

Abstract
Many patients with traumatic spinal cord injury (SCI) report pain that persists indefinitely and is resistant to available therapeutic approaches. We recently showed that microglia become activated after experimental SCI and dynamically maintain hyperresponsiveness of spinal cord nociceptive neurons and pain-related behaviors. Mechanisms of signaling between microglia and neurons that help to maintain abnormal pain processing are unknown. In this study, adult male Sprague Dawley rats underwent T9 spinal cord contusion injury. Four weeks after injury when lumbar dorsal horn multireceptive neurons became hyperresponsive and when behavioral nociceptive thresholds to mechanical and thermal stimuli were decreased, we tested the hypothesis that prostaglandin E2 (PGE2) contributes to signaling between microglia and neurons. Immunohistochemical data showed specific localization of phosphorylated extracellular signal-regulated kinase 1/2 (pERK1/2), an upstream regulator of PGE2 release, to microglial cells and a neuronal localization of the PGE2 receptor E-prostanoid 2 (EP2). Enzyme immunoassay analysis showed that PGE2 release was dependent on microglial activation and ERK1/2 phosphorylation. Pharmacological antagonism of PGE2 release was achieved with the mitogen-activated protein kinase kinase 1/2 (MEK1/2) inhibitor PD98059 [2-(2-amino-3-methyoxyphenyl)-4H-1-benzopyran-4-one] and the microglial inhibitor minocycline. Cyclooxygenase-2 expression in microglia was similarly reduced by MEK1/2 inhibition. PD98059 and EP2 receptor blockade with AH6809 (6-isopropoxy-9-oxoxanthene-2-carboxylic acid) resulted in a decrease in hyperresponsiveness of dorsal horn neurons and partial restoration of behavioral nociceptive thresholds. Selective targeting of dorsal horn microglia with the Mac-1–synapse-associated protein (SAP) immunotoxin resulted in reduced microglia staining, reduction in PGE2 levels, and reversed pain-related behaviors. On the basis of these observations, we propose a PGE2-dependent, ERK1/2-regulated microglia–neuron signaling pathway that mediates the microglial component of pain maintenance after injury to the spinal cord.
Keywords: microglia, PGE2, dorsal horn, pain, spinal cord injury, hypersensitivity
Introduction
Spinal cord injury (SCI) is a devastating event that can result in the development of chronic pain as well as paralysis. Sixty to 80% of persons who have sustained SCI experience clinically significant pain after injury (Finnerup et al., 2001; Siddall et al., 2003); post-SCI pain can increase with time after injury and is often refractory to treatment (Rintala et al., 1998).
Pain after SCI has traditionally been thought to arise from the dysfunction of neurons along the pain-signaling pathway. Neuroimmune alterations have been implicated in the initiation of peripheral injury-induced pain (Watkins et al., 2001; DeLeo et al., 2006). Activation of spinal microglia after peripheral injury can be induced by nerve ligation (Coyle, 1998; Jin et al., 2003), Formalin injection (Fu et al., 1999), and sciatic inflammation (Ledeboer et al., 2005). Resident microglial cells also become activated in the spinal cord after contusive SCI (Popovich et al., 1997; Hains et al., 2003b; Sroga et al., 2003; Nesic et al., 2005; Zai and Wrathall, 2005; Crown et al., 2006). We recently demonstrated a role for microglia in the maintenance of post-SCI chronic pain; microglia in the lumbar dorsal horn dramatically and dynamically maintain, in real time, hyperresponsiveness of pain-processing neurons and pain-related behaviors (Hains and Waxman, 2006).
The signaling mechanisms between activated microglia and neurons that underlie enhanced postsynaptic excitability and changes in sensory processing after SCI have not yet been established. Microglia are known to produce a number of neuroactive substances and cytokines (McMahon et al., 2005) that can influence the excitability of neurons, including interleukin-1β (IL-1β) (Ferrari et al., 1997), BDNF (Coull et al., 2005), and prostaglandin E2 (PGE2) (Akundi et al., 2005; Ikeda-Matsuo et al., 2005; Inoue, 2006). PGE2 in particular has been implicated in the induction of central sensitization of spinal neurons (Minami et al., 1999; Ji et al., 2003), and extracellular signal-regulated kinase 1/2 (ERK1/2), an upstream effector of PGE2 biosynthesis, is activated in stimulated microglia (Akundi et al., 2005).
Given evidence that inhibition of PGE2 release can reduce behavioral signs of pain after SCI (Hains et al., 2001) and peripheral nerve injury (McMahon et al., 2005), we tested the hypothesis that activated microglia modulate the activity of dorsal horn sensory neurons through a PGE2 signaling mechanism and that PGE2 synthesis is regulated by an ERK1/2-dependent mechanism after SCI. Here, we identify a putative microglia–neuron signaling pathway involving PGE2 that contributes to chronic pain after SCI and show that microglial PGE2 release is dependent on activation of an upstream ERK1/2 mitogen-activated protein kinase (MAPK) cascade.
Materials and Methods
Animal care.
Experiments were performed in accordance with National Institutes of Health guidelines for the care and use of laboratory animals; all animal protocols were approved by the Yale University Institutional Animal Use Committee. Adult male Sprague Dawley rats (200–225 g) were used for this study. Animals were housed under a 12 h light/dark cycle in a pathogen-free area with access to water and food ad libitum.
Surgical groups.
Rats were deeply anesthetized with ketamine/xylazine (80 and 5 mg/kg, i.p., respectively). SCI was produced (n = 53 rats) at spinal segment T9 using the Multicenter Animal Spinal Cord Injury Study/New York University impact injury device (Gruner, 1992). A 10 g, 2.0 mm diameter rod was released from a 25 mm height onto the exposed spinal cord. For sham surgery, animals (“intact”; n = 24) underwent laminectomy and placement into the vertebral clips of the impactor without impact injury. After SCI or sham surgery, the overlying muscles and skin were closed in layers with 4-0 silk sutures and staples, respectively, and the animal was allowed to recover on a 30°C heating pad. Postoperative treatments included saline (2.0 cc, s.c.) for rehydration and Baytril (0.3 cc, 22.7 mg/ml, s.c., twice daily) to prevent urinary tract infection. Bladders were manually expressed twice daily until reflex bladder emptying returned, typically by 10 d after injury. After surgery, animals were maintained under the same preoperative conditions and fed ad libitum.
A second group of animals was used to show that abnormal activation of phosphorylated p38 (P-p38) and/or phosphorylated ERK1/2 (pERK1/2) in dorsal root ganglion neurons, caused by SCI, could drive increased dorsal horn levels of PGE2. In these animals (n = 6), the right sciatic nerve was exposed at midthigh level, ligated with 4-0 silk sutures, transected, and placed in a silicon cuff to prevent regeneration. To clearly identify transected neurons, a retrogradely transported fluorescent label (hydroxystilbamine methanesulfonate, 4% w/v; Invitrogen, Carlsbad, CA) was placed in the cuff before stump insertion (Waxman et al., 1994). These animals did not receive intrathecal catheters or drugs and were used for immunohistochemical experiments only.
Intrathecal catheterization and drug delivery.
Twenty-eight days after SCI or sham surgery, under ketamine/xylazine (80 and 5 mg/kg, i.p., respectively) anesthesia, a sterile premeasured 32 gauge intrathecal catheter (ReCathCo, Allison Park, PA) was introduced through a slit in the atlanto-occipital membrane, threaded down to the lumbar enlargement, secured to the neck musculature with suture, and heat sealed to prevent infection and leakage of CSF. The catheters easily slid past the SCI impact site (T9) in all animals. Verification of the lumbar location of the terminal end of the catheter was done at the time the animals were killed by injecting methylene blue dye through the catheter.
Three days after catheter placement, under brief (<1 min) halothane sedation (3% by facial mask), intrathecal infusion of artificial CSF (aCSF) vehicle (n = 4), Mac-1–synapse-associated protein (SAP) (n = 6; 36 μg), minocycline (n = 4; 100 μg), PD98059 [2-(2-amino-3-methyoxyphenyl)-4H-1-benzopyran-4-one] (n = 13; 10 μg), or AH6809 (6-isopropoxy-9-oxoxanthene-2-carboxylic acid) (n = 12; 166 μg) began in SCI animals. Intact animals received fractalkine (n = 4; 30 ng) at the same time points. Mac-1–SAP, a chemical conjugate of mouse monoclonal antibody to CD11b and the ribosome-inactivating protein saporin (Advanced Targeting Systems, San Diego, CA), was used to selectively kill microglia in the dorsal horn (Dommergues et al., 2003). The tetracycline antibiotic minocycline has been shown to potently downregulate the activity of microglia in vivo (Raghavendra et al., 2003; Hua et al., 2005; Ledeboer et al., 2005). 7-Dimethylamino-6-demethyl-6-deoxytetracycline [minocycline hydrochloride; molecular weight (MW) of 493.9; Sigma, St. Louis, MO] was used based on previous reports (Ledeboer et al., 2005; Hains and Waxman, 2006). Recombinant rat CX3CL1/fractalkine (chemokine domain; amino acids 25–100; MW of 8.8 kDa; R & D Systems, Minneapolis, MN), a chemokine that induces glial activation (Harrison et al., 1998; Milligan et al., 2005), was used based on the literature (Milligan et al., 2004) and pilot studies. PD98059 has been shown to selectively target the upstream ERK kinase mitogen-activated protein kinase kinase 1/2 (MEK1/2); because ERK1 and ERK2 are the only known substrates of MEK, MEK1/2 inhibition by PD98059 results in selective inhibition of ERK phosphorylation and thus activation (Alessi et al., 1995; Dudley et al., 1995; Zhuang et al., 2005). PD98059 (MW of 267.28; Sigma) was used based on previous reports (Zhuang et al., 2005) and preliminary experiments. AH6809 (MW of 298.29; Sigma), a stable synthetic antagonist of PGE2 that blocks the activity of the E-prostanoid 2 (EP2) receptor (but not EP1) in rats (Woodward et al., 1995), was used based on previous reports (Haupt et al., 2000) and preliminary experiments. For 3 d, injections were performed twice daily in 5 μl of aCSF (in mm: 1.3 CaCl2·2H2O, 2.6 KCl, 0.9 MgCl, 21.0 NaHCO3, 2.5 Na2HPO4·7H2O, and 125.0 NaCl, prepared in sterile H2O), followed by 10 μl of aCSF flush. As a vehicle control, aCSF was injected during the same time points in separate animals.
Immunohistochemistry.
Tissue was collected from the lumbar enlargement (L4 spinal segment) and dorsal root ganglia (DRG) (L4–L5) from animals that had received SCI 31 d earlier (n = 6). Lumbar DRG tissue was also collected from animals that had undergone unilateral sciatic nerve ligation and transection 10 d earlier (n = 6). Rats were deeply anesthetized with ketamine/xylazine (80 mg and 5 kg, i.p., respectively) and perfused intracardially with 0.01 m PBS, followed by 4% cold, buffered paraformaldehyde. Tissue was postfixed for 15 min in 4% paraformaldehyde and cryopreserved overnight at 4°C in 30% sucrose PBS. Thin (8 μm) cryosections (n = 6 sections per animal) from each treatment group were processed simultaneously.
Slides were incubated at room temperature in the following: (1) blocking solution (PBS containing 5% NGS, 2% BSA, 0.1% Triton X-100, and 0.02% sodium azide) for 30 min; (2) primary antibody: mouse anti-CD11b/c OX-42 clone raised against complement receptor 3 (1:250; BD Biosciences, San Jose, CA), mouse anti-neuron-specific nuclear protein NeuN (1:500; Chemicon, Temecula, CA), rabbit anti-pERK1/2 MAP kinase (1:500; Cell Signaling Technology, Danvers, MA), rabbit anti-PGE2 receptor EP2 (1:1000; Chemicon), rabbit anti-P-p38 (1:50; Cell Signaling Technology), rabbit anti-cyclooxygenase-2 (COX-2) (1:500; Santa Cruz Biotechnology, Santa Cruz, CA), or mouse anti-GFAP (1:500; Chemicon), overnight in blocking solution at 4°C; (3) PBS, six times for 5 min each; (4) either goat anti-rabbit Alexa 546 (1:1500; Invitrogen) or donkey anti-mouse 488 (1:1500; Invitrogen), in blocking solution, 2 h; and (5) PBS, six times for 5 min each. Control experiments were performed without primary or secondary antibodies that yielded only background levels of signal.
Quantitative image analysis.
Images were captured with a Nikon (Tokyo, Japan) Eclipse E800 light microscope equipped with epifluorescence and Nomarski optics, using a Photometrics CoolSnap HQ camera (Roper Scientific, Tucson, AZ) and MetaVue version 6.2r6 software (Universal Imaging Corporation, Downingtown, PA). Quantitative analysis was performed by a blinded observer using MetaVue and IPLab Spectrum version 3.0 software (Scanalytics, Fairfax, VA). Signal intensity in DRG neurons (<50 μm diameter) or activated microglia (n = 26–38 per group) was determined by manually tracing the outline of the cell and allowing the software to compute signal intensity. Percentage of field analysis was used to provide a quantitative estimate (proportional area) of changes in the activation state of glial cells in the spinal cord and coexpression of pERK1/2 and Cd11b/c (Popovich et al., 1997; Hains and Waxman, 2006; Kigerl et al., 2006). Resting and activated astroglia and microglia were classified based on the following criteria. resting glia displayed small compact somata bearing long thin ramified processes. Activated glia exhibited marked cellular hypertrophy and retraction of processes such that the process length was less than the diameter of the soma compartment. Measurement of signal colocalization was performed with MetaMorph (version 7.0, Molecular Devices, Sunnyvale, CA). Cells were sampled only if the nucleus was visible within the plane of section and if cell profiles exhibited distinctly delineated borders. Background levels of signal were subtracted, and control and experimental conditions were evaluated in identical manners.
Tissue PGE2 determination.
Tissue levels of PGE2 in the spinal cord dorsal horn were assayed using enzyme immunoassay (EIA) (Prostaglandin E2 EIA kit, Monoclonal; Cayman Chemical, Ann Arbor, MI), from the following groups: intact (n = 4), intact + fractalkine (n = 4), SCI (n = 4), SCI + Mac-1–SAP (n = 3), SCI + minocycline (n = 4), SCI + PD98059 (n = 5), and SCI + AH6809 (n = 4). Drug groups received intrathecal injections of fractalkine, minocycline, PD98059, or AH6809 for 3 d as described above. Animals were anesthetized with an overdose of pentobarbital (75 mg/kg, i.p.) and decapitated. The lumbar spinal cord (L4–L5) was taken out within 30–60 s after decapitation, and the dorsal horn was rapidly microdissected from the ventral horn, weighed, and flash frozen in liquid nitrogen for storage at −80°C. Tissue was homogenized in ice-cold lysis buffer [0.1 m phosphate, pH 7.4, 1 mm EDTA, 10 μm indomethacin (Cayman Chemical)] using a tube pestle. Acetone was added (2× sample volume), and samples were centrifuged at 1500 × g for 10 min. The supernatants were then stored at −80°C. Samples were run in triplicate based on supplied instructions. The PGE2 monoclonal EIA kit demonstrates sensitivity from 10 to 1000 pg/ml and demonstrates little cross reactivity between structurally related PGE3 and PGE1. Absorbance (412 nm) values of standards and samples were corrected by subtraction of the background value to correct for absorbance caused by nonspecific binding.
Electrophysiologic procedures.
Extracellular unit recordings were obtained from dorsal horn sensory neurons 30 d after SCI. Acute spinal drug delivery was performed by soaking drug solutions onto pledgets (2 mm2), which were placed centered on the dorsal surface of the spinal segment in which cells were isolated, covering both ipsilateral and contralateral dorsal horns (Qin et al., 1999; Hains et al., 2003a). Working doses of PD98059 and AH6809 were determined by behavioral experiments. Both drugs were dissolved in 5 μl of aCSF, pH 7.4. Mineral oil was drawn off and replaced before and immediately after pledget application. Start time of recordings were based on predetermined onset and offset efficacy evaluations. aCSF vehicle control pledgets were applied in the same manner before or after drug application to ensure continuity of response. Unit responses were recorded after drug washout (30–60 min) to assess recovery of hyperresponsiveness.
Animals underwent extracellular single-unit recording according to established methods (Hains et al., 2003a,b). The activity of three to seven units per animal (n = 4 per group from SCI + VEH, SCI + PD98059, or SCI + AH6809) were recorded for each experiment, yielding 12–28 cells per group. The experimenter was blinded to drug treatment for all animals. Rats were initially anesthetized with sodium pentobarbital (40 mg/kg, i.p.) and supplemented (5 mg · kg−1 · h−1) intravenously through a catheter in the jugular vein. Rectal temperature was maintained at 37°C by a thermostatically controlled heating blanket. A T12–L6 laminectomy was done before fixing the head and the vertebral column on a stereotaxic apparatus (David Kopf Instruments, Tujunga, CA). The exposed spinal cord was covered with warm (37°C) mineral oil. Units were isolated from L3–L5 medially near the dorsal root entry zone up to a depth of 1000 μm. Recordings were made with a low-impedance 5 MΩ tungsten-insulated microelectrode (A-M Systems, Carlsborg, WA). Electrical signals were amplified and filtered at 300–3000 Hz (DAM80; World Precision Instruments, Sarasota, FL), processed by a data collection system (CED 1401+; Cambridge Electronics Design, Cambridge, UK), and stored on a computer (Latitude D800; Dell Computer Company, Austin, TX). The stored digital record of individual unit activity was retrieved and analyzed off-line with Spike2 software (version 5.03, Cambridge Electronics Design).
After a cell was identified and its receptive field was mapped, natural stimuli were applied: (1) phasic brush (PB) stimulation of the skin with a cotton applicator, (2) stimulation with calibrated von Frey filaments of increasing force (0.39, 1.01, and 20.8 g), (3) compressive pressure, by attaching a large arterial clip with a weak grip to a fold of the skin (144 g/mm2), and (4) compressive pinch, by applying a small arterial clip with a strong grip to a fold of skin (583 g/mm2). Multireceptive (MR) neurons were identified by their relative magnitude of responsiveness to all stimuli. Because functional phenotype shifts can occur after SCI, such that more units assume a multireceptive functional classification, our search paradigm ensured that in all groups we sampled multireceptive units. Stimulation was applied with the experimenter blinded to the output of the cell during stimulation. Background activity was recorded for 20 s, and stimuli were applied serially for 20 s, separated by another 20 s of spontaneous activity without stimulation. Care was taken to ensure that the responses were maximal, that each stimulus was applied to the primary receptive field of the cell, and that isolated units remained intact for the duration of each experiment using Spike2 template-matching routines. Based on previously published statistical analysis of evoked discharge rates in intact control and SCI animals (Christensen and Hulsebosch, 1997; Hains et al., 2003a,b,c), neurons were considered to be hyperresponsive if evoked discharge rates were >150% of control levels.
Behavioral testing.
All behavioral testing was performed by a blinded observer. Testing began on day 28 after SCI to confirm that SCI animals had developed chronic pain-related behaviors (for all experiments, we used only animals that demonstrated the development of chronic pain) before intrathecal drug administration of aCSF vehicle, Mac-1–SAP, PD98059, or AH6809. Daily testing resumed after intrathecal catheterization on day 30 (n = 8 animals per group). Drugs were administered from days 31 to 33, followed by 2 additional days of testing (until day 35). On the first day of behavioral testing after SCI, motor performance of rats with SCI recovered well enough to yield reliable withdrawal reflex measures, as shown in previous studies (Hains et al., 2001).
Locomotor function was recorded using the Basso, Beattie, and Bresnahan (BBB) rating scale (Basso et al., 1995) to ensure reliability of hindlimb somatosensory testing, as well as to assess the motor effects of delivered compounds. Briefly, the BBB is a 21-point ordinal scale ranging from 0 (no discernable hindlimb movement) to 21 (consistent and coordinated gait with parallel paw placement of the hindlimb and consistent trunk stability). Scores from 0 to 7 rank the early phase of recovery with return of isolated movements of three joints (hip, knee, and ankle); scores from 8 to 13 describe the intermediate recovery phase with return of paw placement, stepping, and forelimb–hindlimb coordination; and scores from 14 to 21 rank the late phase of recovery with return of toe clearance during the step phase, predominant paw position, trunk stability, and tail position.
Mechanical nociceptive thresholds were determined by paw withdrawal to application of a series of calibrated von Frey filaments (Stoelting, Wood Dale, IL) to the glabrous surface of the hindpaws. Before testing, animals were acclimatized to the testing area for 30 min. After application of von Frey filaments (0.4–26 g) with enough force to cause buckling of the filament, a modification of the up–down method of Dixon (1980) was used to determine the value at which paw withdrawal occurred 50% of the time (Chaplan et al., 1994), interpreted to be the mechanical nociceptive threshold.
After acclimation to the test chamber, thermal nociceptive thresholds were assessed by measuring the latency of paw withdrawal in response to a radiant heat source (Dirig et al., 1997). Animals were placed in Plexiglas boxes on an elevated glass plate (37°C) under which a radiant heat source (5.14 A) was applied to the glabrous surface of the paw through the glass plate. The heat source was turned off automatically by a photocell during limb lift, allowing the measurement of paw-withdrawal latency. If no response was detected, the heat source was automatically shut off at 20 s. Three minutes were allowed between each trial, and four trials were averaged for each limb. Although not providing direct measures of the experience of pain by experimental animals, these methods provide measures of pain-related behaviors that coincide with anatomical and physiological observations.
Statistical analysis.
All statistical tests were performed at the α level of significance of 0.05 by two-tailed analyses using parametric tests. Data were tested for significance using one-way ANOVA, followed by Bonferroni's post hoc analysis. Tests of factors including pairwise comparisons were applied with either the paired Student's t test or the two-sample Student's t test. Linear regression analysis was performed on pERK1/2 and Cd11b/c signal intensities. Data management and statistical analyses were performed using SAS (1992) statistical procedures with Jandel SigmaStat (version 1.0; SPSS, Chicago, IL) and graphed using Jandel SigmaPlot (version 7.0; SPSS) as mean ± SD.
Results
pERK1/2 and EP2 localization
Imaging experiments were performed to identify the cellular localization of phosphorylated ERK1/2 MAP kinase (pERK1/2) and the PGE2 receptor EP2 in the lumbar dorsal horn after SCI. Within the dorsal horn, NeuN, a marker of neurons, is present in cells that exhibit a typical neuronal morphology and size (Fig. 1A). pERK1/2 was observed in the intact spinal cord (data not shown), but signal levels were very low, as were the number of pERK1/2-positive cells. After SCI, there was a marked increase in pERK1/2 in all laminas within the lumbar dorsal horn. pERK1/2 signal was observed in both white and gray matter after SCI. The distribution was uniform throughout the dorsal horn gray matter. pERK1/2-positive cells demonstrated round nuclei and compact processes (Fig. 1B). NeuN and pERK1/2 showed very little colocalization (Fig. 1C), which is confirmed in higher-magnification panels. Antibodies against Cd11b/c (OX-42) revealed the presence of microglia in both white and gray matter of the spinal cord after SCI (Fig. 1D), which exhibited an activated morphology: marked cellular hypertrophy and retraction of cytoplasmic processes. pERK1/2 staining was robust (Fig. 1E) and tightly colocalized with Cd11b/c signal in these cells (Fig. 1F). Percentage of field analysis showed a 3.1 ± 0.8% colocalization between NeuN and pERK1/2 and 85.6 ± 6.7% colocalization between Cd11b/c and pERK1/2, indicating that pERK1/2 is almost exclusively located within activated microglia after SCI.
Figure 1.

Identification of the cellular location of pERK1/2 after SCI. A, Thirty-one days after T9 SCI, NeuN immunostaining revealed typically distributed neuronal morphologies in the spinal cord lumbar dorsal horn. B, pERK1/2 signal was also present after SCI. pERK1/2-positive cells demonstrated round nuclei and compact processes. C, NeuN and pERK1/2 showed very little colocalization, which is confirmed in higher-magnification panels. D, Cd11b/c-positive cells exhibited morphological features of activated microglia: cell bodies were small, and several slender branched processes emerged from the soma. E, F, pERK1/2 (E) was strongly colocalized to Cd11b/c (F). Percentage of field analysis showed that pERK1/2 is almost exclusively located within activated microglia after SCI.
Figure 2 shows NeuN (Fig. 2A) and PGE2 EP2 receptor localization in the lumbar dorsal horn after SCI. EP2 signal was strong and was confined to cells exhibiting morphological characteristics consistent with neurons: larger rounded cell bodies with few processes (Fig. 2B). Colocalization revealed strong correspondence between NeuN and EP2 signal (Fig. 2C). Cd11b/c signal showed the expected activation of microglia after SCI (Fig. 2D). EP2 (Fig. 2E) did not colocalize with activated microglia (Fig. 2F). Percentage of field analysis showed a 75.02 ± 4.2% colocalization between NeuN and EP2 but only 4.2 ± 1.3% colocalization between Cd11b/c and EP2, indicating that EP2 is preferentially expressed in neurons after SCI.
Figure 2.

Identification of the cellular localization of the PGE2 receptor EP2 after SCI. A, B, EP2 signal was strong (B) and was confined to cells exhibiting morphological characteristics consistent with neurons (A). C, Colocalization with NeuN revealed strong correspondence between NeuN and EP2 signal. D, Cd11b/c staining showed the expected activation of microglia after SCI. E, F, EP2 immunostaining (E) did not colocalize with activated microglia (F). Percentage of field analysis showed a strong colocalization between NeuN and EP2 but a weak correspondence between Cd11b/c and EP2, indicating that EP2 is preferentially expressed in neurons after SCI.
Analysis of the correspondence of the activation state of microglia and pERK1/2 in the spinal cord after injury is shown in Figure 3. Alteration in morphological features permit field area analysis of activated microglia and pERK1/2 signal in SCI animals (Fig. 3A). Proportional field area of pERK1/2 was plotted against field area of Cd11b/c, showing a strong relationship between increasing pERK1/2 signal and Cd11b/c signal after SCI (Fig. 3B). Regression analysis revealed a positive correlation between pERK1/2 and Cd11b/c (y = 1.38x; r2 = 0.64). Border plot histograms indicate a biphasic clustering of activated microglia and pERK1/2 signal. Data points were partitioned into two groups that likely represented inactivated and activated status.
Figure 3.
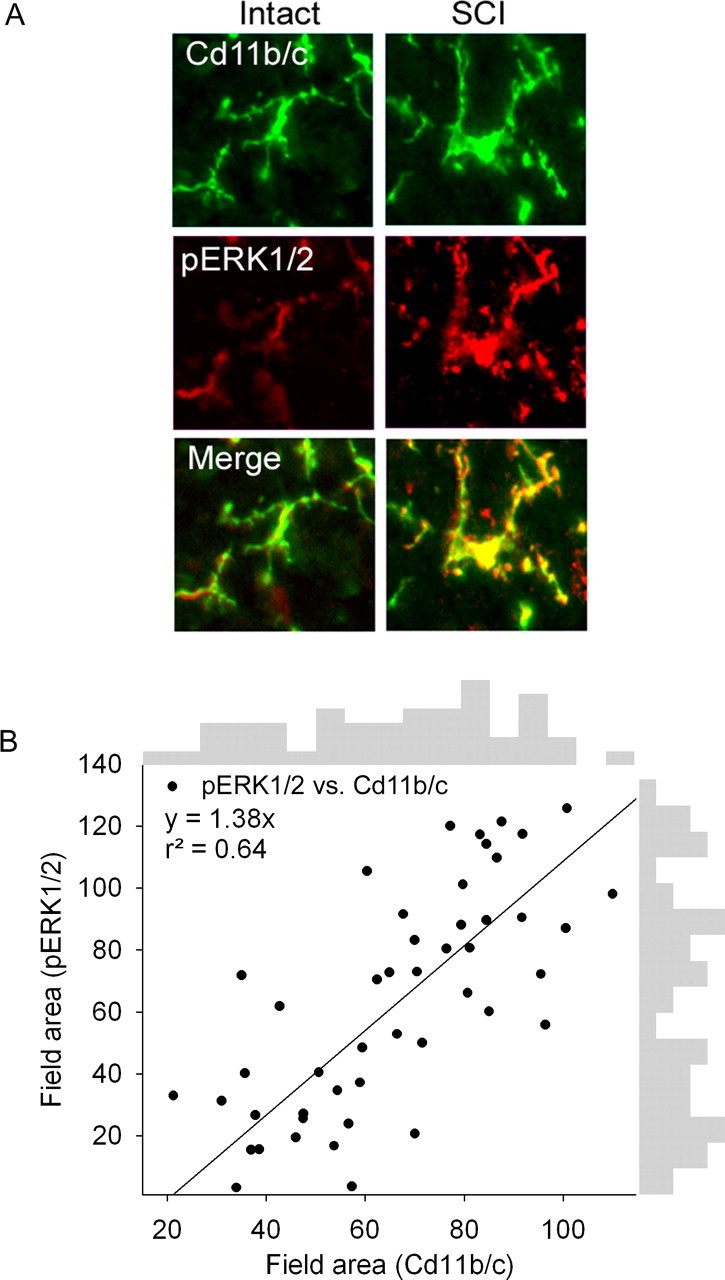
A, Immunostaining showing correspondence of the activation state of microglia (Cd11b/c field area) and pERK1/2 activation (field area) in intact animals and 4 weeks after SCI. B, Proportional field area of pERK1/2 was plotted against field area of Cd11b/c, showing a strong relationship between increasing pERK1/2 signal and Cd11b/c signal 31 d after SCI. Regression analysis revealed a positive correlation between pERK1/2 and Cd11b/c. Boarder plot histograms indicate a biphasic clustering of activated microglia and pERK1/2 signal. Data points were partitioned into two groups that likely represented inactivated and activated status.
To eliminate the possibility that agents used in our pharmacological agents acted on DRG neurons after SCI and to show that SCI does not induce changes in P-p38 or pERK1/2 activation in lumbar DRG neurons that might feedforward into the dorsal horn and increase PGE2 release, which could have secondary effects on dorsal horn neurons, we measured EP2, P-p38, and pERK1/2 levels in lumbar DRG neurons after SCI and after sciatic nerve axotomy and encapsulation (Fig. 4). EP2 signal was very low on both contralateral (17.9 ± 5.9 arbitrary units) (Fig. 4A) and backfilled fluorogold-positive (blue) ipsilateral (13.3 ± 4.6) (Fig. 4B) sides after axotomy. After SCI, EP2 signal was barely detectable in DRG neurons (12.0 ± 4.7) (Fig. 4C). P-p38 was present at low levels in contralateral DRG (50.7 ± 11.4) (Fig. 4D). After axotomy of the sciatic nerve, P-p38 levels were significantly (p < 0.05) increased in fluorogold-positive DRG neurons (108.5 ± 18.7) (Fig. 4E,K). After SCI, P-p38 levels were equivalent to those in uninjured DRG neurons (56.5 ± 13.1) (Fig. 4F). Levels of pERK1/2 were low in contralateral DRG neurons (41.5 ± 10.4) (Fig. 4G), whereas after axotomy there was a significant increase in pERK1/2 signal in fluorogold backfilled neurons (65.8 ± 12.4) (Fig. 4H,L). After SCI, pERK1/2 signal was low, equivalent to uninjured DRG neurons (Fig. 4I).
Figure 4.

EP2, P-p38, and pERK1/2 levels in lumbar DRG neurons after sciatic nerve axotomy or SCI. A, B, EP2 signal was very low on both contralateral (Cont) (A) and backfilled fluorogold-positive (FG, blue) ipsilateral (Ipsi) (B) sides 10 d after axotomy. C, Thirty-one days after SCI, EP2 signal was barely detectable in DRG neurons. D, P-p38 was present at low levels in contralateral DRG. After sciatic axotomy, P-p38 levels were significantly (K) increased in fluorogold-positive DRG neurons (E). F, After SCI, P-p38 levels were equivalent to those in uninjured DRG neurons. G, H, L, Levels of pERK1/2 were low in contralateral DRG neurons (G), whereas after axotomy there was a significant increase in pERK1/2 signal in fluorogold backfilled neurons (H, L). I, After SCI, pERK1/2 signal was low, equivalent to uninjured DRG neurons. J, Quantification revealed no significant differences between axotomy contralateral side (A-c), axotomy ipsilateral side (A-i), or SCI.
Immunocytochemistry for detection of GFAP, a marker for astroglia, in the lumbar enlargement revealed baseline expression in intact animals (Fig. 5A). GFAP-positive astroglia demonstrated typical slender processes and were distributed throughout both white and gray matter laminas. Twenty-eight days after SCI, astroglia possessed a swollen appearance consistent with activation and a significant increase in percentage field area of GFAP signal (Fig. 5F). As assessed on day 33, treatment of SCI animals with vehicle (Fig. 5B), Mac-1–SAP (Fig. 5C), PD98059 (Fig. 5D), or AH6809 (Fig. 5E) did not significantly reduce astroglial activation (Fig. 5F).
Figure 5.
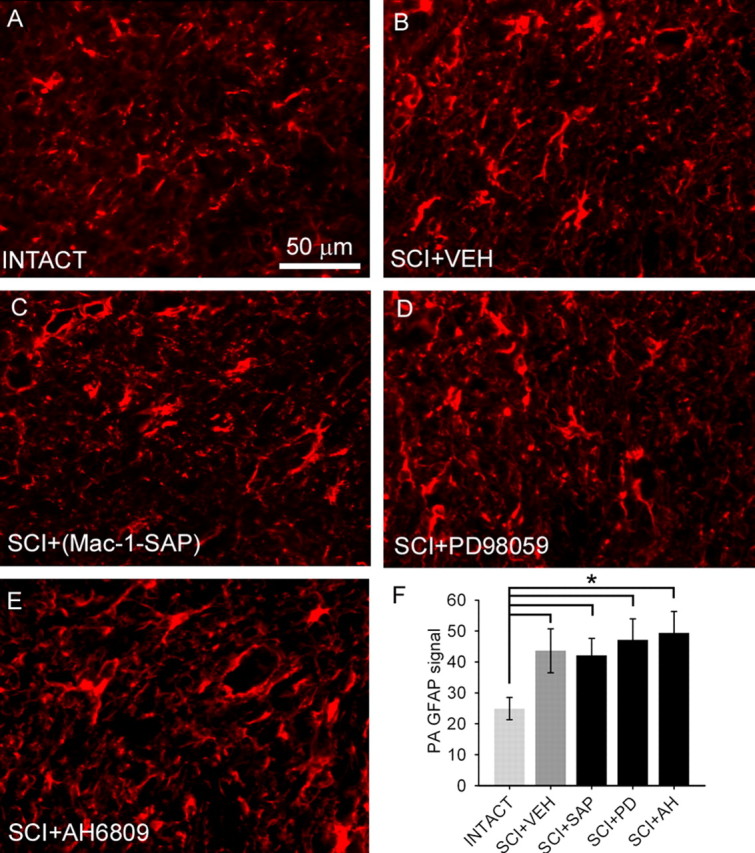
GFAP staining for normal and activated astroglia from the lumbar dorsal horn revealed the presence of resting astroglia in intact (A) animals. Four weeks after SCI and intrathecal administration of aCSF vehicle (VEH) (B), astroglia demonstrated an activated morphology. Administration of the microglial immunotoxin Mac-1–SAP (C), the ERK1/2 inhibitor PD98059 (D), or EP2 receptor antagonist AH6809 (E) did not have a significant effect on astroglial activation as measured by percentage of field analysis, which was significantly (*p < 0.05) elevated after SCI compared with intact animals (F).
Imaging of dorsal horn levels of microglia with the Cd11b/c antibody revealed robust presence of microglia throughout the dorsal horn, as we have shown previously (Fig. 6A). Microglial cells demonstrated cellular morphologies consistent with activation. Selective targeting of dorsal horn microglia with the Mac-1–SAP immunotoxin resulted in a dramatic decrease in the level of Cd11b/c signal after 3 d of treatment (Fig. 6C) when compared with animals administered vehicle for the same duration (Fig. 6B).
Figure 6.

A, Immunostaining for dorsal horn microglia after SCI revealed strong activation within the lumbar enlargement 4 weeks after SCI (A). B, C, Selective targeting of dorsal horn microglia with Mac-1–SAP resulted in a robust decrease in the amount of Cd11b/c signal after 3 d of treatment (C) when compared with animals administered vehicle (VEH) for the same duration (B).
Determination of PGE2 levels
In intact animals, PGE2 levels (314.2 ± 9.8 pg/mg tissue) in the lumbar dorsal horn were determined using an enzyme immunoassay (Fig. 7). In intact animals, intrathecal administration of the microglial activator fractalkine resulted in a significant (p < 0.05) increase in dorsal horn PGE2 levels (484.6 ± 13.8 pg/mg). Thirty days after T9 contusion SCI, PGE2 levels in the lumbar dorsal horn were significantly increased (476.8 ± 16.2 pg/mg) compared with intact animals. After SCI, the levels of PGE2 were not significantly different from those measured from intact animals given fractalkine. Elimination of dorsal horn microglia with Mac-1–SAP (353.9 ± 19.8 pg/mg) and inhibition of microglial activation with minocycline (350.4 ± 20.1 pg/mg) resulted in a significant reduction in PGE2 levels after SCI. Similarly, inhibition of downstream ERK1/2 activation with the MEK1/2 inhibitor PD98059 resulted in significantly lowered levels of PGE2 after SCI (392.9 ± 10.2 pg/mg). AH6809, the PGE2 EP2 receptor antagonist, had no effect on PGE2 levels after SCI (446.1 ± 13.5 pg/mg).
Figure 7.

Determination of PGE2 levels in the lumbar dorsal horn. In intact animals, intrathecal administration of the microglial activator fractalkine (INT+FKN) resulted in a significant increase (*p < 0.05) in dorsal horn PGE2 levels. Thirty days after T9 contusion SCI, PGE2 levels in the lumbar dorsal horn were significantly increased compared with intact animals. Elimination of microglia with Mac-1–SAP immunotoxin (SCI+SAP) and inhibition of microglial activation with minocycline (SCI+MINO) significantly reduced (+p < 0.05) PGE2 levels after SCI. Similarly, inhibition of downstream ERK1/2 activation with the MEK1/2 inhibitor PD98059 (SCI+PD) resulted in significantly lowered levels of PGE2 after SCI. AH6809, the PGE2 EP2 receptor antagonist, had no effect on PGE2 levels after SCI (SCI+AH).
Similarly, inhibition of pERK1/2 with PD98059 after SCI led to a reduction in COX-2 immunostaining in microglial cells (Fig. 8B), whereas with aCSF vehicle administration (Fig. 8A), COX-2 signal was strong in both microglia and neurons after SCI.
Figure 8.
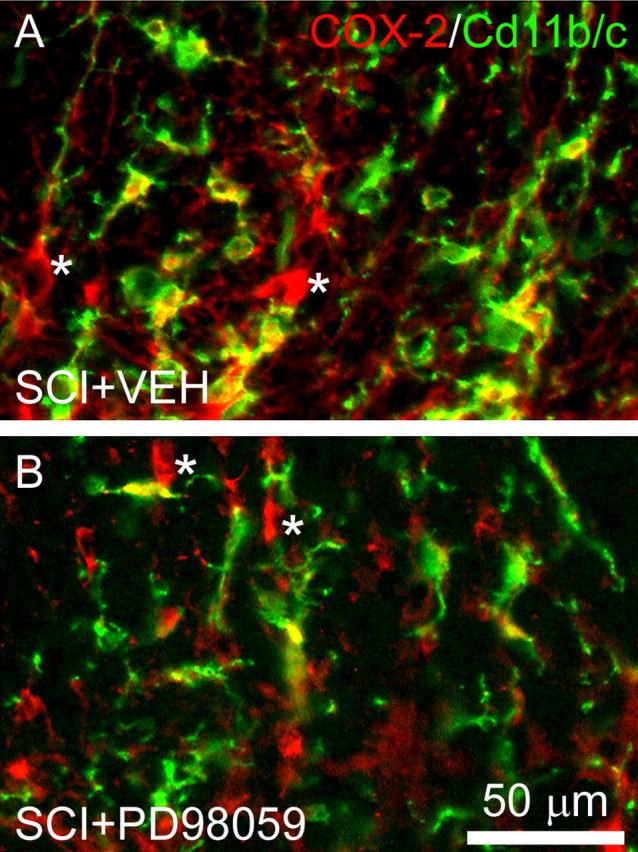
COX-2 immunostaining after SCI is reduced in microglia after administration of the MEK1/2 inhibitor PD98059. A, In animals receiving vehicle (VEH) injections after SCI, COX-2 signal colocalizes with activated microglia (green) and is observed in cells exhibiting a neuronal morphology (indicated by asterisk). B, After PD98059 treatment, however, COX-2 signal is reduced in microglia but persists in neurons.
Effects of microglial PGE2 on evoked unit activity
Dorsal horn multireceptive units were sampled in the lumbar enlargement in intact animals and after SCI and/or acute drug administration on day 30. Recordings from drug efficacy time course studies were also performed at this time point (data not shown).
SCI resulted in the development of evoked hyperresponsiveness and sensitization of dorsal horn neurons in the spinal cord lumbar enlargement consistent with previous observations. Unit discharge activity was elevated in response to application of natural stimuli to identified peripheral receptive fields (Fig. 9A). Evoked responses to phasic brush, increasing strength von Frey filaments, compressive press, and compressive pinch stimuli were all significantly (p < 0.05) increased (between 100 and 300%) when compared with intact animals (Fig. 9D). Peristimulus time histograms show that, after SCI, evoked discharge rates were between 35 and 90 Hz. Acutely administered PD98059 had no effect on spontaneous firing of MR units after SCI (3.2 ± 1.9 vs 4.1 ± 2.5 Hz). PD98059 did result in decreased evoked responses to all peripheral stimuli except the lowest intensity von Frey filament (0.39 g) (Fig. 9B). Evoked responses ranged from 10 to 50 Hz. Predrug unit responses (Fig. 9A) are superimposed on postdrug responses (Fig. 9B). Examples of unit activity of MR units in response to press before and after PD98059 administration are shown (Fig. 9C). Waveform overdraws ensure that the same unit was recorded in both instances, and its depth in the dorsal horn is shown. Quantification of the number of spikes per second revealed that inhibition of microglial ERK1/2 activation significantly reduced neuronal hyperresponsiveness after SCI to all stimuli except the 0.39 g von Frey stimulus compared with vehicle injections (Fig. 9D).
Figure 9.

Effects of inhibition of pERK1/2 on peripherally evoked activity of dorsal horn multireceptive units from SCI animals. A, C, Thirty days after SCI (A), significantly increased (+p < 0.05) evoked responses to natural stimuli (PB, 0.39 g, 1.01 g, 20.8 g, 144 g/mm2, and 583 g/mm2 refer to phasic brush, von Frey filaments of increasing intensities, pressure, and pinch, applied for 20 s) are observed when compared with intact animals (C, dotted line). After SCI, evoked discharge rates were between 35 and 90 Hz. Phasic brush stimulation as well as compressive press and pinch stimuli resulted in high-frequency discharge. von Frey filament stimulation resulted in graded increases in responsiveness of sampled units. B, Topical administration of the ERK1/2 inhibitor PD98059 resulted in decreased evoked responses. Predrug unit responses are overlaid in gray on the SCI+PD98059 histogram. Example waveforms of unit activity to press stimulation for SCI, SCI + PD98059, and after SCI + PD98059 washout (C) illustrate the effect of ERK1/2 inhibition, which attenuated the post-SCI hyperresponsiveness. Waveform overdraws show that the same unit was recorded in both instances, and its depth in the dorsal horn is shown. PD98059 significantly reduced (*p < 0.05) the evoked responses to all peripheral stimuli except the 0.39 g von Frey filament after SCI compared with vehicle (SCI+VEH, D). dlf, Dorsolataeral fasciculus; I–IV, laminas I–IV.
Similarly, antagonism of the EP2 receptor with AH6809 resulted in reduced hyperresponsiveness. Unit recordings from a representative MR neuron after SCI show increased responsiveness to peripheral stimulation (Fig. 10A). Acute administration of AH6809 caused a reduction in the responsiveness of this unit to the same stimuli (Fig. 10B). Examples of unit spiking in response to press stimulation before and after AH6809 administration clearly illustrate the attenuation in post-SCI hyperresponsiveness (Fig. 10C). In SCI animals, inhibition of the neuronal PGE2 receptor EP2 with AH6809 significantly reduced the evoked responses to all peripheral stimuli when compared with vehicle (Fig. 10D).
Figure 10.

Antagonism of the PGE2 receptor EP2 and evoked responsiveness of dorsal horn neurons. A, After SCI, significantly increased (+p < 0.05) evoked responses to natural stimuli (PB, 0.39 g, 1.01 g, 20.8 g, 144 g/mm2, and 583 g/mm2 refer to phasic brush, von Frey filaments of increasing intensities, pressure, and pinch, applied for 20 s) are observed in SCI animals. Phasic brush stimulation, stimulation with von Frey filaments, and compressive press and pinch stimuli resulted in high-frequency discharges. B, Topical administration of the EP2 receptor antagonist AH6809 reduced the evoked firing rate to peripherally applied stimuli. Predrug unit responses are overlaid in gray. Sample waveforms in response to press stimulation are shown for SCI, SCI + AH6809, and after SCI + AH6809 washout (C), to illustrate the effect of EP2 antagonism. Waveform overdraws show that the same unit was recorded in both instances, and its depth in the dorsal horn is shown. D, In SCI animals, when compared with vehicle-treated animals (SCI + VEH), inhibition of the neuronal PGE2 receptor EP2 with AH6809 significantly reduced (+p < 0.05) the evoked responses to all peripheral stimuli.
PGE2 and pain-related behaviors
Intact and pre-SCI animals demonstrated expected levels of locomotor function (group mean of 21.0 ± 0.0) (Fig. 11A). Thirty days after SCI, BBB scores had plateaued at 9.5 ± 1.1. At this level of recovery, animals are capable of withdrawing their paws in response to noxious stimuli. Intact animals did not demonstrate changes in BBB scores throughout the duration of intrathecal administration of Mac-1–SAP, PD98059, or AH6809. Similarly, SCI animals did not show changes in BBB scores throughout the period of drug administration. After cessation of drug delivery, no changes were observed. Vehicle injection did not result in changes in BBB scores (mean of 9.6 ± 1.3).
Figure 11.
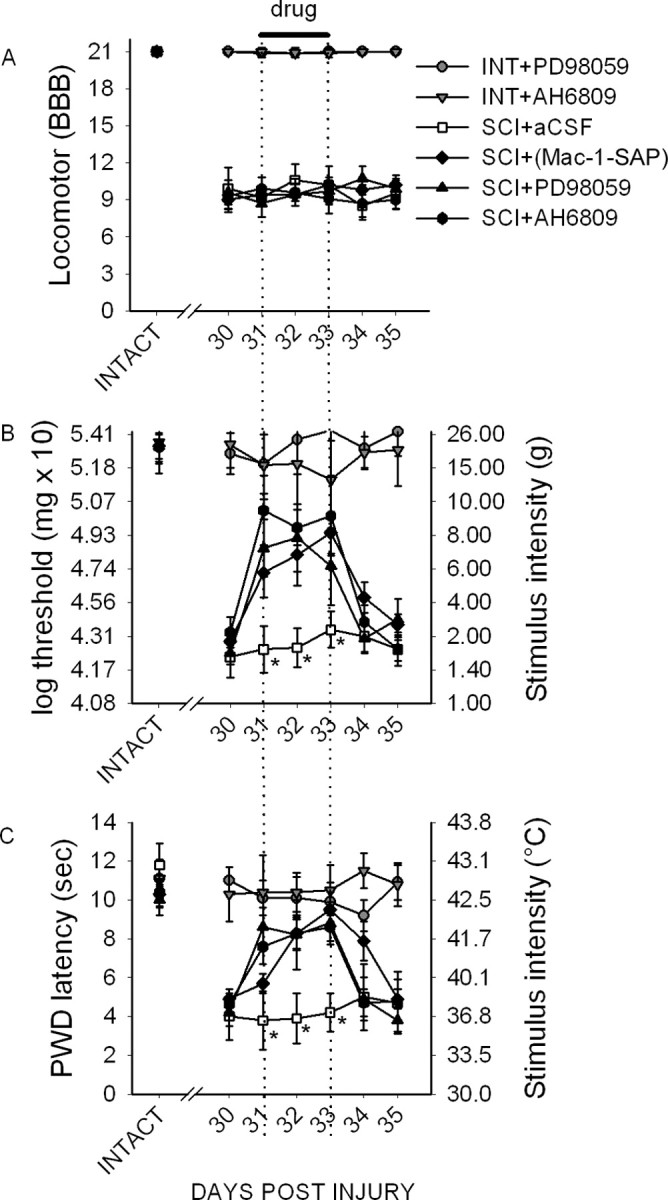
Behavioral analysis of locomotor function and pain-related behaviors. A, Intact (INT) and SCI animals demonstrated expected levels of locomotor function as measured by the BBB scale (A), which permitted testing of nociceptive thresholds. In intact animals, intrathecal delivery of the pERK1/2 inhibitor PD98059 or EP2 receptor antagonist AH6809 had no significant effect during the period of administration or for 2 d after cessation of administration, indicating no activation or depression of motor function. B, After SCI, mechanical paw-withdrawal thresholds were significantly decreased in all groups when compared with intact animals. Both PD98059 and AH6809 resulted in an immediate increase in mechanical thresholds, which persisted for the duration of administration. This effect was significant for Mac-1–SAP, PD98059, and AH6809 compared with aCSF vehicle (*p < 0.05). Immediately after cessation of administration (day 33), mechanical thresholds returned to predrug levels, which were equivalent to untreated SCI animals. C, Thermal paw-withdrawal (PWD) latencies were significantly lowered after SCI. Mac-1–SAP, PD98059, and AH6809 resulted in a significant increase in paw-withdrawal latencies compared with vehicle. After cessation of Mac-1–SAP, PD98059, or AH6809, latencies returned to predrug levels, which persisted for the duration of the experiment.
Before SCI, and in intact animals, the mean mechanical nociceptive threshold was 20.4 ± 1.4 g. Thirty days after SCI, mechanical thresholds had significantly (p < 0.05) decreased to 2.1 ± 1.9 g, demonstrating the development of mechanical allodynia. In intact animals, intrathecal administration of PD98059 had no effect on paw-withdrawal threshold to von Frey filament stimulation at any time point (Fig. 11B). In SCI animals receiving Mac-1–SAP, mechanical thresholds were significantly increased starting on day 1 of drug administration to an average of 6.9 ± 2.9 g for all days when compared with predrug levels and vehicle-treated animals (2.5 ± 1.5 g). Immediately after cessation of delivery of PD98059, mechanical thresholds returned to predrug levels, 1.8 ± 0.9 g. PD98059 resulted in a significantly increase in mechanical thresholds as well, to an average of 7.8 ± 2.6 g for all days when compared with predrug levels and vehicle. Immediately after cessation of delivery, mechanical thresholds returned to predrug levels, 2.8 ± 1.6 g. AH6809 had a similar effect, significantly increasing mechanical thresholds to 9.2 ± 2.4 g when compared with predrug levels and vehicle-treated animals. After AH6809 withdrawal, thresholds returned to predrug levels, 3.1 ± 1.9 g.
Baseline thermal paw-withdrawal latencies for intact and pre-SCI animals was 10.6 ± 0.8 s (Fig. 11C). Thirty days after injury, withdrawal latencies significantly decreased to 4.4 ± 0.8 s for all SCI animals, indicating the development of thermal hyperalgesia. Neither PD98059 nor AH6809 had an effect on withdrawal latencies in intact animals. After SCI, however, Mac-1–SAP administration resulted in significantly increased paw-withdrawal latencies to an average of 8.3 ± 0.9 s compared with vehicle-treated animals (4.3 ± 1.4 s), which decreased after cessation of administration to 4.9 ± 0.5 s. PD98059 also resulted in an immediate restoration in paw-withdrawal latency to 8.5 ± 1.3 s. This was significantly increased compared with vehicle-treated animals. After drug delivery was stopped, latencies returned to predrug levels, 4.3 ± 0.9 s. Compared with vehicle, AH6809 also resulted in a significant increase in withdrawal latencies to 8.2 ± 0.8 s, which was reversed after drug delivery was stopped, to 4.8 ± 1.1 s. Thus, pharmacological inhibition of the elevated microglial release of PGE2 and antagonism of its neuronal receptor reduced mechanical allodynia and thermal hyperalgesia after SCI.
Discussion
There is a dramatic shift in microglial status from a resting to an activated state in the lumbar dorsal horn after SCI at a time when dorsal horn sensory neurons exhibited hyperresponsiveness to peripheral stimulation, and pain-related behaviors were evident (Hains and Waxman, 2006). Pharmacological downregulation of microglial activation resulted in a return to resting morphological phenotype as well as reductions in electrophysiologic and behavioral concomitants of pain, suggesting a new role for activated microglia after SCI. The finding that microglia are actively involved in the maintenance of ongoing pain phenomenology suggests mechanistic differences from peripheral injury in which microglial activation is related to the induction phase of pain. It is not completely unexpected that microglia are involved in the maintenance of changes in sensory processing in the injured cord because, in most models of injury, the process is dynamic and continuous as the injury progresses and reinvents itself over time attributable to the positive feedback of the injury cascade.
The current study builds on these findings and identifies a putative signaling mechanism by which activated microglia activate dorsal horn sensory neurons after SCI. This is the first demonstration of a direct microglia–neuron signaling pathway in the injured spinal cord. We show that PGE2 is a central molecule in microglia-mediated chronic pain. Using morphological, biochemical, electrophysiological, and behavioral methodologies, we demonstrate that pERK1/2 MAP kinase-mediated release from microglia of PGE2, which binds to the EP2 receptor located on dorsal horn neurons, is sufficient to induce changes in their excitability state, which poises them to inappropriately amplify innocuous and noxious sensory stimuli. These changes contribute to the expression of abnormal pain-related behaviors after injury.
We should note, however, that changes in signaling molecules after injury should not be thought of as activity dependent. The changes that are observed are injury dependent and have the same long-range impact as with peripheral injury in which changes in signaling molecules are believed to be dependent on input from the periphery.
ERK activation (phosphorylation) has been documented in the spinal cord after peripheral nerve injury (Ji et al., 1999; Ma and Quirion, 2002; Ciruela et al., 2003; Katsura et al., 2006) and inflammation (Galan et al., 2002). Spinal nerve ligation-induced ERK activation occurs sequentially in dorsal horn neurons, microglia, and astrocytes and is thought to contribute through different mechanisms in different cell types to pain sensitivity (Zhuang et al., 2005). Nociceptor activity leads to microglia-specific ERK activation (Tsuda et al., 2005), possibly through stimulation by nitric oxide, IL-1β, tumor necrosis factor-α, and a variety of cytokines (Ji and Strichartz, 2004; Marchand et al., 2005). After excitotoxic (Yu and Yezierski, 2005) or contusive (Crown et al., 2006) SCI, ERK activation occurs at both acute and chronic time points near the lesion epicenter by Western blot analysis, but its cellular localization has not been demonstrated. Yu and Yezierski (2005) showed that PD98059 prevented the development of at-level excessive grooming behavior after excitotoxic SCI. We localized pERK1/2 specifically to activated microglia after SCI and show a positive relationship between the degree of microglial activation and pERK1/2 levels. Furthermore, we observed upstream activation of this effector at the same time that levels of PGE2 were increased in spinal parenchyma.
In vitro studies indicate that microglia are indeed able to synthesize and release PGE2 (Minghetti et al., 1998; Hoozemans et al., 2002; Ajmone-Cat et al., 2003; Ikeda-Matsuo et al., 2005; Zhang et al., 2006). PGE2 has been implicated in the induction of central sensitization of spinal neurons (Minami et al., 1999; Samad et al., 2001; Ji et al., 2003). Microglial PGE2 may therefore play an important role in the generation of central sensitization after SCI. Reductions in behavioral indicators of pain after SCI are possible through inhibition of COX-2-mediated PGE2 production (Hains et al., 2001). COX-mediated PGE2 release may be possible through a number of cell types. COX-1 and COX-2 are present in DRG and dorsal and ventral spinal cord (Yaksh et al., 2001), and COX-2 is present within neurons (Beiche et al., 1998), astrocytes (Hirst et al., 1999; Falsig et al., 2004), and microglia (Akundi et al., 2005). Our data show that upstream inhibition of ERK1/2 activation results in reductions in COX-2 staining within activated microglia, whereas neuronal COX-2 is unaffected.
ERK1/2 activation has been shown to be an upstream effector of the PGE2 biosynthetic pathway in microglia (Akundi et al., 2005). Similarly, p38 MAPK activation may also induce PGE2 release, but it is not definitively known whether the source of PGE2 is neuronal or glial (Svensson et al., 2003, 2005). Additionally, activation of p38 MAPK is mostly involved in posttranscriptional regulation of COX-2 mRNA stability, whereas the pERK1/2 pathway is essential for COX-2 gene transcription (Chun and Surh, 2004) that leads to PGE2 synthesis and expression (Akundi et al., 2005). For this reason, we chose to target pERK1/2 because it may specifically regulate microglial PGE2 release.
Here we show that direct pharmacological activation of spinal microglia results in increased PGE2 production in the spinal cord dorsal horn. This level of increase is similar to that measured 30 d after SCI. After SCI, we reduced spinal PGE2 levels with agents that specifically inhibited microglia or the ERK1/2 signaling cascade. PGE2 levels were reduced by ∼27% by microglial inhibition but by only 18% with pERK1/2 inhibition. This incomplete reduction in PGE2 release could indicate an additional neuronal or glial source of PGE2 synthesis and/or release. In microglia, the ceramide–p38 MAPK or phosphatidylcholine–phospholipase C pathways might act as additional activators of PGE2 biosynthesis (Akundi et al., 2005). Our behavioral data indicate statistically equivalent effectiveness of both PD98059 and AH6809, however, suggesting that EP2 binding of PGE2 is maximal.
Four major subtypes of the PGE2 EP receptors exist (Vanegas and Schaible, 2001). EP1, EP3, and EP4 are localized to DRG neurons and primary afferents (Oida et al., 1995), and it is probable that microglial PGE2 modulates nociceptive transmission via actions on central terminals of primary afferent fibers (Vasko, 1995). We and others have shown that the EP2 receptor is robustly localized to postsynaptic dorsal horn neurons (Kawamura et al., 1997). PGE2, acting through the EP2 receptor, directly depolarizes spinal neurons (Baba et al., 2001) and contributes to inflammation-induced spinal hyperexcitability (Vasquez et al., 2001; Reinold et al., 2005). We show that the EP2 receptor is not localized to microglia after SCI; however, there are reports suggesting that EP1 and EP2 receptors can be present in cultured microglia (Caggiano and Kraig, 1999).
It is not likely that SCI induces increases in PGE2 production by DRG neurons that project into the dorsal horn. Our data do not show activation of either p38 or ERK1/2 within corresponding lumbar DRG cell bodies. However, because we do not completely eliminate PGE2 in the dorsal horn, PGE2 release by primary afferent remains a possibility.
In conclusion, we identified a putative microglia–neuron signaling mechanism whereby PGE2 released by activated microglia contributes to the sensitization of spinal neurons after SCI. We demonstrate the key role of PGE2 by both interrupting its release at the source and by blocking the binding to its target. Specific activation of microglia with fractalkine or by chronic SCI causes the release of PGE2 in the dorsal horn. Pharmacological blockade of upstream effectors of microglial PGE2 release, as well as inhibition of microglial activation, results in decreased spinal PGE2 levels. Both inhibition of microglial-mediated PGE2 release and blockade of the neuronal PGE2 receptor EP2 result in attenuated neuronal hyperresponsiveness and reductions in pain-related behaviors. Targeting of this signaling mechanism may offer hope for successful management of pain after SCI.
Footnotes
This work was supported in part by grants from the Medical Research Service and Rehabilitation Research Service, Department of Veterans Affairs, and the National Multiple Sclerosis Society. The Center for Neuroscience and Regeneration Research is a Collaboration of the Paralyzed Veterans of America and the United Spinal Association. B.C.H. was funded by Pfizer (Scholar's Grant in Pain Medicine) and The Dana Foundation. We thank Dr. Joel Black for valuable experimental advice.
References
- Ajmone-Cat MA, Nicolini A, Minghetti L. Prolonged exposure of microglia to lipopolysaccharide modifies the intracellular signaling pathways and selectively promotes prostaglandin E2 synthesis. J Neurochem. 2003;87:1193–1203. doi: 10.1046/j.1471-4159.2003.02087.x. [DOI] [PubMed] [Google Scholar]
- Akundi RS, Candelario-Jalil E, Hess S, Hull M, Lieb K, Gebicke-Haerter PJ, Fiebich BL. Signal transduction pathways regulating cyclooxygenase-2 in lipopolysaccharide-activated primary rat microglia. Glia. 2005;51:199–208. doi: 10.1002/glia.20198. [DOI] [PubMed] [Google Scholar]
- Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- Baba H, Kohno T, Moore KA, Woolf CJ. Direct activation of rat spinal dorsal horn neurons by prostaglandin E2. J Neurosci. 2001;21:1750–1756. doi: 10.1523/JNEUROSCI.21-05-01750.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso DM, Beattie MS, Bresnahan JC. A sensitive and reliable locomotor rating scale for open field testing in rats. J Neurotrauma. 1995;12:1–21. doi: 10.1089/neu.1995.12.1. [DOI] [PubMed] [Google Scholar]
- Beiche F, Klein T, Nusing R, Neuhuber W, Goppelt-Struebe M. Localization of cyclooxygenase-2 and prostaglandin E2 receptor EP3 in the rat lumbar spinal cord. J Neuroimmunol. 1998;89:26–34. doi: 10.1016/s0165-5728(98)00061-7. [DOI] [PubMed] [Google Scholar]
- Caggiano AO, Kraig RP. Prostaglandin E receptor subtypes in cultured rat microglia and their role in reducing lipopolysaccharide-induced interleukin-1beta production. J Neurochem. 1999;72:565–575. doi: 10.1046/j.1471-4159.1999.0720565.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Christensen MD, Hulsebosch CE. Chronic central pain after spinal cord injury. J Neurotrauma. 1997;14:517–537. doi: 10.1089/neu.1997.14.517. [DOI] [PubMed] [Google Scholar]
- Chun KS, Surh YJ. Signal transduction pathways regulating cyclooxygenase-2 expression: potential molecular targets for chemoprevention. Biochem Pharmacol. 2004;68:1089–1100. doi: 10.1016/j.bcp.2004.05.031. [DOI] [PubMed] [Google Scholar]
- Ciruela A, Dixon AK, Bramwell S, Gonzalez MI, Pinnock RD, Lee K. Identification of MEK1 as a novel target for the treatment of neuropathic pain. Br J Pharmacol. 2003;138:751–756. doi: 10.1038/sj.bjp.0705103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–1021. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- Coyle DE. Partial peripheral nerve injury leads to activation of astroglia and microglia which parallels the development of allodynic behavior. Glia. 1998;23:75–83. [PubMed] [Google Scholar]
- Crown ED, Ye Z, Johnson KM, Xu GY, McAdoo DJ, Hulsebosch CE. Increases in the activated forms of ERK 1/2, p38 MAPK, and CREB are correlated with the expression of at-level mechanical allodynia following spinal cord injury. Exp Neurol. 2006;199:397–407. doi: 10.1016/j.expneurol.2006.01.003. [DOI] [PubMed] [Google Scholar]
- DeLeo JA, Tawfik VL, LaCroix-Fralish ML. The tetrapartite synapse: path to CNS sensitization and chronic pain. Pain. 2006;122:17–21. doi: 10.1016/j.pain.2006.02.034. [DOI] [PubMed] [Google Scholar]
- Dirig DM, Salami A, Rathbun ML, Ozaki GT, Yaksh TL. Characterization of variables defining hindpaw withdrawal latency evoked by radiant thermal stimuli. J Neurosci Methods. 1997;76:183–191. doi: 10.1016/s0165-0270(97)00097-6. [DOI] [PubMed] [Google Scholar]
- Dixon WJ. Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol. 1980;20:441–462. doi: 10.1146/annurev.pa.20.040180.002301. [DOI] [PubMed] [Google Scholar]
- Dommergues MA, Plaisant F, Verney C, Gressens P. Early microglial activation following neonatal excitotoxic brain damage in mice: a potential target for neuroprotection. Neuroscience. 2003;121:619–628. doi: 10.1016/s0306-4522(03)00558-x. [DOI] [PubMed] [Google Scholar]
- Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci USA. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falsig J, Latta M, Leist M. Defined inflammatory states in astrocyte cultures: correlation with susceptibility towards CD95-driven apoptosis. J Neurochem. 2004;88:181–193. doi: 10.1111/j.1471-4159.2004.02144.x. [DOI] [PubMed] [Google Scholar]
- Ferrari D, Chiozzi P, Falzoni S, Dal Susino M, Melchiorri L, Baricordi OR, Di Virgilio F. Extracellular ATP triggers IL-1 beta release by activating the purinergic P2Z receptor of human macrophages. J Immunol. 1997;159:1451–1458. [PubMed] [Google Scholar]
- Finnerup NB, Johannesen IL, Sindrup SH, Bach FW, Jensen TS. Pain and dysesthesia in patients with spinal cord injury: a postal survey. Spinal Cord. 2001;39:256–262. doi: 10.1038/sj.sc.3101161. [DOI] [PubMed] [Google Scholar]
- Fu KY, Light AR, Matsushima GK, Maixner W. Microglial reactions after subcutaneous formalin injection into the rat hind paw. Brain Res. 1999;825:59–67. doi: 10.1016/s0006-8993(99)01186-5. [DOI] [PubMed] [Google Scholar]
- Galan A, Lopez-Garcia JA, Cervero F, Laird JM. Activation of spinal extracellular signaling-regulated kinase-1 and -2 by intraplantar carrageenan in rodents. Neurosci Lett. 2002;322:37–40. doi: 10.1016/s0304-3940(02)00078-2. [DOI] [PubMed] [Google Scholar]
- Gruner JA. A monitored contusion model of spinal cord injury in the rat. J Neurotrauma. 1992;9:123–126. doi: 10.1089/neu.1992.9.123. [DOI] [PubMed] [Google Scholar]
- Hains BC, Waxman SG. Activated microglia contribute to the maintenance of chronic pain after spinal cord injury. J Neurosci. 2006;26:4308–4317. doi: 10.1523/JNEUROSCI.0003-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hains BC, Yucra JA, Hulsebosch CE. Reduction of pathological and behavioral deficits following spinal cord contusion injury with the selective cyclooxygenase-2 inhibitor NS-398. J Neurotrauma. 2001;18:409–423. doi: 10.1089/089771501750170994. [DOI] [PubMed] [Google Scholar]
- Hains BC, Willis WD, Hulsebosch CE. Serotonin receptors 5-HT1A and 5-HT3 reduce hyperexcitability of dorsal horn neurons after chronic spinal cord hemisection injury in rat. Exp Brain Res. 2003a;149:174–186. doi: 10.1007/s00221-002-1352-x. [DOI] [PubMed] [Google Scholar]
- Hains BC, Klein JP, Saab CY, Craner MJ, Black JA, Waxman SG. Upregulation of sodium channel Nav1.3 and functional involvement in neuronal hyperexcitability associated with central neuropathic pain after spinal cord injury. J Neurosci. 2003b;23:8881–8892. doi: 10.1523/JNEUROSCI.23-26-08881.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hains BC, Johnson KM, Eaton MJ, Willis WD, Hulsebosch CE. Serotonergic neural precursor cell grafts attenuate bilateral hyperexcitability of dorsal horn neurons after spinal hemisection in rat. Neuroscience. 2003c;116:1097–1110. doi: 10.1016/s0306-4522(02)00729-7. [DOI] [PubMed] [Google Scholar]
- Harrison JK, Jiang Y, Chen S, Xia Y, Maciejewski D, McNamara RK, Streit WJ, Salafranca MN, Adhikari S, Thompson DA, Botti P, Bacon KB, Feng L. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc Natl Acad Sci USA. 1998;95:10896–10901. doi: 10.1073/pnas.95.18.10896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haupt W, Jiang W, Kreis ME, Grundy D. Prostaglandin EP receptor subtypes have distinctive effects on jejunal afferent sensitivity in the rat. Gastroenterology. 2000;119:1580–1589. doi: 10.1053/gast.2000.20337. [DOI] [PubMed] [Google Scholar]
- Hirst WD, Young KA, Newton R, Allport VC, Marriott DR, Wilkin GP. Expression of COX-2 by normal and reactive astrocytes in the adult rat central nervous system. Mol Cell Neurosci. 1999;13:57–68. doi: 10.1006/mcne.1998.0731. [DOI] [PubMed] [Google Scholar]
- Hoozemans JJ, Veerhuis R, Janssen I, van Elk EJ, Rozemuller AJ, Eikelenboom P. The role of cyclo-oxygenase 1 and 2 activity in prostaglandin E(2) secretion by cultured human adult microglia: implications for Alzheimer's disease. Brain Res. 2002;951:218–226. doi: 10.1016/s0006-8993(02)03164-5. [DOI] [PubMed] [Google Scholar]
- Hua XY, Svensson CI, Matsui T, Fitzsimmons B, Yaksh TL, Webb M. Intrathecal minocycline attenuates peripheral inflammation-induced hyperalgesia by inhibiting p38 MAPK in spinal microglia. Eur J Neurosci. 2005;22:2431–2440. doi: 10.1111/j.1460-9568.2005.04451.x. [DOI] [PubMed] [Google Scholar]
- Ikeda-Matsuo Y, Ikegaya Y, Matsuki N, Uematsu S, Akira S, Sasaki Y. Microglia-specific expression of microsomal prostaglandin E2 synthase-1 contributes to lipopolysaccharide-induced prostaglandin E2 production. J Neurochem. 2005;94:1546–1558. doi: 10.1111/j.1471-4159.2005.03302.x. [DOI] [PubMed] [Google Scholar]
- Inoue K. ATP receptors of microglia involved in pain. Novartis Found Symp. 2006;276:263–272. [PubMed] [Google Scholar]
- Ji RR, Strichartz G. Cell signaling and the genesis of neuropathic pain. Science STKE. 2004;252:re14. doi: 10.1126/stke.2522004re14. [DOI] [PubMed] [Google Scholar]
- Ji RR, Baba H, Brenner GJ, Woolf CJ. Nociceptive-specific activation of ERK in spinal neurons contributes to pain hypersensitivity. Nat Neurosci. 1999;2:1114–1119. doi: 10.1038/16040. [DOI] [PubMed] [Google Scholar]
- Ji RR, Kohno T, Moore KA, Woolf CJ. Central sensitization and LTP: do pain and memory share similar mechanisms? Trends Neurosci. 2003;26:696–705. doi: 10.1016/j.tins.2003.09.017. [DOI] [PubMed] [Google Scholar]
- Jin SX, Zhuang ZY, Woolf CJ, Ji RR. p38 mitogen-activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. J Neurosci. 2003;23:4017–4022. doi: 10.1523/JNEUROSCI.23-10-04017.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsura H, Obata K, Mizushima T, Sakurai J, Kobayashi K, Yamanaka H, Dai Y, Fukuoka T, Sakagami M, Noguchi K. Activation of Src-family kinases in spinal microglia contributes to mechanical hypersensitivity after nerve injury. J Neurosci. 2006;26:8680–8690. doi: 10.1523/JNEUROSCI.1771-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura T, Yamauchi T, Koyama M, Maruyama T, Akira T, Nakamura N. Expression of prostaglandin EP2 receptor mRNA in the rat spinal cord. Life Sci. 1997;61:2111–2116. doi: 10.1016/s0024-3205(97)00884-9. [DOI] [PubMed] [Google Scholar]
- Kigerl KA, McGaughy VM, Popovich PG. Comparative analysis of lesion development and intraspinal inflammation in four strains of mice following spinal contusion injury. J Comp Neurol. 2006;494:578–594. doi: 10.1002/cne.20827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledeboer A, Sloane EM, Milligan ED, Frank MG, Mahony JH, Maier SF, Watkins LR. Minocycline attenuates mechanical allodynia and proinflammatory cytokine expression in rat models of pain facilitation. Pain. 2005;115:71–83. doi: 10.1016/j.pain.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Ma W, Quirion R. Partial sciatic nerve ligation induces increase in the phosphorylation of extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) in astrocytes in the lumbar spinal dorsal horn and the gracile nucleus. Pain. 2002;99:175–184. doi: 10.1016/s0304-3959(02)00097-0. [DOI] [PubMed] [Google Scholar]
- Marchand F, Perretti M, McMahon SB. Role of the immune system in chronic pain. Nat Rev Neurosci. 2005;6:521–532. doi: 10.1038/nrn1700. [DOI] [PubMed] [Google Scholar]
- McMahon SB, Cafferty WB, Marchand F. Immune and glial cell factors as pain mediators and modulators. Exp Neurol. 2005;192:444–462. doi: 10.1016/j.expneurol.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Milligan ED, Zapata V, Chacur M, Schoeniger D, Biedenkapp J, O'Connor KA, Verge GM, Chapman G, Green P, Foster AC, Naeve GS, Maier SF, Watkins LR. Evidence that exogenous and endogenous fractalkine can induce spinal nociceptive facilitation in rats. Eur J Neurosci. 2004;20:2294–2302. doi: 10.1111/j.1460-9568.2004.03709.x. [DOI] [PubMed] [Google Scholar]
- Milligan E, Zapata V, Schoeniger D, Chacur M, Green P, Poole S, Martin D, Maier SF, Watkins LR. An initial investigation of spinal mechanisms underlying pain enhancement induced by fractalkine, a neuronally released chemokine. Eur J Neurosci. 2005;22:2775–2782. doi: 10.1111/j.1460-9568.2005.04470.x. [DOI] [PubMed] [Google Scholar]
- Minami T, Okuda-Ashitaka E, Hori Y, Sakuma S, Sugimoto T, Sakimura K, Mishina M, Ito S. Involvement of primary afferent C-fibres in touch-evoked pain (allodynia) induced by prostaglandin E2. Eur J Neurosci. 1999;11:1849–1856. doi: 10.1046/j.1460-9568.1999.00602.x. [DOI] [PubMed] [Google Scholar]
- Minghetti L, Polazzi E, Nicolini A, Levi G. Opposite regulation of prostaglandin E2 synthesis by transforming growth factor-beta1 and interleukin 10 in activated microglial cultures. J Neuroimmunol. 1998;82:31–39. doi: 10.1016/S0165-5728(97)00185-9. [DOI] [PubMed] [Google Scholar]
- Nesic O, Lee J, Johnson KM, Ye Z, Xu GY, Unabia GC, Wood TG, McAdoo DJ, Westlund KN, Hulsebosch CE, Perez-Polo JR. Transcriptional profiling of spinal cord injury-induced central neuropathic pain. J Neurochem. 2005;95:998–1014. doi: 10.1111/j.1471-4159.2005.03462.x. [DOI] [PubMed] [Google Scholar]
- Oida H, Namba T, Sugimoto Y, Ushikubi F, Ohishi H, Ichikawa A, Narumiya S. In situ hybridization studies of prostacyclin receptor mRNA expression in various mouse organs. Br J Pharmacol. 1995;116:2828–2837. doi: 10.1111/j.1476-5381.1995.tb15933.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovich PG, Wei P, Stokes BT. Cellular inflammatory response after spinal cord injury in Sprague-Dawley and Lewis rats. J Comp Neurol. 1997;377:443–464. doi: 10.1002/(sici)1096-9861(19970120)377:3<443::aid-cne10>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Qin C, Chandler MJ, Miller KE, Foreman RD. Chemical activation of cervical cell bodies: effects on responses to colorectal distension in lumbosacral spinal cord of rats. J Neurophysiol. 1999;82:3423–3433. doi: 10.1152/jn.1999.82.6.3423. [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Tanga F, DeLeo JA. Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. J Pharmacol Exp Ther. 2003;306:624–630. doi: 10.1124/jpet.103.052407. [DOI] [PubMed] [Google Scholar]
- Reinold H, Ahmadi S, Depner UB, Layh B, Heindl C, Hamza M, Pahl A, Brune K, Narumiya S, Muller U, Zeilhofer HU. Spinal inflammatory hyperalgesia is mediated by prostaglandin E receptors of the EP2 subtype. J Clin Invest. 2005;115:673–679. doi: 10.1172/JCI200523618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rintala DH, Loubser PG, Castro J, Hart KA, Fuhrer MJ. Chronic pain in a community-based sample of men with spinal cord injury: prevalence, severity, and relationship with impairment, disability, handicap, and subjective well-being. Arch Phys Med Rehabil. 1998;79:604–614. doi: 10.1016/s0003-9993(98)90032-6. [DOI] [PubMed] [Google Scholar]
- Samad TA, Moore KA, Sapirstein A, Billet S, Allchorne A, Poole S, Bonventre JV, Woolf CJ. Interleukin-1beta-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature. 2001;410:471–475. doi: 10.1038/35068566. [DOI] [PubMed] [Google Scholar]
- Siddall PJ, McClelland JM, Rutkowski SB, Cousins MJ. A longitudinal study of the prevalence and characteristics of pain in the first 5 years following spinal cord injury. Pain. 2003;103:249–257. doi: 10.1016/S0304-3959(02)00452-9. [DOI] [PubMed] [Google Scholar]
- Sroga JM, Jones TB, Kigerl KA, McGaughy VM, Popovich PG. Rats and mice exhibit distinct inflammatory reactions after spinal cord injury. J Comp Neurol. 2003;462:223–240. doi: 10.1002/cne.10736. [DOI] [PubMed] [Google Scholar]
- Svensson CI, Hua XY, Protter AA, Powell HC, Yaksh TL. Spinal p38 MAP kinase is necessary for NMDA-induced spinal PGE(2) release and thermal hyperalgesia. NeuroReport. 2003;14:1153–1157. doi: 10.1097/00001756-200306110-00010. [DOI] [PubMed] [Google Scholar]
- Svensson CI, Hua XY, Powell HC, Lai J, Porreca F, Yaksh TL. Prostaglandin E2 release evoked by intrathecal dynorphin is dependent on spinal p38 mitogen activated protein kinase. Neuropeptides. 2005;39:485–494. doi: 10.1016/j.npep.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Inoue K, Salter MW. Neuropathic pain and spinal microglia: a big problem from molecules in “small” glia. Trends Neurosci. 2005;28:101–107. doi: 10.1016/j.tins.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Vanegas H, Schaible HG. Prostaglandins and cyclooxygenases [correction of cycloxygenases] in the spinal cord. Prog Neurobiol. 2001;64:327–363. doi: 10.1016/s0301-0082(00)00063-0. [DOI] [PubMed] [Google Scholar]
- Vasko MR. Prostaglandin-induced neuropeptide release from spinal cord. Prog Brain Res. 1995;104:367–380. doi: 10.1016/s0079-6123(08)61801-4. [DOI] [PubMed] [Google Scholar]
- Vasquez E, Bar KJ, Ebersberger A, Klein B, Vanegas H, Schaible HG. Spinal prostaglandins are involved in the development but not the maintenance of inflammation-induced spinal hyperexcitability. J Neurosci. 2001;21:9001–9008. doi: 10.1523/JNEUROSCI.21-22-09001.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins LR, Milligan ED, Maier SF. Glial activation: a driving force for pathological pain. Trends Neurosci. 2001;24:450–455. doi: 10.1016/s0166-2236(00)01854-3. [DOI] [PubMed] [Google Scholar]
- Waxman SG, Kocsis JD, Black JA. Type III sodium channel mRNA is expressed in embryonic but not adult spinal sensory neurons, and is reexpressed following axotomy. J Neurophysiol. 1994;72:466–472. doi: 10.1152/jn.1994.72.1.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward DF, Pepperl DJ, Burkey TH, Regan JW. 6-Isopropoxy-9-oxoxanthene-2-carboxylic acid (AH 6809), a human EP2 receptor antagonist. Biochem Pharmacol. 1995;50:1731–1733. doi: 10.1016/0006-2952(95)02035-7. [DOI] [PubMed] [Google Scholar]
- Yaksh TL, Dirig DM, Conway CM, Svensson C, Luo ZD, Isakson PC. The acute antihyperalgesic action of nonsteroidal, anti-inflammatory drugs and release of spinal prostaglandin E2 is mediated by the inhibition of constitutive spinal cyclooxygenase-2 (COX-2) but not COX-1. J Neurosci. 2001;21:5847–5853. doi: 10.1523/JNEUROSCI.21-16-05847.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu CG, Yezierski RP. Activation of the ERK1/2 signaling cascade by excitotoxic spinal cord injury. Mol Brain Res. 2005;138:244–255. doi: 10.1016/j.molbrainres.2005.04.013. [DOI] [PubMed] [Google Scholar]
- Zai LJ, Wrathall JR. Cell proliferation and replacement following contusive spinal cord injury. Glia. 2005;50:247–257. doi: 10.1002/glia.20176. [DOI] [PubMed] [Google Scholar]
- Zhang J, Fujii S, Wu Z, Hashioka S, Tanaka Y, Shiratsuchi A, Nakanishi Y, Nakanishi H. Involvement of COX-1 and up-regulated prostaglandin E synthases in phosphatidylserine liposome-induced prostaglandin E2 production by microglia. J Neuroimmunol. 2006;172:112–120. doi: 10.1016/j.jneuroim.2005.11.008. [DOI] [PubMed] [Google Scholar]
- Zhuang ZY, Gerner P, Woolf CJ, Ji RR. ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. Pain. 2005;114:149–159. doi: 10.1016/j.pain.2004.12.022. [DOI] [PubMed] [Google Scholar]