells, brains of E13 electroporated rats were harvested at postnatal day 30. At this late time the cortex is fully developed, and control (GFP only) electroporated cells had fully migrated and differentiated (Fig. 4 B,D). In contrast, cells electroporated with APP shRNA-active remained trapped below the cortical plate and formed a macroscopically visible heterotopia (Fig. 4 A,C). These cells took on an abnormal cuboidal morphology (Fig. 4 E). The cells did not express GFAP, but GFAP-positive cells were interspersed throughout the heterotopia (Fig. 4 G,I). In addition, these cells did not express significant amounts of Prominin-1, an ependymal marker, and they lost the high levels of expression of MAP2 observed at earlier time points (Fig. 4, compare H, F, labeling in cortical plate). Additional characterization of these cells showed that cells trapped in the heterotopia expressed both βIII-tubulin and Uch-L1, a highly expressed neuronal deubiquitinating enzyme (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Interestingly, in brains electroporated at E13 and harvested at postnatal day 5, APP shRNA electroporated cells trapped in the intermediate zone strongly expressed MAP2, NeuN, and Oct6, a transcription factor expressed by layer III and V neurons (data not shown). Thus, during and immediately after the window of cortical cell migration, cells receiving APP shRNA that are trapped in the intermediate zone express neuronal markers. However, between P5 and P30, cells forming a heterotopia change morphology and lose expression of some neuronal markers.
Figure 4.

Cells electroporated with active APP shRNA remain trapped in the intermediate zone and form a heterotopia. Cortices were coelectroporated at E13 with GFP and APP shRNA-active (A, C, E, F, G, H, I) or with control DNA (B, D) and harvested 30 d postnatally. Brightfield image of a coronal section of a cortex electroporated with active APP shRNA (A). Arrows point to a heterotopia only present in the electroporated hemisphere. B–I, Electroporated cells are shown in green. Location and morphology of control electroporated cells in the CP (B, D) and of APP shRNA-active electroporated cells in the IZ (C, E). MAP2 (red) and NeuN (blue) immunostaining of APP shRNA electroporated cells in the IZ (H) and CP (F) at P30. G, I, GFAP immunostaining (red) in the heterotopia.
Rescue of the APP shRNA phenotype with full-length human APP
To confirm that the effects observed after APP knock-down were specific to the observed loss of APP expression, rescue experiments, were performed by coelectroporating full-length human APP with APP shRNA-active and GFP. Because of interspecies variation in the shRNA target sequence we had selected, APP shRNA-active does not effectively target human APP, as confirmed in a CHO transfection assay with Western blot analysis (data not shown). Embryonic day 13 cortices were again electroporated and harvested 6 d later (E19). Brains were fixed, sectioned coronally and immunostained for MAP2 (Fig. 5 A,B) and NeuN (data not shown) to delineate the boundaries of the cortical plate, as before. The number of GFP-positive electroporated cells in the cortical plate and intermediate zone were counted in a blinded manner after electroporation with GFP alone (no shRNA) or with GFP plus APP shRNA-active plus the designated rescue construct (Fig. 5 D). As imaged in Figure 2, >90% of control (GFP) electroporated cells enter the cortical plate 6 d after the electroporation at E13. In contrast, with APP shRNA-active electroporation, only 19% (SD, ±8.2%) enter the cortical plate (Fig. 5 D). Coelectroporation of full-length human APP 695 is able to effectively rescue this defect at doses of either 1.5 μg/μl of DNA (88.5 ± 2.9% in the cortical plate) or 3.0 μg/μl (89.7 ± 5.6%). Full-length human APP 751, a splice variant of APP that has an additional Kunitz protease inhibitory domain, also efficiently rescued the APP shRNA effect (Fig. 5 D). Like full-length APP, full-length constructs encoding APLP1 or APLP2 each rescued APP knock-down (Fig. 5 D).
Figure 5.
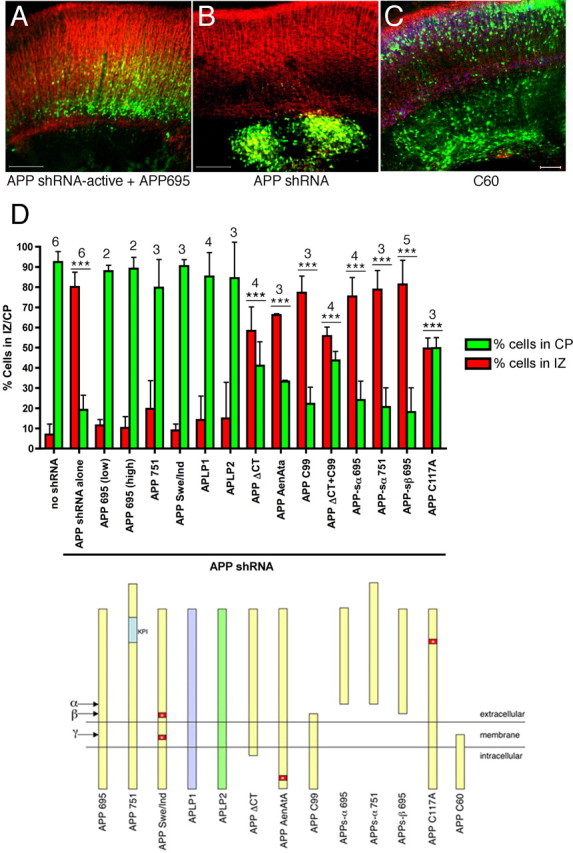
Rescue of APP shRNA-induced migration defects. Embryos were electroporated at E13, harvested after 6 d in utero, and brain sections immunostained for MAP2 (red). A, B, Representative sections from APP shRNA-active plus APP695 (A) and APP shRNA-active alone (B) are shown. Scale bars, 200 μm. C, Image of section from an E13 embryo electroporated with only the C-terminal 60 aa of APP (C60) and GFP (green). Immunostaining for MAP2 shown in red and for NeuN in blue. Scale bar, 100 μm. D, Quantification (top graph) of cells in the IZ and CP for each of the rescue constructs diagramed below. Error bars indicate SD; ***p < 0.001, percentage of cells in IZ and CP relative to the corresponding values with control DNA (no shRNA). The numbers above the bars show the number of independent brains analyzed for each condition.
Analysis of domains of APP required for cortical cell migration
APP is a type-I transmembrane protein that has a large extracellular N-terminal domain and a short intracellular C-terminal domain. Each of these domains has been proposed to play roles in the function of APP (for review, see Reinhard et al., 2005). To test whether different regions of APP are necessary for the above described rescue function of APP to confer proper neuronal migration in vivo, expression plasmids were made from the human APP 695 construct and coelectroporated at the “high” concentration (3.0 μg/μl) with the active APP shRNA. Before doing so, each plasmid had also been transfected into COS cells, and Western blotting had confirmed the correct expression, size and expected processing of each APP construct (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). After electroporation, immunofluorescent staining for APP was performed on brain sections to confirm that each construct had been expressed above endogenous levels (supplemental Fig. 5, available at www.jneurosci.org as supplemental material).
Constructs encoding the full extracellular and transmembrane domains but lacking the intracellular domain (APP ΔCT) or else containing mutations of just three critical amino acids in the YENPTY motif in the intracellular domain (APP AENATA) were unable to efficiently rescue the shRNA defect (Fig. 5 D). A construct encoding solely the transmembrane and intracellular domains (i.e., the naturally occurring APP C99 fragment, which begins at the β-secretase cleavage site) had no rescue ability (Fig. 5 D). Electroporation of both the APP ΔCT and C99 constructs together also did not efficiently rescue the APP shRNA phenotype (Fig. 5 D), indicating that the intact APP holoprotein is necessary to rescue the APP shRNA phenotype.
Previous studies have shown that γ-secretase substrates can compete with one another for cleavage when overexpressed in cells (LaVoie and Selkoe, 2003; Lleo et al., 2003). Thus, it is possible that removal of a substrate, such as occurs with APP knock-down, may increase the cleavage of other substrates by γ-secretase. However, we do not believe that the APP shRNA phenotype that we observe is attributable to an indirect effect on γ-secretase activity, as expressing C99 or APP Aenata (which are both efficiently cleaved by γ-secretase) did not rescue the phenotype. In addition, shRNA induced knock-down of APP in primary neurons did not affect γ-secretase cleavage of Notch, as detected by Western blot analysis (data not shown).
APPs is a naturally occurring secreted product of holoAPP. APPs-α results from α-secretase cleavage, whereas APPs-β derives from β-secretase cleavage. APPs has been shown to have biological activity in a variety of in vitro and in vivo assays (for review, see Reinhard et al., 2005; Ring et al., 2007). However, we found that APPs-α 695, APPs-α 751 and APPs-β 695 were each unable to rescue the APP shRNA migration defect (Fig. 5 D).
The extracellular domain of APP has been proposed to have at least three disulfide bridges that help determine its secondary structure (Rossjohn et al., 1999). Mutation of one of the disulfide bridge cysteines, C117, to alanine, which has been reported to alter the structure of the ectodomain (Rossjohn et al., 1999), impaired the ability of full-length APP to rescue (Fig. 5 D).
FAD can be caused by one of several missense mutations in APP immediately flanking the Aβ region. These mutations increase Aβ generation and/or increase the ratio of Aβ42 to Aβ40 (Hardy and Selkoe, 2002). Consistent with the hypothesis that these are not loss-of-function mutations, an APP695 construct harboring two of these FAD-causing mutations [K595N/M596 (“Swedish”) (Mullan et al., 1992) and V642F (“Indiana”) (Murrell et al., 1991)] was able to rescue the APP shRNA phenotype with the full efficiency of wild-type APP (Fig. 5 D).
The intracellular domain of APP has been shown to be necessary for proper endocytosis of APP and to interact with a number of cytoplasmic factors through its NPTY motif (Fiore et al., 1995; Trommsdorff et al., 1998; Howell et al., 1999; Perez et al., 1999). It has been postulated that full-length APP may function by recruiting these factors to the cell surface, and that this recruitment is regulated via cleavage of APP by γ-secretase (Cao and Sudhof, 2004; Hass and Yankner, 2005). Thus, overexpression of the free APP intracellular domain might act as a dominant negative by competing with endogenous, full-length APP for cytoplasmic binding partners. To assess the effects of overexpressing the intracellular domain in the absence of shRNA, nonmembrane tethered APP intracellular domain (C60) was coelectroporated with GFP at E13 and harvested 6 d later. In these brains, although the majority of electroporated cells were able to enter the cortical plate normally, subsets of neurons in focal regions of the electroporated region were retarded in the intermediate zone (n = 4). In contrast, electroporation of C60 harboring two mutations in the NPTY motif (to NATA) did not alter correct neuronal migration into the cortical plate (n = 6) (data not shown). Thus, not only was the intracellular domain of APP unable to rescue the effects of APP knock-down (Fig. 5 D), but overexpression of the free intracellular domain alone, in the absence of APP knock-down, induced a migration defect in a subset of cells (Fig. 5 C).
The mechanism of APP in neuronal migration involves Disabled-1 as a downstream mediator
It was shown previously that Dab1 physically binds to the NPTY motif of APP (Trommsdorff et al., 1998; Homayouni et al., 1999; Howell et al., 1999), and we have confirmed this data here (supplemental Fig. 6, available at www.jneurosci.org as supplemental material). Previous studies have demonstrated that knock-down of Dab1 using in utero electroporation prevents proper neuronal migration through the cortical plate (Ware et al., 1997; Dulabon et al., 2000; Olson et al., 2006). Based on this knowledge, we directly compared the previous findings on Dab1 with results with APP knock-down. Dab1 shRNA was electroporated into cortices of E13 embryos, and the brains were harvested 3 or 6 d later. Neuronal precursors receiving Dab1 shRNA were unable to enter the cortical plate and were arrested in the intermediate zone, reminiscent of the effects observed with APP knock-down (Figs. 3 K, 6 A,C,E). Moreover, simultaneous knock-down of both APP and Dab1 resulted in an enhanced phenotype, with essentially no cells entering the cortical plate after 6 d (Fig. 6 D,E). Importantly, overexpression of Dab1 (as a Dab1-GFP fusion protein) was able to rescue the phenotype of APP knock-down, although not completely (Fig. 6 B,E). In accord, the accelerated migration observed on wild-type APP overexpression 3 d post electroporation (i.e., at E16) was eliminated either by mutating the YENPTY-motif of APP (Fig. 3 J,K), or by coelectroporating wild-type APP with Dab1 shRNA (Fig. 3 K). The latter result also reveals that simultaneous overexpression of APP cannot rescue the effects of Dab1 knock-down. Together, these findings indicate that Dab1 acts downstream of APP in mediating proper neuronal migration.
Figure 6.

APP and Disabled-1 functionally interact to affect neuronal migration in the cortical plate. A–D, Embryos were electroporated at E13, harvested after 6 d in utero, and brain sections immunostained for MAP2 (blue) and Tbr1 (red). E, Quantification of cells in the IZ and CP for each of the indicated constructs; n = 3 independent brains for each condition. Error bars indicate SD; **p < 0.01.
Discussion
We used in utero electroporation to uncover a role for APP in neuronal cell migration into the cortical plate. Knock-down of endogenous APP expression using shRNA in newly postmitotic cells of the cortical ventricular zone prevents their entrance into the cortical plate. These cells prematurely differentiate into MAP2-positive cells in the intermediate zone and later lose their expression of neuronal markers and form a permanent heterotopia just below the cortical plate. Our observations are shown to be specific for a loss of APP through the use of two independent shRNAs to knock down APP and through the rescue of the APP shRNA phenotype with full-length human APP. Overexpression of APP in neuronal precursor cells accelerates their migration into the cortex, supporting the hypothesis that APP plays a quantitative role in the proper migration of neuronal precursor cells in the developing cortex.
Two features of our approach provided unique advantages for the detection of this APP phenotype that are not achievable with genomic disruption. First, acute knock-down of APP in migrating cells of the cortex bypasses compensatory mechanisms that are implemented with a chronic germline deletion. Second, in APP knock-out mice, all neurons and glia in the brain lack APP and its cleavage products. In contrast, in our experimental paradigm only a subset of cortical cells lack APP, generating a cortex that is mosaic for APP expression. We hypothesize that it is precisely this juxtaposition of APP-expressing and nonexpressing cells that allowed for the detection of this hitherto unknown function for APP in cortical plate entry.
APP and its two mammalian family members, APLP1 and APLP2, are known to compensate functionally for one another, as evidenced by the perinatal lethality of APP/APLP2 and APLP1/APLP2 double knock-out mice that is not observed in deletions of the individual genes (Zheng et al., 1995; von Koch et al., 1997; Heber et al., 2000). We found that overexpression of APLP1 or APLP2 rescued the loss of APP function in cells receiving APP shRNA, providing direct evidence that these proteins can functionally compensate for a loss of APP in cortical plate entry. We hypothesize that endogenous APLP1 and APLP2 are not expressed appropriately to be able to “rescue” APP knock-down in the ∼80% of cells trapped in the intermediate zone. However, ∼20% of cells receiving APP shRNA are able to cross the cortical plate border, and this may be because of endogenous expression of APLP1 and APLP2 in these cells. With a chronic loss of APP, we hypothesized that APLP1 and/or APLP2 may be upregulated to compensate for APP loss. To address this question, we performed an Affymetrix analysis on E16 cortex from genomic APP knock-out versus wild-type littermates. We found that APLP2 RNA is upregulated approximately twofold in APP knock-out animals. Conversely, Affymetrix analysis of APP shRNA electroporated versus control electroporated cortical cells shows no change in APLP2 levels (T. L. Young-Pearse and D. J. Selkoe, unpublished observation). These findings support the hypothesis that other APP family members can compensate for a chronic loss of APP activity, whereas acute disruption of APP expression bypasses this compensatory mechanism.
Triple deletion of all three genes in mice results in perinatal lethality and an over-migration of subsets of neurons in highly focal regions of the embryonic cortex (Herms et al., 2004). This is in contrast to our result, which shows that acute knock-down of APP results in a defect in the ability of cells to enter the cortical plate. As discussed above, there are critical differences between these two experimental paradigms, which may account for the illumination of two different but related roles for APP family members in cortical cell migration: acute versus chronic deletion, and mosaic expression versus complete loss of expression. With a germline deletion, the observation that a limited number of cells were affected by a loss of all three family members while the majority of cells migrated correctly indicates that other molecules can compensate functionally for, or play redundant roles with, APP family members. Type I transmembrane domain proteins with similar structures and binding partners to APP would be potential candidates for this functional compensation.
The APP family members share many similarities to the low-density lipoprotein receptor (LDL-R) family of proteins. Members of both families are type I transmembrane domain proteins that share several binding partners at their common NPXY cytoplasmic motif (Asn-Pro-X-Tyr). APP is known to interact biochemically with at least two members of the LDL-R family, apolipoprotein receptor 2 (ApoER2) and LDL-R related protein, although the functional consequence of these interactions is not understood in vivo. In addition, similar to the phenotype we observed after APP knock-down, members of the LDL-R family are known to play key roles in neuronal migration. Very low-density lipoprotein receptor or ApoER2 knock-out mice do not show any overt phenotype individually, but in combination display a disruption of cortical cell migration similar to the well characterized Reelin and Dab1 phenotype (for review, see Tissir and Goffinet, 2003). Additional studies are required to address whether members of the LDL-R family could be compensating in the germline deletion of APP family members, and whether they functionally interact with APP in cortical plate entry.
APP extracellular and intracellular domains are both necessary for proper entry into the cortical plate
The studies presented here show that, after acute reduction of APP, neuronal precursors can migrate out of the ventricular zone and into the intermediate zone, but accumulate just below the cortical plate in a tightly packed manner (Fig. 2 H). These cells displayed a multipolar morphology without radially aligned processes (Fig. 3, compare B, D). Our finding that APP knock-down cells are able to migrate along the lateral cortical stream, but then cannot enter the cortical plate argues against a general defect in motility. Rather, together these data point to a specific defect in entry into the cortical plate after APP deletion. The cortical plate is defined molecularly by the presence of extracellular factors distinct from those in the intermediate zone, and we hypothesize that it may be the binding of the APP ectodomain to these factors that influences differential adhesion between the IZ and the CP and that, in turn, mediates migration past this boundary.
The rescue studies presented in Figure 5 provide evidence that the APP holoprotein is required for proper migration of neuronal precursors. Deletion of the APP extracellular domain or a point mutation important for its structure (C117A) prevented efficient rescue of APP knock-down. In accord with its demonstrated ability to act as an adhesion molecule that mediates cell-cell and cell-cell matrix attachment, APP has been described to bind to several extracellular factors in vitro, including heparan sulfate proteoglycans (Schubert et al., 1989; Narindrasorasak et al., 1991), laminin (Narindrasorasak et al., 1992; Kibbey et al., 1993), collagen (Breen, 1992; Narindrasorasak et al., 1995), F-spondin (Ho and Sudhof, 2004), and APP family members themselves (Soba et al., 2005). All of these proteins are present in the developing cortex and are candidates to interact with the extracellular domain of APP in migrating cells.
Although the cleaved extracellular domain of APP (APPs-α or -β) alone was unable to rescue APP shRNA in neural precursor migration, this result does not preclude an essential role for APPs in cortical plate entry. Recently, a knock-in of APPs-α into the APP genomic locus has shown that APPs-α can rescue many of the defects observed in the APP knock-out mice, such as in grip strength, body and brain weight, and long term potentiation (Ring et al., 2007). However, other functions for APP, such as its role in neurite outgrowth and neuronal migration into the cortical plate, are not rescued by APPs-α/β. We hypothesize that the presence of APPs-α/β may actually exacerbate certain APP loss-of-function phenotypes. This hypothesis is based on the effects of APP in neurite outgrowth assays: both the deletion of APP in mixed neuronal and glial cultures and the addition of APPs-α to cultured neurons independently increased neurite outgrowth in vitro (Araki et al., 1991; Milward et al., 1992; Ohsawa et al., 1995, 1997; Perez et al., 1997; Wallace et al., 1997). Studies from our lab suggest that this activity of APPs in the neurite outgrowth assay may result from its competition with cell surface APP for interaction with binding partners (T. L. Young-Pearse and D. J. Selkoe, unpublished observation). APPs can bind to several of the same extracellular factors as full-length APP (Reinhard et al., 2005), supporting the hypothesis that it may play a key role in regulating the activity of cell surface APP.
The intact intracellular domain of APP, specifically the YENPTY motif, was necessary for the efficient rescue of failed cortical plate entry. The YENPTY motif of APP is well established to bind to Dab1, Fe65, and X11/MINT-1 (munc18-interacting protein 1) (for review, see Van Gassen et al., 2000). The physical interactions between APP and these proteins may have regulatory activities on signal transduction pathways as well as on the rate of APP endocytosis. The interaction between APP and Dab1 is of particular interest, as the phosphorylation of Dab1 has been well established to play an essential role in migration (for review, see Tissir and Goffinet, 2003). In this regard, our results suggest that Dab1 acts downstream of APP: Dab1 expression rescued the APP knock-down phenotype, APP expression did not rescue Dab1 knock-down, and Dab1 needed to be present for APP overexpression to accelerate neuronal migration. Binding of Dab1, Fe65, and X11/Mint-1 to the NPTY domain of APP is known to regulate APP endocytosis, its cleavage by α- and β-secretases and, thus, the amount of full-length APP available at the cell surface for binding to extracellular cues (Mueller et al., 2000; Guenette et al., 2002; Hoe et al., 2006; Parisiadou and Efthimiopoulos, 2006). Considering that the half-life of APP is relatively short (Perez et al., 1999) and that APP has been shown to localize to sites of dynamic adhesion (Yamazaki et al., 1997; Sabo et al., 2001), proper regulation of the amount of holoAPP at the cell surface may be essential for the dynamic adhesion of motile regions of migrating neurons, in particular to radial glial surface proteins and the extracellular matrix proteins of the cortical plate.
Conclusions
The function of APP in proper neuronal migration into the cortical plate described here is likely to generalize to other cell types, as suggested by in vitro studies describing a role for APP in the migration of Madin-Darby canine kidney cells (Sabo et al., 2001). We speculate that the activity of holoAPP that we show here could be biochemically related to its function in the adult brain. For example, APP is upregulated in the adult brain in response to injury (for review, see Koistinaho and Koistinaho, 2005), and the function of this upregulation may be to help mediate the migration of various cell types to the site of injury. In this context, the further dissection of the pathways by which APP mediates early neuronal migration may reveal specific biochemical functions that should be known in order to anticipate potential side effects of chronic inhibition of APP processing in humans with Alzheimer's disease or mild cognitive impairment.
Neuronal migration disorders in humans range in severity and can lead to epilepsy, mental retardation and premature death. Identification of the genes involved in these disorders has provided key insights about normal cortical cell migration. Using the technique of in utero electroporation, we have identified a new participant involved in correct neuronal positioning in the cortex. The unique advantages of in utero electroporation will now allow us and others to systematically examine additional factors to learn which can compensate for APP loss-of-function, and which exacerbate the phenotype when knocked down or overexpressed. In this manner, the protein–protein interaction pathways that mediate APP function in neuronal migration can be elucidated.
Footnotes
This work was supported by National Institutes of Health Grants F32 NS053320-01A1 (T.L.Y) and ROI AG06173 (D.J.S.). We thank C. Marquez for technical assistance, M. Lavoie, J. Trimarchi, and R. Pearse for critical reading of this manuscript and members of the Selkoe laboratory for helpful discussions.
References
- Anliker B, Muller U. The functions of mammalian amyloid precursor protein and related amyloid precursor-like proteins. Neurodegener Dis. 2006;3:239–246. doi: 10.1159/000095262. [DOI] [PubMed] [Google Scholar]
- Araki W, Kitaguchi N, Tokushima Y, Ishii K, Aratake H, Shimohama S, Nakamura S, Kimura J. Trophic effect of β-amyloid precursor protein on cerebral cortical neurons in culture. Biochem Biophys Res Commun. 1991;181:265–271. doi: 10.1016/s0006-291x(05)81412-3. [DOI] [PubMed] [Google Scholar]
- Bai J, Ramos RL, Ackman JB, Thomas AM, Lee RV, LoTurco JJ. RNAi reveals doublecortin is required for radial migration in rat neocortex. Nat Neurosci. 2003;6:1277–1283. doi: 10.1038/nn1153. [DOI] [PubMed] [Google Scholar]
- Bayer SA, Altman J. Development of the endopiriform nucleus and the claustrum in the rat brain. Neuroscience. 1991;45:391–412. doi: 10.1016/0306-4522(91)90236-h. [DOI] [PubMed] [Google Scholar]
- Bayer SA, Altman J, Russo RJ, Dai XF, Simmons JA. Cell migration in the rat embryonic neocortex. J Comp Neurol. 1991;307:499–516. doi: 10.1002/cne.903070312. [DOI] [PubMed] [Google Scholar]
- Breen KC. APP-collagen interaction is mediated by a heparin bridge mechanism. Mol Chem Neuropathol. 1992;16:109–121. doi: 10.1007/BF03159964. [DOI] [PubMed] [Google Scholar]
- Cao X, Sudhof TC. Dissection of amyloid-beta precursor protein-dependent transcriptional transactivation. J Biol Chem. 2004;279:24601–24611. doi: 10.1074/jbc.M402248200. [DOI] [PubMed] [Google Scholar]
- Dulabon L, Olson EC, Taglienti MG, Eisenhuth S, McGrath B, Walsh CA, Kreidberg JA, Anton ES. Reelin binds α3β1 integrin and inhibits neuronal migration. Neuron. 2000;27:35–44. doi: 10.1016/s0896-6273(00)00007-6. [DOI] [PubMed] [Google Scholar]
- Ferreira A, Caceres A, Kosik K. Intraneuronal compartments of the amyloid precursor protein. J Neurosci. 1993;13:3112–3123. doi: 10.1523/JNEUROSCI.13-07-03112.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiore F, Zambrano N, Minopoli G, Donini V, Duilio A, Russo T. The regions of the Fe65 protein homologous to the phosphotyrosine interaction/phosphotyrosine binding domain of Shc bind the intracellular domain of the Alzheimer's amyloid precursor protein. J Biol Chem. 1995;270:30853–30856. doi: 10.1074/jbc.270.52.30853. [DOI] [PubMed] [Google Scholar]
- Guenette SY, Chang Y, Hyman BT, Tanzi RE, Rebeck GW. Low-density lipoprotein receptor-related protein levels and endocytic function are reduced by overexpression of the FE65 adaptor protein, FE65L1. J Neurochem. 2002;82:755–762. doi: 10.1046/j.1471-4159.2002.01009.x. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hass MR, Yankner BA. A γ-secretase-independent mechanism of signal transduction by the amyloid precursor protein. J Biol Chem. 2005;280:36895–36904. doi: 10.1074/jbc.M502861200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heber S, Herms J, Gajic V, Hainfellner J, Aguzzi A, Rulicke T, von Kretzschmar H, von Koch C, Sisodia S, Tremml P, Lipp HP, Wolfer DP, Müller U. Mice with combined gene knock-outs reveal essential and partially redundant functions of amyloid precursor protein family members. J Neurosci. 2000;20:7951–7963. doi: 10.1523/JNEUROSCI.20-21-07951.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herms J, Anliker B, Heber S, Ring S, Fuhrmann M, Kretzschmar H, Sisodia S, Muller U. Cortical dysplasia resembling human type 2 lissencephaly in mice lacking all three APP family members. EMBO J. 2004;23:4106–4115. doi: 10.1038/sj.emboj.7600390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho A, Sudhof TC. Binding of F-spondin to amyloid-β precursor protein: a candidate amyloid-β precursor protein ligand that modulates amyloid-β precursor protein cleavage. Proc Natl Acad Sci USA. 2004;101:2548–2553. doi: 10.1073/pnas.0308655100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoe HS, Tran TS, Matsuoka Y, Howell BW, Rebeck GW. DAB1 and Reelin effects on amyloid precursor protein and ApoE receptor 2 trafficking and processing. J Biol Chem. 2006;281:35176–35185. doi: 10.1074/jbc.M602162200. [DOI] [PubMed] [Google Scholar]
- Homayouni R, Rice DS, Sheldon M, Curran T. Disabled-1 binds to the cytoplasmic domain of amyloid precursor-like protein 1. J Neurosci. 1999;19:7507–7515. doi: 10.1523/JNEUROSCI.19-17-07507.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell BW, Lanier LM, Frank R, Gertler FB, Cooper JA. The disabled 1 phosphotyrosine-binding domain binds to the internalization signals of transmembrane glycoproteins and to phospholipids. Mol Cell Biol. 1999;19:5179–5188. doi: 10.1128/mcb.19.7.5179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kibbey MC, Jucker M, Weeks BS, Neve RL, Van Nostrand WE, Kleinman HK. β-Amyloid precursor protein binds to the neurite-promoting IKVAV site of laminin. Proc Natl Acad Sci USA. 1993;90:10150–10153. doi: 10.1073/pnas.90.21.10150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimberly WT, Zheng JB, Town T, Flavell RA, Selkoe DJ. Physiological regulation of the beta-amyloid precursor protein signaling domain by c-Jun N-terminal kinase JNK3 during neuronal differentiation. J Neurosci. 2005;25:5533–5543. doi: 10.1523/JNEUROSCI.4883-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koistinaho M, Koistinaho J. Interactions between Alzheimer's disease and cerebral ischemia—focus on inflammation. Brain Res Brain Res Rev. 2005;48:240–250. doi: 10.1016/j.brainresrev.2004.12.014. [DOI] [PubMed] [Google Scholar]
- LaVoie MJ, Selkoe DJ. The Notch ligands, Jagged and Delta, are sequentially processed by alpha-secretase and presenilin/gamma-secretase and release signaling fragments. J Biol Chem. 2003;278:34427–34437. doi: 10.1074/jbc.M302659200. [DOI] [PubMed] [Google Scholar]
- Lleo A, Berezovska O, Ramdya P, Fukumoto H, Raju S, Shah T, Hyman BT. Notch1 competes with the amyloid precursor protein for gamma-secretase and down-regulates presenilin-1 gene expression. J Biol Chem. 2003;278:47370–47375. doi: 10.1074/jbc.M308480200. [DOI] [PubMed] [Google Scholar]
- Lorent K, Overbergh L, Moechars D, De Strooper B, Van Leuven F, Van den Berghe H. Expression in mouse embryos and in adult mouse brain of three members of the amyloid precursor protein family, of the α-2-macroglobulin receptor/low density lipoprotein receptor-related protein and of its ligands apolipoprotein E, lipoprotein lipase, α-2-macroglobulin and the 40,000 molecular weight receptor-associated protein. Neuroscience. 1995;65:1009–1025. doi: 10.1016/0306-4522(94)00555-j. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Cellular actions of β-amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiol Rev. 1997;77:1081–1132. doi: 10.1152/physrev.1997.77.4.1081. [DOI] [PubMed] [Google Scholar]
- Milward EA, Papadopoulos R, Fuller SJ, Moir RD, Small D, Beyreuther K, Masters CL. The amyloid protein precursor of Alzheimer's disease is a mediator of the effects of nerve growth factor on neurite outgrowth. Neuron. 1992;9:129–137. doi: 10.1016/0896-6273(92)90228-6. [DOI] [PubMed] [Google Scholar]
- Mueller HT, Borg JP, Margolis B, Turner RS. Modulation of amyloid precursor protein metabolism by X11alpha/Mint-1. A deletion analysis of protein-protein interaction domains. J Biol Chem. 2000;275:39302–39306. doi: 10.1074/jbc.M008453200. [DOI] [PubMed] [Google Scholar]
- Mullan M, Crawford F, Houlden H, Axelman K, Lilius L, Winblad B, Lannfelt L. A pathogenic mutation for probable Alzheimer's disease in the APP gene at the N-terminus of β-amyloid. Nat Genet. 1992;1:345–347. doi: 10.1038/ng0892-345. [DOI] [PubMed] [Google Scholar]
- Murrell J, Farlow M, Ghetti B, Benson MD. A mutation in the amyloid precursor protein associated with hereditary Alzheimer's disease. Science. 1991;254:97–99. doi: 10.1126/science.1925564. [DOI] [PubMed] [Google Scholar]
- Narindrasorasak S, Lowery D, Gonzalez-DeWhitt P, Poorman RA, Greenberg B, Kisilevsky R. High affinity interactions between the Alzheimer's β-amyloid precursor proteins and the basement membrane form of heparan sulfate proteoglycan. J Biol Chem. 1991;266:12878–12883. [PubMed] [Google Scholar]
- Narindrasorasak S, Lowery DE, Altman RA, Gonzalez-DeWhitt PA, Greenberg BD, Kisilevsky R. Characterization of high affinity binding between laminin and Alzheimer's disease amyloid precursor proteins. Lab Invest. 1992;67:643–652. [PubMed] [Google Scholar]
- Narindrasorasak S, Altman RA, Gonzalez-DeWhitt P, Greenberg BD, Kisilevsky R. An interaction between basement membrane and Alzheimer amyloid precursor proteins suggests a role in the pathogenesis of Alzheimer's disease. Lab Invest. 1995;72:272–282. [PubMed] [Google Scholar]
- Ohsawa I, Hirose Y, Ishiguro M, Imai Y, Ishiura S, Kohsaka S. Expression, purification, and neurotrophic activity of amyloid precursor protein-secreted forms produced by yeast. Biochem Biophys Res Commun. 1995;213:52–58. doi: 10.1006/bbrc.1995.2097. [DOI] [PubMed] [Google Scholar]
- Ohsawa I, Takamura C, Kohsaka S. The amino-terminal region of amyloid precursor protein is responsible for neurite outgrowth in rat neocortical explant culture. Biochem Biophys Res Commun. 1997;236:59–65. doi: 10.1006/bbrc.1997.6903. [DOI] [PubMed] [Google Scholar]
- Olson EC, Kim S, Walsh CA. Impaired neuronal positioning and dendritogenesis in the neocortex after cell-autonomous Dab1 suppression. J Neurosci. 2006;26:1767–1775. doi: 10.1523/JNEUROSCI.3000-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisiadou L, Efthimiopoulos S. Expression of mDab1 promotes the stability and processing of amyloid precursor protein and this effect is counteracted by X11alpha. Neurobiol Aging. 2006 doi: 10.1016/j.neurobiolaging.2005.12.015. [DOI] [PubMed] [Google Scholar]
- Perez RG, Zheng H, Van der Ploeg LH, Koo EH. The β-amyloid precursor protein of Alzheimer's disease enhances neuron viability and modulates neuronal polarity. J Neurosci. 1997;17:9407–9414. doi: 10.1523/JNEUROSCI.17-24-09407.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez RG, Soriano S, Hayes JD, Ostaszewski BL, Xia W, Selkoe DJ, Chen X, Stokin GB, Koo EH. Mutagenesis identifies new signals for β-amyloid precursor protein endotycosis, turnover and the generation of secreted fragments, including A42. J Biol Chem. 1999;274:18851–18856. doi: 10.1074/jbc.274.27.18851. [DOI] [PubMed] [Google Scholar]
- Priller C, Bauer T, Mitteregger G, Krebs B, Kretzschmar HA, Herms J. Synapse formation and function is modulated by the amyloid precursor protein. J Neurosci. 2006;26:7212–7221. doi: 10.1523/JNEUROSCI.1450-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhard C, Hebert SS, De Strooper B. The amyloid-β precursor protein: integrating structure with biological function. EMBO J. 2005;24:3996–4006. doi: 10.1038/sj.emboj.7600860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring S, Weyer SW, Kilian SB, Waldron E, Pietrzik CU, Filippov MA, Herms J, Buchholz C, Eckman CB, Korte M, Wolfer DP, Müller UC. The secreted β-amyloid precursor protein ectodomain APPs alpha is sufficient to rescue the anatomical, behavioral, and electrophysiological abnormalities of APP-deficient mice. J Neurosci. 2007;27:7817–7826. doi: 10.1523/JNEUROSCI.1026-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossjohn J, Cappai R, Feil SC, Henry A, McKinstry WJ, Galatis D, Hesse L, Multhaup G, Beyreuther K, Masters CL, Parker MW. Crystal structure of the N-terminal, growth factor-like domain of Alzheimer amyloid precursor protein. Nat Struct Biol. 1999;6:327–331. doi: 10.1038/7562. [DOI] [PubMed] [Google Scholar]
- Sabo SL, Ikin AF, Buxbaum JD, Greengard P. The Alzheimer amyloid precursor protein (APP) and FE65, an APP-binding protein, regulate cell movement. J Cell Biol. 2001;153:1403–1414. doi: 10.1083/jcb.153.7.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabo SL, Ikin AF, Buxbaum JD, Greengard P. The amyloid precursor protein and its regulatory protein, FE65, in growth cones and synapses in vitro and in vivo . J Neurosci. 2003;23:5407–5415. doi: 10.1523/JNEUROSCI.23-13-05407.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert D, Jin LW, Saitoh T, Cole G. The regulation of amyloid β protein precursor secretion and its modulatory role in cell adhesion. Neuron. 1989;3:689–694. doi: 10.1016/0896-6273(89)90237-7. [DOI] [PubMed] [Google Scholar]
- Slunt HH, Thinakaran G, Von Koch C, Lo ACY, Tanzi RE, Sisodia SS. Expression of a ubiquitous, cross-reactive homologue of the mouse β-amyloid precursor protein (APP) J Biol Chem. 1994;269:2637–2644. [PubMed] [Google Scholar]
- Small DH, Nurcombe V, Reed G, Clarris H, Moir R, Beyreuther K, Masters CL. A heparin-binding domain in the amyloid protein precursor of Alzheimer's disease is involved in the regulation of neurite outgrowth. J Neurosci. 1994;14:2117–2127. doi: 10.1523/JNEUROSCI.14-04-02117.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soba P, Eggert S, Wagner K, Zentgraf H, Siehl K, Kreger S, Lower A, Langer A, Merdes G, Paro R, Masters CL, Müller U, Kins S, Beyreuther K. Homo- and heterodimerization of APP family members promotes intercellular adhesion. EMBO J. 2005;24:3624–3634. doi: 10.1038/sj.emboj.7600824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabata H, Nakajima K. Efficient in utero gene transfer system to the developing mouse brain using electroporation: visualization of neuronal migration in the developing cortex. Neuroscience. 2001;103:865–872. doi: 10.1016/s0306-4522(01)00016-1. [DOI] [PubMed] [Google Scholar]
- Tissir F, Goffinet AM. Reelin and brain development. Nat Rev Neurosci. 2003;4:496–505. doi: 10.1038/nrn1113. [DOI] [PubMed] [Google Scholar]
- Trommsdorff M, Borg JP, Margolis B, Herz J. Interaction of cytosolic adaptor proteins with neuronal apolipoprotein E receptors and the amyloid precursor protein. J Biol Chem. 1998;273:33556–33560. doi: 10.1074/jbc.273.50.33556. [DOI] [PubMed] [Google Scholar]
- Van Gassen G, Annaert W, Van Broeckhoven C. Binding partners of Alzheimer's disease proteins: are they physiologically relevant? Neurobiol Dis. 2000;7:135–151. doi: 10.1006/nbdi.2000.0306. [DOI] [PubMed] [Google Scholar]
- von Koch CS, Zheng H, Chen H, Trumbauer M, Thinakaran G, van der Ploeg LH, Price DL, Sisodia SS. Generation of APLP2 KO mice and early postnatal lethality in APLP2/APP double KO mice. Neurobiol Aging. 1997;18:661–669. doi: 10.1016/s0197-4580(97)00151-6. [DOI] [PubMed] [Google Scholar]
- Wallace WC, Akar CA, Lyons WE. Amyloid precursor protein potentiates the neurotrophic activity of NGF. Brain Res Mol Brain Res. 1997;52:201–212. doi: 10.1016/s0169-328x(97)00258-1. [DOI] [PubMed] [Google Scholar]
- Ware ML, Fox JW, Gonzalez JL, Davis NM, Lambert de Rouvroit C, Russo CJ, Chua SC, Jr, Goffinet AM, Walsh CA. Aberrant splicing of a mouse disabled homolog, mdab1, in the scrambler mouse. Neuron. 1997;19:239–249. doi: 10.1016/s0896-6273(00)80936-8. [DOI] [PubMed] [Google Scholar]
- Wasco W, Gurubhagavatula S, Paradis MD, Romano DM, Sisodia SS, Hyman BT, Neve RL, Tanzi RE. Isolation and characterization of the human APLP2 gene encoding a homologue of the Alzheimer's associated amyloid β protein precursor. Nat Genet. 1993;5:95–99. doi: 10.1038/ng0993-95. [DOI] [PubMed] [Google Scholar]
- Wolfe MS, De Los Angeles J, Miller DD, Xia W, Selkoe DJ. Are presenilins intramembrane-cleaving proteases? Implications for the molecular mechanism of Alzheimer's disease. Biochemistry. 1999;38:11223–11230. doi: 10.1021/bi991080q. [DOI] [PubMed] [Google Scholar]
- Yamazaki T, Koo EH, Selkoe DJ. Cell surface amyloid beta-protein precursor colocalizes with beta 1 integrins at substrate contact sites in neural cells. J Neurosci. 1997;17:1004–1010. doi: 10.1523/JNEUROSCI.17-03-01004.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Jiang M, Trumbauer ME, Sirinathsinghji DJ, Hopkins R, Smith DW, Heavens RP, Dawson GR, Boyce S, Conner MW, Stevens KA, Slunt HH, Sisoda SS, Chen HY, Van der Ploeg LH. β-amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell. 1995;81:525–531. doi: 10.1016/0092-8674(95)90073-x. [DOI] [PubMed] [Google Scholar]