

Abstract
Although cholesterol is a major component of the CNS, there is little information on how or whether a change in sterol flux across the blood–brain barrier might alter neurodegeneration. In Niemann-Pick type C (NPC) disease, a mutation in NPC1 protein causes unesterified cholesterol to accumulate in the lysosomal compartment of every cell, including neurons and glia. Using the murine model of this disease, we used genetic and pharmacologic approaches in an attempt to alter cholesterol homeostasis across the CNS. Genetic deletion of the sterol transporters ATP-binding cassette transporter A1 (ABCA1) and low-density lipoprotein receptor in the NPC1 mouse did not affect sterol balance or longevity. However, deletion of the nuclear receptor, liver X receptor β (LXRβ), had an adverse effect on progression of the disease. We therefore tested the effects of increasing LXR activity by oral administration of a synthetic ligand for this transcription factor. Treatment with this LXR agonist increased cholesterol excretion out of brain from 17 to 49 μg per day, slowed neurodegeneration, and prolonged life. This agonist did not alter synthesis of cholesterol or expression of genes associated with the formation of 24(S)-hydroxycholesterol or neurosteroids such as CYP46A1, 3αHSD, and CYP11A1. However, levels of the sterol transporters ABCA1 and ATP-binding cassette transporter G1 were increased. Concomitantly, markers of neuroinflammation, CD14, MAC1, CD11c, and inducible nitric oxide synthase, were reduced, and microglia reverted from their amoeboid, active form to a ramified, resting configuration. Thus, LXR activation resulted in increased cholesterol excretion from the brain, decreased neuroinflammation, and deactivation of microglia to slow neurodegeneration and extend the lifespan of the NPC1 mouse.
Keywords: 24(S)-hydroxycholesterol, microglia, astroglia, blood–brain barrier, nuclear receptors, Purkinje cells
Introduction
The richest collection of cholesterol in the body is found in the brain. Although the pool of cholesterol in the whole body is essentially the same in all mammalian species (∼2100 mg/kg body weight), the CNS accounts for ∼23% of this pool (∼490 mg/kg) in the adult human, but only 16% (∼335 mg/kg) in the mouse (Dietschy and Turley, 2004). In the mouse, as in the human, the turnover of this cholesterol in the CNS is much slower (0.4%/d) than that in the remaining tissues of the body (8%/d). The sources for the cholesterol driving this turnover are complex (Quan et al., 2003). Although the brain does not have direct access to cholesterol carried in lipoproteins in the plasma, apparently all cells, including neurons and glia, have the capacity to synthesize this essential molecule. Astrocytes, in particular, synthesize both cholesterol and apolipoprotein E (apoE) and secrete these two molecules complexed together into the interstitial fluid of the brain (Pfrieger, 2003a; Vance et al., 2005). This complex is then presumably taken up by other glial cells and by neurons using receptor-mediated endocytosis (Mauch et al., 2001). During normal turnover, a subset of these neurons metabolize part of this cholesterol to hydroxylated derivatives, particularly 24(S)-hydroxycholesterol (Lund et al., 2003; Wahrle et al., 2004). Thus, in the mature mouse, ∼1.4 mg/kg body weight of the pool of cholesterol in the CNS is lost from the brain each day. Approximately 60% of this sterol reaches the plasma as 24(S)-hydroxycholesterol, whereas the remainder is excreted as either cholesterol itself or some other unidentified hydroxysterol (Xie et al., 2003).
This turnover of sterol in the brain must be as tightly regulated as is that in all other organs of the body. Important in this regard may be various transcription factors like the liver X receptor (LXR) and retinoid X receptor (RXR), which control critical steps in sterol metabolism (Repa and Mangelsdorf, 2000; Wang et al., 2002). In particular, two subtypes of LXR, LXRα and LXRβ, are ligand-activated transcription factors expressed in many tissues, including the brain, that appear to respond to expansion of the metabolically active pool of cholesterol in the cytosol of many cells. Such activation leads to increased transcription of genes encoding the ATP-binding cassette transporters A1 (ABCA1) and G1 (ABCG1), which are involved in the movement of cholesterol across the plasma membrane of certain cells. In addition, genes that transcribe the apolipoproteins D (apoD) and E are also targets of these nuclear receptors. Although several of these genes in the CNS have been shown to respond to LXR agonists (Whitney et al., 2002), there is currently no information on whether such treatment actually alters net cholesterol flux in this organ or modifies the level of nerve cell death in any model of neurodegeneration.
One of the best characterized models of progressive neurodegeneration is the mouse having a mutation in the gene encoding NPC1, a protein necessary for the movement of unesterified cholesterol from the lysosomal compartment of cells to the metabolically active pool in the cytosol (Loftus et al., 1997). As a consequence, nearly every cell in the body accumulates unesterified cholesterol that is derived from the receptor-mediated or bulk-phase uptake of lipoproteins containing either apoB100 or apoE (Liu et al., 2007). This accumulation of sterol in the late endosomal/lysosomal compartment of cells leads to the clinical syndrome known as Niemann-Pick type C (NPC) disease, where there is significant hepatic, pulmonary, and neurological damage (Pentchev et al., 1995). Because disordered cholesterol metabolism is a major part of this syndrome, the NPC mouse is an ideal model in which to explore the relationship between sterol movement across the blood–brain barrier (BBB) and the progressive neurodegeneration characteristic of this disease. The present studies, therefore, were undertaken to determine whether activation of the target genes of LXR leads to a change in cholesterol balance across the brain and, additionally, whether this change affects the degree of nerve cell death.
Materials and Methods
Animals and diets.
Heterozygous NPC1 mice on a BALB/c background (Loftus et al., 1997) were bred to generate the control (npc1+/+) and homozygous (npc1−/−) mice used in all studies, except those shown in Figure 1. Mice were genotyped at weaning and placed on the appropriate diet. For the mice used in studies depicted in Figure 1, npc1+/− animals were bred with ldlr−/− (Ishibashi et al., 1993), abca1−/− (Christiansen-Weber et al., 2000), or lxrβ −/− (Repa et al., 2000) mice to produce animals of the desired genotypes. Littermate npc1+/+ and npc1−/− mice were used as controls in all of these studies to minimize variation introduced by the mixed strain backgrounds. All mice were kept in a facility with 12 h light/dark cycles, and tissues were harvested at the end of the dark cycle when the mice were in a postprandial state. Mice were fed ad libitum a low-cholesterol mouse diet (no. 7001; Harlan Teklad, Madison, WI) containing 0.02% cholesterol, or this same diet supplemented with T0901317 (0.025% w/w; Cayman Chemical, Ann Arbor, MI). This compound is hereafter abbreviated T1317. In one experiment, the basal diet was supplemented with 1% cholesterol. Mice were weighed on alternate days, and motor coordination was evaluated by testing on an accelerating Rotarod apparatus (Rotamex 4/8; Columbus Instruments, Columbus, OH) (Li et al., 2005). These studies were approved by the Institutional Animal Care and Use Committee of the University of Texas Southwestern Medical Center.
Figure 1.

Effect of cholesterol feeding or functional deletion of cholesterol trafficking-related genes on lifespan and cholesterol metabolism in the npc1−/− mouse. A, Survival rates of npc1−/− mice fed the 1% cholesterol diet (n = 76) from weaning compared with npc1−/− animals fed the 0.02% low-cholesterol diet (n = 72; p > 0.05). B, Survival rates of npc1−/−/ldlr−/− mice (n = 111) compared with npc1−/− animals (n = 57) when both groups were fed the low-cholesterol diet (p > 0.05). C, Survival rates of npc1−/−/abca1−/−mice (n = 24) compared with npc1−/− animals (n = 32) when both groups were fed the low-cholesterol diet (p > 0.05). D, Survival rates of npc1−/−/lxrβ −/− mice (n = 16) compared with npc1−/− animals (n = 16) when both groups were fed the low-cholesterol diet (p < 0.01). E, Cholesterol concentration (mg/g wet weight) in tissues of five groups of mice 7–8 weeks of age and maintained on the low-cholesterol diet except for group 3, which was fed the 1% cholesterol diet from weaning (n = 6–24). Different letters indicate results that were significantly different at p < 0.05. F, Cholesterol synthesis (nmol of 3H2O incorporated per hour per gram wet weight) in tissues of the animals described above in E (n = 6–18). Different letters indicate results that were significantly different at p < 0.05.
Plasma and tissue lipid concentrations and cholesterol synthesis rates.
Blood was withdrawn from the inferior vena cava and anticoagulated with heparin. Plasma total cholesterol and triacylglycerol (TAG) concentrations were measured as described previously (Repa et al., 2005; Beltroy et al., 2007). Tissue total cholesterol levels were determined using gas chromatography (Repa et al., 2005). The rate of cholesterol synthesis in multiple organs was determined in vivo using [3H]water as described previously (Repa et al., 2005).
Gene expression.
Quantitative real-time (qRT)-PCR was performed using an Applied Biosystem 7900HT sequence detection system and SYBR-green chemistry (Kurrasch et al., 2004). Total RNA was isolated from whole cerebella using RNA STAT-60 (Tel-Test, Friendswood, TX), and 2 μg of total RNA was treated with RNase-free DNase (Roche Molecular Biochemicals, Indianapolis, IN) then reverse transcribed with random hexamers using SuperScript II (Invitrogen, San Diego, CA). Gene-specific primers were designed using Primer Express Software (PerkinElmer, Emeryville, CA) and validated by analysis of template titration and dissociation curves (Valasek and Repa, 2005). Primer sequences are provided in a supplementary table. Ten microliter qRT-PCR reaction volumes contained 25 ng of reverse-transcribed RNA, each primer (150 nm), and 5 μl of 2× SYBR Green PCR master mix (Applied Biosystems, Foster City, CA). Results of qRT-PCR were evaluated by the comparative cycle number determined at threshold (Ct) method (user bulletin no. 2; PerkinElmer) using cyclophilin as the invariant control gene.
Immunohistochemistry.
Under deep anesthesia, mice were perfused with 0.1 m PBS containing 0.1% glucose and 1 U heparin/ml, followed by perfusion with 4% paraformaldehyde (PFA) in PBS, pH 7.4. Brains were postfixed in 4% PFA, removed, cryoprotected in 20% sucrose/PBS, and then embedded in tissue mounting medium for freezing (Triangle Biomedical Science, Durham, NC). Sequential sagittal sections were cut of each cerebellar vermis at 16 μm on a Leica CM 1850 cryostat. For Purkinje cell identification, cerebellar sections were immunostained for calbindin (Frank et al., 2003). Briefly, sections were permeabilized in 0.3% Triton X-100, then endogenous peroxidases were quenched with 3% H2O2, and sections were blocked with 5% goat serum. Sections were incubated with anti-calbindin D-28K (1:2000; Chemicon International, Temecula, CA), followed by biotinylated goat anti-rabbit (1:400; Vector Laboratories, Burlingame, CA) and NeutrAvidin-HRP (1:5000; Pierce, Rockford, IL). The tissue-bound peroxidase activity was revealed by incubation in 0.024% DAB (Sigma, St. Louis, MO) and 0.006% H2O2 for 20 min. Tissue sections were dehydrated, immersed in xylene, and coverslipped with vectamount (Vector Laboratories).
Staining of unesterifed cholesterol.
Cerebellar sections were labeled with biotinylated perfringolysin peptide (BCθ) at 15 μg/ml in 0.1 m PBS containing 1 mg/ml BSA for 20 min (Iwamoto et al., 1997; Reid et al., 2004). The tissue-bound biotin was further labeled with FITC-tagged NeutrAvidin (1:500; Pierce) for 30 min, and then sections were coverslipped with Prolong anti-fade (Invitrogen, Eugene, OR). For microglia characterization, frozen cerebellum sections were treated with 0.2 m glycine in PBS, pH 7.4, for 1 h at room temperature to quench autofluorescence. Sections were then permeabilized in 0.3% Triton X-100 with 1% goat serum in 20 mm TBS. Sections were incubated with anti-CD14 (1:100; Santa Cruz Biotechnology, Santa Cruz, CA), followed by goat anti-rabbit Alexa 594 (1:1000; Invitrogen) or F4/80 (1:150; Serotec, Indianapolis, IN), followed by goat anti-rat Alexa 488 (1:1000; Invitrogen). After washes, sections were coverslipped with Biomedia Gel/Mount aqueous mounting media with anti-fade (Biomedia, Foster City, CA). For computer photocapture, sections of cerebellar lobules II and III are featured and oriented with anterior (left) and posterior (right).
Data analysis.
All data are expressed as the mean ± SEM, with the number of mice indicated in the figure legends. Statistical analyses were performed using GraphPad Prism5 software (GraphPad Software, San Diego, CA). Experiments involving multiple groups were compared using two-way ANOVA with genotype and drug treatment as factors, except for the data shown in Figure 1, E and F, which were analyzed by one-way ANOVA. Newman-Keuls post hoc test was used, and groups designated with different letters are statistically different. Significant differences in survival curves were determined by Wilcoxon–Gehar and Log-rank analyses. (Supplemental data showing the primer sequences used to determine the mRNA levels for the various proteins in this study are available at www.jneurosci.org as supplemental material. This supplemental table provides the definitions for all abbreviations.)
Results
Changes in systemic cholesterol homeostasis and longevity in npc1−/− mice
Initial experiments were done to determine whether manipulations that potentially alter systemic cholesterol balance, sterol movement across the BBB, or cholesterol homeostasis within the brain itself might alter longevity of the npc1−/− mouse. Expansion of chylomicron cholesterol content through cholesterol feeding elevated the concentration of cholesterol in the liver (Fig. 1E), a change known to worsen the liver disease of the npc1−/− mouse (Beltroy et al., 2005, 2007). However, this dietary manipulation did not change either the rate of synthesis or the concentration of cholesterol in the brain and did not alter the lifespan of the animals (Fig. 1A,E,F). Two membrane cholesterol transporters, the low-density lipoprotein receptor (LDLR) and ABCA1, are reported to be expressed in brain capillary endothelial cells and might be involved in sterol homeostasis across the BBB. Deletion of LDLR activity, as in the npc1−/−/ldlr−/− animals, raised the plasma LDL cholesterol level 15-fold and significantly increased the concentration of cholesterol in the spleen and lung (Fig. 1E), a change known to worsen the lung disease of the npc1−/− mice (Liu et al., 2007). In contrast, deletion of ABCA1 activity, as in the npc1−/−/abca1−/− mice, markedly lowered the plasma cholesterol concentration but had no significant effect on either the rate of synthesis or concentration of cholesterol in most tissues (Fig. 1E,F). Despite these marked changes in systemic cholesterol metabolism, neither deletion of LDLR nor ABCA1 function significantly altered cholesterol concentration or synthesis in the brain or changed survival in these animals (Fig. 1B,C,E,F). Thus, in keeping with previous studies, these three manipulations that markedly altered different aspects of cholesterol metabolism in the whole animal had no apparent effect on sterol metabolism in the CNS or on the age at death of the npc1−/− animals from neurodegeneration. However, in a final manipulation, LXRβ was deleted in the npc1−/− mouse, and the resulting npc1−/−/lxrβ −/− animals died at a significantly earlier age (Fig. 1D). This nuclear receptor is known to be expressed in the brain and to control several critical steps in sterol metabolism (Repa and Mangelsdorf, 2002). This result, therefore, raised the possibility that LXR was normally driving a portion of cholesterol excretion from the CNS so that when this efflux was lost, the neurological disease worsened.
Effect of the LXR agonist, T1317, on growth, neurological function, and cholesterol concentrations in the npc1−/− mice
This latter finding also suggested that activating LXR with an agonist might increase cholesterol excretion from the brain and improve neurological function. To test this hypothesis, npc1+/+ and npc1−/− pups were placed on diets at 21 d of age that contained amounts of the LXR agonist T1317 (Schultz et al., 2000) sufficient to deliver a daily oral dose of ∼50 mg/kg. Daily administration of this agonist to the npc1+/+ animals had no effect on growth or body weight (Fig. 2A); however, T1317 significantly reduced body weight gain in the npc1−/− pups (Fig. 2B). Nevertheless, neurological function, as measured by the time animals remained on the Rotarod apparatus, was improved to a modest, but significant, degree (Fig. 2C). There was no effect on the performance of the npc+/+ animals. Most importantly, the lifespan of the T1317 treated animals was significantly prolonged (Fig. 2D). It should be noted that the average age at death increased from ∼69 d in the npc1−/−/lxrβ−/− mice to ∼100 d in the animals where LXR was activated by administration of the agonist. Despite these favorable changes, the concentration of cholesterol in most tissues of the treated mice was changed only slightly or not at all, and, in particular, there was no alteration in the mean concentration of sterol in the brains of the npc1−/− mice (Fig. 2E). This prolonged T1317 administration resulted in hepatomegaly and steatosis (data not shown); however, the plasma TAG concentration was reduced in contrast to the hypertriglyceridemia commonly observed after acute exposure to this drug (Schultz et al., 2000). Plasma cholesterol concentration in both the npc1+/+ and npc1−/− animals (Fig. 2F) was increased by T1317 treatment, as observed previously in mice (Repa et al., 2000).
Figure 2.
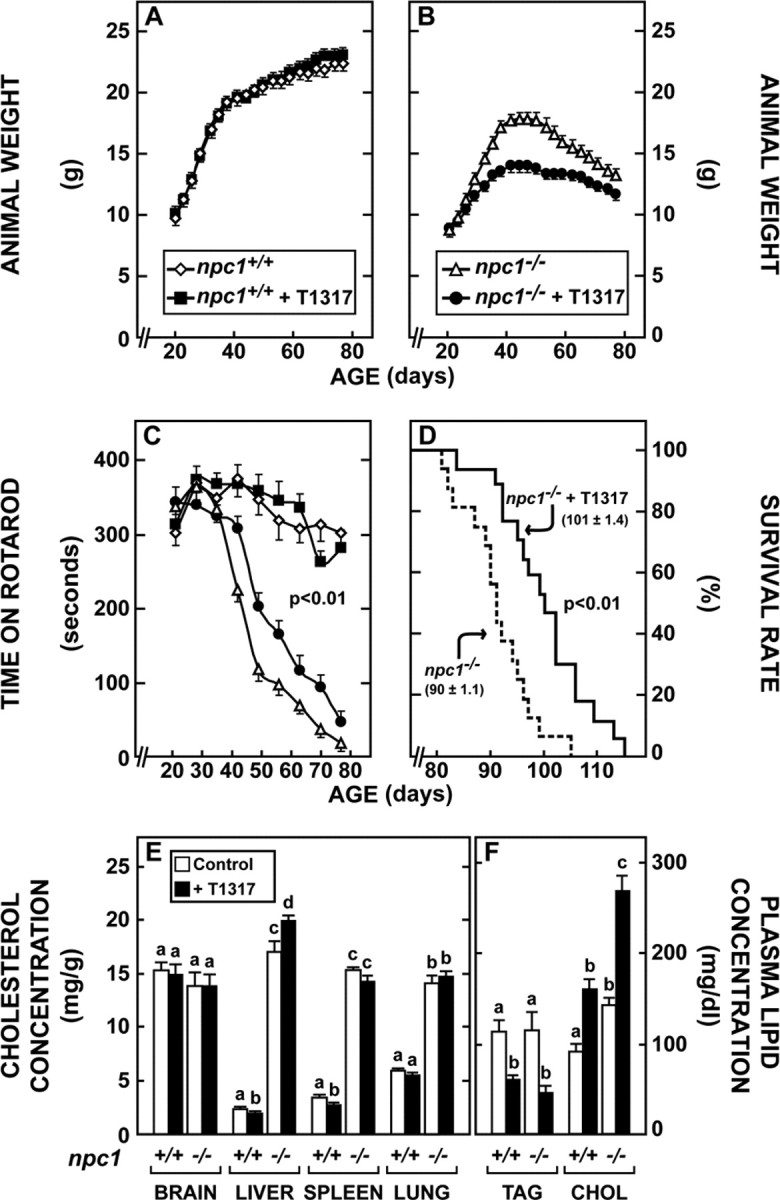
Effect of the LXR agonist T1317 on animal weight, neurological function, longevity, and cholesterol metabolism. A, B, Both npc1+/+ and npc1−/− mice were fed diets from the time of weaning that were either the low-cholesterol diet alone or this diet to which was added an amount of T1317 sufficient to deliver a daily dose of ∼50 mg/kg body weight (n = 8 in each of 4 groups). The animals were then weighed at intervals over 60 d. C, These same four groups of animals described in A and B were periodically tested on the Rotarod apparatus. Although treatment with T1317 had no effect on the performance of the npc1+/+ mice, the drug did increase neurological function of the npc1−/− animals (p < 0.01). D, Survival rates of npc1−/− mice treated with T1317 (n = 16) compared with untreated npc1−/− animals (n = 16; p < 0.01). The mean age at death is also shown. E, F, Cholesterol (CHOL) and triacylglycerol (TAG) concentrations in 49-d-old npc1+/+ mice and in npc1−/− mice receiving either no drug or T1317 from the time of weaning (n = 6 in each group). Different letters identify results that were significantly different at p < 0.05.
Effect of T1317 on cerebellar morphology and mRNA levels for various nuclear receptors
To determine whether these changes in function and longevity were reflected in improvement in brain morphology, histological sections were prepared, and RNA was extracted from the cerebellum, because this brain region is most profoundly affected by the NPC mutation (German et al., 2001). Sections of cerebellum stained for calbindin, a marker of Purkinje cells, revealed that administration of T1317 to npc1+/+ mice had no effect on the morphology or number of these large metabolically active neurons (Fig. 3A,B). As reported previously, the 49-d-old npc1−/− animals had marked loss (68%) of Purkinje cells (Fig. 3C) in a striated pattern (Sarna et al., 2003), but this loss was much less severe (39%) in the animals treated with T1317 (Fig. 3D). Thus, the improved neurological function and lifespan appeared to correlate with less severe nerve cell loss.
Figure 3.
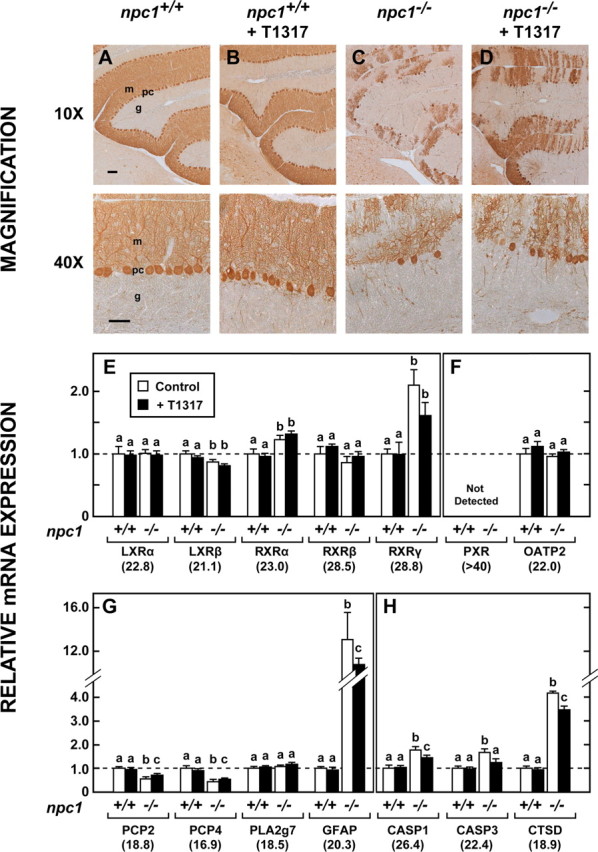
Cerebellar morphology and mRNA levels for proteins reflecting nuclear receptors, cell number, and apoptosis. A–D, Representative sections of cerebellum stained for calbindin from both npc1+/+ and npc1−/− mice 7 weeks of age that had been fed either the low-cholesterol diet alone or this diet containing T1317. In these photomicrographs, m and g identify the molecular and granular layers, respectively, whereas pc represents Purkinje cells. Scale bars: 10×, 100 μm; 40×, 50 μm. E, F, mRNA levels for various nuclear receptors found in the cerebellum except for OATP2, which represents a target gene of PXR (n = 6). The value beneath each gene is the cycle number at threshold by qRT-PCR and provides a measure of the relative mRNA levels of each gene. Different letters identify results that were significantly different at p < 0.05. G, H, Relative mRNA levels for proteins that reflect cell number, cell activation, and apoptosis.
A survey of relevant nuclear receptors revealed that LXRβ mRNA was most abundant in mouse cerebellum, whereas LXRα and RXRα and particularly RXRβ and RXRγ, were present at much lower RNA levels (Fig. 3E). mRNA for PXR was essentially undetectable in this brain region (Fig. 3F) and recently confirmed (Gofflot et al., 2007), although that does not rule out expression of this receptor in a minor cell population to account for its purported activities as a therapeutic target for treatment of NPC1 disease (Langmade et al., 2006). Although there were marginal differences in some of these mRNA levels in the npc1−/− mice compared with the npc1+/+ animals, in no case did the administration of T1317 significantly change these levels in either group of animals. Additionally, this agent did not alter the mRNA level for OATP2 (organic anion transport protein 2), a known target gene of PXR. This suggests that although T1317 can activate human, and to a lesser extent, mouse PXR (Mitro et al., 2007), it appears that LXR rather than PXR is responsible for these gene expression changes observed in the mouse cerebellum.
The complement of various cell types in the cerebellum during disease progression was estimated by the mRNA levels of cell-specific markers, as done previously (Li et al., 2005). mRNA levels for Purkinje cell protein 2 (PCP2) and PCP4 closely paralleled changes observed in Purkinje cell number by calbindin staining (Fig. 3G). The astrocyte-specific gene PLA2g7 (Bachoo et al., 2004) exhibited similar RNA levels in all experimental groups, whereas GFAP mRNA was markedly increased in the npc1−/− mouse, in agreement with reports of astrogliosis in the CNS of this animal model (Baudry et al., 2003). The mRNA levels for caspases 1 and 3 (CASP1, CASP3), and cathepsin D (CTSD) were also elevated in the mutant mice (Fig. 3H). Importantly, treatment with T1317 significantly reduced the mRNA levels for GFAP, CASP1, CASP3, and CTSD, a finding that was consistent with the delay in neurodegeneration observed in the histological sections from similarly treated animals (Fig. 3D). It should be noted that this disease progresses from the anterior to posterior portions of the cerebellum. Thus, these large changes observed with Purkinje cell staining (which focused on the anterior cerebellum) are less apparent in the mRNA measurements (which reflect the whole cerebellum).
Cholesterol accretion, synthesis, and excretion rates in the whole brain and cerebellum
The next major question was whether these favorable alterations in histopathology induced by T1317 were associated with actual changes in the net excretion of cholesterol from the CNS. Because there were subtle differences in brain size in the npc1+/+ and npc1−/− mice, these measurements required quantifying cholesterol content in the brains of a large number of animals at exactly the same age. Brain weight was found to increase from 439 mg at 42 d of age to 448 mg at 56 d of age in the npc1+/+ mice, but from only 396 to 398 mg in the npc1−/− animals over the same interval (Fig. 4A). Treatment with T1317 led to a decrease in brain mass in both groups, and in the npc1−/− animals, this value fell from 370 to 339 mg over this 14 d interval. Similar decreases were seen in the cerebellum (Fig. 4B). The absolute content of cholesterol in the whole brain of the npc1+/+ mice increased from 6.50 to 7.01 mg between 42 and 56 d of age, but from only 5.36 to 5.66 mg in the mutant mice. This rate of accumulation, however, was significantly reduced in both the npc1+/+ and npc1−/− mice receiving T1317 in the diet (Fig. 4C). In fact, in the npc1−/− mice receiving this agonist, there was actually net loss of cholesterol from the whole brain and cerebellum over this 14 d interval (Fig. 4C,D).
Figure 4.
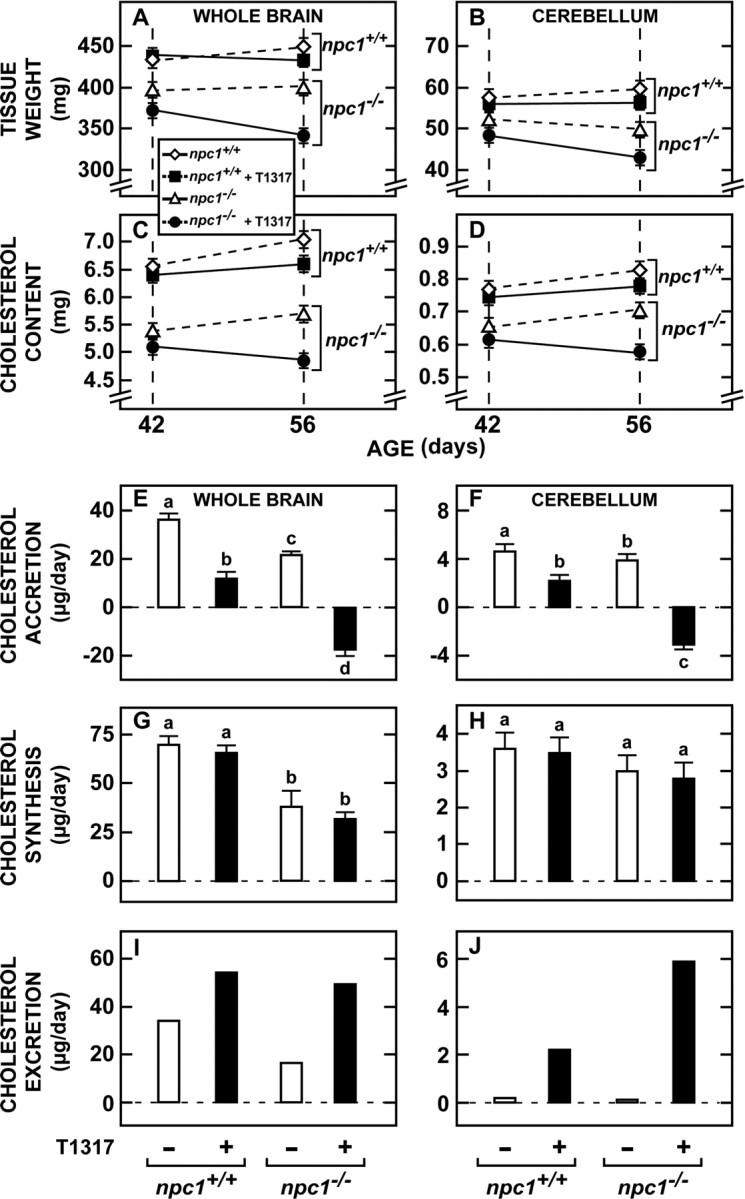
Cholesterol accretion, synthesis, and excretion in the whole brain and cerebellum. A, B, Both npc1+/+ and npc1−/− mice were fed the low-cholesterol diet alone or with added T1317 from weaning. Groups of these mice were killed at either 42 or 56 d of age, and the whole brain and cerebellum from each mouse was weighed (n = 10 in each of the 8 groups). C, D, The absolute content of cholesterol in the whole brain and cerebellum of these groups was quantified (n = 10 in each of the 8 groups). E, F, From these values for cholesterol content (C, D), the mean daily accretion rates at 49 d of age were calculated for each group and are expressed as the microgram of cholesterol accreted by the brain and cerebellum each day. G, H, In a companion group of animals, 49 d of age, the absolute rates of cholesterol synthesis were measured, and these are expressed as the microgram of cholesterol synthesized each day (n = 10 in each of the 4 groups). I, J, The rates of cholesterol excretion were calculated as the difference between the observed rates of synthesis and the absolute rates of accretion and are also expressed as the microgram of cholesterol leaving the brain and cerebellum each day.
From these data, the mean accretion rates of cholesterol in the brain of animals 49 d of age could be determined. Because the CNS of mice of this age was still growing, the whole brain was accreting 36 μg/d cholesterol in the npc1+/+ mice, but only 22 μg/d in the mutant animals. However, with T1317 treatment, these accretion rates were markedly reduced in both the whole brain and cerebellum in both groups of animals and, in the case of the npc1−/− mice, these rates became negative (i.e., the whole brain and cerebellum were actually losing cholesterol with age) (Fig. 4E,F). Using animals 49 d of age, absolute rates of cholesterol synthesis were next quantified. Although the rates of synthesis were again higher in the whole brains of the npc1+/+ mice (70 μg/d) compared with the npc1−/− animals (39 μg/d), in this case, treatment with T1317 did not significantly alter the values found in the two groups either in the whole brain or cerebellum (Fig. 4G,H).
From these two sets of data, the rates of net cholesterol excretion from the whole brain and cerebellum could be calculated. Net loss of sterol from the CNS in the npc1+/+ mice equaled 34 μg/d or ∼1.65 mg/d per kg body weight, a value nearly identical to that previously reported in control mice (Xie et al., 2003). Administration of T1317 increased this rate of excretion to 54 μg/d in the normal animals and from 17 to 49 μg/d (3.55 mg/d/kg) in the npc1−/− animals (Fig. 4I,J). Excretion from the cerebellum increased from 0.1 to 5.9 μg/d in the mutant animals. Thus, the improvement in neurological function, lifespan, and histopathology seen with administration of T1317 was associated with a marked increase in cholesterol excretion from the CNS.
Cellular cholesterol accumulation and mRNA levels for proteins involved in cholesterol metabolism in the cerebellum
Sections of cerebellum stained with BCθ, a probe for detection of unesterified cholesterol (Reid et al., 2004), revealed many cholesterol-filled cells in the npc1−/− mice that were not evident in the npc1+/+ animals. Although some of these were clearly Purkinje cells, most were randomly distributed throughout the molecular and granular layers, suggesting they were glia or other neuronal cell types (Fig. 5A,B). Treatment with T1317 appeared to reduce the number of such cells (Fig. 5C). To elucidate the molecular mechanisms associated with this loss of CNS cholesterol, the expression of genes involved in cholesterol homeostasis was next determined. As has been reported previously (Li et al., 2005), despite this abnormal accumulation of intracellular cholesterol in the late endosomal/lysosomal compartment of cerebellar cells in the npc1−/− mice, there was no change in expression of the LXR target genes, ABCA1 and ABCG1, although these mutant animals did reveal significant elevations of the mRNA levels for apoE, apoD, Lip1, and NPC2 (Fig. 5D). Treatment with the agonist did, however, activate ABCA1 and ABCG1 but did not further elevate the mRNA levels of apoE, Lip1, or NPC2. There was a modest T1317-mediated increase in apoE mRNA evident with 2 weeks of treatment at 5 weeks of age (data not shown); however, by 7 weeks of age, the apoE mRNA levels in the npc1−/− mice were so high, that additional effects of T1317 were no longer evident. Only the mRNA level of apoD was differentially elevated in the npc1−/− animals after administration of T1317.
Figure 5.
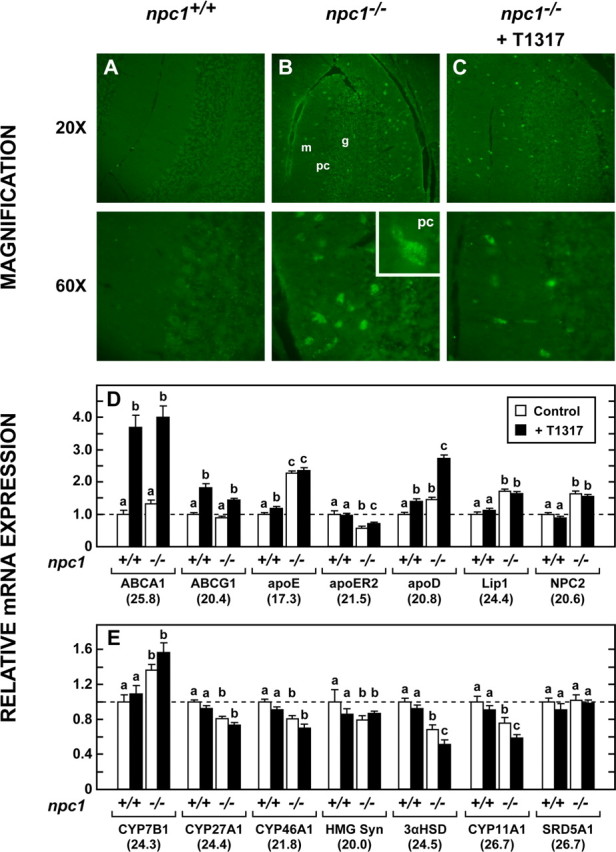
Cholesterol accumulation in cerebellum and mRNA levels for proteins involved in cholesterol metabolism. A–C, The cerebellum was removed from npc1+/+ mice 49 d of age that had been given the low-cholesterol diet since weaning and from npc1−/− animals given this diet alone or with T1317. Frozen sections of this region were stained with biotinylated perfringolysin peptide (BCθ) to identify sequestration of unesterified cholesterol in the cells of the npc1−/− animals. m and g identify the molecular and granular layers, respectively, whereas pc represents Purkinje cells. D, Relative mRNA levels for proteins potentially involved in the cellular trafficking of cholesterol (n = 6). E, Relative mRNA levels for proteins potentially involved in the synthesis or degradation of cholesterol (n = 6).
In theory, this marked increase in sterol excretion from the brain might reflect an increase in the movement of cholesterol itself or increased production and excretion of one of the hydroxylated sterols. CYP46A1 is responsible for the production of 24(S)-hydroxycholesterol, the known major pathway for cholesterol excretion from the brain (Björkhem et al., 1998; Lund et al., 2003). However, mRNA levels of CYP46A1 declined significantly in the npc1−/− mice and did not increase with T1317 administration (Fig. 5E). Similarly, there were no increases in the mRNA levels of CYP7B1 or CYP27A1, two other sterol hydroxylases. Although quantitatively of less importance, it was also conceivable that formation of increased amounts of neurosteroids could account for some of the increased sterol loss from the brain or neuroprotection (Griffin et al., 2004; Langmade et al., 2006). Again, however, the mRNAs for the major enzymes in this pathway, 3αHSD, CYP11A1, and SRD5A1 (Compagnone and Mellon, 2000), were unchanged with T1317 administration. Thus, the major increase in sterol excretion that was seen in the treated npc1−/− mice (3.55 mg/d/kg) compared with the untreated control animals (1.65 mg/d/kg) likely occurred through the movement of cholesterol itself rather than a hydroxylated sterol.
Immunological responses of the cells of the cerebellum to net cholesterol loss
A final set of studies investigated the potential anti-inflammatory effects of LXR agonist to elicit neuroprotection. Immunostaining of sections of brain with CD14 revealed many activated glial cells in the cerebellum (Fig. 6A,B). Treatment with T1317 appeared to reduce the number of such cells (Fig. 6C). More striking was the change in morphology of these cells with treatment. In sections stained with F4/80, the microglia in the npc1−/− mice were scattered throughout the molecular and granular layers and had the typical “amoeboid” morphology of activated cells (Fig. 6E). In the npc1−/− mice treated with T1317, the microglia mostly reverted to the “ramified” resting configuration (Fig. 6F).
Figure 6.
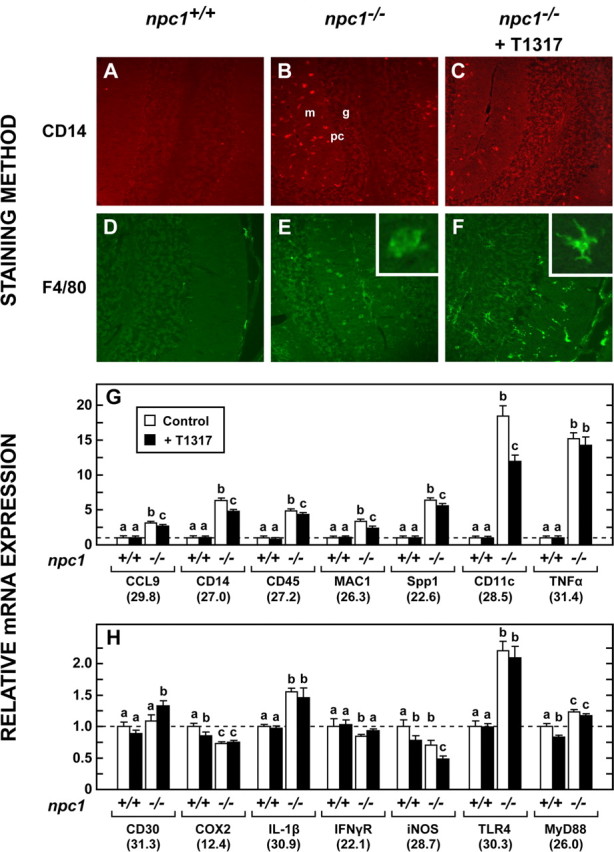
Activated microglia and mRNA levels of proteins involved in cerebellar inflammation. A–C, Activated microglia were identified by immunohistochemistry in 49-d-old npc1+/+ mice, npc1−/− animals, and npc1−/− mice receiving T1317. Images are presented at 20× magnification. m and g identify the molecular and granular layers, respectively, whereas pc represents Purkinje cells. D–F, Adjacent sections were stained with F4/80 to better delineate the morphology of microglial cells in the cerebellum. G, H, Relative mRNA levels for various proteins potentially involved in neuroinflammation in the cerebellum.
This qualitative assessment of microglia status was confirmed by measuring mRNA levels for a number of inflammatory markers in the cerebellum of these npc1−/− animals. In particular, those for CD11c and tumor necrosis factor α (TNFα) were markedly increased, and there were lesser, but significant, increases in Spp1, CD14, CD45, CCL9, Toll-like receptor 4 (TLR4), and interleukin-1B (IL-1β) (Fig. 6G,H). Importantly, treatment with T1317 decreased the mRNA levels for a number of these inflammatory markers, including CD11c, CD14, and MAC1; however, although reduced, their values were still elevated above those seen in the npc1+/+ mice. Notably, treatment with the agonist did not reduce the mRNA levels of TNFα or TLR4. Thus, LXR activation and the enhanced loss of cholesterol brought about by the administration of T1317 were associated with significant reductions in the mRNA levels of several inflammatory markers and with a change in the morphology of the activated microglia to their resting configuration.
Discussion
These studies provide the first description of a treatment that enhances net sterol excretion from the CNS and reduces the mass of cholesterol in the brain. The CNS of the mouse cannot use plasma cholesterol; thus, during the first 3 weeks of life, it synthesizes ∼250 μg/d of sterol and uses this entire amount for expansion of the cellular content of the brain and for myelination (Quan et al., 2003). By 49 d of age, however, synthesis declines to 70 μg/d, and the rate of accretion slows to 36 μg/d so that 34 μg of sterol must be excreted each day (Fig. 4). Approximately 65% of this excretion is as 24(S)-hydroxycholesterol, whereas the remainder presumably is as cholesterol itself (Xie et al., 2003). As illustrated in Figure 7, both neurons and glia synthesize sterol and, in the case of astrocytes, apoE (Pfrieger, 2003b). A portion of the apoE-cholesterol complex secreted by these cells is taken up through receptor-mediated endocytosis by other glia and, importantly, by neurons where it plays an important role in axon growth and synapse remodeling (de Chaves et al., 1997; Mauch et al., 2001; Vance et al., 2005). Daily turnover of cholesterol in a subset of large neurons is probably reflected in the rate of excretion of the 24(S)-hydroxycholesterol (22 μg/d), which moves into both the CSF and venous outflow from the brain (Björkhem et al., 1998). In contrast, sterol turnover in glial cells, including oligodendrocytes, is likely reflected in the rate of excretion of the cholesterol (12 μg/d) from the CNS. Treatment of npc1+/+ animals with the LXR agonist increases this excretion of total sterol from 34 to 54 μg/d under circumstances where the rate of synthesis is unchanged and reduces the rate of cholesterol accretion in the brain from 36 to 12 μg/d (Fig. 4). Although this treatment also increases the mRNA levels for ABCA1 and ABCG1, but not that of CYP46A1 (Fig. 5), it is not currently known whether it is the enhanced activity of one of these sterol transporters (X in Fig. 7) that might account for the observed increase in cholesterol excretion induced by T1317.
Figure 7.

Diagrammatic summary of the major sterol compartments and fluxes in the brain of a 49-d-old npc1+/+ mouse. The interstitial fluid bathing the glial and nerve cells of the brain is separated from the blood and CSF by a blood–brain barrier and an ependymal barrier, respectively. The pool of cholesterol in the brain as well as the rates of accretion and the rates of excretion as cholesterol (C) and 24(S)-hydroxycholesterol (24OH-C) are shown. This diagram implies that all cells both synthesize cholesterol from acetyl-CoA (AcCoA) and take up sterol bound to apoE through receptor-mediated endocytosis. The nature of the transport proteins, if any, involved in the movement of cholesterol (x) and 24(S)-hydroxycholesterol (y) across the blood–brain and ependymal barriers is not currently known. The term “lyso” refers to the late endosomal/lysosomal compartment of these cells, from which unesterified cholesterol is transported under the influence of the NPC1 protein.
In the face of mutational inactivation of NPC1, astrocytes continue to synthesize and secrete the apoE-cholesterol complex in normal amounts (Karten et al., 2005). However, when this complex is taken up by receptor-mediated endocytosis, it becomes trapped in the late endosomal/lysosomal compartment so that both glia and neurons begin to accumulate sterol, even in animals only a few days of age (Xie et al., 2000; Reid et al., 2004). Similar cholesterol accumulation takes place in every other organ in the body (Xie et al., 1999). However, the pathophysiology of the clinical disease manifest in each of these organs is clearly more complex than can probably be accounted for merely by this accumulation of cholesterol in parenchymal cells. In the liver, for example, there is an age-dependent accumulation of cholesterol in hepatocytes that is accompanied by infiltration of the liver by lipid-laden macrophages. The level of sterol accumulation in these hepatocytes, the magnitude of macrophage infiltration, and the severity of liver dysfunction are all correlated and directly related to the mass of chylomicron remnant cholesterol delivered to this organ (Beltroy et al., 2005, 2007). Similarly, the magnitude of cholesterol accumulating in the lung, the degree of macrophage infiltration in the pulmonary septae, and the severity of the lung disease are also related to the amount of cholesterol carried in LDL particles and delivered to this organ (Liu et al., 2007). Identical findings are seen in the present study in the brain where uptake of cholesterol complexed to apoE leads not only to lipid accumulation in neurons but also to an increase in microglia number, likely resulting from infiltration of peripheral monocytes into the CNS, as has been described previously for other neurodegenerative diseases (El Khoury et al., 2007). Clearly, both astrocytes and microglia are activated in response to the NPC1 mutation as evidenced by the marked increase seen in mRNA levels for GFAP, CD14, CD11C, TNF- α, IL-1B, TLR4, and other inflammatory proteins. In addition, the microglia assume their characteristic, activated “amoeboid” form (Figs. 3, 6). Thus, in all three organ systems, there is apparently a detrimental interaction of parenchymal cells and infiltrating macrophages (microglia) that depends on the amount of cholesterol reaching these cells.
Such an exaggerated damage-response mechanism has been described in other lysosomal storage diseases (Proia and Wu, 2004). In the face of subtle neuronal damage brought about by abnormal lysosomal storage, activation of an inflammatory response by microglia would be anticipated and appropriate. However, in these genetic disorders, metabolism within the lysosomes of the phagocytic cells also is abnormal so that the inflammatory response may be more aggressive and inappropriate, leading to accelerated neurodegeneration. If this is the case, then normalizing the metabolic defect in the microglia, but not in the neurons, would be expected to slow the neurodegeneration and improve the clinical syndrome. Such improvement has been described, for example, in mice with both Sandhoff disease (Wada et al., 2000) and metachromatic leukodystrophy (Biffi et al., 2004), where the metabolic integrity of the microglia was restored by bone marrow transplantation. Notably, in Sandhoff disease, restoration of normal lysosomal function to the microglia markedly reduced the level of neurodegeneration, improved neurological function, and prolonged life, although no change in the level of GM2 ganglioside storage in neurons was detected (Wada et al., 2000). Certainly, the findings in the present study are also consistent with this putative aggressive role of microglia in driving the neurodegeneration. In the NPC mouse, microglial activation occurs very early just as destruction of Purkinje cells begins (Li et al., 2005). Treatment with the LXR agonist restores microglia to their resting configuration (Fig. 6), decreases mRNA levels for a number of inflammatory factors (as has been elucidated in macrophages) (Ghisletti et al., 2007), slows neurodegeneration (Fig. 3), improves neurological function, and prolongs survival (Fig. 2). Thus, it seems very likely that the metabolic effect of T1317 may, in part, be exerted through improvement in microglial function.
At least four different molecular events might explain this improvement. First, it has been reported previously that administration of the neurosteroid, allopregnanolone, along with T1317, also leads to improved neurological function and prolonged lifespan in the npc1−/− mouse (Langmade et al., 2006). From these observations, it was suggested that the allopregnanolone and T1317 were exerting this favorable physiological effect by activating some PXR-dependent process. However, as shown in this study (Fig. 3) and previously (Bookout et al., 2006; Gofflot et al., 2007), the cerebellum of the mouse has no detectable PXR activity and, additionally, we have shown that it is the vehicle in which the allopregnanolone is administered that brings about the improvement in clinical characteristics of the npc1−/− animal (our unpublished observations).
Second, a more likely explanation is that LXR plays the critical role in regulating cholesterol flux from the CNS. Because the increased excretion of cholesterol from 17 to 49 μg/d occurred without an increase in synthesis (Fig. 4), less of the apoE-cholesterol complex would be available for uptake by glia and neurons, resulting in less of an inflammatory response and neurodegeneration. That the rate of cholesterol excretion from the brain is under control of LXR would also explain why deletion of this activity shortens the lifespan of the NPC1 animal, whereas driving the activity of this receptor prolongs life (Figs. 1, 2). In both cases, the severity of the clinical disease presumably is related to the availability of cholesterol for uptake and storage in the late endosomal/lysosomal compartment of these cells. This explanation also would be consistent with what is known about the liver and lung disease seen in these same animals. Reduction of uptake of cholesterol by both the liver and lung diminishes the level of macrophage infiltration into these two organs and reduces the magnitude of parenchymal cell damage (Beltroy et al., 2007; Liu et al., 2007).
A third, although less likely, possibility is that T1317 treatment partially corrects the metabolic defect by enhancing the movement of cholesterol from the late endosomal/lysosomal compartment to the cytosol, where it can be used normally. Notably, the cerebellum of the NPC1 mouse has an increase in mRNA levels of NPC2 and Lip1, two other lysosomal proteins that also are involved in the processing of lipoprotein sterol. Similarly, the mRNA level of apoD, another lysosomal protein, is elevated (Suresh et al., 1998; Zhang et al., 2001). This protein is expressed in glial cells and pericytes (Hu et al., 2001) and is the only mRNA that increases differentially in the npc1−/− mice in response to treatment with the LXR agonist (Fig. 5). Although the precise role of apoD in cholesterol metabolism is currently unknown, it is conceivable that an increase in this protein might partially bypass the NPC1 transport defect. Fourth, it is also possible that T1317 has an effect directly on some aspect of the immune response. It is reported, for example, that this agonist inhibits the apoptotic response of macrophages to M-CSF (Valledor et al., 2004). Furthermore, such compounds attenuate the inflammatory response of glial cells to fibrillar amyloid β peptide (Zelcer et al., 2007) and of macrophages to lipopolysaccharide (LPS) (Joseph et al., 2003). The mechanism for the potent anti-inflammatory action of LXR has been attributed most recently to receptor-mediated transrepression of nuclear factor κB-regulated gene expression (Ghisletti et al., 2007).
Thus, the sterol nuclear receptors LXRα and LXRβ appear to regulate cholesterol homeostasis across the CNS. In the presence of NPC1 disease, activation of these receptors increases cholesterol loss from the brain, limits the availability of sterol for uptake into glia and neurons, reduces neuroinflammation, slows neurodegeneration, and prolongs lifespan. Such findings suggest the possibility that these compounds may also have a role in treatment of other neurodegenerative disorders.
Footnotes
This work was supported by U.S. Public Health Service Grants R01-HL09610 (J.M.D., S.D.T.) and T32-GM07062 (M.A.V.) and by grants from the Moss Heart Fund (J.M.D.) and the Ara Parseghian Medical Research Foundation (J.J.R.). We gratefully acknowledge the kind gift of BCθ from Dr. Yoshiko Ohno-Iwashita (Tokyo Metropolitan Institute of Gerontology) and the excellent technical support of Heather Waddell, Brian Griffith, and Chunmei Yang.
References
- Bachoo RM, Kim RS, Ligon KL, Maher EA, Brennan C, Billings N, Chan S, Li C, Rowitch DH, Wong WH, DePinho RA. Molecular diversity of astrocytes with implications for neurological disorders. Proc Natl Acad Sci USA. 2004;101:8384–8389. doi: 10.1073/pnas.0402140101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudry M, Yao Y, Simmons D, Liu J, Bi X. Postnatal development of inflammation in a murine model of Niemann-Pick type C disease: immunohistochemical observations of microglia and astroglia. Exp Neurol. 2003;184:887–903. doi: 10.1016/S0014-4886(03)00345-5. [DOI] [PubMed] [Google Scholar]
- Beltroy EP, Richardson JA, Horton JD, Turley SD, Dietschy JM. Cholesterol accumulation and liver cell death in mice with Niemann-Pick type C disease. Hepatology. 2005;42:886–893. doi: 10.1002/hep.20868. [DOI] [PubMed] [Google Scholar]
- Beltroy EP, Liu B, Dietschy JM, Turley SD. Lysosomal unesterified cholesterol content correlates with liver cell death in murine Niemann-Pick type C disease. J Lipid Res. 2007;48:869–881. doi: 10.1194/jlr.M600488-JLR200. [DOI] [PubMed] [Google Scholar]
- Biffi A, De Palma M, Quattrini A, Del Carro U, Amadio S, Visigalli I, Sessa M, Fasano S, Brambilla R, Marchesini S, Bordignon C, Naldini L. Correction of metachromatic leukodystrophy in the mouse model by transplantation of genetically modified hematopoietic stem cells. J Clin Invest. 2004;113:1118–1129. doi: 10.1172/JCI19205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björkhem I, Lütjohann D, Diczfalusy U, Ståhle L, Ahlborg G, Wahren J. Cholesterol homeostasis in human brain: turnover of 24S-hydroxycholesterol and evidence for a cerebral origin of most of this oxysterol in the circulation. J Lipid Res. 1998;39:1594–1600. [PubMed] [Google Scholar]
- Bookout AL, Jeong Y, Downes M, Yu RT, Evans RM, Mangelsdorf DJ. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell. 2006;126:789–799. doi: 10.1016/j.cell.2006.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiansen-Weber TA, Voland JR, Wu Y, Ngo K, Roland BL, Nguyen S, Peterson PA, Fung-Leung W-P. Functional loss of ABCA1 in mice causes severe placental malformation, aberrant lipid distribution, and kidney glomerulonephritis as well as high-density lipoprotein cholesterol deficiency. Am J Pathol. 2000;157:1017–1029. doi: 10.1016/S0002-9440(10)64614-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compagnone NA, Mellon SH. Neurosteroids: biosynthesis and function of these novel neuromodulators. Front Neuroendocrinol. 2000;21:1–56. doi: 10.1006/frne.1999.0188. [DOI] [PubMed] [Google Scholar]
- de Chaves EIP, Rusiñol AE, Vance DE, Campenot RB, Vance JE. Role of lipoproteins in the delivery of lipids to axons during axonal regeneration. J Biol Chem. 1997;272:30766–30773. doi: 10.1074/jbc.272.49.30766. [DOI] [PubMed] [Google Scholar]
- Dietschy JM, Turley SD. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J Lipid Res. 2004;45:1375–1397. doi: 10.1194/jlr.R400004-JLR200. [DOI] [PubMed] [Google Scholar]
- El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, Luster AD. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. 2007;13:432–438. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- Frank TC, Nunley MC, Sons HD, Ramon R, Abbott LC. Fluoro-jade identification of cerebellar granule cell and Purkinje cell death in the α-1A calcium ion channel mutant mouse, leaner. Neuroscience. 2003;118:667–680. doi: 10.1016/s0306-4522(03)00019-8. [DOI] [PubMed] [Google Scholar]
- German DC, Quintero EM, Liang C-L, Ng B, Punia S, Xie C, Dietschy JM. Selective neurodegeneration, without neurofibrillary tangles, in a mouse model of Niemann-Pick C disease. J Comp Neurol. 2001;433:415–425. doi: 10.1002/cne.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghisletti S, Huang W, Ogawa S, Pascual G, Lin M-E, Willson TM, Rosenfeld MG, Glass CK. Parallel SUMOylation-dependent pathways mediate gene- and signal-specific transrepression by LXRs and PPARγ. Mol Cell. 2007;25:57–70. doi: 10.1016/j.molcel.2006.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gofflot F, Chartoire N, Vasseur L, Heikkinen S, Dembele D, Le Merrer J, Auwerx J. Systematic gene expression mapping clusters nuclear receptors according to their function in the brain. Cell. 2007;131:405–418. doi: 10.1016/j.cell.2007.09.012. [DOI] [PubMed] [Google Scholar]
- Griffin LD, Gong W, Verot L, Mellon SH. Niemann-Pick type C disease involves disrupted neurosteroidogenesis and responds to allopregnanolone. Nat Med. 2004;10:704–711. doi: 10.1038/nm1073. [DOI] [PubMed] [Google Scholar]
- Hu C-Y, Ong W-Y, Sundaram RK, Chan C, Patel SC. Immunocytochemical localization of apolipoprotein D in oligodendrocyte precursor-like cells, perivascular cells, and pericytes in the human cerebral cortex. J Neurocytol. 2001;30:209–218. doi: 10.1023/a:1012797623620. [DOI] [PubMed] [Google Scholar]
- Ishibashi S, Brown MS, Goldstein JL, Gerard RD, Hammer RE, Herz J. Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. J Clin Invest. 1993;92:883–893. doi: 10.1172/JCI116663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto M, Morita I, Fukuda M, Murota S-i, Ando S, Ohno-Iwashita Y. A biotinylated perfringolysin O derivative: a new probe for detection of cell surface cholesterol. Biochim Biophys Acta. 1997;1327:222–230. doi: 10.1016/s0005-2736(97)00061-8. [DOI] [PubMed] [Google Scholar]
- Joseph SB, Castrillo A, Laffitte BA, Mangelsdorf DJ, Tontonoz P. Reciprocal regulation of inflammation and lipid metabolism by liver x receptors. Nat Med. 2003;9:213–219. doi: 10.1038/nm820. [DOI] [PubMed] [Google Scholar]
- Karten B, Hayashi H, Francis GA, Campenot RB, Vance DE, Vance JE. Generation and function of astroglial lipoproteins from Niemann-Pick type C1-deficient mice. Biochem J. 2005;387:779–788. doi: 10.1042/BJ20041694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurrasch DM, Huang J, Wilkie TM, Repa JJ. Quantitative real-time polymerase chain reaction measurement of regulators of G-protein signaling mRNA levels in mouse tissues. Methods Enzymol. 2004;389:3–15. doi: 10.1016/S0076-6879(04)89001-3. [DOI] [PubMed] [Google Scholar]
- Langmade SJ, Gale SE, Frolov A, Mohri I, Suzuki K, Mellon SH, Walkley SU, Covey DF, Schaffer JE, Ory DS. Pregnane x receptor (PXR) activation: a mechanism for neuroprotection in a mouse model of Niemann-Pick C disease. Proc Natl Acad Sci USA. 2006;103:13807–13812. doi: 10.1073/pnas.0606218103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Repa JJ, Valasek MA, Beltroy EP, Turley SD, German DC, Dietschy JM. Molecular, anatomical, and biochemical events associated with neurodegeneration in mice with Niemann-Pick type C disease. J Neuropathol Exp Neurol. 2005;64:323–333. doi: 10.1093/jnen/64.4.323. [DOI] [PubMed] [Google Scholar]
- Liu B, Xie C, Richardson JA, Turley SD, Dietschy JM. Receptor-mediated and bulk-phase endocytosis cause macrophage and cholesterol accumulation in Niemann-Pick C disease. J Lipid Res. 2007;48:1710–1723. doi: 10.1194/jlr.M700125-JLR200. [DOI] [PubMed] [Google Scholar]
- Loftus SK, Morris JA, Carstea ED, Gu JZ, Cummings C, Brown A, Ellison J, Ohno K, Rosenfeld MA, Tagle DA, Pentchev PG, Pavan WJ. Murine model of Niemann-Pick C disease: mutation in a cholesterol homeostasis gene. Science. 1997;277:232–235. doi: 10.1126/science.277.5323.232. [DOI] [PubMed] [Google Scholar]
- Lund EG, Xie C, Kotti T, Turley SD, Dietschy JM, Russell DW. Knockout of the cholesterol 24-hydroxylase gene in mice reveals a brain-specific mechanism of cholesterol turnover. J Biol Chem. 2003;278:22980–22988. doi: 10.1074/jbc.M303415200. [DOI] [PubMed] [Google Scholar]
- Mauch DH, Nägler K, Schumacher S, Göritz C, Müller E-C, Otto A, Pfrieger FW. CNS synaptogenesis promoted by glia-derived cholesterol. Science. 2001;294:1354–1357. doi: 10.1126/science.294.5545.1354. [DOI] [PubMed] [Google Scholar]
- Mitro N, Vargas L, Romeo R, Koder A, Saez E. T0901317 is a potent PXR ligand: implications for the biology ascribed to LXR. FEBS Lett. 2007;581:1721–1726. doi: 10.1016/j.febslet.2007.03.047. [DOI] [PubMed] [Google Scholar]
- Pentchev PG, Vanier MT, Suzuki K, Patterson MC. Niemann-Pick disease type C: a cellular cholesterol lipidosis. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Stanbury JB, Wyngaarden JB, Fredrickson DS, editors. The metabolic and molecular bases of inherited diseases. Ed 7. New York: McGraw-Hill; 1995. pp. 2625–2639. [Google Scholar]
- Pfrieger FW. Outsourcing in the brain: do neurons depend on cholesterol delivery by astrocytes? BioEssays. 2003a;25:72–78. doi: 10.1002/bies.10195. [DOI] [PubMed] [Google Scholar]
- Pfrieger FW. Cholesterol homeostasis and function in neurons of the central nervous system. Cell Mol Life Sci. 2003b;60:1158–1171. doi: 10.1007/s00018-003-3018-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proia RL, Wu Y-P. Blood to brain to the rescue. J Clin Invest. 2004;113:1108–1110. doi: 10.1172/JCI21476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan G, Xie C, Dietschy JM, Turley SD. Ontogenesis and regulation of cholesterol metabolism in the central nervous system of the mouse. Dev Brain Res. 2003;146:87–98. doi: 10.1016/j.devbrainres.2003.09.015. [DOI] [PubMed] [Google Scholar]
- Reid PC, Sakashita N, Sugii S, Ohno-Iwashita Y, Shimada Y, Hickey WF, Chang T-Y. A novel cholesterol stain reveals early neuronal cholesterol accumulation in the Niemann-Pick type C1 mouse brain. J Lipid Res. 2004;45:582–591. doi: 10.1194/jlr.D300032-JLR200. [DOI] [PubMed] [Google Scholar]
- Repa JJ, Mangelsdorf DJ. The role of orphan nuclear receptors in the regulation of cholesterol homeostasis. Annu Rev Cell Dev Biol. 2000;16:459–481. doi: 10.1146/annurev.cellbio.16.1.459. [DOI] [PubMed] [Google Scholar]
- Repa JJ, Mangelsdorf DJ. The liver X receptor gene team: potential new players in atherosclerosis. Nat Med. 2002;8:1243–1248. doi: 10.1038/nm1102-1243. [DOI] [PubMed] [Google Scholar]
- Repa JJ, Turley SD, Lobaccaro J-MA, Medina J, Li L, Lustig K, Shan B, Heyman RA, Dietschy JM, Mangelsdorf DJ. Regulation of absorption and ABC1-mediated efflux of cholesterol by RXR heterodimers. Science. 2000;289:1524–1529. doi: 10.1126/science.289.5484.1524. [DOI] [PubMed] [Google Scholar]
- Repa JJ, Turley SD, Quan G, Dietschy JM. Delineation of molecular changes in intrahepatic cholesterol metabolism resulting from diminished cholesterol absorption. J Lipid Res. 2005;46:779–789. doi: 10.1194/jlr.M400475-JLR200. [DOI] [PubMed] [Google Scholar]
- Sarna JR, Larouche M, Marzban H, Sillitoe RV, Rancourt DE, Hawkes R. Patterned Purkinje cell degeneration in mouse models of Niemann-Pick type C disease. J Comp Neurol. 2003;456:279–291. doi: 10.1002/cne.10522. [DOI] [PubMed] [Google Scholar]
- Schultz JR, Tu H, Luk A, Repa JJ, Medina JC, Li L, Schwendner S, Wang S, Thoolen M, Mangelsdorf DJ, Lustig KD, Shan B. Role of LXRs in control of lipogenesis. Genes Dev. 2000;14:2831–2838. doi: 10.1101/gad.850400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suresh S, Yan Z, Patel RC, Patel YC, Patel SC. Cellular cholesterol storage in the Niemann-Pick disease type C mouse is associated with increased expression and defective processing of apolipoprotein D. J Neurochem. 1998;70:242–251. doi: 10.1046/j.1471-4159.1998.70010242.x. [DOI] [PubMed] [Google Scholar]
- Valasek MA, Repa JJ. The power of real-time PCR. Adv Physiol Educ. 2005;29:151–159. doi: 10.1152/advan.00019.2005. [DOI] [PubMed] [Google Scholar]
- Valledor AF, Hsu L-C, Ogawa S, Sawka-Verhelle D, Karin M, Glass CK. Activation of liver X receptors and retinoid X receptors prevents bacterial-induced macrophage apoptosis. Proc Natl Acad Sci USA. 2004;101:17813–17818. doi: 10.1073/pnas.0407749101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance JE, Hayashi H, Karten B. Cholesterol homeostasis in neurons and glial cells. Semin Cell Dev Biol. 2005;16:193–212. doi: 10.1016/j.semcdb.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Wada R, Tifft CJ, Proia RL. Microglial activation precedes acute neurodegeneration in Sandhoff disease and is suppressed by bone marrow transplantation. Proc Natl Acad Sci USA. 2000;97:10954–10959. doi: 10.1073/pnas.97.20.10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahrle SE, Jiang H, Parsadanian M, Legleiter J, Han X, Fryer JD, Kowalewski T, Holtzman DM. ABCA1 is required for normal central nervous system apoE levels and for lipidation of astrocyte-secreted apoE. J Biol Chem. 2004;279:40987–40993. doi: 10.1074/jbc.M407963200. [DOI] [PubMed] [Google Scholar]
- Wang L, Schuster GU, Hultenby K, Zhang Q, Andersson S, Gustafsson J-Å. Liver x receptors in the central nervous system: from lipid homeostasis to neuronal degeneration. Proc Natl Acad Sci USA. 2002;99:13878–13883. doi: 10.1073/pnas.172510899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitney KD, Watson MA, Collins JL, Benson WG, Stone TM, Numerick MJ, Tippin TK, Wilson JG, Winegar DA, Kliewer SA. Regulation of cholesterol homeostasis by the liver X receptors in the central nervous system. Mol Endocrinol. 2002;16:1378–1385. doi: 10.1210/mend.16.6.0835. [DOI] [PubMed] [Google Scholar]
- Xie C, Turley SD, Pentchev PG, Dietschy JM. Cholesterol balance and metabolism in mice with loss of function of Niemann-Pick C protein. Am J Physiol. 1999;276:E336–E344. doi: 10.1152/ajpendo.1999.276.2.E336. [DOI] [PubMed] [Google Scholar]
- Xie C, Burns DK, Turley SD, Dietschy JM. Cholesterol is sequestered in the brains of mice with Niemann-Pick type C disease but turnover is increased. J Neuropathol Exp Neurol. 2000;59:1106–1117. doi: 10.1093/jnen/59.12.1106. [DOI] [PubMed] [Google Scholar]
- Xie C, Lund EG, Turley SD, Russell DW, Dietschy JM. Quantitation of two pathways for cholesterol excretion from the brain in normal mice and mice with neurodegeneration. J Lipid Res. 2003;44:1780–1789. doi: 10.1194/jlr.M300164-JLR200. [DOI] [PubMed] [Google Scholar]
- Zelcer N, Khanlou N, Clare R, Jiang Q, Reed-Geaghan EG, Landreth GE, Vinters HV, Tontonoz P. Attenuation of neuroinflammation and Alzheimer's disease pathology by liver X receptors. Proc Natl Acad Sci USA. 2007;104:10601–10606. doi: 10.1073/pnas.0701096104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Dwyer NK, Neufeld EB, Love DC, Cooney A, Comly M, Patel SC, Watari H, Strauss JF, III, Pentchev PG, Hanover JA, Blanchette-Mackie EJ. Sterol-modulated glycolipid sorting occurs in Niemann-Pick C1 late endosomes. J Biol Chem. 2001;276:3417–3425. doi: 10.1074/jbc.M005393200. [DOI] [PubMed] [Google Scholar]