

Abstract
Brain-derived neurotrophic factor (BDNF) and its receptor TrkB are expressed in several hypothalamic and hindbrain nuclei involved in regulating energy homeostasis, developmentally and in the adult animal. Their depletion during the fetal or early postnatal periods when developmental processes are still ongoing elicits hyperphagic behavior and obesity in mice. Whether BDNF is a chief element in appetite control in the mature brain remains controversial. The required sources of this neurotrophin are also unknown. We show that glucose administration rapidly induced BDNF mRNA expression, mediated by Bdnf promoter 1, and TrkB transcription in the ventromedial hypothalamus (VMH) of adult mice, consistent with a role of this pathway in satiety. Using viral-mediated selective knock-down of BDNF in the VMH and dorsomedial hypothalamus (DMH) of adult mice, we were able to elucidate the physiological relevance of BDNF in energy balance regulation. Site-specific mutants exhibited hyperphagic behavior and obesity but normal energy expenditure. Furthermore, intracerebroventricular administration of BDNF triggered an immediate neuronal response in multiple hypothalamic nuclei in wild-type mice, suggesting that its anorexigenic actions involve short-term mechanisms. Locomotor, aggressive, and depressive-like behaviors, all of which are associated with neural circuits involving the VMH, were not altered in VMH/DMH-specific BDNF mutants. These findings demonstrate that BDNF is an integral component of central mechanisms mediating satiety in the adult mouse and, moreover, that its synthesis in the VMH and/or DMH is required for the suppression of appetite.
Keywords: BDNF, VMH, DMH, satiety, obesity, adult
Introduction
Obesity and its related medical complications are reaching epidemic proportions. They are triggered by alterations in the modulation of energy balance, the finely regulated equilibrium between caloric intake and expenditure. Recently, brain-derived neurotrophic factor (BDNF), a neuronal survival, differentiation, and plasticity factor, emerged as an essential constituent of central neural circuits involved in regulating energy homeostasis. Fetal or early-postnatal depletion of this neurotrophin or its receptor, TrkB, in mice results in hyperphagic behavior and dramatic obesity (Lyons et al., 1999; Kernie et al., 2000; Rios et al., 2001; Xu et al., 2003). In humans, similar alterations were linked to BDNF haploinsufficiency (Gray et al., 2006) and to a de novo missense mutation in the TrkB gene (Yeo et al., 2004). However, it remains to be elucidated whether BDNF acts as a required satiety factor in the adult brain and/or as a developmental facilitator of neural circuits that regulate appetite.
BDNF is expressed in several energy balance centers in the hypothalamus and hindbrain in the developing and mature brain (Conner et al., 1997; Sugiyama et al., 2003; Tran et al., 2003; Xu et al., 2003; Bariohay et al., 2005). Within the hypothalamus, it is most abundant in the ventromedial hypothalamus (VMH), a critical subpopulation of neurons important for the regulation of food intake. The VMH contains receptors for signals denoting energy status, including leptin, insulin, melanin-concentrating hormone (MCH), and orexin (Storlien et al., 1975; Mercer et al., 1996; Trivedi et al., 1998; Kokkotou et al., 2001). Furthermore, bilateral lesions to this region in rodents triggered hyperphagia and obesity (Penicaud et al., 1983). BDNF is also expressed in other brain areas associated with appetite control, including the dorsomedial hypothalamus (DMH), paraventricular nucleus (PVN), and lateral hypothalamic area (LH) (Castren et al., 1995; Xu et al., 2003). The essential hypothalamic sources of BDNF and triggers of its activity for the regulation of energy homeostasis remain to be elucidated.
Previous reports suggest that signaling through the BDNF/TrkB pathway promotes satiety in the adult animal. For example, infusion of BDNF into the brain of mature rats significantly reduces food intake (Lapchak and Hefti, 1992). Additionally, BDNF mRNA levels are reduced by fasting in the VMH and nucleus of the solitary tract (Xu et al., 2003; Bariohay et al., 2005; Tran et al., 2006). BDNF expression in these regions is also regulated by the peripheral satiety factors leptin and cholecystokinin (Bariohay et al., 2005; Komori et al., 2006).
Here, we describe studies designed to differentiate the role of BDNF in the adult animal from its developmental functions impacting appetite control. We show that glucose, a caloric signal, directs transcription of BDNF and TrkB in the VMH and that BDNF induces neuronal activity in hypothalamic energy balance centers. Furthermore, we demonstrate that specific deletion of Bdnf in the VMH and DMH of adult mice results in hyperphagic behavior without affecting energy expenditure and behaviors associated with these hypothalamic regions. These findings demonstrate that BDNF is a required satiety signal in the adult brain and that the VMH/DMH regions comprise an essential source.
Materials and Methods
Animals.
All of the following procedures were approved by the Institutional Animal Care and Use Committee at Tufts University and were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Every effort was made to minimize the number of animals used in these studies and their suffering. BDNF2L/2L mice were generated as previously described and were in a hybrid background with C57BL/6 and 129 strain contributions (Rios et al., 2001). Sex-matched littermates were used in every experiment to reduce genetic background differences. All animals were between 10 and 14 weeks of age at the start of each experiment. All mice used in long-term studies were fed ad libitum, unless otherwise noted, and housed individually in the Tufts University animal care unit on a 12 h light/dark cycle.
In situ hybridization analysis.
To measure BDNF mRNA levels in the brain after glucose or saline treatment, wild-type mice (10–14 weeks of age) were fasted for 48 h followed by a single intraperitoneal injection of glucose (2 mg/kg) or saline. Fresh brains were collected at 0, 15, 30, 60, 120, or 360 min after treatment and immediately frozen on dry ice. The experiments were timed appropriately so that all mice were killed between 3:00 and 4:00 P.M. to avoid potential circadian fluctuations of BDNF mRNA. Twelve-micrometer-thick sections containing VMH and DMH from each animal were hybridized for 16 h at 60°C with a 35S-labeled, antisense riboprobe representing bases 507–833 of the BDNF cDNA. Specificity of this riboprobe was confirmed by lack of hybridization to tissue from BDNF2L/2LCamk-Cre mutant mice (Rios et al., 2001). After the hybridization step, sections were stringently washed and placed on x-ray film for 12 d. Densitometry of BDNF mRNA signal was conducted using the Kodak 1D Image Analysis software. Coronal level was confirmed by staining adjacent sections with cresyl violet. A similar protocol was used to measure BDNF mRNA levels in AAV2/8-GFP and AAV2/8-Cre-treated mice.
Laser-capture microdissection.
Wild-type mice that were fasted for 48 h received a single dose of saline or glucose (2 mg/kg) and were killed 30 min later. Their brains were extracted and immediately frozen. Twelve-micrometer-thick sections representing two independent coronal levels of the VMH were obtained and dehydrated in a series of ethanols, stained in cresyl violet, followed by a 1 min incubation in histoclear. Sections were then dried in a desiccator, and bilateral laser microdissection of cresyl violet-stained cells in the VMH was performed using the Arcturus Pixcell IIe LCM system. Total RNA was extracted from captured cells using the Picopure RNA extraction kit (Arcturus, Sunnyvale, CA), DNased, and amplified (two rounds) using the MessageAmp II RNA amplification kit (Ambion, Austin, TX) as per the manufacturer's specifications.
Quantitative reverse transcription-PCR.
RNA extracted from laser-captured VMH cells was treated with DNase, amplified as described earlier, and tested for genomic DNA contamination in PCRs. Reverse transcription (RT) to generate cDNA was conducted with 2 μg of RNA and using 200 U of Superscript II reverse transcriptase (Invitrogen, Carlsbad, CA) and 150 ng of random hexamers (Invitrogen) in a 20 μl reaction. Real-time PCR amplification was performed using a MX-4000 Stratagene (La Jolla, CA) cycler and SYBR green PCR master mix (Qiagen, Valencia, CA). For each primer set, the specificity of the product amplification was confirmed by dissociation curve analysis and agarose gel electrophoresis. Furthermore, curves were created using serial dilutions, and the efficiencies for each primer set was calculated. The amplification efficiency for all the primers used in this study was between 90 and 100%. For each target primer set, a validation experiment was performed to demonstrate that the PCR efficiencies were approximately equal to those of the reference gene. A two-step protocol was used: 95°C for 10 min and 45–50 cycles with 95°C for 30 s, 58 or 60°C for 30 s, and 72°C for 30 s. GAPDH and actin were used as normalizers. The following intron-spanning primers were used for detecting each of the BDNF transcript isoforms: I: forward (F), 5′-CCTGCATCTGTTGGGGAGAC-3′; reverse (R), 5′-GCCTTGTCCGTGGACGTTTA-3′; II: F, 5-′CTAGCCACCGGGGTGGTGTAA-3′; R, 5′-AGGATGGTCATCACTCTTCTC-3′; III: F, 5′-CAGGAGTACATATCGGCCAC-3′; R, 5′-AGGATGGTCATCACTCTTCTC-3′; IV: F, 5′-CAGAGCAGCTGCCTTGATGTT-3′; R, 5′-GCCTTGTCCGTGGACGTTTA-3′; V: F, 5′-TTGGGGCAGACGAGAAAGCGC-3′; R, 5′-AGGATGGTCATCACTCTTCTC-3′. All samples were analyzed in triplicates, and nontemplate controls were included to ascertain any level of contamination. Amplification products ranged from 100 to 124 bp. Data obtained were analyzed using the comparative Ct method. Every experiment was repeated at least once.
Intracerebroventricular administration of glucose and insulin.
Wild-type mice (10–14 weeks of age) were fasted for 48 h, anesthetized with pentobarbital (50 mg/kg), and stereotaxically administered 100 μg of glucose (Acros Organics, Geel, Belgium), 1 μU of insulin (Humulin R) (Eli Lilly, Indianapolis, IN), or saline vehicle control into the dorsal third ventricle. Thirty minutes after injection, fresh brains were collected, immediately frozen on dry ice, and processed for in situ hybridization as previously described. Densitometry of BDNF mRNA signal was conducted using the Kodak 1D Image Analysis software. All coronal levels were confirmed by staining adjacent sections with cresyl violet.
Immunohistochemistry.
To identify the hypothalamic centers activated by BDNF, 10 μg of recombinant human BDNF (Millipore, Billerica, MA) or saline vehicle control was stereotaxically delivered into the dorsal third ventricle of wild-type mice anesthetized with pentobarbital (50 mg/kg). Forty-five minutes after BDNF injection, mice were transcardially perfused with 20 ml of cold saline followed by 50 ml of 4% paraformaldehyde (PFA). Brains were immediately removed, postfixed in 4% PFA overnight at 4°C, cryoprotected in a 30% sucrose solution, and frozen in mounting media (Tissue-Tek, Torrance, CA) until further use. Twenty-five-micrometer-thick coronal sections representing the whole rostrocaudal extent of the hypothalamus were obtained and c-fos immunohistochemistry was performed using standard methods. In brief, endogenous peroxidase activity was quenched in free-floating sections with 0.5% H2O2 in 1× PBS for 15 min, permeabilized with 0.5% Triton X-100 in 1× PBS for 20 min, and blocked in 10% normal horse serum for 30 min at room temperature. Sections were then incubated for 48 h at 4°C with a rabbit primary antibody against c-fos (1:50,000; Ab5; Oncogene Science, Cambridge, MA), followed by washes and incubation with a biotinylated secondary antibody (1:200) and an avidin–biotin peroxidase complex as per the manufacturers instructions (ABC, Vector Elite Kit; Vector Laboratories, Burlingame, CA). Sections were then incubated with substrate solution (0.025% diaminobenzidine/0.15% nickel ammonium sulfate/0.0036% H2O2 in 0.05 m Tris buffer) for 5 min. c-fos-positive neurons were counted in rostral, medial, and caudal levels of the VMH, DMH, PVN, LH, and arcuate nucleus (Arc). The locations of cells were confirmed in adjacent sections stained with cresyl violet. Data presented represent total number of c-fos+ cells per nuclei from unilateral counts.
Site-specific deletion of Bdnf.
AAV2/8 vectors were produced, purified, and titered as previously described (Broekman et al., 2006). Cre recombinase cDNA carrying an N-terminal nuclear localization signal was cloned into the AAV2-CBA-W vector plasmid. Adeno-associated virus (1.25 ml; 1 × 1013 infectious units/ml) encoding Cre recombinase (AAV2/8-Cre) or green fluorescence protein (AAV2/8-GFP) was delivered to the VMH and DMH of anesthetized floxed BDNF mice (10–14 weeks of age, males) using the following coordinates: anteroposterior, −1.5 mm; mediolateral, ±0.5 mm; dorsoventral, −5.4 mm. Virus was administered bilaterally using a 10 μl Hamilton syringe with a 33-gauge needle attached to a digital stereotaxic apparatus (Benchmark, St. Louis, MO) and an infusion microsyringe nanopump (kd Scientific, Holliston, MA) at a rate of 0.1 μl/min. The needle was held in place for 10 min after injection to permit diffusion of the virus and to minimize backflow after needle retraction. Mice were then given 1 week to recover from surgery before any experiments were conducted. Coordinates for targeting the VMH/DMH were determined using the mouse atlas (Paxinos and Franklin, 2001) and by pilot experiments involving injection of AAV2/8-Cre virus into ROSA reporter mice (Soriano, 1999). Accurate targeting was confirmed in AAV2/8-GFP-treated mice by analysis of GFP signal and in AAV2/8-Cre-treated mice by measurements of BDNF mRNA expression.
Food intake and body weight measurements.
Food intake and body weights of AAV2/8-GFP and AAV2/8-Cre-injected mice were measured starting 1 week after surgery. Mice were housed individually and given a premeasured amount of food. The changes in food and body weight were measured on the same day and time each week.
Pair feeding studies.
To assess deficits in energy expenditure as a result of BDNF depletion from the medial hypothalamus, site-specific injections of AAV were performed as previously described. After surgery, mice were fed ad libitum until AAV2/8-Cre-treated mice exhibited hyperphagic behavior and increased body weight (∼6.5 weeks after surgery), after which they were pair fed to AAV2/8-GFP-injected mice. Body weight was then monitored weekly for the following 9 weeks.
Body temperature measurements.
Core body temperature for pair-fed AAV2/8-GFP and AAV2/8-Cre mice (6–7 months of age) was measured using the CMA/150 Temperature Controller (CMA Microdialysis, Solna, Sweden) fitted with a mouse rectal probe (part # 831 1130). All measurements were conducted between 3:00 and 4:00 P.M. and were recorded when the temperature readout stabilized for >5 s.
Serum analysis.
Serum levels of leptin and insulin were determined using enzyme immunoassay kits from Linco Research (Billerica, MA), respectively. All procedures were performed according to the manufacturer's instructions. To determine blood glucose levels, mice were fasted for 16 h and bled, and measurements were taken using the Freestyle blood glucose monitoring system. The lean/obese labels refer to the body weights of the AAV2/8-Cre-injected mice at the time the study was conducted. For the insulin and glucose measurements, lean refers to AAV2/8-Cre-injected mice that were already hyperphagic but not yet significantly heavier than AAV2/8-GFP-treated mice. For the leptin measurements, lean refers to measurements performed after pair feeding.
Locomotor activity.
For monitoring of locomotor activity, AAV2/8-GFP and AAV2/8-Cre (pair fed and of normal body weight) mice were individually housed in standard 15 × 24 cm plastic cages. Activity was measured as beam breaks using the SmartFrame Activity System (Hamilton/Kinder, Poway, CA) after a 6 d adjustment period. Briefly, the setup consisted of 12 PC-interfaced horizontal beams (8 in y-axis × 4 in x-axis, spaced 1.5 cm apart), which surrounded the perimeter of the cage. Data obtained were analyzed using the MotorMonitor software (Hamilton/Kinder).
Resident intruder test.
To assess isolation-induced aggression as a result of BDNF depletion from the VMH and DMH, all AAV2/8-GFP and AAV2/8-Cre-treated male mice were maintained in their individual cages and subjected to the resident intruder test (Winslow and Miczek, 1983). Cages were not changed for 1 week before testing. On the test day, one wild-type male mouse, serving as the intruder, was placed in the cage of a resident AAV2/8-GFP or AAV2/8-Cre mouse. Aggressive behavior was monitored over the following 5 min and scored as latency to the first biting attack and total number of attacks by the resident animal. Each intruder was used only once in these experiments. Failure of a resident mouse to attack the intruder was scored as 5 min.
Tail suspension test.
The tail suspension test was modified from the method described previously (Steru et al., 1985). AAV2/8-GFP and AAV2/8-Cre-injected mice were suspended by their tail, which was taped to the edge of a table 80 cm from the floor. The test was performed for 6 min and scored for latency to immobility and total immobility. Mice were considered immobile when hanging motionless with their limbs tucked into their body.
Statistical analysis.
Measurements for food intake, body weight, serum analysis, and locomotor activity were analyzed using an unpaired t test. Quantification of in situ hybridizations and quantitative RT-PCR were analyzed by one-way ANOVA. Data were considered statistically significant when p < 0.05, and all values represent mean ± SEM.
Results
Glucose administration rapidly induces BDNF mRNA and TrkB mRNA expression in the VMH
A common feature of appetite-regulating factors is their modulation by energy status signals. Work by others has shown that extended fasting significantly decreases BDNF mRNA content in the VMH (Xu et al., 2003; Tran et al., 2006), consistent with a role of BDNF in regulating feeding behavior. Because this change could be part of a stress response to prolonged food deprivation, it was important to determine whether expression of BDNF in the VMH was sensitive to peripheral macronutrients. As a first step toward distinguishing actions of BDNF in the mature brain from those in the developing brain that influence energy balance, we asked whether caloric signals modulated its hypothalamic expression. To address this, we investigated the effect of glucose on BDNF mRNA expression in the VMH and DMH. For this, wild-type mice fasted over a period of 48 h received a single intraperitoneal dose of glucose (2 mg/kg) or saline, and BDNF mRNA expression was measured 15, 30, 60, 120, and 360 min after treatment. Consistent with previous findings (Xu et al., 2003; Tran et al., 2006), a 58% decrease in levels of BDNF mRNA was observed in the VMH as a response to fasting (Fig. 1A,B) (n = 5; *p = 0.01). Levels of BDNF transcripts in the DMH were not significantly altered by low energy levels (data not shown). Moreover, glucose treatment triggered a rapid elevation of BDNF mRNA content in the VMH as early as 30 min after treatment relative to saline administration (Fig. 1C,D). At this time point, there was a near twofold increase in BDNF transcript levels, which remained significantly elevated 2 h after treatment (Fig. 1D). In agreement with a role in feeding behavior regulation, the increase in BDNF mRNA was only transient. By 6 h after glucose administration, content of BDNF mRNA returned to fasting levels. No changes in BDNF mRNA expression in response to glucose were observed in other brain regions, including the DMH, cortex, and hippocampus (data not shown).
Figure 1.

Glucose rapidly induces BDNF mRNA expression in the VMH. A–C, BDNF mRNA in situ hybridization analysis of representative coronal sections from fed (A), fasted (B), and fasted then glucose-treated (systemic treatment; 30 min after treatment) (C) wild-type mice showing BDNF mRNA expression in the VMH (arrows). D, Time course of BDNF mRNA induction after systemic treatment with saline (open bars) or glucose (closed bars) (n = 4–8 per group; *p = 0.0003; **p = 0.01; ***p = 0.05). E, Densitometry of BDNF mRNA signal in the VMH of wild-type mice 30 min after intracerebroventricular (ICV) treatment with saline (S), glucose (G), or insulin (I) (n = 4; *p = 0.005).
To distinguish whether glucose acts centrally and/or peripherally to modulate BDNF transcription, we measured levels of BDNF mRNA in the VMH of wild-type mice that were administered glucose or saline via intracerebroventricular injection. Mice that received intracerebroventricular glucose exhibited a 51% increase in levels of BDNF transcripts 30 min after treatment compared with saline controls (n = 4; p = 0.005) (Fig. 1E). This result shows that glucose acts directly in the brain to positively regulate expression of BDNF mRNA. To further delve into peripheral actions of glucose influencing BDNF expression, we investigated the effects of intracerebroventricular insulin administration. Insulin is released into the circulation in response to glucose, and insulin receptors are located within the VMH (Storlien et al., 1975). We found that transcript levels of BDNF in the VMH were not altered in response to insulin (Fig. 1E).
Next, we sought to elucidate the molecular mechanisms mediating the rapid induction of BDNF mRNA in the VMH by peripheral administration of glucose. The mouse Bdnf gene comprises at least five untranslated exons (I–V), each of which splices to a single coding exon (VI) to generate several transcript isoforms (Tsankova et al., 2006). Each untranslated exon is associated with an unique promoter. To investigate which Bdnf promoters were activated in the VMH after glucose treatment, cells from this region in glucose or saline-treated (30 min after treatment) wild-type mice were isolated by laser-capture microdissection (Fig. 2A,B). RNA extracted from captured cells was amplified and reverse transcribed for BDNF isoform-specific quantitative PCR analysis. We found that content of transcripts containing exon 1 was significantly increased in response to glucose (p = 0.03; n = 4) (Fig. 2C). Moreover, there was a trend toward a significant increase in transcripts containing exon IV (p = 0.058; n = 4) (Fig. 2C). These results are consistent with a previous report (Tran et al., 2006), which showed that exon I and IV-containing isoforms are enriched in the VMH under basal conditions. Surprisingly, the level of transcripts containing exon V was significantly reduced in the VMH by glucose treatment (p = 0.02; n = 4) (Fig. 2C).
Figure 2.
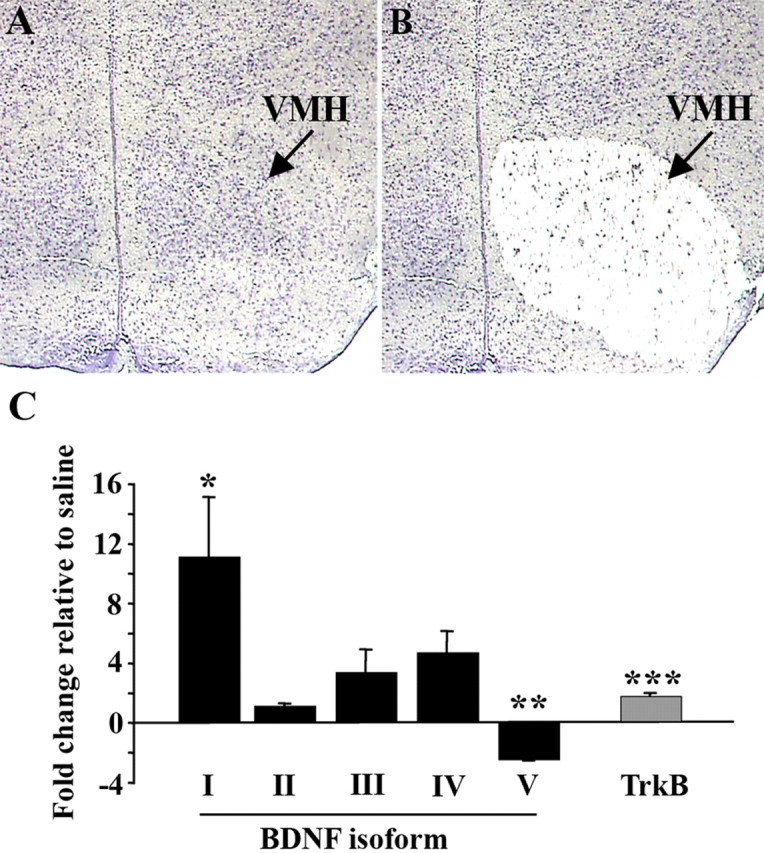
Systemic administration of glucose induces transcription of BDNF directed by promoter 1 in the VMH. A, B, Representative coronal section stained with cresyl violet and containing VMH before (A) and after (B) laser-capture microdissection. C, Content of BDNF mRNA isoforms and full-length TrkB mRNA in glucose-treated fasted mice relative to saline-treated fasted mice using laser-captured VMH tissue determined by quantitative RT-PCR analysis (n = 4–7 per group; *p = 0.03; **p = 0.02; ***p = 0.004). Roman numerals indicate untranslated exon contained in BDNF mRNA isoform. Data are expressed as fold difference of glucose-treated mice relative to saline-treated animals. The mean value for saline-treated mice was set at 1. p values were calculated based on 2ΔΔCt values for each sample.
To further ascertain how energy signals might impact the BDNF signaling pathway, we investigated whether glucose (30 min after treatment) also regulated levels of full-length TrkB receptor (TrkB.FL) in the VMH. An 80% elevation in the levels of TrkB.FL mRNA was detected in the VMH of glucose-treated animals when compared with saline controls (p = 0.003; n = 7) (Fig. 2C). Our observation that glucose, an energy signal, rapidly induces both BDNF and TrkB.FL mRNA expression in the VMH supports a role for BDNF signaling in satiety.
Exogenous BDNF rapidly activates neurons in hypothalamic appetite-regulating centers
Because BDNF can be secreted from dendrites or axonal terminals, it can act presynaptically or postsynaptically (Gottschalk et al., 1998; Xu et al., 2000; Chakravarthy et al., 2006). There are also examples of this neurotrophin acting in an autocrine manner in the literature (Barnabe-Heider and Miller, 2003; Kim et al., 2004). In the hypothalamus, extensive expression of TrkB in the Arc, VMH, PVN, DMH, and LH suggests many potential target sites for BDNF action influencing food intake behavior (Yan et al., 1997; Kernie et al., 2000). To further delve into the mechanisms facilitating the effects of BDNF in the adult animal, we assessed its ability to rapidly activate neurons contained in these discrete appetite-regulating hypothalamic nuclei. Exogenous BDNF (10 μg) or saline was stereotaxically delivered to the third ventricle of wild-type mice. Forty-five minutes after treatment, hypothalamic expression of c-fos, an immediate early response gene and marker of neuronal activation, was measured. BDNF rapidly induced c-fos expression in several hypothalamic nuclei including the VMH (204.1 ± 8.78 vs 53.5 ± 3.5; p < 0.01), DMH (146.7 ± 3.8 vs 62.8 ± 8.4; p < 0.01), PVN (209.3 ± 27.0 vs 61.6 ± 4.7; p < 0.01), and LH (91.8 ± 4.2 vs 41.0 ± 4.9; p < 0.01) compared with saline-treated mice (n = 4 per group) (Fig. 3A–E). In the PVN, VMH, DMH, and LH, the number of c-fos+ cells was increased by 285, 237, 132, and 124%, respectively, compared with saline controls (Fig. 3E). However, no significant changes in the number of c-fos-expressing cells were observed in the Arc. These data show that BDNF functions as a fast-acting signal that induces neuronal activity within several hypothalamic nuclei.
Figure 3.

Administration of BDNF into the third ventricle (3V) rapidly induces neuronal activity in several hypothalamic nuclei. A–D, Representative coronal sections showing c-fos signal in the hypothalami of wild-type mice treated with saline (A, C) or BDNF (B, D) 45 min after treatment. Arrows denote c-fos-containing cells in the PVN in D. E, Quantification of total number of c-fos+ neurons in the PVN, VMH, DMH, LH, and Arc of wild-type mice treated with saline (open bars) or BDNF (closed bars) (n = 4; *p < 0.01). Counts represent total number of c-fos+ cells in three independent coronal levels of each of the studied regions determined from unilateral counts.
Selectively deleting Bdnf in the ventromedial and dorsomedial hypothalamus of adult mice results in hyperphagic behavior and body weight gain
BDNF is expressed in various energy balance-regulating centers in the hypothalamus and hindbrain in the developing and mature brain (Conner et al., 1997; Sugiyama et al., 2003; Tran et al., 2003; Xu et al., 2003; Bariohay et al., 2005). Mouse models of global or central depletion of BDNF and TrkB exhibit increases in food intake and dramatic obesity (Lyons et al., 1999; Kernie et al., 2000; Rios et al., 2001; Xu et al., 2003). However, it remains unclear what proportion of these alterations are caused by perturbed developmental or maintenance functions of BDNF. Furthermore, the essential sources of BDNF for the regulation of appetite remain unknown. Because we found that nutritional signals modulate expression of BDNF mRNA in the VMH, we hypothesized that BDNF derived from this region acts as an essential satiety signal in the adult animal. To test this, we selectively deleted Bdnf alleles in the VMH and DMH of adult male mice (10–14 weeks of age). Adeno-associated virus encoding Cre recombinase (AAV2/8-Cre) or green fluorescence protein (AAV2/8-GFP) was stereotaxically and bilaterally delivered to the VMH of mice homozygous for floxed Bdnf alleles (Bdnf 2L/2L mice) (Rios et al., 2001). Because of its proximity to the VMH, cells in the DMH were also targeted. Untreated BDNF2L/2L mice have normal body weight and food intake behavior and express BDNF normally unless Cre recombinase is present to mediate the elimination of the single coding exon of Bdnf (Rios et al., 2001). AAV2/8-GFP-treated mice were used as controls for the effects of the surgery and the viral infection. A time course of BDNF mRNA depletion using this approach revealed that this neurotrophin was not maximally depleted in the VMH and DMH of AAV2/8-Cre-treated mice until 4 weeks after injection (data not shown). This finding is consistent with other reports using similar viral vectors (Berton et al., 2006).
First, we investigated whether there were toxicity effects associated with the viral infection. Cell counts in cresyl violet-stained sections obtained from AAV2/8-GFP-treated, AAV2/8-Cre-treated, and untreated wild-type mice did not reveal any changes in cell content in the VMH or DMH at 16 weeks after viral injection (data not shown). We then quantified the degree and specificity of BDNF mRNA depletion in AAV2/8-Cre-injected mice compared with AAV2/8-GFP-treated mice, throughout the entire rostrocaudal extent of the hypothalamus. In situ hybridization analysis revealed that on average, AAV2/8-Cre-treated mice exhibited bilateral 87 and 81% reductions in BDNF mRNA in the VMH and DMH, respectively (Fig. 4A–E). Depletion of this neurotrophin was evident in all segments of these hypothalamic nuclei. No significant changes in levels of BDNF transcripts were observed in the PVN, cortex, or hippocampus, indicating that targeting was specific to the VMH and DMH and that the majority of cells in the targeted region had been depleted of BDNF. Only data obtained from AAV2/8-Cre-injected mice that were confirmed to have bilateral depletion of BDNF mRNA from the VMH and DMH >70% through post hoc examination were included in each study.
Figure 4.

AAV2/8-Cre-treated mice exhibit significant depletion of BDNF mRNA from the basomedial hypothalamus compared with AAV2/8-GFP-treated controls. A–D, In situ hybridization analysis of representative coronal sections obtained from floxed BDNF mice treated with AAV2/8-GFP (A, C) or AAV2/8-Cre (B, D) showing extensive depletion of BDNF mRNA in the VMH (arrows) and DMH (arrowheads) of AAV2/8-Cre-treated mice. C, D, Higher-magnification views of VMH and DMH areas. E, Quantification of BDNF mRNA in situ hybridizations by densitometry demonstrates significant reductions in BDNF mRNA content in the VMH and DMH of AAV2/8-Cre-treated mice (closed bars) relative to AAV2/8-GFP-treated mice (open bars). BDNF mRNA content in the PVN, cortex (Ctx), and hippocampus (Hipp) was comparable in both experimental groups (n = 7; *p = 0.0007; **p = 0.0001).
Already at 4 weeks after surgery when BDNF depletion became maximal, animals that received AAV2/8-Cre consumed significantly more food than AAV2/8-GFP-injected mice (Fig. 5A) (p = 0.05). Hyperphagic behavior persisted throughout the remainder of the study and by 10 weeks after surgery, AAV2/8-Cre injected mice exhibited a 27% increase in daily food intake compared with AAV2/8-GFP injected mice (4.87 ± 0.11 and 3.83 ± 0.14 g/d, respectively; p = 0.00009; n = 7) (Fig. 5A). Moreover, they became significantly heavier at 7 weeks after surgery (Fig. 5B). Sixteen weeks after viral treatment, AAV2/8-Cre injected mice were, on average, 41% heavier than AAV2/8-GFP injected mice (38.8 ± 1.94 and 27.4 ± 2.16 g, respectively; n = 7; p = 0.0001) (Fig. 5B). These studies demonstrate that BDNF acts as a satiety factor in the adult brain and that its synthesis in the VMH and DMH is required for normal energy balance.
Figure 5.

Depletion of BDNF in the VMH and DMH induces hyperphagia and significant body weight gain. A, Daily food intake of AAV2/8-GFP- (open triangles) and AAV2/8-Cre (closed triangles)-injected mice. B, Body weight of AAV2/8-GFP- (open triangles) and AAV2/8-Cre (closed triangles)-injected mice after surgery (n = 7; *p < 0.05).
Mice with depletion of VMH- and DMH-derived BDNF develop hyperleptinemia, hyperinsulinemia, and hyperglycemia
In our previous studies, we found that mice with central depletion of BDNF (BDNF2L/2LCamK-Cre mice) had elevated circulating levels of leptin, insulin, and glucose (Rios et al., 2001). To investigate whether these physiological changes were elicited by depletion of BDNF in the VMH and DMH, serum analysis was conducted. Serum samples were obtained from AAV2/8-Cre-treated mice that exhibited hyperphagic behavior and that had normal or increased body weights and compared with their AAV2/8-GFP-treated counterparts. AAV2/8-Cre-injected mice that had not yet become obese had normal circulating levels of leptin, insulin, and glucose relative to AAV2/8-GFP controls (Fig. 6A–C). In contrast, obese AAV2/8-Cre injected mice exhibited fourfold, 6.3-fold, and 30% increases in serum levels of leptin (n = 7; p = 0.0002), insulin (n = 7; p = 0.008), and glucose (n = 7; p = 0.001), respectively, compared with AAV2/8-GFP mice (Fig. 6A–C). These results demonstrate that VMH/DMH-specific BDNF mutants develop leptin and insulin resistance and hyperglycemia because of their body weight gain and not as a result of BDNF depletion.
Figure 6.

Obese VMH/DMH-specific BDNF mutants have elevated circulating levels of leptin, insulin and glucose. A, Serum leptin levels of AAV2/8-GFP (G) and AAV2/8-Cre (C) mice (n = 7 per group; *p = 0.002). B, Serum insulin levels of AAV2/8-GFP (G) and AAV2/8-Cre (C) mice (n = 7; *p = 0.008). C, Fasted blood glucose levels of AAV2/8-GFP (G) and AAV2/8-Cre (C) mice (n = 7; *p = 0.001). Lean and obese labels refer to body weights of AAV2/8-cre-treated mice at the time of analysis.
Limiting caloric intake restores normal body weight in VMH/DMH-site specific BDNF mutants
The VMH and DMH influence energy expenditure through the modulation of sympathetic outflow to brown adipose tissue and thermogenesis (Perkins et al., 1981; Takahashi and Shimazu, 1982; Kelly and Bielajew, 1991; Zaretskaia et al., 2002). Therefore, we sought to elucidate whether depleting BDNF in these areas resulted in alterations in energy expenditure. For this, we investigated whether caloric restriction was enough to restore body weight levels of the site-specific BDNF mutants to those of the controls. After confirming that AAV2/8-Cre-injected mice exhibited hyperphagic behavior and obesity, we limited their caloric intake to that of AAV2/8-GFP-injected mice. Pair feeding induced a progressive loss of body weight in AAV2/8-Cre-treated mice (Fig. 7). Indeed, after 5 weeks of pair feeding, there was no significant difference in body weight between the two groups. Additionally, body temperatures of pair-fed AAV2/8-Cre-injected mice were similar to those of AAV2/8-GFP-injected mice, consistent with normal thermogenesis (data not shown). These results suggest that the obesity triggered by BDNF depletion in the VMH and DMH is caused by increased food intake, not by changes in energy expenditure.
Figure 7.

Mice with BDNF depletion from the VMH and DMH do not exhibit alterations in energy expenditure. Pair feeding AAV2/8-Cre-treated mice (closed triangles) with AAV2/8-GFP-treated mice (open triangles) progressively reduced their body weights until they became comparable with those of the controls (n = 5; *p < 0.05).
Mice with depleted BDNF in the VMH and DMH do not exhibit the behavioral alterations associated with central deletion of Bdnf
Reported lines of BDNF mutant mice display behavioral abnormalities, including hyperaggression, depressive-like behavior, and hyperactivity (Lyons et al., 1999; Kernie et al., 2000; Monteggia et al., 2004, 2007; Chan et al., 2006). Electrical stimulation of neural circuits involving the VMH elicits increases in aggressive behavior, and stimulation of kainate-type glutamate receptors in the VMH of rats induces running behavior (Adamec, 1991; Narita et al., 2002). Moreover, the VMH has reciprocal connections with the amygdala, a limbic structure associated with depression (Krieger et al., 1979). Therefore, we asked whether behavioral deficits observed in reported lines of BDNF insufficiency were triggered by lack of BDNF in the VMH. Locomotor activity of AAV2/8-Cre and AAV2/8-GFP mice in their home cages was monitored over six continuous 24 h periods. No significant differences in total activity or in activity during the light or dark cycles were observed between the two groups (Fig. 8).
Figure 8.

AAV2/8-Cre-injected mice display normal levels of locomotor activity. Total, light cycle, and dark cycle levels of activity of AAV2/8-Cre mutants (closed bars) and AAV2/8-GFP controls (open bars) over a period of 6 d calculated based on beam interruptions in their home cages (n = 7).
Next, we tested AAV2/8-GFP and AAV2/8-Cre-treated mice in the resident intruder behavioral paradigm to assess changes in aggressive behavior. No significant changes in the latency to the first biting attack (AAV2/8-GFP, 243 ± 14 s; AAV2/8-Cre, 256 ± 15 s) or in the total number of attacks (AAV2/8-GFP, 1.88 ± 0.39 attacks; AAV2/8-Cre, 2.34 ± 0.9 attacks) were observed between both groups of mice. Finally, we subjected AAV2/8-Cre and AAV2/8-GFP mice to the tail suspension test, a behavioral paradigm designed to assess depressive-like behavior in rodents. Both experimental groups exhibited similar total immobility times (AAV2/8-GFP, 91.2 ± 17.8 s; AAV2/8-Cre, 78.4 ± 12.7 s) and latency to immobility (AAV2/8-GFP, 112.4 ± 12.9 s; AAV2/8-cre, 119.8 ± 5.8 s). In summary, these studies show that BDNF expression in the VMH and DMH is not required for the regulation of locomotor, aggressive, or depressive-like behavior.
Discussion
In this study, we demonstrate for the first time that BDNF signaling in the mature brain is required for the fine control of energy balance and that the VMH and/or DMH are essential sources of it in mediating satiety. In support of this, we also showed that glucose rapidly and robustly induced expression of both BDNF mRNA and TrkB mRNA in the VMH. Additionally, BDNF was able to rapidly induce neuronal activity in key hypothalamic nuclei involved in body weight regulation. In contrast, BDNF expression in the VMH and DMH was not required for the modulation of energy expenditure or aggressive, depressive-like, or locomotor behavior. Together, these findings highlight the prominent and direct role of BDNF in the regulation of energy homeostasis in adult animals and bring us closer to elucidating the neural circuits that rely on BDNF signaling to modulate appetite.
BDNF regulates appetite in the adult animal
BDNF and TrkB have well illustrated roles facilitating neuronal survival, differentiation, and connectivity during development of the nervous system (Patapoutian and Reichardt, 2001). Some of these functions could extend to the hypothalamus, where BDNF mRNA expression is already evident during the prenatal period (Sugiyama et al., 2003). Our examination of mice in which Bdnf was selectively deleted in the adult VMH and DMH demonstrates that BDNF acts as a critical satiety factor in adult mice. However, these findings do not preclude the possibility that it also serves essential roles during development of energy balance-regulating neural circuits. Relevant to this assertion, it is important to note that the degree of hyperphagic behavior and obesity exhibited by VMH/DMH-specific BDNF mutants was not as pronounced as that of our previously reported BDNF2L/2LCamK-Cre mice (Rios et al., 2001). Whereas the site-specific mutant males described here exhibited a 27% increase in food intake, BDNF2L/2LCamK-Cre mice ate 75% more than wild types. This discrepancy is unlikely to be caused by differential levels of BDNF depletion, because they are comparable in both lines of mutants. Instead, the more severe hyperphagia of BDNF2L/2LCamK-Cre mice could be attributable to the additive effects of perturbing developmental and maintenance functions of BDNF. In that line of mutants, BDNF depletion begins during the first postnatal week and is completed 2 weeks later. Because hypothalamic cell proliferation, exit from the cell cycle, and migration are completed before BDNF levels are reduced in BDNF2L/2LCamK-Cre mice, these processes are expected to be intact (Shimada and Nakamura, 1973; Rodier, 1980; McClellan et al., 2006). However, deficient hypothalamic cell differentiation, axonal projections, and synapse formation might be features of these mutants, because these are ongoing developmental processes during the early postnatal period. Interestingly, steroidogenic factor 1 (SF-1), a paramount element in VMH differentiation, induces BDNF expression in this region (Tran et al., 2006). However, selective depletion of BDNF in SF-1 neurons did not elicit obesity in mice (Tong et al., 2007). It is worth noting that SF-1 is expressed in only 60% of BDNF-containing cells in the VMH (Tran et., 2006). Therefore, the cumulative data indicate that whereas BDNF expression by SF-1 neurons might not be required for normal energy homeostasis, its secretion by other cells within the VMH is necessary.
It is important to consider additional lines of evidence that argue against BDNF having a required role in the development of energy balance centers. For example, our previous examination of BDNF2L/2LCamK-Cre mutants failed to uncover defects in the overall organization of the hypothalamus or in the hypothalamic pattern of distribution of NPY, MCH, orexin, AGRP (agouti-related protein), α-MSH, and TRH (thyrotropin-releasing hormone)-containing cell bodies and fibers (Rios et al., 2001). Moreover, BDNF−/− mice did not exhibit alterations in cell migration and positioning in the VMH (McClellan et al., 2006). Finally, central infusion of exogenous BDNF restored normal body weights in BDNF+/− mutant mice (Kernie et al., 2000). The latter observation, in particular, is consistent with BDNF acting to facilitate the physiological function of weight-regulating neurons. An alternative explanation for the more severe obesity of BDNF2L/2LCamK-Cre mutants is the additive effects of depleting BDNF in additional energy balance-regulating regions of the brain. BDNF is vastly depleted across the brain of BDNF2L/2LCamK-Cre mice, the cerebellum exempted (Rios et al., 2001). Based on the functions ascribed to the PVN previously, depleting BDNF from this area could have deleterious effects on the regulation of food intake, metabolic function, and sympathetic outflow (Swanson and Sawchenko, 1983; Sims and Lorden, 1986). Accordingly, administration of exogenous BDNF into the PVN reduced energy intake and increased body temperature and expression of UCP1 (uncoupling protein 1) in brown adipose tissue (Wang et al., 2007a,b). Lack of BDNF in the dorsal vagal complex is also likely to contribute to the obesity of BDNF2L/2LCamK-Cre mutants. Whereas fasting elicits a reduction in neurotrophin expression in this hindbrain area, refeeding after extended fasting increases its content of BDNF (Bariohay et al., 2005). The involvement of BDNF in the mesolimbic pathway also warrants consideration, because this circuit was associated previously with the hedonistic aspects of food intake regulation (Georgescu et al., 2005; Fulton et al., 2006).
Regardless of the importance of its expression elsewhere, it is clear from our studies that depleting BDNF stores from the VMH and DMH significantly hinders the regulation of appetite. The high level of technical difficulty of the studies described here prevented targeting Bdnf exclusively in the VMH or DMH. We propose that between these two areas, the VMH is more likely to be an essential source of this neurotrophin for the short-term regulation of food intake. In agreement, energy status signals influence BDNF mRNA expression in the VMH but not in the DMH (Xu et al., 2003; Tran et al., 2006; our observations). Moreover, we showed here that glucose induces BDNF mRNA expression in the VMH through central mechanisms. A direct mechanism might involve glucose-mediated calcium influx in glucose-sensing neurons in the VMH with a subsequent stimulation of calcium-responsive elements present in promoters 1 and 4 of the Bdnf gene. A surprising finding was the concomitant reduction in promoter V-directed BDNF mRNA expression induced by glucose. This change could reflect a feedback inhibitory mechanism designed to maintain physiological levels of BDNF transcripts in this hypothalamic region.
Targets and mechanisms of action of VMH/DMH-derived BDNF
We report here that BDNF administration leads to rapid neuronal activation in several hypothalamic nuclei, including the VMH. This suggests that VMH-derived BDNF might exert its satiety effects by acting locally within this nucleus in an autocrine or paracrine manner. In support of this, it was reported recently that BDNF infusion into the VMH triggered reductions in food consumption and body weight (Wang et al., 2007c). BDNF produced in the VMH could also interact with several intrahypothalamic or extrahypothalamic regions to signal for satiety, including the DMH, Arc, PVN, amygdala, nucleus accumbens, and nucleus of the solitary tract (Canteras et al., 1994). No changes in the number of c-fos+ cells were observed in the Arc in response to BDNF administration albeit having a high content of TrkB receptors, suggesting that BDNF might exert both excitatory and inhibitory effects on neurons residing there.
The findings that BDNF rapidly activates neurons in hypothalamic nuclei and that glucose induces a fast increase in BDNF and TrkB content in the VMH support the idea that this signaling pathway regulates energy balance through short-term mechanisms. These features are consistent with the involvement of BDNF in meal termination signaling, perhaps through the modulation of other weight-regulating factors. However, the effects of this neurotrophin could also entail longer-term mechanisms involving interpretation of other satiety signals. In the postnatal brain, plasticity of hypothalamic neurons is a recently identified and likely important element in body weight regulation. Indeed, excitatory input from the VMH to appetite-inhibiting proopiomelanocortin (POMC) neurons in the arcuate nucleus is dynamically modulated by nutritional cues (Sternson et al., 2005). Leptin, an anorexigenic factor, is also a participant in hypothalamic synaptic plasticity. In addition to modulating the expression of POMC and NPY and the activity of neurons containing them in the Arc (Mizuno et al., 1998; Sahu, 1998; Ahima et al., 1999; Elias et al., 1999), it facilitates rewiring of circuits in that hypothalamic region (Pinto et al., 2004). After considering BDNF's well established role in synaptic plasticity elsewhere in the brain (Korte et al., 1995; Carter et al., 2002) and the ability of leptin to induce its expression in the VMH (Komori et al., 2006), it is reasonable to speculate about its potential involvement with the remodeling of feeding circuits. Future research should focus on elucidating whether BDNF produced in the VMH contributes to these reported forms of hypothalamic plasticity.
In summary, we conclude that BDNF is a pivotal element in the regulation of energy homeostasis in the adult brain and that its secretion from the VMH and/or DMH is required for normal appetite control. Future investigations will aim to further pinpoint cellular and molecular targets of BDNF action in this context.
Footnotes
This work was supported by National Institutes of Health Grants MH67817 and DK073311 (M.R.). We thank the Genomics and Imaging core facilities at Tufts University for facilitating these studies. These cores are supported by the Center for Neuroscience Research (P30 NS047243) and the Center for Gastroenterology Research on Absorptive and Secretory Processes (P30 DK34928).
References
- Adamec RE. Partial kindling of the ventral hippocampus: identification of changes in limbic physiology which accompany changes in feline aggression and defense. Physiol Behav. 1991;49:443–453. doi: 10.1016/0031-9384(91)90263-n. [DOI] [PubMed] [Google Scholar]
- Ahima RS, Kelly J, Elmquist JK, Flier JS. Distinct physiologic and neuronal responses to decreased leptin and mild hyperleptinemia. Endocrinology. 1999;140:4923–4931. doi: 10.1210/endo.140.11.7105. [DOI] [PubMed] [Google Scholar]
- Bariohay B, Lebrun B, Moyse E, Jean A. Brain-derived neurotrophic factor plays a role as an anorexigenic factor in the dorsal vagal complex. Endocrinology. 2005;146:5612–5620. doi: 10.1210/en.2005-0419. [DOI] [PubMed] [Google Scholar]
- Barnabe-Heider F, Miller FD. Endogenously produced neurotrophins regulate survival and differentiation of cortical progenitors via distinct signaling pathways. J Neurosci. 2003;23:5149–5160. doi: 10.1523/JNEUROSCI.23-12-05149.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berton O, McClung C, Dileone RJ, Krishnan V, Renthal W, Russo SJ, Graham D, Tsankova NM, Bolanos C, Rios M, Monteggia LM, Self DW, Nestler EJ. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science. 2006;311:864–868. doi: 10.1126/science.1120972. [DOI] [PubMed] [Google Scholar]
- Broekman ML, Comer LA, Hyman BT, Sena-Esteves M. Adeno-associated virus vectors serotyped with AAV8 capsid are more efficient than AAV-1 or -2 serotypes for widespread gene delivery to the neonatal mouse brain. Neuroscience. 2006;138:501–510. doi: 10.1016/j.neuroscience.2005.11.057. [DOI] [PubMed] [Google Scholar]
- Canteras NS, Simerly RB, Swanson LW. Organization of projections from the ventromedial nucleus of the hypothalamus: a Phaseolus vulgaris-leucoagglutinin study in the rat. J Comp Neurol. 1994;348:41–79. doi: 10.1002/cne.903480103. [DOI] [PubMed] [Google Scholar]
- Carter AR, Chen C, Schwartz PM, Segal RA. Brain-derived neurotrophic factor modulates cerebellar plasticity and synaptic ultrastructure. J Neurosci. 2002;22:1316–1327. doi: 10.1523/JNEUROSCI.22-04-01316.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castren E, Thoenen H, Lindholm D. Brain-derived neurotrophic factor messenger RNA is expressed in the septum, hypothalamus and in adrenergic brain stem nuclei of adult rat brain and is increased by osmotic stimulation in the paraventricular nucleus. Neuroscience. 1995;64:71–80. doi: 10.1016/0306-4522(94)00386-j. [DOI] [PubMed] [Google Scholar]
- Chakravarthy S, Saiepour MH, Bence M, Perry S, Hartman R, Couey JJ, Mansvelder HD, Levelt CN. Postsynaptic TrkB signaling has distinct roles in spine maintenance in adult visual cortex and hippocampus. Proc Natl Acad Sci USA. 2006;103:1071–1076. doi: 10.1073/pnas.0506305103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J, Unger TJ, Byrnes J, Rios M. Examination of behavioral deficits triggered by targeting Bdnf in fetal or postnatal brains of mice. Neuroscience J. 2006;142:49–58. doi: 10.1016/j.neuroscience.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Conner JM, Lauterborn JC, Yan Q, Gall CM, Varon S. Distribution of brain-derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat CNS: evidence for anterograde axonal transport. J Neurosci. 1997;17:2295–2313. doi: 10.1523/JNEUROSCI.17-07-02295.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias CF, Aschkenasi C, Lee C, Kelly J, Ahima RS, Bjorbaek C, Flier JS, Saper CB, Elmquist JK. Leptin differentially regulates NPY and POMC neurons projecting to the lateral hypothalamic area. Neuron. 1999;23:775–786. doi: 10.1016/s0896-6273(01)80035-0. [DOI] [PubMed] [Google Scholar]
- Fulton S, Pissios P, Manchon RP, Stiles L, Frank L, Pothos EN, Maratos-Flier E, Flier JS. Leptin regulation of the mesoaccumbens dopamine pathway. Neuron. 2006;51:811–822. doi: 10.1016/j.neuron.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Georgescu D, Sears RM, Hommel JD, Barrot M, Bolanos CA, Marsh DJ, Bednarek MA, Bibb JA, Maratos-Flier E, Nestler EJ, DiLeone RJ. The hypothalamic neuropeptide melanin-concentrating hormone acts in the nucleus accumbens to modulate feeding behavior and forced-swim performance. J Neurosci. 2005;25:2933–2940. doi: 10.1523/JNEUROSCI.1714-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottschalk W, Pozzo-Miller LD, Figurov A, Lu B. Presynaptic modulation of synaptic transmission and plasticity by brain-derived neurotrophic factor in the developing hippocampus. J Neurosci. 1998;18:6830–6839. doi: 10.1523/JNEUROSCI.18-17-06830.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray J, Yeo GS, Cox JJ, Morton J, Adlam AL, Keogh JM, Yanovski JA, El Gharbawy A, Han JC, Tung YC, Hodges JR, Raymond FL, O'Rahilly S, Farooqi IS. Hyperphagia, severe obesity, impaired cognitive function, and hyperactivity associated with functional loss of one copy of the brain-derived neurotrophic factor (BDNF) gene. Diabetes. 2006;55:3366–3371. doi: 10.2337/db06-0550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly L, Bielajew C. Ventromedial hypothalamic regulation of brown adipose tissue. NeuroReport. 1991;2:41–44. doi: 10.1097/00001756-199101000-00010. [DOI] [PubMed] [Google Scholar]
- Kernie SG, Liebl DJ, Parada LF. BDNF regulates eating behavior and locomotor activity in mice. EMBO J. 2000;19:1290–1300. doi: 10.1093/emboj/19.6.1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Li Q, Hempstead BL, Madri JA. Paracrine and autocrine functions of brain-derived neurotrophic factor (BDNF) and nerve growth factor (NGF) in brain-derived endothelial cells. J Biol Chem. 2004;279:33538–33546. doi: 10.1074/jbc.M404115200. [DOI] [PubMed] [Google Scholar]
- Kokkotou EG, Tritos NA, Mastaitis JW, Slieker L, Maratos-Flier E. Melanin-concentrating hormone receptor is a target of leptin action in the mouse brain. Endocrinology. 2001;142:680–686. doi: 10.1210/endo.142.2.7981. [DOI] [PubMed] [Google Scholar]
- Komori T, Morikawa Y, Nanjo K, Senba E. Induction of brain-derived neurotrophic factor by leptin in the ventromedial hypothalamus. Neuroscience. 2006;139:1107–1115. doi: 10.1016/j.neuroscience.2005.12.066. [DOI] [PubMed] [Google Scholar]
- Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci USA. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieger MS, Conrad LC, Pfaff DW. An autoradiographic study of the efferent connections of the ventromedial nucleus of the hypothalamus. J Comp Neurol. 1979;183:785–815. doi: 10.1002/cne.901830408. [DOI] [PubMed] [Google Scholar]
- Lapchak PA, Hefti F. BDNF and NGF treatment in lesioned rats: effects on cholinergic function and weight gain. NeuroReport. 1992;3:405–408. doi: 10.1097/00001756-199205000-00007. [DOI] [PubMed] [Google Scholar]
- Lyons WE, Mamounas LA, Ricaurte GA, Coppola V, Reid SW, Bora SH, Wihler C, Koliatsos VE, Tessarollo L. Brain-derived neurotrophic factor-deficient mice develop aggressiveness and hyperphagia in conjunction with brain serotonergic abnormalities. Proc Natl Acad Sci USA. 1999;96:15239–15244. doi: 10.1073/pnas.96.26.15239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClellan KM, Parker KL, Tobet S. Development of the ventromedial nucleus of the hypothalamus. Front Neuroendocrinol. 2006;27:193–209. doi: 10.1016/j.yfrne.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Mercer JG, Hoggard N, Williams LM, Lawrence CB, Hannah LT, Trayhurn P. Localization of leptin receptor mRNA and the long form splice variant (Ob-Rb) in mouse hypothalamus and adjacent brain regions by in situ hybridization. FEBS Lett. 1996;387:113–116. doi: 10.1016/0014-5793(96)00473-5. [DOI] [PubMed] [Google Scholar]
- Mizuno TM, Kleopoulos SP, Bergen HT, Roberts JL, Priest CA, Mobbs CV. Hypothalamic pro-opiomelanocortin mRNA is reduced by fasting and [corrected] in ob/ob and db/db mice, but is stimulated by leptin. Diabetes. 1998;47:294–297. doi: 10.2337/diab.47.2.294. [Erratum (1998) 47:696] [DOI] [PubMed] [Google Scholar]
- Monteggia LM, Barrot M, Powell CM, Berton O, Galanis V, Gemelli T, Meuth S, Nagy A, Greene RW, Nestler EJ. Essential role of brain-derived neurotrophic factor in adult hippocampal function. Proc Natl Acad Sci USA. 2004;101:10827–10832. doi: 10.1073/pnas.0402141101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteggia LM, Luikart B, Barrot M, Theobold D, Malkovska I, Nef S, Parada LF, Nestler EJ. Brain-derived neurotrophic factor conditional knockouts show gender differences in depression-related behaviors. Biol Psychiatry. 2007;61:187–197. doi: 10.1016/j.biopsych.2006.03.021. [DOI] [PubMed] [Google Scholar]
- Narita K, Murata T, Honda K, Nishihara M, Takahashi M, Higuchi T. Subthalamic locomotor region is involved in running activity originating in the rat ventromedial hypothalamus. Behav Brain Res. 2002;134:275–281. doi: 10.1016/s0166-4328(02)00041-4. [DOI] [PubMed] [Google Scholar]
- Patapoutian A, Reichardt LF. Trk receptors: mediators of neurotrophin action. Curr Opin Neurobiol. 2001;11:272–280. doi: 10.1016/s0959-4388(00)00208-7. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. Ed 2. San Diego: Academic; 2001. [Google Scholar]
- Penicaud L, Larue-Achagiotis C, Le Magnen J. Endocrine basis for weight gain after fasting or VMH lesion in rats. Am J Physiol. 1983;245:E246–E252. doi: 10.1152/ajpendo.1983.245.3.E246. [DOI] [PubMed] [Google Scholar]
- Perkins MN, Rothwell NJ, Stock MJ, Stone TW. Activation of brown adipose tissue thermogenesis by the ventromedial hypothalamus. Nature. 1981;289:401–402. doi: 10.1038/289401a0. [DOI] [PubMed] [Google Scholar]
- Pinto S, Roseberry AG, Liu H, Diano S, Shanabrough M, Cai X, Friedman JM, Horvath TL. Rapid rewiring of arcuate nucleus feeding circuits by leptin. Science. 2004;304:110–115. doi: 10.1126/science.1089459. [DOI] [PubMed] [Google Scholar]
- Rios M, Fan G, Fekete C, Kelly J, Bates B, Kuehn R, Lechan RM, Jaenisch R. Conditional deletion of brain-derived neurotrophic factor in the postnatal brain leads to obesity and hyperactivity. Mol Endocrinol. 2001;15:1748–1757. doi: 10.1210/mend.15.10.0706. [DOI] [PubMed] [Google Scholar]
- Rodier PM. Chronology of neuron development: animal studies and their clinical implications. Dev Med Child Neurol. 1980;22:525–545. doi: 10.1111/j.1469-8749.1980.tb04363.x. [DOI] [PubMed] [Google Scholar]
- Sahu A. Evidence suggesting that galanin (GAL), melanin-concentrating hormone (MCH), neurotensin (NT), proopiomelanocortin (POMC) and neuropeptide Y (NPY) are targets of leptin signaling in the hypothalamus. Endocrinology. 1998;139:795–798. doi: 10.1210/endo.139.2.5909. [DOI] [PubMed] [Google Scholar]
- Shimada M, Nakamura T. Time of neuron origin in mouse hypothalamic nuclei. Exp Neurol. 1973;41:163–173. doi: 10.1016/0014-4886(73)90187-8. [DOI] [PubMed] [Google Scholar]
- Sims JS, Lorden JF. Effect of paraventricular nucleus lesions on body weight, food intake and insulin levels. Behav Brain Res. 1986;22:265–281. doi: 10.1016/0166-4328(86)90071-9. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain [letter] Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Sternson SM, Shepherd GM, Friedman JM. Topographic mapping of VMH→arcuate nucleus microcircuits and their reorganization by fasting. Nat Neurosci. 2005;8:1356–1363. doi: 10.1038/nn1550. [DOI] [PubMed] [Google Scholar]
- Steru L, Chermat R, Thierry B, Simon P. The tail suspension test: a new method for screening antidepressants in mice. Psychopharmacology (Berl) 1985;85:367–370. doi: 10.1007/BF00428203. [DOI] [PubMed] [Google Scholar]
- Storlien LH, Bellingham WP, Martin GM. Localization of CNS glucoregulatory insulin receptors within the ventromedial hypothalamus. Brain Res. 1975;96:156–160. doi: 10.1016/0006-8993(75)90590-9. [DOI] [PubMed] [Google Scholar]
- Sugiyama N, Kanba S, Arita J. Temporal changes in the expression of brain-derived neurotrophic factor mRNA in the ventromedial nucleus of the hypothalamus of the developing rat brain. Brain Res Mol Brain Res. 2003;115:69–77. doi: 10.1016/s0169-328x(03)00184-0. [DOI] [PubMed] [Google Scholar]
- Swanson LW, Sawchenko PE. Hypothalamic integration: organization of the paraventricular and supraoptic nuclei. Annu Rev Neurosci. 1983;6:269–324. doi: 10.1146/annurev.ne.06.030183.001413. [DOI] [PubMed] [Google Scholar]
- Takahashi A, Shimazu T. Hypothalamic regulation of lipid metabolism in the rat: effect of hypothalamic stimulation on lipogenesis. J Auton Nerv Syst. 1982;6:225–235. doi: 10.1016/0165-1838(82)90053-4. [DOI] [PubMed] [Google Scholar]
- Tong Q, Ye C, McCrimmon RJ, Dhillon H, Choi B, Kramer MD, Yu J, Yang Z, Christiansen LM, Lee CE, Choi CS, Zigman JM, Shulman GI, Sherwin RS, Elmquist JK, Lowell BB. Synaptic glutamate release by ventromedial hypothalamic neurons is part of the neurocircuitry that prevents hypoglycemia. Cell Metab. 2007;5:383–393. doi: 10.1016/j.cmet.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran PV, Lee MB, Marin O, Xu B, Jones KR, Reichardt LF, Rubenstein JR, Ingraham HA. Requirement of the orphan nuclear receptor SF-1 in terminal differentiation of ventromedial hypothalamic neurons. Mol Cell Neurosci. 2003;22:441–453. doi: 10.1016/S1044-7431(03)00027-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran PV, Akana SF, Malkovska I, Dallman MF, Parada LF, Ingraham HA. Diminished hypothalamic bdnf expression and impaired VMH function are associated with reduced SF-1 gene dosage. J Comp Neurol. 2006;498:637–648. doi: 10.1002/cne.21070. [DOI] [PubMed] [Google Scholar]
- Trivedi P, Yu H, MacNeil DJ, Van der Ploeg LH, Guan XM. Distribution of orexin receptor mRNA in the rat brain. FEBS Lett. 1998;438:71–75. doi: 10.1016/s0014-5793(98)01266-6. [DOI] [PubMed] [Google Scholar]
- Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci. 2006;9:519–525. doi: 10.1038/nn1659. [DOI] [PubMed] [Google Scholar]
- Wang C, Bomberg E, Billington C, Levine A, Kotz CM. Brain-derived neurotrophic factor in the hypothalamic paraventricular nucleus increases energy expenditure by elevating metabolic rate. Am J Physiol Regul Integr Comp Physiol. 2007a;293:R992–R1002. doi: 10.1152/ajpregu.00516.2006. [DOI] [PubMed] [Google Scholar]
- Wang C, Bomberg E, Billington C, Levine A, Kotz CM. Brain-derived neurotrophic factor in the hypothalamic paraventricular nucleus reduces energy intake. Am J Physiol Regul Integr Comp Physiol. 2007b;293:R1003–R1012. doi: 10.1152/ajpregu.00011.2007. [DOI] [PubMed] [Google Scholar]
- Wang C, Bomberg E, Levine A, Billington C, Kotz CM. Brain-derived neurotrophic factor in the ventromedial nucleus of the hypothalamus reduces energy intake. Am J Physiol Regul Integr Comp Physiol. 2007c;293:R1037–R1045. doi: 10.1152/ajpregu.00125.2007. [DOI] [PubMed] [Google Scholar]
- Winslow JT, Miczek KA. Habituation of aggression in mice: pharmacological evidence of catecholaminergic and serotonergic mediation. Psychopharmacology (Berl) 1983;81:286–291. doi: 10.1007/BF00427564. [DOI] [PubMed] [Google Scholar]
- Xu B, Gottschalk W, Chow A, Wilson RI, Schnell E, Zang K, Wang D, Nicoll RA, Lu B, Reichardt LF. The role of brain-derived neurotrophic factor receptors in the mature hippocampus: modulation of long-term potentiation through a presynaptic mechanism involving TrkB. J Neurosci. 2000;20:6888–6897. doi: 10.1523/JNEUROSCI.20-18-06888.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Goulding EH, Zang K, Cepoi D, Cone RD, Jones KR, Tecott LH, Reichardt LF. Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat Neurosci. 2003;6:736–742. doi: 10.1038/nn1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Q, Radeke MJ, Matheson CR, Talvenheimo J, Welcher AA, Feinstein SC. Immunocytochemical localization of TrkB in the central nervous system of the adult rat. J Comp Neurol. 1997;378:135–157. [PubMed] [Google Scholar]
- Yeo GS, Connie Hung CC, Rochford J, Keogh J, Gray J, Sivaramakrishnan S, O'Rahilly S, Farooqi IS. A de novo mutation affecting human TrkB associated with severe obesity and developmental delay. Nat Neurosci. 2004;7:1187–1189. doi: 10.1038/nn1336. [DOI] [PubMed] [Google Scholar]
- Zaretskaia MV, Zaretsky DV, Shekhar A, DiMicco JA. Chemical stimulation of the dorsomedial hypothalamus evokes non-shivering thermogenesis in anesthetized rats. Brain Res. 2002;928:113–125. doi: 10.1016/s0006-8993(01)03369-8. [DOI] [PubMed] [Google Scholar]