

Abstract
Deafness is the most common form of sensory impairment in the human population and is frequently caused by recessive mutations. To obtain animal models for recessive forms of deafness and to identify genes that control the development and function of the auditory sense organs, we performed a forward genetics screen in mice. We identified 13 mouse lines with defects in auditory function and six lines with auditory and vestibular defects. We mapped several of the affected genetic loci and identified point mutations in four genes. Interestingly, all identified genes are expressed in mechanosensory hair cells and required for their function. One mutation maps to the pejvakin gene, which encodes a new member of the gasdermin protein family. Previous studies have described two missense mutations in the human pejvakin gene that cause nonsyndromic recessive deafness (DFNB59) by affecting the function of auditory neurons. In contrast, the pejvakin allele described here introduces a premature stop codon, causes outer hair cell defects, and leads to progressive hearing loss. We also identified a novel allele of the human pejvakin gene in an Iranian pedigree that is afflicted with progressive hearing loss. Our findings suggest that the mechanisms of pathogenesis associated with pejvakin mutations are more diverse than previously appreciated. More generally, our findings demonstrate that recessive screens in mice are powerful tools for identifying genes that control the development and function of mechanosensory hair cells and cause deafness in humans, as well as generating animal models for disease.
Keywords: ENU, inner ear, hearing loss, gasdermin, deafness, genetics
Introduction
Hair cells in the cochlea of the inner ear transduce sound-induced mechanical stimuli into electrical signals to provide our sense of hearing (Gillespie and Walker, 2001). Cochlear defects lead to sensorineuronal deafness, the most common form of deafness in humans (Friedman and Griffith, 2003). The inaccessibility of the cochlea within the bony labyrinth poses challenges for studying the mechanisms that control the development and function of the auditory end organ by conventional molecular approaches. Genetic approaches provide an alternative method for defining the molecules that are essential for auditory perception. By means of positional cloning strategies, genes with different functions, such as transcription factors and myosin motors, have been linked to inherited forms of deafness in humans. Importantly, mutations in orthologous human and mouse genes frequently cause deafness, which suggests that Mus musculus is a useful model for studying cochlear impairment relevant to human disease (Friedman and Griffith, 2003; Quint and Steel, 2003).
In recent years, phenotype-based forward genetics screens have generated mouse lines with auditory defects. For the most part, the published screens have identified dominant mutations that affect the development of the auditory system (Hrabe de Angelis et al., 2000; Nolan et al., 2000; Kiernan et al., 2001; Tsai et al., 2001; Vreugde et al., 2002; Curtin et al., 2003; Hardisty et al., 2003; Rhodes et al., 2003, 2004; Marcotti et al., 2006). Such mutations are less informative for studying cochlear defects not associated with gross morphological changes. In addition, the affected mouse lines frequently exhibit vestibular defects that manifest as circling behavior or abnormal head posture. However, many forms of deafness in humans are not associated with obvious vestibular defects and are frequently caused by point mutations that are inherited as recessive traits (Friedman and Griffith, 2003).
To obtain animal models for recessive forms of deafness, we performed a forward genetics screen in mice. We chose N-ethyl-N-nitrosourea (ENU) as a mutagen, because it randomly induces point mutations with high efficiency (Ashby et al., 1997; Shelby and Tindall, 1997). Here, we describe the identification of 19 mouse lines that are afflicted with hearing loss, six of which also show vestibular defects. All of the lines analyzed thus far show morphological or functional defects in cochlear hair cells. We mapped several of the affected genomic loci and identified point mutations in four genes. One mutation introduces a premature stop codon into the DFNB59 gene (also referred to as the pejvakin gene). The mutation differs from previously described missense mutations, which cause deafness by affecting the function of auditory neurons (Delmaghani et al., 2006). Unlike these missense mutations, the pejvakin allele reported here causes progressive hearing loss, which is associated with defects in outer hair cell function. Consistent with this finding, we show by in situ hybridization that pejvakin is expressed in hair cells. Finally, we identified in an Iranian pedigree a novel pejvakin allele that also leads to progressive hearing loss. We conclude that pejvakin is essential for auditory hair cell function, and that some pejvakin alleles cause progressive hearing loss. More generally, our findings demonstrate that screens for recessive auditory phenotypes in mice are useful for defining the genetic program that controls cochlear function and for providing new mechanistic insights into the molecular pathogenesis of deafness.
Materials and Methods
ENU mutagenesis screen and phenotyping.
The ENU mutagenesis protocol and primary phenotypic screen has been described previously (Reijmers et al., 2006). All experiments were performed with littermates. Age-matched wild-type mice were used to establish baseline parameters. For auditory startle response (ASR) measurements, mice (8 weeks of age) in SR-LAB startle response chambers (San Diego Instruments, San Diego, CA) were exposed to a 5 min acclimation period of white noise (70 dB) followed by six initial startle response trials (120 dB). Immediately after these startle response trials, mice were exposed to five blocks of six different types of stimuli presented in pseudorandom order, including a 120 dB startle stimulus, a no-stimulus trial, and a 120 dB prepulse stimulus paired with a prepulse at one of four different intensity levels (78, 84, 86, and 90 dB) 100 ms before the onset of the startle stimulus. At the conclusion of the session, six startle response trials were again presented to measure habituation of the startle response. Startle responses were measured over 100 ms, by recording the peak startle amplitude and the time required to reach the peak were recorded. For auditory brainstem response (ABR) measurements, mice (8–12 weeks of age) were anesthetized and transferred into a sound-attenuation chamber. An EC1 speaker [Tucker Davis Technologies (TDT), Gainesville, FL] was coupled to the external ear canal, and electrodes were inserted at the vertex and pinna while a ground was inserted near the tail. 12.1-balanced click stimuli were applied per second, each with a duration of 0.1 ms, starting at 90 dB and decreasing stepwise in intensity. Recordings were performed with a TDT System 3 auditory evoked response workstation. The number of acquisition trials was set at 512 (256 rarefaction and 256 condensation stimuli); the mean ± SD were determined. Auditory thresholds were analyzed for both ears. Testing of wild-type C57BL/6J determined the baseline threshold to be 35 ± 15 dB. This baseline was stable for up to at least 9 months of age, as reported previously (Zheng et al., 1999). Using the TDT workstation, the cubic (2f1-f2) distortion product otoacoustic emission (DPOAE) thresholds were determined at a range of frequencies (6–28 kHz), evoked using two equal intensity primary stimuli about the test frequency at ratios of 0.894 and 1.118, applied to the ear canal using two EC1 speakers coupled via an ER10B+ microphone probe (Etymotic Research, Elk Grove Village, IL). Stimuli were increased in intensity from 0 to 70 dB in 5 dB increments to establish thresholds. To assess vestibular function, mice were placed for 10 min in an open field (43 × 43 cm), and activity was measured using infrared beams and software (Med Associates, Lafayette, IN). The total distance traveled and the number of small-diameter rotations (<2.75 cm radius) were measured [baseline value for wild-type mice (mean ± SE), 3855 ± 117 cm distance traveled; 10 ± 5 small-diameter rotations/min]. Mice were considered affected when they scored three or more SDs above the mean. For swim tests, mice were placed in a glass cylinder (diameter, 18 cm) filled with water (25°C). Over a period of 6 min, all movements were registered with a video tracking system. Histology, whole mount staining, and scanning electron microscopy (SEM) were performed as described previously (Belvindrah et al., 2006; Senften et al., 2006). In situ hybridization was performed on 12-μm-thick cryosections as described previously (Tiveron et al., 1996). Antisense RNA probes were as follows: Otof, bp 799–1503 of AF183183; Mass1, bp 2653–3084 of NM054053; sDFNA5 [full-length coding sequence (1059 bp) corresponding to the gene model 1001 (National Center for Biotechnology Information), LOC381375].
Linkage analysis and DNA sequencing.
Affected mice were bred with 129S1/SvImJ and/or BALB/cByJ mice. The F1 offspring were intercrossed to obtain F2 mice, and tail DNA was prepared for linkage mapping using single nucleotide polymorphism (SNP) markers (Wiltshire et al., 2003). Map Manager QTX (Manly et al., 2001) was used to calculate logarithm of the odds scores and perform interval mapping. Exons and exon–intron boundaries of genes in the mapped intervals were sequenced. Primers for PCR amplification and DNA sequencing were designed with Primer 3 software (Massachusetts Institute of Technology, Cambridge, MA).
Mutation screening by denaturing HPLC.
The mouse amino acid sequence of sDFNA5 was backtranslated and aligned with human chr2:179143644–179151505 by BLAST (basic local alignment search tool). Intron/exon boundaries were defined, and intronic primers were designed to screen each of the six coding exons of sDFNA5 for mutations. Affected probands (177), born of a consanguineious marriage, were amplified by PCR using 20 μg of genomic DNA in a 30 μl reaction contain-ing 3 μl of 10× buffer [160 mm (NH4)2SO4, 670 mm Tris-HCl, pH 8.8, 0.1% Tween 20], 0.99 μl (50 mm MgCl2), 1.5 μl of dNTP mixture (2.5 mm each dATP, dCTP, dTTP, and dGTP), 20 pmol each forward and reverse primer, and 0.12 U DNA polymerase (Bioline, Springfield, NJ). Conventional amplification conditions were used; samples were first denatured at 94°C for 4 min, underwent 40 cycles of 94°C for 30 s, 56°C for 30 s, 72°C for 30 s, and a final extension at 72°C for 10 min. The first half of the samples were heteroduplexed with the second half of the samples and ran on denaturing HPLC at 56 and 57.5°C. Samples producing allele variant profiles were identified and directly sequenced to determine the particular variant. One of the 177 affected probands producing a mutant chromatogram was sequenced and found to have a deletion of an Adenine at the 12 s nucleotide from the start codon (ATG) of the first exon creating a premature stop codon at amino acid 58 (see Figs. 2, 3).
Figure 2.

Inheritance tests. F2 mice obtained from crosses between affected mice on the C57BL/6J background with mice from the indicated inbred strains were phenotyped by ABR measurements. The total number of animals analyzed and the number of affected animals (gray bars) are indicated. Mice that inherit their phenotype recessively are expected to show the phenotype in ∼25% of the offspring.
Figure 3.

Analysis of the ABR response in rueda, twist, and pachanga mice. A–C, Click-evoked ABRs at different sound intensities. The peaks of waves I–IV are indicated. Averaged recordings from 2-month-old mutant (red) and age-matched wild-type (black) animals are shown. Note that the same traces are shown in all panels for wild-type mice, which represent the average of all recordings collected with 2-month-old wild-type animals. All mouse lines showed thresholds at or above 90 dB. Note that some rueda mice showed residual activity between 60 and 90 dB. D, Rueda mice carry a point mutation in Mass1 that introduces a G-to-T transversion leading to a premature stop-codon. Predicted proteins (VLGR1a-e, Mass1.1–1.3) generated from alternatively spliced Mass1 transcripts are shown (Nikkila et al., 2000; Skradski et al., 2001; McMillan et al., 2002; McMillan and White, 2004; Yagi et al., 2005). The mutation in rueda affects only some Mass1 variants. EC, Extracellular domain; TM, transmembrane domain. E, A point mutation in the Myo6 gene creates a stop-codon. HD, Head domain; IQ, IQ domain; CC, coiled coil; TD, tail domain. F, A point mutation introduces an Asp-to-Gly substitution in the most C-terminal C2 domain of otoferlin. TM, Transmembrane domain.
Histology, immunohistochemistry, and scanning electron microscopy.
Hematoxylin/eosin staining of sections was performed as described previously (Farinas et al., 1996; Müller et al., 1997). Whole-mount staining and scanning electron microscopy of cochlear sensory epithelia were performed as described previously (Senften et al., 2006).
Results
Identification of mouse lines with auditory defects
Male C57BL/6J mice were treated with ENU and bred with wild-type C57BL/6J females. The resulting G1 males were bred with wild-type C57BL/6J females to produce G2 females, which were backcrossed to their G1 fathers. The resulting G3 offspring were phenotyped (Fig. 1 A). In total, we analyzed ∼850 pedigrees in a variety of assays. To obtain the most interesting mouse lines for studying the processing of auditory signals, we inspected the mice visually and performed behavioral tests to exclude lines with obvious developmental defects or general nervous system dysfunction (Fig. 1 A). At 4 weeks of age, mice were analyzed for morphological abnormalities such as reduced body size, abnormal posture, or distorted facial features. Mice that did not show obvious defects were further analyzed in behavioral assays that were performed between 8 and 12 weeks of age: open-field test to analyze locomotor behavior and anxiety; ASR to analyze locomotor activity in response to an auditory stimulus; prepulse inhibition to analyze the ability to filter environmental information; fear conditioning to analyze amygdala and hippocampal-based memory; wheel running to analyze circadian behavior; hot plate and tail flick assay to analyze pain perception; in some mice, Morris water maze and object recognition tests to assess spatial learning and memory (Fig. 1 A).
Figure 1.
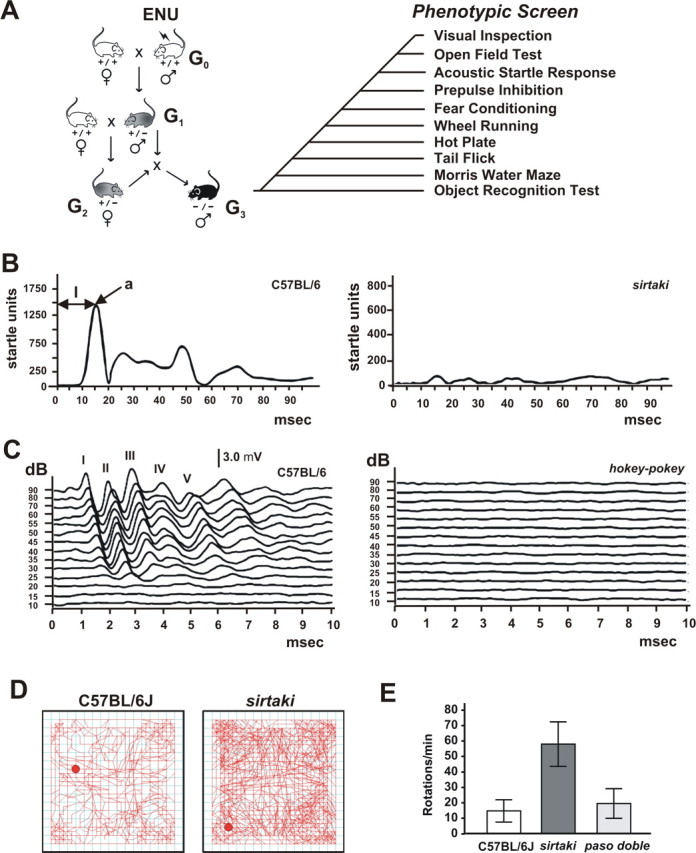
Screen design. A, Male C57BL/6J mice were treated with ENU and bred with wild-type C57BL/6J females. Male G1 offspring were bred with wild-type C57BL/6J females to produce G2 females, which were backcrossed to their G1 fathers. The resulting G3 offspring were phenotyped as indicated. As a reference, the experiments were also performed with age-matched wild-type mice. Note that for some lines, some of the tests (such as swim tests) were not performed with G3 animals but with animals from subsequent generations. B, Representative startle traces from an ASR recording. The C57BL/6J wild-type mouse shows a maximal startle amplitude (a) of 1500 relative startle units with a latency time (l) of 15 ms (msec). No startle response was observed in one mouse line from the screen, which we named sirtaki. C, Representative click-evoked ABR waveforms at different sound intensities. ABR waves I–V are indicated for recordings obtained from a wild-type control mouse. One affected mouse line from the screen, which we named hokey-pokey, is shown as an example. hokey-pokey mice did not respond to sound intensities up to 90 dB and were considered deaf. D, Representative open-field recording. The red dot indicates the position of the mouse at the start of the recording, and the red line traces the movement. Sirtaki mice are hyperactive. E, Analysis of the movement in the open field shows that some lines such as sirtaki show increased small-diameter rotations (<2.75 cm radius). Others, such as paso-doble, are not affected. Values are given as mean ± SD.
Mice that score positive in the ASR (Fig. 1 B, Table 1) can be hearing impaired or otherwise show defects elsewhere in the nervous system or in motor function. To identify among the lines with startle defects those with auditory phenotypes, we next measured the ABR (Fig. 1 C, Table 1). The ABR assay establishes auditory thresholds and provides a measure of the sound transduction sensitivity of cochlear hair cells and the degree of synchronization among discharging afferent neurons (Zheng et al., 1999). Click stimuli were applied to mice (8 weeks of age) starting at 90 dB and then decreasing in intensity. Testing of wild-type C57BL/6J determined the baseline ABR threshold to be 35 ± 15 dB. Of ∼850 pedigrees analyzed, 51 pedigrees (6%) had a low ASR. In the ABR test, 16 of the 51 pedigrees (31%) did not respond to sound levels up to 90 dB and were considered severely hearing impaired or deaf. Mice in three pedigrees (6%) showed elevated ABR thresholds and were considered moderately hearing impaired (Table 1).
Table 1.
Mouse lines with auditory and vestibular phenotypes
| Mouse line | Locus abbreviation | Gene/chromosome/map position (mB) | Inheritance | ASR | ABR (dB) | Circling | Swimming |
|---|---|---|---|---|---|---|---|
| Rueda | Rda | Mass1 | Recessive | Low | 90 | No | Normal |
| Pachanga | Pga | Otoferlin | Recessive | Low | >90 | No | Normal |
| Twist | Twt | Myo6 | Recessive | Low | >90 | Yes | Disoriented |
| Sirtaki | Sti | Pejvakin | Recessive | Low | >90 | Yes | In circles |
| Salsa | Sal | Chr 10 (56.1–60.6) | Recessive | Low | >90 | No | Normal |
| Salsa Rueda | Salr | Chr 10 (50.2–67.0) | Recessive | Low | >70 | No | Normal |
| Pogo | Pgo | Chr 11 (37–64) | Recessive | Low | >90 | Yes | Disoriented |
| Polka | Pok | Chr 7 (75.3–100.2) | Recessive | Low | >90 | Yes | Disoriented |
| Hokey Pokey | Hkp | Chr 2 (6–67) | Recessive | Low | >90 | Yes | Disoriented |
| Merengue | Mge | IP | Recessive | Low | >90 | No | Normal |
| Samba | Sba | IP | Recessive | Low | >90 | No | Normal |
| Mambo | Mbo | IP | Recessive | Low | >90 | No | Normal |
| Cha-cha | Cha | IP | Recessive | Low | >90 | No | Normal |
| Paso doble | Pado | IP | IP | Low | >90 | No | Normal |
| Rumba | Rba | IP | Recessive | Low | >90 | Yes | Disoriented |
| Bossa Nova | Bona | IP | IP | Low | >90 | No | IP |
| Jive | Jve | IP | IP | Low | >90 | No | IP |
| Lambada | Lda | IP | IP | Low | >70 | No | IP |
| Bolero | Bro | IP | IP | Low | >70 | No | IP |
mB, Megabase; Chr., chromsome; IP, in progress.
Auditory defects are frequently associated with vestibular defects. Therefore, mice with elevated ABR thresholds were tested for hyperactivity and circling behavior in an open-field chamber equipped with infrared detection connected to a computer for automated data analysis (Fig. 1 D). Several parameters were recorded over 10 min, including the total distance traveled and the number of small-diameter rotations (Fig. 1 D,E). Baseline values for wild-type mice were established: 3855 ± 117 cm for distance traveled, and 10 ± 5 rotations (maximum 2.75 cm radius). Mice were considered affected if they scored ≥3 SDs above the mean. Among the 19 lines with auditory defects, six lines were hyperactive and circling. To identify among mouse lines that were hyperactivity/circling unambiguously those with balance defects, and to search for a more subtle vestibular defect in the remaining mouse lines, we next performed forced swim tests. The same six lines that were hyperactive/circling were affected to varying degrees. Mice from five lines were disoriented, unable to maintain an upright position, and had to be rescued from drowning. Mice from one additional line swam in tight circles (Table 1). The remaining analyzed lines were unaffected in their vestibular function. Lines with only an auditory phenotype were named in accordance with Latin dances, whereas lines with auditory and vestibular defects received names of folk dances (Table 1).
We next determined the heritability of the auditory/vestibular phenotypes. We crossed affected G3 mice to C57BL/6J mice and intercrossed the F1 offspring to obtain the F2 generation. Both F1 and F2 animals were phenotyped. Lines that inherit their auditory/vestibular defects as dominant traits are predicted to give rise to affected animals in F1. Lines that inherit their phenotype recessively are predicted to generate 25% affected offspring in F2. Among the 19 lines analyzed, 14 lines inherited their phenotype recessively (Table 1) (see below). Heritability testing is in progress for the remaining lines, but preliminary analysis indicates that two of the lines (paso doble and bossa nova) may inherit their phenotype dominantly (Table 1).
Positional cloning
To map and positionally clone the affected genes, we outcrossed affected mice to 129S1/SvImJ or BALB/cByJ mice. We intercrossed the F1 mice and phenotyped the F2 progeny by ABR and activity/circling tests. All tested lines inherited their phenotype stably on the mixed genetic background and maintained the recessive mode of inheritance (Fig. 2). Next, we isolated tail DNA from the F2 mice to map the affected genomic loci using SNP markers as described previously (Wiltshire et al., 2003) (for SNP markers, see http://snp.gnf.org). Thus far, we have mapped mutations to individual chromosomes or genomic intervals in nine affected mouse lines (Figs. 3, 5; Table 1). The mutations map to loci throughout the genome with no apparent preference for a particular chromosome or genomic region on a chromosome.
Figure 5.
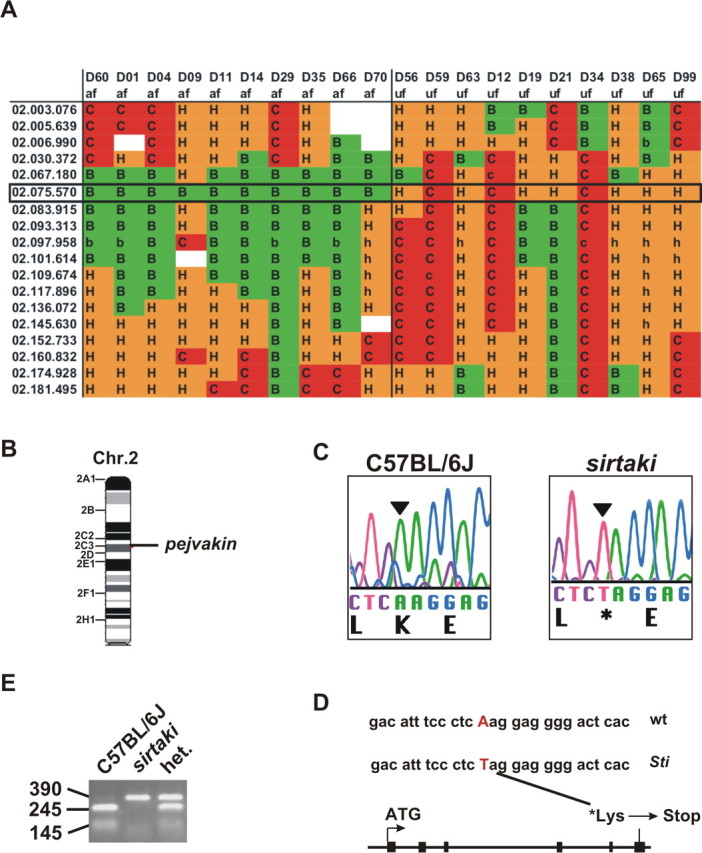
Mapping of the mutation in sirtaki mice. A, The diagram shows the fine mapping data linking the phenotype in sirtaki mice to a region on chromosome 2. A box delineates the affected interval. Markers in the first column indicate chromosome and megabase position. The mouse identification numbers (D) and the auditory phenotypes (af, affected; uf, unaffected) are indicated in the top two rows. The genotype for each SNP marker is represented as follows: B, homozygous C57BL/6J; C, homozygous BALB/cByJ; H, heterozygous; and blanks indicate SNPs for which the data collection was incomplete. B, The mutation in sirtaki mice maps to an interval on chromosome 2 that contains the pejvakin gene. C, D, We identified a point mutation in the sixth coding exon (black boxes) of the pejvakin gene, which is predicted to introduce a stop-codon at amino acid 290. E, The point mutation eliminates a Sml1 restriction site, facilitating genotyping of the mice. An agarose gel picture is shown where a genomic PCR fragment from a wild-type, heterozygous, and homozygous sirtaki mouse was resolved after cutting with SmlI (wild type, 245 and 145 bp; mutant, 390 bp).
To identify mutations by DNA sequencing, we prioritized our efforts based on several criteria. First, we focused on mouse lines that were obtained early in the screen and where we had sufficient numbers of affected offspring available for additional phenotypic characterization. Second, in mouse lines in which the affected interval contained a candidate gene that has been linked previously to deafness, we sequenced all exons and exon–intron borders of the candidate genes and of neighboring genes as well. By identifying mutations in known “deafness” genes, we would be able to verify our screen and demonstrate its usefulness for obtaining mouse models carrying new alleles of known human disease genes. Third, for mouse lines in which the genomic interval had not been linked to deafness, we designed primers to sequence the exons and exon–intron boundaries of the predicted genes in the interval. As outlined below, we identified single nucleotide changes in four mouse lines. In each case, the mutation is predicted to introduce a premature stop codon or change single amino acids in functionally important protein domains. Consistent with the recessive mode of inheritance, unaffected parents carry the mutation in the heterozygous state, whereas the mutation is homozygous in all affected offspring but not in their unaffected littermates.
Analysis of rueda, twist, and pachanga mice
Our initial positional cloning efforts and additional phenotypic characterization focused on rueda, twist, and pachanga mice. Measurement of the ABR in 3-week-old and 2-month-old animals revealed that the mouse lines are severely hearing impaired, with auditory thresholds of 90 dB (rueda) or >90 dB (twist, pachanga) (Fig. 3 A–C, Table 1). Only twist mice show hyperactivity and circling behavior consistent with vestibular defects (Table 1). A general histological survey using sagittal sections did not reveal obvious morphological defects in the inner ear or brain (data not shown). Because the ABR depends on hair cell function, we next evaluated hair cell morphology in postnatal day 5 (P5) cochlear whole mounts by staining with phalloidin to label F-actin in stereocilia. Hair cell morphology was also examined by SEM. Remarkably, the three mouse lines show defects in the hair bundles of cochlear hair cells (Fig. 4). In rueda mice, the width of hair bundles in outer hair cells is irregular. Bundles are organized asymmetrically with one side of the bundle being longer than the other (Fig. 4 B,G,H, arrows). Kinocilia in hair cells are shifted to ectopic positions, and the polarization of the hair bundles is perturbed (Fig. 4 G,H, arrowheads). In twist mice, hair bundles are disorganized, fragmented, and frequently contain malformed stereocilia in reduced numbers (Fig. 4 C, arrows). In pachanga mice, hair bundles appeared mostly normal when analyzed by light microscopy, but we occasionally observed hair cells with hair bundles that appeared slightly distorted (Fig. 4 D, arrows). Consistent with this finding, SEM analysis of the cochlea of pachanga mice revealed that hair bundles are also split at times (Fig. 4 I,J, arrows). Together, these findings suggest that the genes mutated in rueda, twist, and pachanga control the development and/or function of hair cells.
Figure 4.

Analysis of hair cell morphology in rueda, twist, and pachanga mice. A–D, Cochlear whole mounts of P5 animals were stained with phalloidin (green). Outer hair cells (OHC) and inner hair cells (IHC) are indicated. Hair bundle morphology and polarity were altered in rueda, twist, and pachanga mice (arrows). E–J, Cochlear whole mounts were analyzed by SEM. Kinocilia in hair bundles of rueda mice were frequently in ectopic positions (G, H, arrowheads), and hair bundles were asymmetric (H, arrow). Hair bundles in pachanga mice were often split (I, J, arrows). Scale bars: (in A) A–D, 8 μm; (in E) E, G, I, 5 μm; (in F) F, H, J, 2 μm.
By SNP mapping, we linked the phenotype in rueda mice to a genomic interval on chromosome 13, in twist mice to an interval on chromosome 9, and in pachanga mice to an interval on chromosome 5 (Table 1). All three intervals contain genes that have been linked to deafness and hair cell function. These are the Mass1 gene (rueda mice; Usher Syndrome Type 2C) (Weston et al., 2004), the Myo6 gene (twist mice; DFNA22, DFNB37) (Melchionda et al., 2001; Ahmed et al., 2003), and the otoferlin gene (pachanga mice, DFNB9) (Migliosi et al., 2002; Rodriguez-Ballesteros et al., 2003; Varga et al., 2003, 2006; Tekin et al., 2005; Wang et al., 2005). In rueda mice, we identified a G-to-T transversion in the Mass1 gene, which is uniquely homozygous in mice with an auditory phenotype (Fig. 3 D). Several splice variants have been described for Mass1. The longest variant encodes VLGR-1 (very large G-coupled receptor-1), a transmembrane protein with homology to G-coupled receptors (Fig. 3 D) (Nikkila et al., 2000; Skradski et al., 2001; McMillan et al., 2002; McMillan and White, 2004; Yagi et al., 2005). The mutation in rueda mice causes a premature stop codon that is predicted to truncate the VLGR-1 protein and also to affect some of the shorter splice variants (Fig. 3 D). Studies in mice have shown that VLGR-1 colocalizes with ankle links at the base of stereocilia (McGee et al., 2006). VLGR-1 is required for the structural integrity of hair bundles in the cochlea but not the vestibule (McGee et al., 2006). Consistent with these findings, rueda mice show hair bundle defects in the cochlea (Fig. 4 B,G,H) and are hearing impaired (Fig. 3 A) but show no balance defects (Table 1).
Twist mice contain a T-to-A transversion in the Myo6 gene that is predicted to replace a tyrosine residue with a stop codon that truncates the tail domain (Fig. 3 E). The mutation is located at an exon/intron boundary that precedes an alternatively spliced exon (Breckler et al., 2000) and could also lead to an internal deletion by affecting splicing. The murine Myo6 gene is expressed in hair cells, and mutations in Myo6 affect hair bundle morphology leading to hearing and balance defects (Avraham et al., 1995). Consistent with this finding, twist mice show hair bundle defects and are deaf and balance impaired (Figs. 3 B, 4 C; Table 1).
In pachanga mice, we identified a point mutation in the otoferlin gene, introducing an amino acid substitution (Asp to Gly) close to the C terminus of the protein (Fig. 3 F). Recent findings have shown that otoferlin is required for the function of ribbon synapses in hair cells (Roux et al., 2006). Consistent with this finding, pachanga mice show ABR thresholds above 90 dB (Fig. 3 C), but the DPOAE is unaffected (data not shown). Otoferlin contains six C2 domains that are implicated in Ca2+ binding, and otoferlin displays Ca2+-dependent interactions with components of the synaptic vesicle release machinery. Importantly, the mutation in pachanga mice affects an Asp residue in the most C-terminal C2 domain. Asp residues in the C2 domain of rat synaptotagmin are important for binding Ca2+ (Sutton et al., 1995), which suggests that the mutation in pachanga affects binding of Ca2+ to otoferlin. Hair bundles of pachanga mice have subtle structural defects (Fig. 4 I,J). No such defects have been reported in other mice with defects in the function of hair cell synapses (Platzer et al., 2000; Brandt et al., 2003; Sidi et al., 2004), which implies that otoferlin may have additional functions beyond a role in controlling synaptic function in hair cells.
Together, our findings show that we have obtained novel alleles of genes from the screen that have been linked previously to hair cell function. Importantly, mutations in two of these genes affect auditory but not vestibular function, which shows that our screen is useful for identifying genes and mutations that specifically affect the cochlea. Our findings also demonstrate that the new alleles are useful for defining protein domains that are important for gene function, such as the C2 domain of otoferlin.
Sirtaki mice carry a point mutation in the pejvakin gene
The mutation in sirtaki maps to a genomic interval on chromosome 2 (Fig. 5 A,B). Genome analysis using public databases (GenBank; University of California, Santa Cruz, Santa Cruz, CA) revealed several genes within the interval. Multiple sequence alignments showed that one of the genes in the interval is a member of the gasdermin family and a close homolog of the gasdermin protein DFNA5 (Fig. 6) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), which is mutated in autosomal dominant nonsyndromic hearing impairment (Van Laer et al., 1998). Sequencing of exons and exon–intron boundaries of the genes in the affected interval revealed only one point mutation, which is present within an exon of the gene with similarity to DFNA5 (Fig. 5 C,D). The mutation, an A-to-T transversion, fortuitously eliminates a restriction site, allowing for easy genotyping (Fig. 5 E). The mutation is uniquely homozygous in all affected sirtaki mice and heterozygous in their unaffected parents, consistent with the recessive mode of inheritance of the auditory phenotype. The mutation creates a stop codon, which leads to the deletion of the last 62 amino acids of the predicted 352 amino acid-containing protein (Figs. 5 D, 6).
Figure 6.
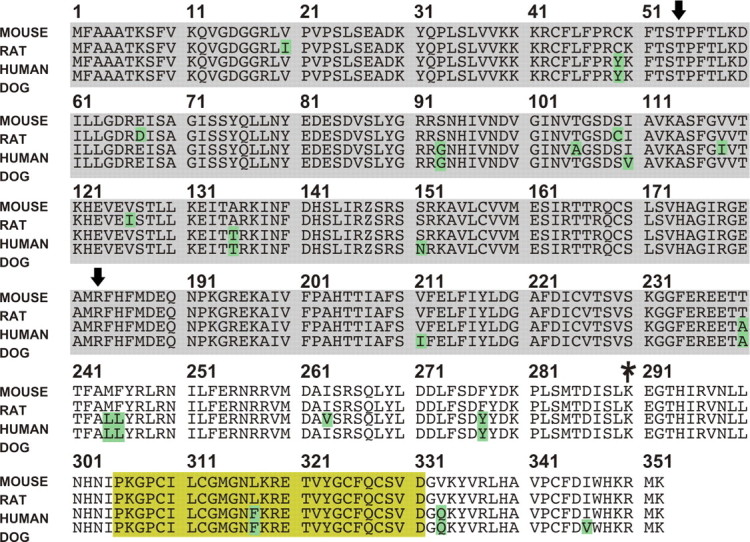
Sequence alignment between pejvakin orthologs. The protein is highly conserved across species. Divergent amino acids are indicated with green boxes. The domain with homology to gasdermin proteins is indicated with a gray box (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). The yellow box outlines a domain with homology to zinc-fingers. The asterisk indicates the position of the stop-codon in sirtaki mice. The arrows indicate the missense mutations in DFNB59 patients.
While our work was in progress, two missense mutations in the gene affected in sirtaki mice were identified in humans afflicted with recessive nonsyndromic deafness (DFNB59) (missense mutations are indicated by arrows in Fig. 6), the gene was named pejvakin, and a mouse model carrying a point mutation mimicking DFNB59 was developed (Delmaghani et al., 2006). The phenotype of the humans and DFNB59 model mice has been characterized as a nonprogressive auditory neuropathy, where the transmission of auditory signals is impaired, while cochlear hair cell function is maintained (Delmaghani et al., 2006). Vestibular function is also maintained in DFNB59 patients and model mice (Delmaghani et al., 2006). In contrast, sirtaki mice show vestibular defects (Fig. 1, Table 1), suggesting that the pejvakin allele in sirtaki mice leads to distinct phenotypic consequences from the previously described alleles. Interestingly, pejvakin contains in its C-terminal portion a domain with weak homology to zinc-fingers (Fig. 6, yellow box). Zinc-finger domains in other proteins mediate interactions with proteins, DNA, or RNA (Brown et al., 2001; Brown, 2005; Sarai and Kono, 2005). The missense mutations associated with DFNB59 are positioned outside the zinc-finger domain, whereas the mutation in sirtaki mice is predicted to truncate pejvakin before the zinc-finger domain, which could account for the phenotypic variability associated with the different mutations (see Discussion).
Sirtaki mice have progressive hearing loss and defects in outer hair cell function
To define the cause of the auditory phenotype in sirtaki mice, we searched for morphological defects. We analyzed sagittal sections from the inner ears and brains of 3-month-old mice but could not detect any obvious morphological defects (Fig. 7) (data not shown). The organ of corti and vestibular sensory epithelia such as the utricle and saccule were unaffected. The neurons of the vestibulocochlear ganglion were also maintained, and we could not find evidence of degenerative changes (Fig. 7) (data not shown). Using in situ hybridization, expression of pejvakin was detected in hair cells of the cochlea and vestibule but not in neurons (Fig. 8 A–E), suggesting that hair cell development or function may be affected. We therefore analyzed the cochlear sensory epithelia in P5 and P30 mice by immunofluorescence analysis and SEM. At both of these ages, there was no detectable defect in the patterning of the cochlear sensory epithelium (Fig. 8 F,G,I,J,L,O). The morphology of cochlear hair bundles was also not affected (Fig. 8 H,K,M,N,P,Q).
Figure 7.
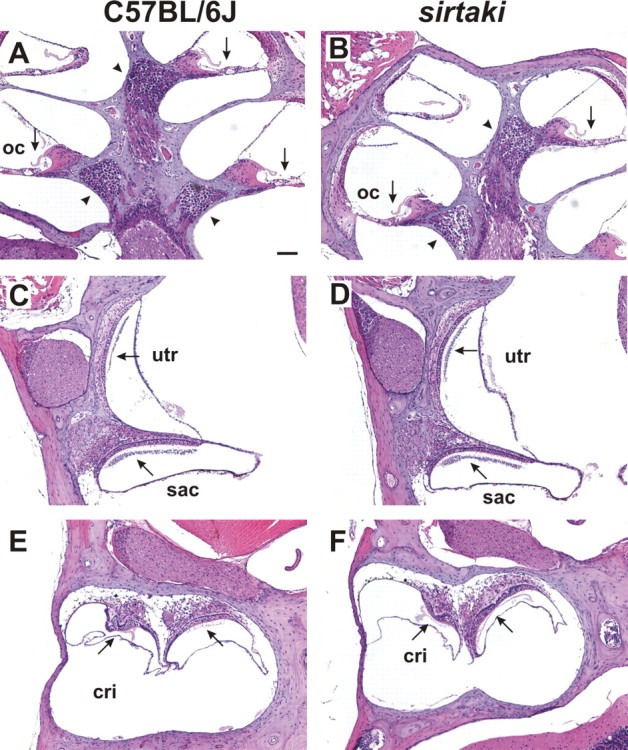
Analysis of inner ears by histology. Sagittal paraffin sections from the ears of C57BL/6J wild-type mice (A, C, E) and sirtaki mice (B, D, F) at 3 months of age were stained with hematoxylin and eosin. The cochlea (A, B) and vestibule (C–F) are shown. The spiral ganglia are marked with an arrowhead. Arrows point to the sensory epithelia. No obvious defects were observed in sirtaki mice. OC, Organ of corti; utr, utricle; cri, crista. Scale bar, 80 μm.
Figure 8.

Pejvakin expression in hair cells and hair cell morphology. A–E, In situ hybridization demonstrates that pejvakin is strongly expressed at P1 and P4 in hair cells of the cochlea (A, B, D) and vestibule (C) (arrows). No signal is seen with the sense control probe (E). F–K, Hair cell morphology at P5. Cochlear whole mounts were processed for staining with phalloidin (green) and an antibody to CDH23 (red) (F, I) or for SEM analysis (G, H, J, K). There were no obvious defects in the patterning of the cochlear sensory epithelium in three rows of OHCs and one row of IHCs, and hair bundle morphology was not affected. L–Q, SEM analysis of cochlear whole mounts (8-week-old animals) revealed no obvious structural defects. Scale bars: A, 200 μm; B, C, 50 μm; D, E, 10 μm; F, I, 8 μm; G, J, 5 μm; L, O, 2.5 μm; H, K, M, N, P, Q, 2 μm.
In the initial ENU screen of 8-week-old G3 animals, ABR thresholds in sirtaki mice in response to click-stimuli were determined to be ≥90 dB (Fig. 1, Table 1). Because the auditory system in wild-type mice is almost completely mature at 3 weeks of age, defects in hearing function in older mice could be the result of progressive hearing loss. We therefore analyzed ABR thresholds in sirtaki mice at earlier time points. At 3 weeks of age, the earliest time point tested, ABR thresholds in sirtaki mice were 74 ± 13 dB, whereas auditory thresholds in control littermates were 35 ± 15 dB (Fig. 9 A,B). In contrast, ABR thresholds in 8-week-old sirtaki mice were ≥90 dB (Fig. 9 A,B), consistent with the data obtained with 8-week-old G3 animals in the initial screen (Table 1). Analysis of auditory thresholds using pure tones between 4 and 32 kHz revealed that sirtaki mice were equally affected across all frequencies (Fig. 9 C). We conclude that the auditory phenotype in sirtaki mice is progressive in nature and affects all tested frequencies.
Figure 9.
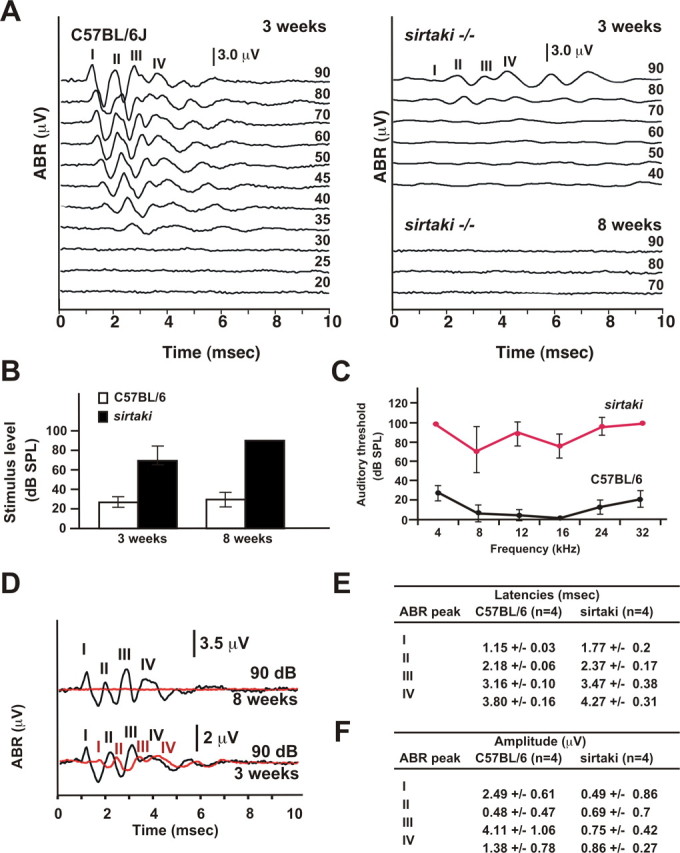
Analysis of sirtaki mice by ABR measurements. A, Representative ABR recordings in response to click stimuli for a 3-week-old wild-type C57BL/6J mouse and a 3-week-old and 8-week-old sirtaki mouse. The auditory threshold in the wild-type mouse was at 35 dB. The 3-week-old sirtaki mouse showed ABR thresholds at 70–80 dB, whereas the 8-week-old sirtaki mouse showed no response even at 90 dB. B, ABR thresholds (click-stimuli) in 3-week-old and 8-week-old mice (3-week-old: wild type n = 4, sirtaki n = 6; 8-week-old: wild type n = 9, sirtaki n = 7; the mean ± SD is indicated). C, ABR thresholds were determined for pure tones between 4 and 32 kHz for 8-week-old mice. Thresholds are elevated in sirtaki mice (red trace) across all analyzed frequencies (n = 6 for both wild-type and sirtaki mice; the mean ± SD is indicated). D, ABR traces (click-stimuli) at 90 dB were superimposed (wild type, black; sirtaki, red). Note that wave I–IV in 3-week-old sirtaki mice showed augmented latencies and reduced peak amplitudes. E, F, The latencies and amplitudes for ABR peak I–IV in 3-week-old mice (n = 4) in response to a click-stimulus at 90 dB were determined. The latency for all waves was significantly augmented in sirtaki mice. The amplitudes of the peaks were also significantly reduced with substantial variation between different sirtaki mice. Values are given as mean ± SD.
Analysis of the ABR waveforms in 3-week-old mice revealed additional defects. An overlay of ABR traces recorded at 90 dB demonstrated that the absolute latencies of all four ABR waveforms were significantly augmented in 3-week-old animals (Fig. 9 D,E). In addition, the amplitude of the peaks was significantly reduced in the mutants and showed considerable variability between animals (Fig. 9 D,F). Importantly, we did not observe any structural defects in the cochlea or nervous system of sirtaki mice (Figs. 7, 8). The findings suggest that the defect is caused by functional perturbations that affect the communication of hair cells with afferent neurons, signal propagation along the auditory nerve, or both.
In the cochlea, we observed strong expression of pejvakin in hair cells, suggesting that the auditory phenotype could be caused at least in part by hair cell defects. One possibility is that the progressive loss of sensitivity in the auditory sense organ could be a consequence of compromised outer hair cell function, which leads to defects in the amplification of acoustic signals. Perturbations that affect outer hair cell function can be determined by measuring the DPOAE. We therefore computed DPOAE response spectra from the acoustic signal recorded from the ear canal in homozygous sirtaki mice and control littermates. The experiments were performed at stimulus frequencies between 6 and 28 kHz and at an intensity range between 0 and 70 dB. Figure 10 A shows representative DPOAE response spectra for an 8-week-old wild-type and mutant mouse for two stimulus conditions. Acoustic signals for the primary stimulus frequencies (f1, f2) and the cubic distortion frequency (2f1-f2) could be recorded from the ear canal of mutant animals at high sound intensities (Fig. 10 A, bottom traces for 70 dB) but not at lower sound intensities (Fig. 10 A, top traces for 25 dB). In Figure 10 B, we plotted DPOAE levels at 16 kHz for stimulus levels between 15 and 70 dB. As reported previously, the DPOAE levels of wild-type mice increase with the stimulus intensity at a given frequency (Paraham et al., 2001). In contrast, DPOAE levels in sirtaki mutants were within noise levels at most stimulus intensities, and only a small response could be recorded at 70 dB. Similar observations were made at all of the frequencies analyzed (6–28 kHz) (Fig. 10 C). We conclude that the mechanical activity of outer hair cells across the entire analyzed frequency spectrum is functionally impaired in sirtaki mice.
Figure 10.
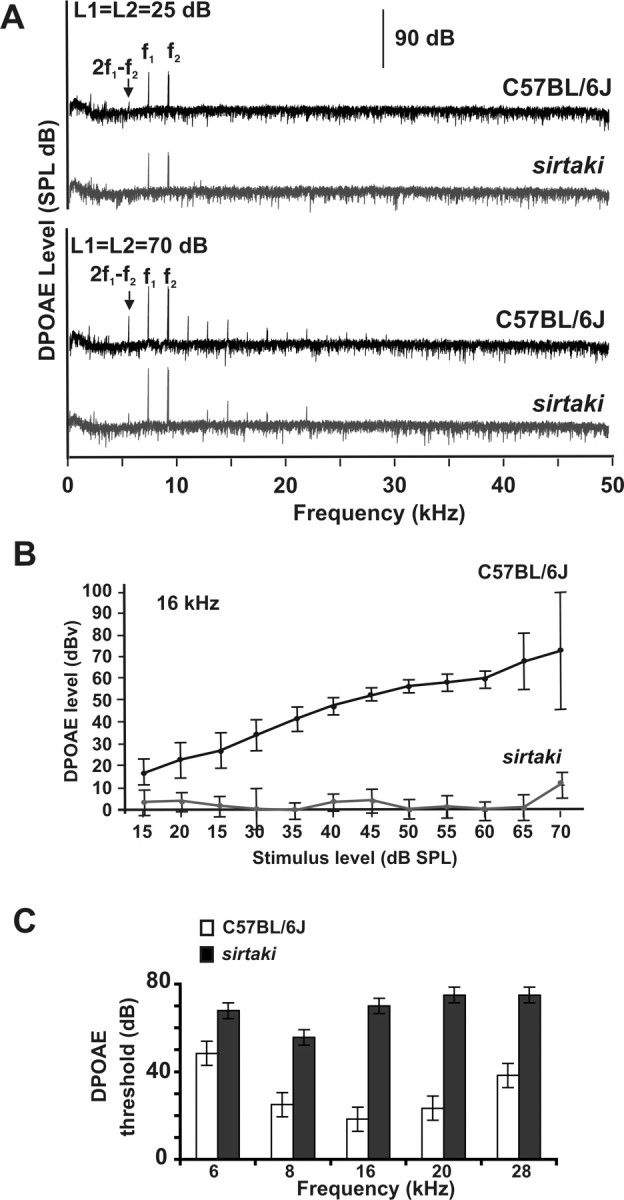
Measurement of the DPOAE. A, Representative DPOAE response spectra for an 8-week-old wild-type and sirtaki mouse at 25 dB (top traces) and 70 dB (bottom traces). Note the 2f1-f2 product, which is absent in recordings with sirtaki mice at 25 dB. B, DPOAE measurements were performed with 8-week-old mice at 16 kHz using stimulus levels between 15 and 70 dB. Sirtaki mice were severely affected at all intensity levels (n = 3, wild type; n = 7, sirtaki; the mean ± SD are indicated). C, DPOAE thresholds in 8-week-old sirtaki mice were elevated at all frequencies analyzed (n = 3, wild-type; n = 7, sirtaki; the mean ± SD are indicated).
Interestingly, sirtaki mice show defects in the latency of ABR waves as well as outer hair cell defects, whereas mice with a missense mutation in pejvakin show delays in ABR waves but no outer hair cell defects (Delmaghani et al., 2006). These findings suggest that the missense mutation specifically affects pejvakin function in neurons, whereas the allele described here is more severe and affects both hair cells and neurons. Notably, the morphology or survival of hair cells and spiral ganglion neurons appears unaffected, which suggests that pejvakin is important for the function but not development or maintenance of the two cell types.
Mutations in the human sDFNA5 gene
Previous findings have associated two missense mutations in the pejvakin gene with DFNB59, a nonsyndromic form of deafness that is nonprogressive (Delmaghani et al., 2006). Based on our findings that hearing loss in sirtaki mice is progressive, we searched for mutations in the human pejvakin gene that are associated with progressive hearing loss. We completed mutation screening of the human ortholog in probands from 177 consanguineous families that segregates autosomal recessive nonsyndromic deafness. We identified one family segregating a 122delA mutation in the pejvakin gene (Fig. 11). In contrast to previously described DFNB59 patients, the affected children show moderate-to-profound deafness, and the phenotype is progressive in nature. The affected individuals demonstrate autozygosity-by-descent to a parental region on chromosome 2 flanked by microsatellite markers D2S2330 and D2S2382 and are homozygous for the 122delA mutation. The unaffected sibling and parents are heterozygous carriers of this mutation (Fig. 11). The deletion is predicted to result in a premature stop-codon at amino acid 58, which most likely results in a functionally null allele, which could explain the progressive nature of the auditory phenotype, a phenotype that is not apparent in DFNB59 patients carrying missense mutations (Delmaghani et al., 2006).
Figure 11.

Family L-998 carries a mutation in the human pejvakin gene. A consanguineous Iranian family in which children with profound deafness demonstrate autozygosity-by-descent to a parental region on chromosome 2 flanked by microsatellite markers D2S2330 and D2S2382. DNA sequencing shows that the deaf children are homozygous for a deletion of an adenosine (122delA); the unaffected sibling and both parents are carriers of this mutation.
Discussion
We demonstrate here that forward genetics screens in mice are powerful tools for studying auditory perception and for providing animal models for inherited forms of deafness in humans. ENU mutagenesis screens in mice have been performed previously with success, but these screens were designed to identify dominant alleles. Many of the identified mutations have gross morphological defects, indicative of perturbations in developmental processes (Hrabe de Angelis et al., 2000; Nolan et al., 2000; Kiernan et al., 2001; Tsai et al., 2001; Vreugde et al., 2002; Curtin et al., 2003; Hardisty et al., 2003; Rhodes et al., 2003, 2004; Marcotti et al., 2006). However, many forms of deafness in humans are caused by point mutations that are inherited as recessive traits and affect cochlear function without causing gross morphological alterations (Friedman and Griffith, 2003). We therefore performed a screen for recessive mutations to increase the repertoire of animal models for the human disease. Our screen was designed to exclude mice with general defects in development and nervous system function, as determined by histological and behavioral analysis. Among 850 pedigrees analyzed, we identified 19 mouse lines that are afflicted with hearing loss. All lines that have been analyzed thus far inherit their phenotype recessively. Thirteen of the 19 mouse lines show auditory defects without vestibular impairment. Importantly, analysis of general tissue morphology and nervous system function revealed no defects outside the inner ear. In addition, the first four genes that we characterized are expressed in mechanosensory hair cells, which show morphological or functional defects in the mutant mice. These findings indicate that hair cells are particularly vulnerable to genetic perturbations, and that the mouse lines will be valuable tools for the research community to study defects in cochlear function that cause deafness. Mapping studies demonstrate that the affected genes are located throughout the genome without any obvious preference, suggesting that there is no particular hot spot for ENU mutagenesis.
From our screen, we identified novel alleles of genes that have been linked previously to deafness in humans, which demonstrates that the screen provides animal models that are relevant for studying disease mechanisms. First, we identified a mutation in the Mass1 gene, which has been linked in humans to USH2C (Weston et al., 2004). The mutation in the Mass1 gene in rueda mice most likely leads to a functional null allele that mimics previously obtained alleles (Skradski et al., 2001; McMillan and White, 2004; Johnson et al., 2005; Yagi et al., 2005). The VLGR-1 protein that is encoded by the Mass1 gene colocalized with the ankle-link in hair cells (McGee et al., 2006), suggesting that the phenotype in rueda mice is caused at least in part by defects in ankle links. Second, we identified in twist mice a mutation in the Myo6 gene, which has been linked to dominant and recessive forms of deafness in humans (DFNA22, DFNB37) (Melchionda et al., 2001; Ahmed et al., 2003). The allele in twist mice is predicted to truncate the tail domain of the molecular motor protein Myo6. Because the Myo6 tail domain is involved in the docking of transport cargo (Buss et al., 2004), defects in this process likely cause deafness in twist mice. Third, in pachanga mice, we obtained a mutation in the otoferlin gene, which has been linked to DFNB9 in humans (Migliosi et al., 2002; Rodriguez-Ballesteros et al., 2003; Varga et al., 2003, 2006; Tekin et al., 2005; Wang et al., 2005). Several lines of evidence suggest that pachanga mice are an excellent model for the human disease. First, the human and mouse otoferlin proteins are highly homologous and contain six putative Ca2+ binding C2 domains (Sutton et al., 1995). In DFNB9 patients, as in pachanga mice, mutations have been identified within the C2 domains (Migliosi et al., 2002; Rodriguez-Ballesteros et al., 2003; Varga et al., 2003, 2006; Tekin et al., 2005; Wang et al., 2005). Importantly, the mutation in pachanga mice affects an Asp residue. Asp residues in C2 domains of rat synaptotagmin are important for binding Ca2+ (Sutton et al., 1995), suggesting that the mutation in pachanga mice (and some human patients) affects binding of Ca2+ to otoferlin. Second, DFNB9 patients have been diagnosed with auditory neuropathy, a form of deafness characterized by impaired auditory function (defects in the ABR) but preserved outer hair cell function (normal otoacoustic emission) (Migliosi et al., 2002; Rodriguez-Ballesteros et al., 2003; Varga et al., 2003, 2006; Tekin et al., 2005; Wang et al., 2005). Similarly, pachanga mice show ABR thresholds above 90 dB, but the DPOAE is unaffected. Recent findings have shown that otoferlin binds in a Ca2+-dependent manner to components of the synaptic vesicle release machinery and is required for ribbon synapse function (Roux et al., 2006). These findings suggest that the C2 domain affected in pachanga mice is important for the function of otoferlin at synapses. However, pachanga mice show subtle defects in the morphology of hair bundles that are not easily explained by synaptic defects. In muscle, the otoferlin homolog dysferlin is required for membrane repair (Bansal et al., 2003). As one possibility, otoferlin in hair cells may control membrane vesicle fusion that is not restricted to synaptic sites and is important for membrane repair. This model can be tested with the availability of the pachanga mouse model.
Our screen has also identified a novel deafness locus, which has been linked in parallel studies to DFNB59 in humans (Delmaghani et al., 2006). The locus encodes pejvakin, a member of the gasdermin protein family, and our studies provide important new insights into the function of pejvakin and potential disease mechanisms. Studies by Delmaghani et al. (2006) have identified two missense mutations in the pejvakin gene that cause deafness DFNB59. In mice that carry a missense mutation that mimics one of the mutations found in human DFNB59 patients, ABR thresholds are elevated, and the absolute latencies of the ABR peaks are augmented, but there is no defect in the DPOAE. These findings suggest that outer hair cell function is maintained, but that afferent auditory neurons are affected (Delmaghani et al., 2006). We now show that sirtaki mice carry a point mutation that introduces a premature stop codon, which is predicted to truncate the pejvakin protein. Similar to the previously reported DFNB59 model mice with a missense mutation, sirtaki mice show an augmentation of the absolute latencies in the ABR peaks. However, unlike the previous mouse model, sirtaki mice suffer from hearing loss that is progressive. DPOAE measurements reveal defects in outer hair cell function across all frequencies, a phenotype that was not observed in the previous mouse model. The allele in sirtaki mice truncates the pejvakin protein before a highly conserved domain with homology to zinc-fingers, which in other proteins mediate interactions with proteins, DNA, or RNA (Brown et al., 2001; Brown, 2005; Sarai and Kono, 2005). One possibility is that the zinc-finger domain may have an essential function in hair cells that is not affected by the missense mutations. Alternatively, the mutation in sirtaki mice may lead to an unstable mRNA or protein and represent a functional null allele of the pejvakin gene. Interestingly, we also identified a point mutation in the human pejvakin gene that likely creates a functional null allele. Unlike previously described alleles in humans and mice, but similar to the sirtaki allele, the mutation causes progressive hearing loss. Together, these findings indicate that the molecular pathogenesis associated with different mutations in pejvakin is far more complex than previously appreciated. Based on our findings and the previous study, we propose that functional null alleles of pejvakin inactivate its function in both hair cells and neurons, whereas reported missense mutations represent hypomorphic alleles that selectively affect its function only in neurons.
Pejvakin is a member of the gasdermin family (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) (Delmaghani et al., 2006). Unfortunately, little is known about the function of gasdermin proteins. We were not able to identify any structural defects in the cochlea and spiral ganglion of sirtaki mice, and SEM analysis confirmed that hair bundle morphology is not affected in the mice. These findings suggest that pejvakin controls the function of hair cells and afferent neurons but is not essential for the development or maintenance of these cell types. Aside from a protein domain with homology to zinc-finger proteins, we have not been able to identify motifs within pejvakin that would shed light on its biochemical function. The closest homolog of pejvakin is DFNA5. Mutations in DFNA5 cause progressive dominant nonsyndromic deafness in humans, but the mechanisms of DFNA5 function is not known (Van Laer et al., 2004). Studies in yeast and in cultured mammalian cells have provided evidence that DFNA5 controls the cell cycle and is involved in the p53-mediated cellular response to DNA damage (Gregan et al., 2003; Masuda et al., 2006). Other members of the gasdermin family have been implicated in alopecia (Runkel et al., 2004; Lunny et al., 2005), but in none of the cases has the biochemical function of gasdermin proteins been defined. Because mutations in pejvakin affect the function of two cell types that are specialized for the generation and propagation of electrical signals (mechanosensory hair cells and sensory neurons), pejvakin may be essential for the signaling properties of these cells. Intriguingly, the human and murine pejvakin alleles reported here as well as previously reported alleles for DFNA5 cause progressive hearing loss. As one possibility, the progressive hearing loss caused by mutations in the two highly homologous genes may be caused by a similar mechanism and includes defects in hair cells. Mutations in DFNA5 are inherited as dominant traits, raising the possibility that the resulting mutation leads to a gain of function phenotype or that the mutant protein acts as a dominant negative (Van Laer et al., 2004). It is tempting to speculate that DFNA5 and pejvakin act in a similar pathway and that the mutant DFNA5 protein may affect the function of the wild-type pejvakin protein.
In summary, we generated mouse lines with mutations that are inherited as recessive traits and affect cochlear function. Unlike previously described screens for dominant deafness traits, the mouse lines described here show defects in mechanosensory hair cells that are not associated with gross morphological changes elsewhere in the inner ear or nervous system. Because sensorineuronal hearing loss is the most common cause of deafness in humans, we anticipate that the mouse lines will be an important resource for the research community to gain new insights into the genetic mechanisms that control cochlear function and to study molecular defects that cause deafness.
Footnotes
This work was supported by the Novartis Research Foundation (L.M.T., T.W.); National Institutes of Health–National Institute on Deafness and Other Communication Disorders Grants DC005965 and DC007704 (U.M.), DC02842 and DC03555 (R.J.H.S.), and DC00139 (A.F.R.); the Research Service of the Veterans Affairs (A.F.R.); the Iranian Deputy of Research and Technology, Ministry of Health and Medical Education Grant P6193 (H.N.); James Cook Fellowship Royal Society of New Zealand (G.D.H.); and a fellowship from the Fondation pour la Recherche Médicale (N.G.). We thank members of the Müller laboratory for discussions and Evelyn York (Scripps Institute of Oceanography) for help with the SEM.
References
- Ahmed ZM, Morell RJ, Riazuddin S, Gropman A, Shaukat S, Ahmad MM, Mohiddin SA, Fananapazir L, Caruso RC, Husnain T, Khan SN, Griffith AJ, Friedman TB, Wilcox ER. Mutations of MYO6 are associated with recessive deafness, DFNB37. Am J Hum Genet. 2003;72:1315–1322. doi: 10.1086/375122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashby J, Gorelick NJ, Shelby MD. Mutation assays in male germ cells from transgenic mice: overview of study and conclusions. Mutat Res. 1997;388:111–122. doi: 10.1016/s1383-5718(96)00107-6. [DOI] [PubMed] [Google Scholar]
- Avraham KB, Hasson T, Steel KP, Kingsley DM, Russell LB, Mooseker MS, Copeland NG, Jenkins NA. The mouse Snell's waltzer deafness gene encodes an unconventional myosin required for structural integrity of inner ear hair cells. Nat Genet. 1995;11:369–375. doi: 10.1038/ng1295-369. [DOI] [PubMed] [Google Scholar]
- Bansal D, Miyake K, Vogel SS, Groh S, Chen CC, Williamson R, McNeil PL, Campbell KP. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature. 2003;423:168–172. doi: 10.1038/nature01573. [DOI] [PubMed] [Google Scholar]
- Belvindrah R, Nalbant P, Ding S, Wu C, Bokoch GM, Muller U. Integrin-linked kinase regulates Bergmann glial differentiation during cerebellar development. Mol Cell Neurosci. 2006;33:109–125. doi: 10.1016/j.mcn.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Brandt A, Striessnig J, Moser T. CaV1.3 channels are essential for development and presynaptic activity of cochlear inner hair cells. J Neurosci. 2003;23:10832–10840. doi: 10.1523/JNEUROSCI.23-34-10832.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breckler J, Au K, Cheng J, Hasson T, Burnside B. Novel myosin VI isoform is abundantly expressed in retina. Exp Eye Res. 2000;70:121–134. doi: 10.1006/exer.1999.0758. [DOI] [PubMed] [Google Scholar]
- Brown RS. Zinc finger proteins: getting a grip on RNA. Curr Opin Struct Biol. 2005;15:94–98. doi: 10.1016/j.sbi.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Brown S, Coghill ID, McGrath MJ, Robinson PA. Role of LIM domains in mediating signaling protein interactions. IUBMB Life. 2001;51:359–364. doi: 10.1080/152165401753366113. [DOI] [PubMed] [Google Scholar]
- Buss F, Spudich G, Kendrick-Jones J. Myosin VI: cellular functions and motor properties. Annu Rev Cell Dev Biol. 2004;20:649–676. doi: 10.1146/annurev.cellbio.20.012103.094243. [DOI] [PubMed] [Google Scholar]
- Curtin JA, Quint E, Tsipouri V, Arkell RM, Cattanach B, Copp AJ, Henderson DJ, Spurr N, Stanier P, Fisher EM, Nolan PM, Steel KP, Brown SD, Gray IC, Murdoch JN. Mutation of Celsr1 disrupts planar polarity of inner ear hair cells and causes severe neural tube defects in the mouse. Curr Biol. 2003;13:1129–1133. doi: 10.1016/s0960-9822(03)00374-9. [DOI] [PubMed] [Google Scholar]
- Delmaghani S, del Castillo FJ, Michel V, Leibovici M, Aghaie A, Ron U, Van Laer L, Ben-Tal N, Van Camp G, Weil D, Langa F, Lathrop M, Avan P, Petit C. Mutations in the gene encoding pejvakin, a newly identified protein of the afferent auditory pathway, cause DFNB59 auditory neuropathy. Nat Genet. 2006;38:770–778. doi: 10.1038/ng1829. [DOI] [PubMed] [Google Scholar]
- Farinas I, Yoshida CK, Backus C, Reichardt LF. Lack of neurotrophin-3 results in death of spinal sensory neurons and premature differentiation of their precursors. Neuron. 1996;17:1065–1078. doi: 10.1016/s0896-6273(00)80240-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman TB, Griffith AJ. Human nonsyndromic sensorineural deafness. Annu Rev Genomics Hum Genet. 2003;4:341–402. doi: 10.1146/annurev.genom.4.070802.110347. [DOI] [PubMed] [Google Scholar]
- Gillespie PG, Walker RG. Molecular basis of mechanosensory transduction. Nature. 2001;413:194–202. doi: 10.1038/35093011. [DOI] [PubMed] [Google Scholar]
- Gregan J, Van Laer L, Lieto LD, Van Camp G, Kearsey SE. A yeast model for the study of human DFNA5, a gene mutated in nonsyndromic hearing impairment. Biochim Biophys Acta. 2003;1638:179–186. doi: 10.1016/s0925-4439(03)00083-8. [DOI] [PubMed] [Google Scholar]
- Hardisty RE, Erven A, Logan K, Morse S, Guionaud S, Sancho-Oliver S, Hunter AJ, Brown SD, Steel KP. The deaf mouse mutant Jeff (Jf) is a single gene model of otitis media. J Assoc Res Otolaryngol. 2003;4:130–138. doi: 10.1007/s10162-002-3015-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrabe de Angelis MH, Flaswinkel H, Fuchs H, Rathkolb B, Soewarto D, Marschall S, Heffner S, Pargent W, Wuensch K, Jung M, Reis A, Richter T, Alessandrini F, Jakob T, Fuchs E, Kolb H, Kremmer E, Schaeble K, Rollinski B, Roscher A, et al. Genome-wide, large-scale production of mutant mice by ENU mutagenesis. Nat Genet. 2000;25:444–447. doi: 10.1038/78146. [DOI] [PubMed] [Google Scholar]
- Johnson KR, Zheng QY, Weston MD, Ptacek LJ, Noben-Trauth K. The Mass1frings mutation underlies early onset hearing impairment in BUB/BnJ mice, a model for the auditory pathology of Usher syndrome IIC. Genomics. 2005;85:582–590. doi: 10.1016/j.ygeno.2005.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiernan AE, Ahituv N, Fuchs H, Balling R, Avraham KB, Steel KP, Hrabe de Angelis M. The Notch ligand Jagged1 is required for inner ear sensory development. Proc Natl Acad Sci USA. 2001;98:3873–3878. doi: 10.1073/pnas.071496998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunny DP, Weed E, Nolan PM, Marquardt A, Augustin M, Porter RM. Mutations in gasdermin 3 cause aberrant differentiation of the hair follicle and sebaceous gland. J Invest Dermatol. 2005;124:615–621. doi: 10.1111/j.0022-202X.2005.23623.x. [DOI] [PubMed] [Google Scholar]
- Manly KF, Cudmore RH, Jr, Meer JM. Map Manager QTX, cross-platform software for genetic mapping. Mamm Genome. 2001;12:930–932. doi: 10.1007/s00335-001-1016-3. [DOI] [PubMed] [Google Scholar]
- Marcotti W, Erven A, Johnson SL, Steel KP, Kros CJ. Tmc1 is necessary for normal functional maturation and survival of inner and outer hair cells in the mouse cochlea. J Physiol (Lond) 2006;574:677–698. doi: 10.1113/jphysiol.2005.095661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda Y, Futamura M, Kamino H, Nakamura Y, Kitamura N, Ohnishi S, Miyamoto Y, Ichikawa H, Ohta T, Ohki M, Kiyono T, Egami H, Baba H, Arakawa H. The potential role of DFNA5, a hearing impairment gene, in p53-mediated cellular response to DNA damage. J Hum Genet. 2006;51:652–664. doi: 10.1007/s10038-006-0004-6. [DOI] [PubMed] [Google Scholar]
- McGee J, Goodyear RJ, McMillan DR, Stauffer EA, Holt JR, Locke KG, Birch DG, Legan PK, White PC, Walsh EJ, Richardson GP. The very large G-protein-coupled receptor VLGR1: a component of the ankle link complex required for the normal development of auditory hair bundles. J Neurosci. 2006;26:6543–6553. doi: 10.1523/JNEUROSCI.0693-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan DR, White PC. Loss of the transmembrane and cytoplasmic domains of the very large G-protein-coupled receptor-1 (VLGR1 or Mass1) causes audiogenic seizures in mice. Mol Cell Neurosci. 2004;26:322–329. doi: 10.1016/j.mcn.2004.02.005. [DOI] [PubMed] [Google Scholar]
- McMillan DR, Kayes-Wandover KM, Richardson JA, White PC. Very large G protein-coupled receptor-1, the largest known cell surface protein, is highly expressed in the developing central nervous system. J Biol Chem. 2002;277:785–792. doi: 10.1074/jbc.M108929200. [DOI] [PubMed] [Google Scholar]
- Melchionda S, Ahituv N, Bisceglia L, Sobe T, Glaser F, Rabionet R, Arbones ML, Notarangelo A, Di Iorio E, Carella M, Zelante L, Estivill X, Avraham KB, Gasparini P. MYO6, the human homologue of the gene responsible for deafness in Snell's waltzer mice, is mutated in autosomal dominant nonsyndromic hearing loss. Am J Hum Genet. 2001;69:635–640. doi: 10.1086/323156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliosi V, Modamio-Hoybjor S, Moreno-Pelayo MA, Rodriguez-Ballesteros M, Villamar M, Telleria D, Menendez I, Moreno F, Del Castillo I. Q829X, a novel mutation in the gene encoding otoferlin (OTOF), is frequently found in Spanish patients with prelingual non-syndromic hearing loss. J Med Genet. 2002;39:502–506. doi: 10.1136/jmg.39.7.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller U, Wang D, Denda S, Meneses JJ, Pedersen RA, Reichardt LF. Integrin alpha8beta1 is critically important for epithelial-mesenchymal interactions during kidney morphogenesis. Cell. 1997;88:603–613. doi: 10.1016/s0092-8674(00)81903-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikkila H, McMillan DR, Nunez BS, Pascoe L, Curnow KM, White PC. Sequence similarities between a novel putative G protein-coupled receptor and Na+/Ca2+ exchangers define a cation binding domain. Mol Endocrinol. 2000;14:1351–1364. doi: 10.1210/mend.14.9.0511. [DOI] [PubMed] [Google Scholar]
- Nolan PM, Peters J, Vizor L, Strivens M, Washbourne R, Hough T, Wells C, Glenister P, Thornton C, Martin J, Fisher E, Rogers D, Hagan J, Reavill C, Gray I, Wood J, Spurr N, Browne M, Rastan S, Hunter J, Brown SD. Implementation of a large-scale ENU mutagenesis program: towards increasing the mouse mutant resource. Mamm Genome. 2000;11:500–506. doi: 10.1007/s003350010096. [DOI] [PubMed] [Google Scholar]
- Paraham K, Xiao-Ming S, Kim DO. Noninvasive assessment of auditory function in mice: auditory brain stem response and distortion product otoacoustic emission. In: Willot JF, editor. Handbook of mouse auditory research. Boca Raton, FL: CRC; 2001. pp. 37–58. [Google Scholar]
- Platzer J, Engel J, Schrott-Fischer A, Stephan K, Bova S, Chen H, Zheng H, Striessnig J. Congenital deafness and sinoatrial node dysfunction in mice lacking class D L-type Ca2+ channels. Cell. 2000;102:89–97. doi: 10.1016/s0092-8674(00)00013-1. [DOI] [PubMed] [Google Scholar]
- Quint E, Steel KP. Use of mouse genetics for studying inner ear development. Curr Top Dev Biol. 2003;57:45–83. doi: 10.1016/s0070-2153(03)57002-8. [DOI] [PubMed] [Google Scholar]
- Reijmers LG, Coats JK, Pletcher MT, Wiltshire T, Tarantino LM, Mayford M. A mutant mouse with a highly specific contextual fear-conditioning deficit found in an N-ethyl-N-nitrosourea (ENU) mutagenesis screen. Learn Mem. 2006;13:143–149. doi: 10.1101/lm.98606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes CR, Parkinson N, Tsai H, Brooker D, Mansell S, Spurr N, Hunter AJ, Steel KP, Brown SD. The homeobox gene Emx2 underlies middle ear and inner ear defects in the deaf mouse mutant pardon. J Neurocytol. 2003;32:1143–1154. doi: 10.1023/B:NEUR.0000021908.98337.91. [DOI] [PubMed] [Google Scholar]
- Rhodes CR, Hertzano R, Fuchs H, Bell RE, De Angelis MH, Steel KP, Avraham KB. A Myo7a mutation cosegregates with stereocilia defects and low-frequency hearing impairment. Mamm Genome. 2004;15:686–697. doi: 10.1007/s00335-004-2344-x. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Ballesteros M, del Castillo FJ, Martin Y, Moreno-Pelayo MA, Morera C, Prieto F, Marco J, Morant A, Gallo-Teran J, Morales-Angulo C, Navas C, Trinidad G, Tapia MC, Moreno F, del Castillo I. Auditory neuropathy in patients carrying mutations in the otoferlin gene (OTOF) Hum Mutat. 2003;22:451–456. doi: 10.1002/humu.10274. [DOI] [PubMed] [Google Scholar]
- Roux I, Safieddine S, Nouvian R, Grati M, Simmler MC, Bahloul A, Perfettini I, Le Gall M, Rostaing P, Hamard G, Triller A, Avan P, Moser T, Petit C. Otoferlin, defective in a human deafness form, is essential for exocytosis at the auditory ribbon synapse. Cell. 2006;127:277–289. doi: 10.1016/j.cell.2006.08.040. [DOI] [PubMed] [Google Scholar]
- Runkel F, Marquardt A, Stoeger C, Kochmann E, Simon D, Kohnke B, Korthaus D, Wattler F, Fuchs H, Hrabe de Angelis M, Stumm G, Nehls M, Wattler S, Franz T, Augustin M. The dominant alopecia phenotypes Bareskin, Rex-denuded, and Reduced Coat 2 are caused by mutations in gasdermin 3. Genomics. 2004;84:824–835. doi: 10.1016/j.ygeno.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Sarai A, Kono H. Protein-DNA recognition patterns and predictions. Annu Rev Biophys Biomol Struct. 2005;34:379–398. doi: 10.1146/annurev.biophys.34.040204.144537. [DOI] [PubMed] [Google Scholar]
- Senften M, Schwander M, Kazmierczak P, Lillo C, Shin JB, Hasson T, Geleoc GS, Gillespie PG, Williams D, Holt JR, Muller U. Physical and functional interaction between protocadherin 15 and myosin VIIa in mechanosensory hair cells. J Neurosci. 2006;26:2060–2071. doi: 10.1523/JNEUROSCI.4251-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelby MD, Tindall KR. Mammalian germ cell mutagenicity of ENU, IPMS and MMS, chemicals selected for a transgenic mouse collaborative study. Mutat Res. 1997;388:99–109. doi: 10.1016/s1383-5718(96)00106-4. [DOI] [PubMed] [Google Scholar]
- Sidi S, Busch-Nentwich E, Friedrich R, Schoenberger U, Nicolson T. gemini encodes a zebrafish L-type calcium channel that localizes at sensory hair cell ribbon synapses. J Neurosci. 2004;24:4213–4223. doi: 10.1523/JNEUROSCI.0223-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skradski SL, Clark AM, Jiang H, White HS, Fu YH, Ptacek LJ. A novel gene causing a mendelian audiogenic mouse epilepsy. Neuron. 2001;31:537–544. doi: 10.1016/s0896-6273(01)00397-x. [DOI] [PubMed] [Google Scholar]
- Sutton RB, Davletov BA, Berghuis AM, Sudhof TC, Sprang SR. Structure of the first C2 domain of synaptotagmin I: a novel Ca2+/phospholipid-binding fold. Cell. 1995;80:929–938. doi: 10.1016/0092-8674(95)90296-1. [DOI] [PubMed] [Google Scholar]
- Tekin M, Akcayoz D, Incesulu A. A novel missense mutation in a C2 domain of OTOF results in autosomal recessive auditory neuropathy. Am J Med Genet A. 2005;138:6–10. doi: 10.1002/ajmg.a.30907. [DOI] [PubMed] [Google Scholar]
- Tiveron MC, Hirsch MR, Brunet JF. The expression pattern of the transcription factor Phox2 delineates synaptic pathways of the autonomic nervous system. J Neurosci. 1996;16:7649–7660. doi: 10.1523/JNEUROSCI.16-23-07649.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai H, Hardisty RE, Rhodes C, Kiernan AE, Roby P, Tymowska-Lalanne Z, Mburu P, Rastan S, Hunter AJ, Brown SD, Steel KP. The mouse slalom mutant demonstrates a role for Jagged1 in neuroepithelial patterning in the organ of Corti. Hum Mol Genet. 2001;10:507–512. doi: 10.1093/hmg/10.5.507. [DOI] [PubMed] [Google Scholar]
- Van Laer L, Huizing EH, Verstreken M, van Zuijlen D, Wauters JG, Bossuyt PJ, Van de Heyning P, McGuirt WT, Smith RJ, Willems PJ, Legan PK, Richardson GP, Van Camp G. Nonsyndromic hearing impairment is associated with a mutation in DFNA5. Nat Genet. 1998;20:194–197. doi: 10.1038/2503. [DOI] [PubMed] [Google Scholar]
- Van Laer L, Vrijens K, Thys S, Van Tendeloo VF, Smith RJ, Van Bockstaele DR, Timmermans JP, Van Camp G. DFNA5: hearing impairment exon instead of hearing impairment gene? J Med Genet. 2004;41:401–406. doi: 10.1136/jmg.2003.015073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga R, Kelley PM, Keats BJ, Starr A, Leal SM, Cohn E, Kimberling WJ. Non-syndromic recessive auditory neuropathy is the result of mutations in the otoferlin (OTOF) gene. J Med Genet. 2003;40:45–50. doi: 10.1136/jmg.40.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga R, Avenarius MR, Kelley PM, Keats BJ, Berlin CI, Hood LJ, Morlet TG, Brashears SM, Starr A, Cohn ES, Smith RJ, Kimberling WJ. OTOF mutations revealed by genetic analysis of hearing loss families including a potential temperature sensitive auditory neuropathy allele. J Med Genet. 2006;43:576–581. doi: 10.1136/jmg.2005.038612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vreugde S, Erven A, Kros CJ, Marcotti W, Fuchs H, Kurima K, Wilcox ER, Friedman TB, Griffith AJ, Balling R, Hrabe De Angelis M, Avraham KB, Steel KP. Beethoven, a mouse model for dominant, progressive hearing loss DFNA36. Nat Genet. 2002;30:257–258. doi: 10.1038/ng848. [DOI] [PubMed] [Google Scholar]
- Wang Q, Li R, Zhao H, Peters JL, Liu Q, Yang L, Han D, Greinwald JH, Jr, Young WY, Guan MX. Clinical and molecular characterization of a Chinese patient with auditory neuropathy associated with mitochondrial 12S rRNA T1095C mutation. Am J Med Genet A. 2005;133:27–30. doi: 10.1002/ajmg.a.30424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston MD, Luijendijk MW, Humphrey KD, Moller C, Kimberling WJ. Mutations in the VLGR1 gene implicate G-protein signaling in the pathogenesis of Usher syndrome type II. Am J Hum Genet. 2004;74:357–366. doi: 10.1086/381685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiltshire T, Pletcher MT, Batalov S, Barnes SW, Tarantino LM, Cooke MP, Wu H, Smylie K, Santrosyan A, Copeland NG, Jenkins NA, Kalush F, Mural RJ, Glynne RJ, Kay SA, Adams MD, Fletcher CF. Genome-wide single-nucleotide polymorphism analysis defines haplotype patterns in mouse. Proc Natl Acad Sci USA. 2003;100:3380–3385. doi: 10.1073/pnas.0130101100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi H, Takamura Y, Yoneda T, Konno D, Akagi Y, Yoshida K, Sato M. Vlgr1 knockout mice show audiogenic seizure susceptibility. J Neurochem. 2005;92:191–202. doi: 10.1111/j.1471-4159.2004.02875.x. [DOI] [PubMed] [Google Scholar]
- Zheng QY, Johnson KR, Erway LC. Assessment of hearing in 80 inbred strains of mice by ABR threshold analyses. Hear Res. 1999;130:94–107. doi: 10.1016/s0378-5955(99)00003-9. [DOI] [PMC free article] [PubMed] [Google Scholar]