

Abstract
We developed a transgenic mouse line that expresses the Gi-coupled RASSL (receptor activated solely by synthetic ligand) Ro1 in astrocytes to study astrocyte–neuronal communication. Surprisingly, we found that all transgenics expressing Ro1 developed hydrocephalus. We analyzed these mice in an effort to develop a new model of hydrocephalus that will further our understanding of the pathophysiology of the disease. Expression of Ro1 was restricted to astrocytes by crossing the transgenic hGFAP-tTA (tet transactivator behind the human glial fibrillary acidic protein promoter) mouse line with the transgenic tetO-Ro1/tetO-LacZ mouse line. This cross produced double-transgenic mice that expressed Ro1 in astrocytes. All double transgenics developed hydrocephalus by postnatal day 15, whereas single-transgenic littermate controls appeared normal. Hydrocephalic Ro1 mice displayed enlarged ventricles, partial denudation of the ependymal cell layer, altered subcommissural organ morphology, and obliteration of the cerebral aqueduct. Severely hydrocephalic mice also had increased levels of phospho-Erk and GFAP expression. Administration of doxycycline to breeding pairs suppressed Ro1 expression and the onset of hydrocephalus in double-transgenic offspring. Ro1 animals maintained on dox did not develop hydrocephalus; however, if taken off doxycycline at weaning, double-transgenic mice developed enlarged ventricles within 7 weeks, indicating that Ro1 expression also induces hydrocephalus in adults. This study discovered a new model of hydrocephalus in which the rate of pathogenesis can be controlled enabling the study of the pathogenesis of both juvenile and adult onset hydrocephalus.
Keywords: astrocyte, hydrocephalus, opioid, Gi, RASSL, SCO, tetracycline
Introduction
Astrocytes carry out numerous functions critical for the CNS to develop and operate normally. Certain of these functions are passive and supportive in nature; for example, astrocytes prevent neural excitotoxicity by buffering extracelluar potassium (Gabriel et al., 1998; Walz, 2000; D'Ambrosio et al., 2002). However, recent studies suggest astrocytes can actively modulate or initiate neuronal signaling (Carmignoto et al., 1997; Araque et al., 2000, 2001, 2002; Pasti et al., 2001; Fiacco and McCarthy, 2004). If astrocytes actively regulate neurophysiology, alterations in astrocyte–neuronal signaling could contribute to the etiology of many CNS disorders. Isolating astrocytic signaling systems to test this hypothesis is difficult because astrocytes and neurons express a similar complement of neuroligand receptors (Porter and McCarthy, 1996). Consequently, neuronal responses to astrocyte signaling under normal or pathological conditions have been difficult to measure directly.
We generated a mouse line expressing the Gi-coupled Ro1 receptor activated solely by synthetic ligands (RASSL) in astrocytes to directly assess the role of astrocytic G-protein-coupled receptors (GPCRs) in neurophysiology. Ro1 is a κ-opioid receptor (KOR) modified by replacing its second extracellular loop with the second extracellular loop of the δ-opioid receptor (Fig. 1) and adding the FLAG epitope tag to the N terminus. Consequently, the affinity of Ro1 for endogenous ligands is greatly reduced while maintaining its ability to bind the synthetic KOR agonist spiradoline (Coward et al., 1998; Redfern et al., 1999). Expressing Ro1 in astrocytes on a KOR knock-out background allows us to activate Gi-coupled receptor signaling in astrocytes while measuring changes in neuronal excitability.
Figure 1.

Ro1 structure [from Coward et al. (1998)]. To create Ro1, the second extracellular loop of the κ-opioid receptor was replaced with the second extracellular loop of the δ-opioid receptor. A FLAG tag was added to the N terminus for detection. Ro1 has a greatly reduced affinity for endogenous ligands but still responds to the small molecule drug spiradoline.
Restricting Ro1 expression to astrocytes required crossing two lines of transgenic mice, the Ro1 line and the tet-transactivator (tTA) line. The Ro1 line carries the Ro1 gene under the control of the tetO promoter; the tTA line carries the tTA gene under the control of a 2.0 kb fragment of the human glial fibrillary acidic protein (hGFAP) promoter. Because Ro1 expression requires tTA and tTA expression is restricted to GFAP-positive cells, double-transgenic progeny will express Ro1 only in astrocytes. Additionally, Ro1 expression levels can be regulated using doxycycline (dox) to bind tTA, preventing it from binding the tetO promoter. Although double-transgenic mice were obtained in normal numbers, they unexpectedly developed severe hydrocephalus when maintained off dox. This is the first indication that activating a Gi-coupled GPCR in astrocytes can lead to hydrocephalus.
Hydrocephalus, the net accumulation of CSF in the ventricular system, is a serious neurological disorder whose pathogenesis is poorly understood. Current treatments carry high risk for additional complications (Blount et al., 1993; Bondurant and Jimenez, 1995; Patwardhan and Nanda, 2005) and neurological problems often persist after treatment (Fernell et al., 1994; Mancao et al., 1998; Del Bigio, 2004). Understanding the causes of hydrocephalus is critical for developing new treatment and prevention options. Our finding suggests aberrant astrocytic signaling may underlie the development of certain forms of hydrocephalus and provides a new model in which the timing and progression of hydrocephalus can be manipulated.
Materials and Methods
Animals.
All experiments were performed in accordance with the Institutional Animal Care and Use Committee at the University of North Carolina at Chapel Hill and federal guidelines. Animals were maintained in climate-controlled housing with a 12 h light/dark cycle and were given food and water ad libitum. The day of birth was defined as postnatal day 0 (P0).
Transgenic mouse lines.
The hGFAP-tTA mouse line was made by isolating the tTA gene from pUHD 15-1, kindly provided by H. Bujard (Universität Heidelberg, Heidelberg, Germany), and cloning it downstream of the 2 kb human GFAP promoter in place of lacZ in the pGFA2lac1 vector (Pascual et al., 2005). The GFAP-tTA-mP1 cassette was placed between four copies of genomic insulators from chicken β-globin gene (Chung et al., 1993). Transgenic mice were obtained by standard methods and backcrossed to C57BL/6J for at least five generations. FVB/N-TgN(tetO-Ro1-LacZ)CONK mice were obtained from The Jackson Laboratory (Bar Harbor, ME) and crossed to GFAP-tTA mice to get double-transgenic mice. KOR knock-out mice (Hough et al., 2000) were obtained courtesy of Dr. John E. Pintar (University of Medicine and Dentistry New Jersey, Piscataway, NJ).
Doxycycline administration.
Selected animals were given 25 μg/ml dox (Sigma, St. Louis, MO) in the drinking water. Amber bottles were used to protect the dox from light and the water was changed once a week. Breeding pairs on dox were given dox water at the time of mating and maintained on dox continuously. Breeding pairs off dox were never exposed to dox.
Immunofluorescence.
Adult (P45–P60; n = 13) mice were deeply anesthetized and transcardially perfused with cold 4% paraformaldehyde/0.1 m NaPO4, pH 7.4. Brains were removed and postfixed overnight at 4°C in the same buffer. To detect the Ro1 receptor, sections were probed for FLAG, the peptide (DYKDDDDK) epitope tag fused to the N terminus of Ro1. For FLAG staining, 50 μm sections were cut on a vibratome, washed in alternating PBS and PBS/0.5% Triton X-100 (Tx-100) washes, and blocked in PBS/0.5% Tx-100/10% NGS/0.2% gelatin/3% milk. Sections were incubated with primary antibodies (rabbit anti-FLAG; Sigma) diluted in PBS/0.5% Tx-100/3% NGS/0.2% gelatin overnight at 4°C with gentle agitation. Sections were extensively washed in PBS and PBS/0.2% Tx-100. Secondary antibodies (goat anti-rabbit IgG conjugated to Alexa-488; Invitrogen, Carlsbad, CA) were diluted in PBS/0.5% Tx-100/5% NGS/0.2% gelatin and applied for 2–4 h at room temperature. For GFAP and NeuN staining, fixed brains were cryoprotected in PBS/30% sucrose overnight at 4°C. Sections (14 μm) were cut on a cryostat, blocked for 2–4 h in PBS/20% normal goat serum/2% BSA/0.2% Tx-100 at room temperature, and incubated overnight with primary antibodies (mouse anti-GFAP; 1:500; Sigma; mouse anti-NeuN; 1:1000; Chemicon, Temecula, CA) diluted in the same buffer. After washing, sections were incubated with secondary antibodies (goat anti-mouse conjugated to Alexa-488 and goat anti-rabbit conjugated to Alexa-594; Invitrogen) diluted 1:400 in blocking buffer for 2–4 h at room temperature.
Xgal histochemistry.
Adult mice (P30–P45; n = 6) were deeply anesthetized and transcardially perfused with cold 4% paraformaldehyde/0.1 m NaPO4, pH 7.4. One hundred micrometer sections were cut on a vibratome. Sections were washed three times (10 min each) at room temperature in Xgal rinse buffer (0.1 m KPO4, pH 7.4, 2 mm MgCl2, 0.01% sodium deoxycholate, 0.2% Igepal CA-630) and stained overnight at 37°C in Xgal stain buffer [rinse buffer plus 1 mg/ml 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside, 5 mm K3Fe(CN)6, and 5 mm K4Fe(CN)6].
Immunoprecipitation/Western blotting.
Fresh brain tissue (P21–P50; n = 15) was homogenized in ice-cold lysis buffer (50 mm Tris-HCl, pH 7.4, 100 mm NaCl, 5 mm EDTA, 50 mm NaF, 10 mm NaPP, 200 μm Na3VO4, 1% Tx-100, 100 μg/ml PMSF, 1 μg/ml leupeptin, 2 μg/ml aprotinin, 1 μg/ml soybean trypsin inhibitor, 1 μg/ml pepstatin A, 10 μg/ml benzamidine) and sonicated on ice. Lysates were incubated on a rotator for 1 h at 4°C. Supernatants were precleared and incubated for 2 h at 4°C with M2 FLAG antibody (Sigma) and protein A/Sepharose beads (Sigma) that had been blocked with BSA. Beads were boiled in Laemmli buffer, run on an SDS-PAGE gel, and blotted onto a nitrocellulose membrane. The blots were blocked in 5% milk–TBS/0.1% Tween 20 (TBST) for 2 h at room temperature and probed overnight at 4°C with a 1:250 dilution of a rabbit anti-FLAG polyclonal antibody (Sigma) in 5% milk/TBST. Membranes were washed in TBST, probed with HRP-conjugated goat anti-rabbit IgG (1:10,000), and processed for ECL. For GFAP, fresh brains (P21–P35; n = 6) were dissected in ice-cold PBS, and tissues were homogenized in a volume of homogenization buffer (50 mm HEPES, pH 7.3, 150 mm NaCl2, 1.5 mm MgCl2, 1 mm EGTA, 10% glycerol, 1% Tx-100 with Complete protease inhibitor (Roche Diagnostics, Mannheim, Germany) equal to 10× tissue weight. The samples were sonicated and centrifuged. Equal amounts of protein were run on a Tris-glycine gel, blotted onto a nitrocellulose membrane, and probed with rabbit anti-GFAP antibody (DakoCytomation; 1:5000) for 48 h at 4°C.
Hematoxylin and eosin staining.
Brains from perfused animals (P30–P66; n = 14) were drop-fixed in 10% formalin overnight, rinsed in distilled water, and stored in 70% ethanol. The fixed brains were taken to the University of North Carolina histology core for paraffin embedding and staining with hematoxylin and eosin. Sections (6 μm) were cut on a sliding microtome.
Immunocytochemistry.
Brains from adult mice (P30–P45; n = 11) were fixed in 4% paraformaldehyde, cryoprotected in PBS/30% sucrose, and cut in 35 μm sections on a cryostat. Sections were rehydrated in PBS and incubated in 100% methanol at −20°C for 10 min. Sections were washed three times (10 min each) in TBST and once in TBS before incubating for 2 h in blocking buffer (5.5% NGS/5.5% BSA/TBST) at room temperature. Avidin/biotin was blocked using an avidin/biotin blocking kit (Vector Laboratories, Burlingame, CA) according to the manufacturer's protocol. Primary antibodies [rabbit anti-phospho-p44/42 mitogen-activated protein (MAP) kinase (Thr202/Tyr204); Cell Signaling Technology, Beverly, MA; 1:250; or mouse anti-GFAP; Sigma; 1:500] were diluted in blocking buffer, and sections were incubated overnight at 4°C. Biotinylated goat anti-rabbit secondary antibody (Vectastain Elite ABC kit; Vector Laboratories) was diluted 1:500 in TBS/3% BSA, and sections were incubated for 2 h at room temperature. Sections were then incubated for exactly 30 min in 0.6% H2O2 at room temperature. ABC reagent was prepared according to kit instructions, and sections were incubated for 2 h at room temperature. DAB reagent (Vector Laboratories) was added, and the reaction was allowed to proceed until sufficient staining developed. Coverslips were mounted using Vectashield with DAPI (4′,6′-diamidino-2-phenylindole) (Vector Laboratories).
Ventricle size.
Brains fixed in 4% paraformaldehyde were embedded in agarose and cut coronally into 500 μm (P15; n = 22) or 375 μm (P0; n = 24) sections on a vibratome (Leica VT 1000S; Leica, Nussloch, Germany). Sequential sections were viewed under a light microscope (Zeiss, Oberkochen, Germany), and images were collected using MetaMorph imaging software. The section at the level of striatum and anterior commissure was selected from each animal, and the size (maximum width) of the lateral ventricles was measured using the MetaMorph software and divided by the maximum brain width. The resulting ratio of ventricle to brain diameter provides a measurement of hydrocephalus severity (Jones et al., 2001a). Hydrocephalic phenotypes were classified as mild (0.20–0.40), moderate (0.41–0.60), or severe (>0.60).
Results
Expression of Ro1
Mice expressing Ro1 in astrocytes were generated by crossing the hGFAP-tTA mouse line to the tetO-Ro1/tetO-LacZ mouse line. This genotype was moved onto a KOR knock-out background (Hough et al., 2000) to prevent spiradoline from activating endogenous κ-opioid receptors. KOR−/−/hGFAP-tTA+/− or KOR−/−/hGFAP-tTA+/+ mice were crossed to KOR−/−/tetO-Ro1/tetO-LacZ+/− mice to produce double-transgenic (hGFAP-tTA+/−/tetO-Ro1/tetO-LacZ+/−) and control (hGFAP-tTA+/−, tetO-Ro1/tetO-LacZ+/−, or neither) littermates, all on the KOR background. Double-transgenic mice (hereafter referred to as Ro1 mice) were produced in expected numbers. To determine that Ro1 expression was restricted to astrocytes, brain sections from Ro1 mice were probed with anti-FLAG (the epitope tag fused to Ro1) plus anti-GFAP (glial fibrillary astrocytic protein, an astrocytic marker) or anti-NeuN (neuronal nuclei, a neuron-specific nuclear protein) antibodies. Positive FLAG staining was apparent in most brain regions, including cortex, hippocampus, cerebellum, and thalamus. FLAG staining was observed in GFAP-positive cells but not in NeuN-positive cells (Fig. 2A,B), confirming that Ro1 expression is astrocytic.
Figure 2.

Ro1 expression is restricted to astrocytes and is regulated by dox. A, B, Hippocampal brain sections were double-stained for FLAG and GFAP (astrocyte marker) or FLAG and NeuN (neuronal marker). A, Positive FLAG staining (left, arrow) colocalized with GFAP (right, arrow) but not with NeuN (B). The arrows point to the same cell. Scale bars, 100 μm. C, Brain lysates from a C57BL/6 control mouse, Ro1 mouse on dox (25 μg/ml), and Ro1 mouse off dox were immunoprecipitated with FLAG monoclonal antibody, and the blots were probed with FLAG polyclonal antibody. Only the Ro1 mouse off dox had positive bands (solid arrows). A nonspecific band was found in all samples (arrowhead). Br, Brain; Hipp, hippocampus; Ce, cerebellum.
Because Ro1 expression is driven by the tetO system, dox can be used to manipulate when Ro1 is expressed. To verify that dox represses Ro1 expression, brain tissue extracts from Ro1 animals (P60) that had been maintained off dox, Ro1 animals that had been maintained on 25 μg/ml dox, and C57BL/6J wild-type control animals were immunoprecipitated with a monoclonal Flag antibody. A band at ∼44 kDa, consistent with the predicted size of the receptor, along with bands representing multimers of the receptor were detected in extracts from animals maintained off dox. No FLAG-specific bands were detected in wild-type mice or Ro1 mice on 25 μg/ml dox, indicating that 25 μg/ml dox represses Ro1 expression (Fig. 2C). Because Ro1 mice also carry a tetO-LacZ transgene, we used Xgal histochemistry to confirm that dox regulates expression (Fig. 3). No Xgal staining was detected in any brain region of sections from Ro1 mice on 25 μg/ml dox. Strong Xgal staining was observed in the hippocampus, thalamus, and brainstem of mice maintained off dox; weak Xgal staining was seen in the cortex.
Figure 3.
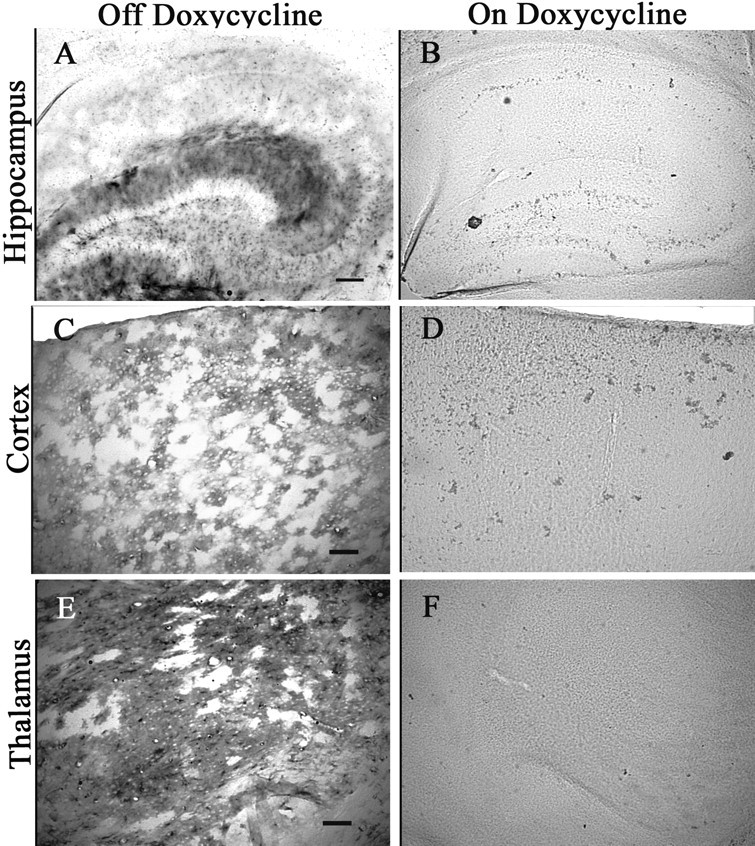
LacZ expression is regulated by dox. Brain sections from Ro1 mice on dox (25 μg/ml) (B, D, F) and Ro1 mice off dox (A, C, E) were stained by Xgal histochemistry. Ro1 animals on dox had no detectable positive cells, whereas robust staining was seen in Ro1 animals off dox, indicating that dox is effectively controlling expression. Scale bars: A, B, 150 μm; C–F, 50 μm.
Ro1 mice develop hydrocephalus
Although Ro1 mice off dox appear grossly normal at birth, by P15, 100% of Ro1 mice off dox begin to exhibit macrocephalus characterized by a swollen, dome-shaped cranium (Fig. 4B). By 12 weeks of age, all Ro1 mice off dox develop severe hydrocephalus and ∼50% die (Fig. 4C). Survival studies were terminated at 12 weeks because of the severe morbidity of the remaining Ro1 animals. Dissection of the brain revealed greatly enlarged ventricles filled with fluid, indicative of hydrocephalus and impaired CSF homeostasis. Blood on the surface of the brain was frequently observed in hydrocephalic mice, and on occasion excess CSF was seen between the skull and brain surface. Control littermates (hGFAP-tTA+/− only or tetO-Ro1/tetO-lacZ+/− only) and Ro1 mice on 25 μg/ml dox appear normal at P15 and do not develop macrocephalus at any age, indicating that the insertion of either or both genes does not cause hydrocephalus.
Figure 4.

Ro1 mice maintained off dox develop hydrocephalus. B, C, All Ro1 pups from breeding pairs maintained off dox (B) develop overt hydrocephalus, and >50% die by 12 weeks of age (C). A, Ro1 mice maintained on dox (25 μg/ml) appear normal. Mice shown are P45.
To further characterize the progression of hydrocephalus in Ro1 mice, the ventricle size of litters off dox were examined at P0 and P15 (Fig. 5). At P0, Ro1 mice (n = 10) and control littermates (n = 14) showed no enlargement of the lateral ventricles. At P15, however, all Ro1 mice (n = 12) exhibited enlarged ventricles with a ventricle-to-brain ratio >0.30, whereas control littermates (n = 10) had an average ratio of 0.10. We observed variation in the severity of the hydrocephalus phenotype (Table 1) as defined in Materials and Methods. The severity of hydrocephalus is not linked to homozygosity because all animals measured were heterozygous for both transgenes. The severity of hydrocephalus continued to progress with age because all Ro1 animals off dox developed severe hydrocephalus by P30 (Fig. 5G) (E. J. Sweger, unpublished observations).
Figure 5.

Ro1 mice develop progressive enlargement of the lateral ventricles. At birth, whole brains and coronal sections of Ro1 mice (b, B) and single-transgenic control littermates (a, A) are indistinguishable. By P15, brains from Ro1 mice (d, e, D, E) are larger than controls (c, C) and have pronounced enlargement of the lateral ventricles. The hydrocephalus in Ro1 mice can be classified as mild (D) (n = 4), moderate (E) (n = 6), or severe (data not shown) (n = 2) cases based on ventricle-to-brain ratio. By P30, Ro1 mice (G) have severe hydrocephalus characterized by greatly enlarged lateral ventricles and extremely thin cortex. Control mice at P30 (F) appear normal. All mice were maintained off dox.
Table 1.
Severity of hydrocephalic phenotype in P15 Ro1 mice
| Phenotype | Average ratio | n |
|---|---|---|
| Mild | 0.31 | 4 |
| Moderate | 0.50 | 6 |
| Severe | 0.67 | 2 |
Brains of four control mice (single transgenic, P30–P125) and five hydrocephalic Ro1 (P30–P68) mice off dox were serially sectioned and stained with hematoxylin and eosin for a more detailed histological analysis (Fig. 6). The most striking feature of the hydrocephalic brains was the greatly enlarged lateral ventricles (Fig. 5G). The third ventricle, optic recess, pineal recess, and intraventricular foramen were also enlarged. The cerebral cortex was reduced in thickness, with the most severely affected animals exhibiting an almost completely atrophied cortex. All cortical layers appeared to be present (data not shown). The septum and retrosplenial cortex–hippocampus transition were disrupted; other brain regions, including the hippocampus, appeared compressed and caudally displaced. Red blood cells indicative of recent hemorrhaging were found throughout the brain (data not shown). The white tract tissue underlying the lateral ventricles was disrupted, and there was an incomplete denudation of ependymal cells lining the lateral (data not shown) and third ventricles (Fig. 6J). In some instances, the ependymal layer appears to be separated from the underlying tissue (Fig. 6F). Although it is possible that the separation is attributable to a fixation artifact, ependymal layer separation was not observed in control mice. Where intact, the ependymal cell layer appeared thinner (Fig. 6B,D,F), and not all ependymal cells were ciliated. The rostral end of the aqueduct of Sylvius was completely obliterated (Fig. 6H), suggesting that hydrocephalus results in part from the blocked flow of CSF between the third and fourth ventricles. No ependymal cells were detected at the site of obstruction and the surrounding tissues appeared vacuolated. Abnormalities in the subcommissural organ (SCO) have been implicated in other models of hydrocephalus (Jones and Bucknall, 1988; Perez-Figares et al., 2001; Bach et al., 2003; Blackshear et al., 2003; Fernandez-Llebrez et al., 2004; Jones et al., 2004; Krebs et al., 2004); likewise, in Ro1 brains, the SCO appeared disorganized (Fig. 6D).
Figure 6.
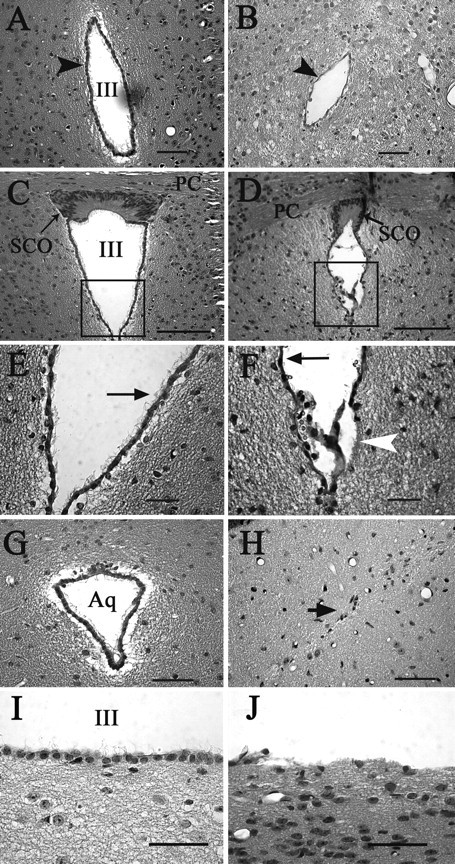
Alterations in the ventricular system of hydrocephalic Ro1 mice. Hematoxylin and eosin staining of coronal sections from single-transgenic control (P45, left column) and Ro1 (P33, right column) mice. A, B, In Ro1 mice (B), the third ventricle at the level of the periventricular hypothalamic nucleus is reduced in size and the ependymal cell layer appears thinner compared with controls (A). D, F, H, J, The subcommissural organ is disorganized in Ro1 mice (D), and there is a partial denudation of ependymal cells lining the ventricular walls (F, arrowhead; J). The remaining ependymal cells have fewer cilia (F, arrow), and the aqueduct is obliterated with no apparent ependymal cell layer (H). C, E, G, I, In control mice, the subcommissural organ (C) and aqueduct (G) show normal morphology. The ependymal layer is intact with multiple cilia per cell (E, arrow; I). E and F are enlargements of the boxes in C and D, respectively. Mice were maintained off dox. Aq, Aqueduct; SCO, subcommissural organ; III, third ventricle; PC, posterior commissure. Scale bars: A, B, G, H, I, J, 50 μm; C, D, 100 μm; E, F, 30 μm.
To determine whether Ro1 must be expressed early in development in order for hydrocephalus to develop, mice were kept on 25 μg/ml dox until weaning (P21) and maintained off dox until P60. Because of hardening of the skull, none of the mice taken off dox developed overt enlargement of the cranium by P60; however, Ro1 mice (n = 4) had enlarged lateral ventricles with an average ventricle-to-brain ratio of 0.30. In comparison, the ventricle-to-brain ratio for littermate controls (n = 2) was 0.11, similar to that of Ro1 mice (n = 8; ratio, 0.15) and controls (n = 7; ratio, 0.13) that were maintained on dox continuously until P60 (Fig. 7). Hydrocephalus appears to develop with Ro1 expression independent of age and thus is not dependent on processes linked to early stages of development.
Figure 7.

Ro1 mice will develop enlarged ventricles if taken off dox at weaning. A, B, In litters taken off dox at weaning (P21), Ro1 mice (B) develop enlarged ventricles when compared with single-transgenic littermate controls (A). C, D, Ro1 mice (D) have normal ventricles when mice were kept on dox continuously compared with single-transgenic littermate controls (C). All mice were processed at P60. A, n = 2; B, n = 4; C, n = 7; D, n = 8.
GFAP expression in Ro1 mice
Increased GFAP expression, an indicator of reactive glia, is observed in most forms of damage to the brain, including hydrocephalus. Increased levels of GFAP staining were seen in hydrocephalic Ro1 mice. The hippocampus and cortex, a region typically low in GFAP immunoreactivity (Fig. 8C), showed the greatest increases in GFAP staining compared with control mice (Fig. 8B). Western blots confirmed that GFAP protein levels were elevated in the cortex and hippocampus of Ro1 mice; GFAP levels in cerebellar tissue extracts from the same mice appeared similar to controls (Fig. 8A).
Figure 8.

GFAP expression is upregulated in hydrocephalic Ro1 mice. Brain lysates from two Ro1 mice with severe hydrocephalus and one littermate control (P22) were separated by SDS-PAGE and probed with anti-GFAP antibody (A). Ro1 mice have increased GFAP levels in the hippocampus and cortex. Brain sections from control (B) and Ro1 (C) mice were stained for GFAP to show increased expression. Cortical sections are shown; pial surface is on the top right corner. Scale bar, 50 μm.
Phospho-Erk is elevated in Ro1 mice
Because Gi-coupled receptors are known to activate the MAP kinase pathway, coronal sections from Ro1 mice were stained for phosphorylated extracellular signal-regulated kinase (Erk) p44/42, the active form of Erk p44/42. Increased phospho-Erk staining was observed in the superficial layers of entorhinal cortex and in the striatum surrounding the lateral ventricles of Ro1 animals off dox, but not in littermate single-transgenic controls (Fig. 9).
Figure 9.
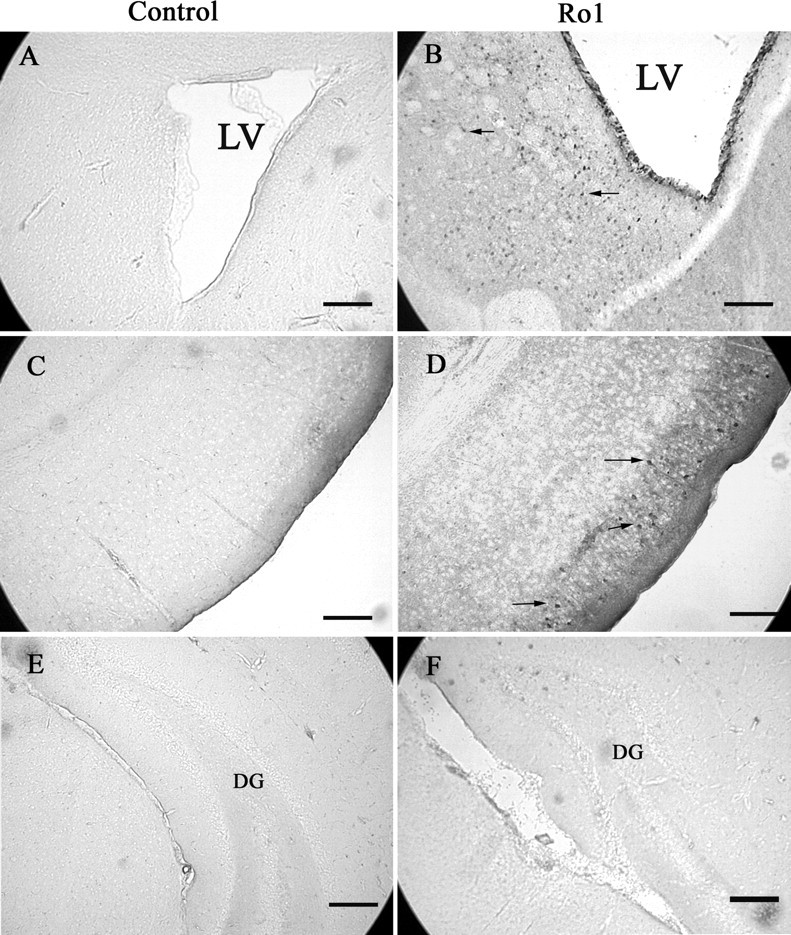
Phospho-Erk p44/42 levels are elevated in hydrocephalic Ro1 mice. Increased phospho-Erk p44/42 staining is observed around the lateral ventricles (B) and in the entorhinal cortex (D) of hydrocephalic Ro1 mice but not in littermate single-transgenic controls (P21) (A, C). No increases in phospho-Erk were observed in the hippocampus (F). Sections shown in A and B are at the level of the anterior commissure and striatum; C–F are at the level of the subcommissural organ. Scale bars, 150 μm. LV, Lateral ventricle; DG, dentate gyrus.
Discussion
To investigate how Gi protein signaling in astrocytes affects brain neurophysiology, we expressed the RASSL Ro1, a Gi-coupled GPCR, in astrocytes using the hGFAP promoter and the tet-off system. Unexpectedly, all mice expressing Ro1 developed noncommunicating triventricular hydrocephalus. Hydrocephalus appears to result from Ro1 expression, because single-transgenic mice do not develop hydrocephalus as would be expected if caused by an insertional defect. Moreover, when Ro1 expression is repressed with dox, hydrocephalus is also inhibited. Ro1 mice taken off dox at weaning (P21) develop enlarged ventricles, further supporting the idea that hydrocephalus is a consequence of Ro1 expression.
At birth, Ro1 mice off dox appeared grossly normal and were indistinguishable from their control littermates, but began to exhibit macrocephaly between P10 and P15. At P15, Ro1 mice have a lateral ventricle-to-brain ratio that exceeds 0.30 and by P30 have developed a severe hydrocephalus phenotype characterized by greatly dilated lateral and third ventricles that caudally displace surrounding brain structures. The rostral end of the Sylvius aqueduct is obliterated, with no lumen and no detectable ependymal cells visible in coronal sections of this region, suggesting a denudation of ependymal cells has occurred. The collicular recess and caudal end of the Sylvius aqueduct, however, remain open. The SCO also appears disorganized in hydrocephalic mice, although it is unclear whether this is attributable to Ro1 expression or whether it is a result of hydrocephalus.
It is unclear how, in the absence of applied ligand, Ro1 expression leads to the development of hydrocephalus. Ro1 is likely to be highly overexpressed relative to other astrocytic receptors. Because G-protein-coupled receptors have a low level of constitutive activity (Costa and Cotecchia, 2005), Ro1 may be expressed at a high enough level that its constitutive activity is sufficient to trigger a series of events that ultimately result in hydrocephalus. Ro1 may be also expressed at a high enough level that dynorphin or another ligand that normally would have a low affinity for Ro1 can bind and activate the receptor. Significantly, mice with Ro1 expression targeted to the heart developed cardiomyopathology when taken off dox. When these mice were given an intermediate dose of dox to reduce Ro1 expression, their heart rates normalized. Treating these mice with pertussis toxin or the KOR antagonist nor-binaltorphimine also restored normal heart rates, demonstrating that Ro1 signaling was responsible for the changes in heart physiology (Redfern et al., 2000). A similar process in which Ro1 overexpression and constitutive activity leads to pathology may be occurring in our mice.
Our finding is, to our knowledge, the first to implicate astrocytic Gi signaling pathways in the development of hydrocephalus. Ro1 expression in astrocytes may contribute to the development of hydrocephalus by altering the extracellular matrix so that more fluid from the extracellular space enters the ventricles. It could also alter the levels of various neurotransmitters, growth factors, or cytokines in the CSF, resulting in a dysregulation of CSF production by the choroid plexus. Astrocytic release of glutamine is known to be downregulated in the kaolin model of hydrocephalus (Kondziella et al., 2003); it is possible that increased Gi signaling in astrocytes may affect glutamine production or release that in turn changes neuronal input into the SCO. It is interesting to note that gene expression analysis in the H-Tx rat, which has primary stenosis of the aqueduct similar to Ro1 mice, has implicated several genes linked to astrocytes and G-protein-coupled receptors, including connexin 30, β-integrin 5, and somatostatin (Jones et al., 2001b; Miller et al., 2006).
Although SCO secretory function could not be determined from our histological examination of Ro1 brains, the observation that the SCO is disorganized in Ro1 mice is significant. The presence of a normal SCO appears to be necessary for the development and maintenance of the aqueduct, because impaired SCO formation and/or function are found in multiple animal hydrocephalus models and in human infantile hydrocephalus (Overholser et al., 1954; Newberne, 1962; Takeuchi and Takeuchi, 1986; Jones et al., 1987; Jones and Bucknall, 1988; Takahashi et al., 1997; Perez-Figares et al., 1998; Takahashi et al., 1998; Louvi and Wassef, 2000; Estivill-Torrus et al., 2001; Sakakibara et al., 2002; Fernandez-Llebrez et al., 2004). The SCO secretes negatively charged glycoproteins, such as SCO-spondin and RF-Gly I, that appear to be critical for maintaining an open aqueduct by their physical presence. The SCO may also regulate CSF formation. Receptors for SCO glycoproteins are found on the choroid plexus (Miranda et al., 2001), suggesting that CSF production could be influenced by SCO activity. It would be interesting to determine whether any of these parameters are altered in Ro1 mice.
We have shown that phospho-Erk levels are increased in hydrocephalic mice. Gi-coupled receptors are known to signal via the MAP kinase pathway; thus, Ro1 signaling may lead to changes in gene expression that contribute to the pathogenesis of hydrocephalus. We cannot rule out, however, the possibility that phospho-Erk is increased in response to hydrocephalus. Elevated phospho-Erk has been associated with astrogliosis (Mandell and VandenBerg, 1999), a pathological response of astrocytes to many types of brain injury, including hydrocephalus (Fukumizu et al., 1996). Increased GFAP expression observed in severely hydrocephalic Ro1 mice is indicative of astrogliosis, particularly because GFAP levels are not elevated in Ro1 mice with mild hydrocephalus (data not shown).
The observed loss of ependymal cells in Ro1 mice is in agreement with other studies that demonstrate a denudation of ependymal cells in the ventricular system (Jimenez et al., 2001; Dominguez-Pinos et al., 2005). In these models, denudation begins embryonically and precedes the closure of the aqueduct and subsequent ventricle enlargement (Wagner et al., 2003). The underlying cause of ependymal detachment is unknown, although defects in certain adhesion molecules, such as L1, have been indicated (Schmid et al., 2000; Itoh et al., 2004). Because a subset of ependymal cells express GFAP (Takano et al., 1996; Rodriguez-Perez et al., 2003), it is possible that Ro1 may be expressed in these cells, causing a defect that leads to detachment. Ependymal cells may also be affected indirectly by changes in levels of secreted cytokines or altered neuronal inputs resulting from Ro1-induced changes in astrocyte signaling.
New treatments for hydrocephalous are clearly needed. Hydrocephalus occurs at a rate of 0.48–3 in 1000 births (Mangano et al., 1998; Fletcher and Northrup, 2000; Ding et al., 2001; Del Bigio, 2004) and is the most common neurological disorder requiring surgery among children (Casey et al., 1997). Mortality rates as high as 96% are reported for children that are left untreated (Laurence and Coates, 1962; Yashon et al., 1965; Eckstein and Macnab, 1966). Shunting off the excess CSF, thereby relieving intracranial pressure and preventing additional tissue damage, is the most effective treatment to date. Yet shunting is not ideal because it does not address underlying causes of hydrocephalus and the procedure itself carries significant health risks. Shunting does not restore normal CSF circulation, which is important for carrying growth factors and cytokines to neuronal and glial precursor cells in the germinal matrix surrounding the lateral ventricles (Miyan et al., 1998). Children treated with shunts frequently have residual neurological deficits (Fernell et al., 1990, 1994; Fletcher et al., 1992; McAllister et al., 1998), including impaired memory (Scott et al., 1998). Shunts are also associated with a high risk for complications such as infection, obstruction, and over drainage (Pople et al., 1990; Blount et al., 1993); nearly 70% of shunts require revision within 10 years.
To develop more effective treatments, a better understanding of hydrocephalus pathogenesis is needed. The Ro1 model of hydrocephalus has several characteristics that will make it valuable for studying hydrocephalus. First, the Ro1 model does not require injection of foreign bodies into the ventricles to cause hydrocephalus, making the Ro1 model easier to work with and more relevant to human cases. Second, hydrocephalic animals are produced reliably; all Ro1 mice will develop hydrocephalus if maintained off dox. Inherited models of hydrocephalus, such as the H-Tx rat, reliably produce hydrocephalic offspring but at a much lower rate, and which animals will develop hydrocephalus cannot be predicted at early ages (Jones et al., 2000, 2001b; Miller et al., 2006). Third, dox can be used to regulate when hydrocephalus will occur. It would be interesting to compare juvenile and adult onset to see whether age changes how hydrocephalus develops. Fourth, the rate of development and the severity of hydrocephalus can be manipulated by giving Ro1 mice a lower concentration of dox. Slowing the development of hydrocephalus allows changes in the brain to be studied more thoroughly and could provide insights into how hydrocephalus progresses. The Ro1 model of hydrocephalus should prove useful in advancing our knowledge of hydrocephalus pathophysiology. Furthermore, the Ro1 model provides new evidence for the involvement of astrocytic G-protein-coupled receptor signaling in the development of hydrocephalus.
Footnotes
This work was supported by grants from the National Institutes of Health (K.D.M.). We thank Kinuko Suzuki for help with histological analysis and John E. Pintar for the gift of the KOR-null mice.
References
- Araque A, Li N, Doyle RT, Haydon PG. SNARE protein-dependent glutamate release from astrocytes. J Neurosci. 2000;20:666–673. doi: 10.1523/JNEUROSCI.20-02-00666.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque A, Carmignoto G, Haydon PG. Dynamic signaling between astrocytes and neurons. Annu Rev Physiol. 2001;63:795–813. doi: 10.1146/annurev.physiol.63.1.795. [DOI] [PubMed] [Google Scholar]
- Araque A, Martin ED, Perea G, Arellano JI, Buno W. Synaptically released acetylcholine evokes Ca2+ elevations in astrocytes in hippocampal slices. J Neurosci. 2002;22:2443–2450. doi: 10.1523/JNEUROSCI.22-07-02443.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach A, Lallemand Y, Nicola MA, Ramos C, Mathis L, Maufras M, Robert B. Msx1 is required for dorsal diencephalon patterning. Development. 2003;130:4025–4036. doi: 10.1242/dev.00609. [DOI] [PubMed] [Google Scholar]
- Blackshear PJ, Graves JP, Stumpo DJ, Cobos I, Rubenstein JL, Zeldin DC. Graded phenotypic response to partial and complete deficiency of a brain-specific transcript variant of the winged helix transcription factor RFX4. Development. 2003;130:4539–4552. doi: 10.1242/dev.00661. [DOI] [PubMed] [Google Scholar]
- Blount JP, Campbell JA, Haines SJ. Complications in ventricular cerebrospinal fluid shunting. Neurosurg Clin N Am. 1993;4:633–656. [PubMed] [Google Scholar]
- Bondurant CP, Jimenez DF. Epidemiology of cerebrospinal fluid shunting. Pediatr Neurosurg. 1995;23:254–258. doi: 10.1159/000120968. discussion 259. [DOI] [PubMed] [Google Scholar]
- Carmignoto G, Pizzorusso T, Tia S, Vicini S. Brain-derived neurotrophic factor and nerve growth factor potentiate excitatory synaptic transmission in the rat visual cortex. J Physiol (Lond) 1997;498:153–164. doi: 10.1113/jphysiol.1997.sp021848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey AT, Kimmings EJ, Kleinlugtebeld AD, Taylor WA, Harkness WF, Hayward RD. The long-term outlook for hydrocephalus in childhood. A ten-year cohort study of 155 patients. Pediatr Neurosurg. 1997;27:63–70. doi: 10.1159/000121229. [DOI] [PubMed] [Google Scholar]
- Chung JH, Whiteley M, Felsenfield G. A 5′ element of the chicken beta-globin domain serves as an insulator in human erythroid cells and protects against position effect in Drosophila. Cell. 1993;74:505–514. doi: 10.1016/0092-8674(93)80052-g. [DOI] [PubMed] [Google Scholar]
- Costa T, Cotecchia S. Historical review: negative efficacy and the constitutive activity of G-protein-coupled receptors. Trends Pharmacol Sci. 2005;26:618–624. doi: 10.1016/j.tips.2005.10.009. [DOI] [PubMed] [Google Scholar]
- Coward P, Wada HG, Falk MS, Chan SD, Meng F, Akil H, Conklin BR. Controlling signaling with a specifically designed Gi-coupled receptor. Proc Natl Acad Sci USA. 1998;95:352–357. doi: 10.1073/pnas.95.1.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Ambrosio R, Gordon DS, Winn HR. Differential role of KIR channel and Na+/K+-pump in the regulation of extracellular K+ in rat hippocampus. J Neurophysiol. 2002;87:87–102. doi: 10.1152/jn.00240.2001. [DOI] [PubMed] [Google Scholar]
- Del Bigio MR. Cellular damage and prevention in childhood hydrocephalus. Brain Pathol. 2004;14:317–324. doi: 10.1111/j.1750-3639.2004.tb00071.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Lai Q, McAllister IJ, Canady AI. Impaired motor learning in children with hydrocephalus. Pediatr Neurosurg. 2001;34:182–189. doi: 10.1159/000056017. [DOI] [PubMed] [Google Scholar]
- Dominguez-Pinos MD, Paez P, Jimenez AJ, Weil B, Arraez MA, Perez-Figares JM, Rodriguez EM. Ependymal denudation and alterations of the subventricular zone occur in human fetuses with a moderate communicating hydrocephalus. J Neuropathol Exp Neurol. 2005;64:595–604. doi: 10.1097/01.jnen.0000171648.86718.bb. [DOI] [PubMed] [Google Scholar]
- Eckstein HB, Macnab GH. Myelomeningocele and hydrocephalus. The impact of modern treatment. Lancet. 1966;1:842–845. doi: 10.1016/s0140-6736(66)90184-x. [DOI] [PubMed] [Google Scholar]
- Estivill-Torrus G, Vitalis T, Fernandez-Llebrez P, Price DJ. The transcription factor Pax6 is required for development of the diencephalic dorsal midline secretory radial glia that form the subcommissural organ. Mech Dev. 2001;109:215–224. doi: 10.1016/s0925-4773(01)00527-5. [DOI] [PubMed] [Google Scholar]
- Fernandez-Llebrez P, Grondona JM, Perez J, Lopez-Aranda MF, Estivill-Torrus G, Llebrez-Zayas PF, Soriano E, Ramos C, Lallemand Y, Bach A, Robert B. Msx1-deficient mice fail to form prosomere 1 derivatives, subcommissural organ, and posterior commissure and develop hydrocephalus. J Neuropathol Exp Neurol. 2004;63:574–586. doi: 10.1093/jnen/63.6.574. [DOI] [PubMed] [Google Scholar]
- Fernell E, Hagberg G, Hagberg B. Infantile hydrocephalus—the impact of enhanced preterm survival. Acta Paediatr Scand. 1990;79:1080–1086. doi: 10.1111/j.1651-2227.1990.tb11387.x. [DOI] [PubMed] [Google Scholar]
- Fernell E, Hagberg G, Hagberg B. Infantile hydrocephalus epidemiology: an indicator of enhanced survival. Arch Dis Child Fetal Neonatal Ed. 1994;70:F123–F128. doi: 10.1136/fn.70.2.f123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiacco TA, McCarthy KD. Intracellular astrocyte calcium waves in situ increase the frequency of spontaneous AMPA receptor currents in CA1 pyramidal neurons. J Neurosci. 2004;24:722–732. doi: 10.1523/JNEUROSCI.2859-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher JM, Francis DJ, Thompson NM, Davidson KC, Miner ME. Verbal and nonverbal skill discrepancies in hydrocephalic children. J Clin Exp Neuropsychol. 1992;14:593–609. doi: 10.1080/01688639208402847. [DOI] [PubMed] [Google Scholar]
- Fletcher JMDM, Northrup H. Hydrocephalus. In: Yeates KO, Ris MD, Taylor HG, editors. Pediatric neuropsychology research, theory, and practice. New York: Guilford; 2000. pp. 25–46. [Google Scholar]
- Fukumizu M, Takashima S, Becker LE. Glial reaction in periventricular areas of the brainstem in fetal and neonatal posthemorrhagic hydrocephalus and congenital hydrocephalus. Brain Dev. 1996;18:40–45. doi: 10.1016/0387-7604(95)00103-4. [DOI] [PubMed] [Google Scholar]
- Gabriel S, Kivi A, Kovacs R, Lehmann TN, Lanksch WR, Meencke HJ, Heinemann U. Effects of barium on stimulus-induced changes in [K+]o and field potentials in dentate gyrus and area CA1 of human epileptic hippocampus. Neurosci Lett. 1998;249:91–94. doi: 10.1016/s0304-3940(98)00420-0. [DOI] [PubMed] [Google Scholar]
- Hough LB, Nalwalk JW, Chen Y, Schuller A, Zhu Y, Zhang J, Menge WM, Leurs R, Timmerman H, Pintar JE. Improgan, a cimetidine analog, induces morphine-like antinociception in opioid receptor-knockout mice. Brain Res. 2000;880:102–108. doi: 10.1016/s0006-8993(00)02776-1. [DOI] [PubMed] [Google Scholar]
- Itoh K, Cheng L, Kamei Y, Fushiki S, Kamiguchi H, Gutwein P, Stoeck A, Arnold B, Altevogt P, Lemmon V. Brain development in mice lacking L1–L1 homophilic adhesion. J Cell Biol. 2004;165:145–154. doi: 10.1083/jcb.200312107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez AJ, Tome M, Paez P, Wagner C, Rodriguez S, Fernandez-Llebrez P, Rodriguez EM, Perez-Figares JM. A programmed ependymal denudation precedes congenital hydrocephalus in the hyh mutant mouse. J Neuropathol Exp Neurol. 2001;60:1105–1119. doi: 10.1093/jnen/60.11.1105. [DOI] [PubMed] [Google Scholar]
- Jones HC, Bucknall RM. Inherited prenatal hydrocephalus in the H-Tx rat: a morphological study. Neuropathol Appl Neurobiol. 1988;14:263–274. doi: 10.1111/j.1365-2990.1988.tb00887.x. [DOI] [PubMed] [Google Scholar]
- Jones HC, Dack S, Ellis C. Morphological aspects of the development of hydrocephalus in a mouse mutant (SUMS/NP) Acta Neuropathol (Berl) 1987;72:268–276. doi: 10.1007/BF00691100. [DOI] [PubMed] [Google Scholar]
- Jones HC, Lopman BA, Jones TW, Carter BJ, Depelteau JS, Morel L. The expression of inherited hydrocephalus in H-Tx rats. Childs Nerv Syst. 2000;16:578–584. doi: 10.1007/s003810000330. [DOI] [PubMed] [Google Scholar]
- Jones HC, Carter BJ, Depelteau JS, Roman M, Morel L. Chromosomal linkage associated with disease severity in the hydrocephalic H-Tx rat. Behav Genet. 2001a;31:101–111. doi: 10.1023/a:1010266110762. [DOI] [PubMed] [Google Scholar]
- Jones HC, Depelteau JS, Carter BJ, Lopman BA, Morel L. Genome-wide linkage analysis of inherited hydrocephalus in the H-Tx rat. Mamm Genome. 2001b;12:22–26. doi: 10.1007/s003350010226. [DOI] [PubMed] [Google Scholar]
- Jones HC, Yehia B, Chen GF, Carter BJ. Genetic analysis of inherited hydrocephalus in a rat model. Exp Neurol. 2004;190:79–90. doi: 10.1016/j.expneurol.2004.06.019. [DOI] [PubMed] [Google Scholar]
- Kondziella D, Qu H, Ludemann W, Brinker T, Sletvold O, Sonnewald U. Astrocyte metabolism is disturbed in the early development of experimental hydrocephalus. J Neurochem. 2003;85:274–281. doi: 10.1046/j.1471-4159.2003.01656.x. [DOI] [PubMed] [Google Scholar]
- Krebs DL, Metcalf D, Merson TD, Voss AK, Thomas T, Zhang JG, Rakar S, O'Bryan MK, Willson TA, Viney EM, Mielke LA, Nicola NA, Hilton DJ, Alexander WS. Development of hydrocephalus in mice lacking SOCS7. Proc Natl Acad Sci USA. 2004;101:15446–15451. doi: 10.1073/pnas.0406870101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurence KM, Coates S. The natural history of hydrocephalus. Detailed analysis of 182 unoperated cases. Arch Dis Child. 1962;37:345–362. doi: 10.1136/adc.37.194.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louvi A, Wassef M. Ectopic engrailed 1 expression in the dorsal midline causes cell death, abnormal differentiation of circumventricular organs and errors in axonal pathfinding. Development. 2000;127:4061–4071. doi: 10.1242/dev.127.18.4061. [DOI] [PubMed] [Google Scholar]
- Mancao M, Miller C, Cochrane B, Hoff C, Sauter K, Weber E. Cerebrospinal fluid shunt infections in infants and children in Mobile, Alabama. Acta Paediatr. 1998;87:667–670. doi: 10.1080/080352598750014085. [DOI] [PubMed] [Google Scholar]
- Mandell JW, VandenBerg SR. ERK/MAP kinase is chronically activated in human reactive astrocytes. NeuroReport. 1999;10:3567–3572. doi: 10.1097/00001756-199911260-00019. [DOI] [PubMed] [Google Scholar]
- Mangano FT, McAllister JP, II, Jones HC, Johnson MJ, Kriebel RM. The microglial response to progressive hydrocephalus in a model of inherited aqueductal stenosis. Neurol Res. 1998;20:697–704. doi: 10.1080/01616412.1998.11740586. [DOI] [PubMed] [Google Scholar]
- McAllister JP, II, Mangano FT, Jones HC, Kriebel RM. The microglial response in experimental infantile hydrocephalus. Eur J Pediatr Surg. 1998;8(Suppl 1):62. [PubMed] [Google Scholar]
- Miller JM, Kumar R, McAllister JP, II, Krause GS. Gene expression analysis of the development of congenital hydrocephalus in the H-Tx rat. Brain Res. 2006;1075:36–47. doi: 10.1016/j.brainres.2005.12.094. [DOI] [PubMed] [Google Scholar]
- Miranda E, Almonacid JA, Rodriguez S, Perez J, Hein S, Cifuentes M, Fernandez-Llebrez P, Rodriguez EM. Searching for specific binding sites of the secretory glycoproteins of the subcommissural organ. Microsc Res Tech. 2001;52:541–551. doi: 10.1002/1097-0029(20010301)52:5<541::AID-JEMT1039>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Miyan JA, Khan MI, Kawarada Y, Sugiyama T, Bannister CM. Cell death in the brain of the HTx rat. Eur J Pediatr Surg. 1998;8(Suppl 1):43–48. doi: 10.1055/s-2008-1071253. [DOI] [PubMed] [Google Scholar]
- Newberne PM. The subcommissural organ of the vitamin B12-deficient rat. J Nutr. 1962;76:393–413. doi: 10.1093/jn/76.4.393. [DOI] [PubMed] [Google Scholar]
- Overholser MD, Whitley JR, O'Dell BL, Hogan AG. The ventricular system in hydrocephalic rat brains produced by a deficiency of vitamin B12 or of folic acid in the maternal diet. Anat Rec. 1954;120:917–933. doi: 10.1002/ar.1091200407. [DOI] [PubMed] [Google Scholar]
- Pascual O, Casper KB, Kubera C, Zhang J, Revilla-Sanchez R, Sul JY, Takano H, Moss SJ, McCarthy K, Haydon PG. Astrocytic purinergic signaling coordinates synaptic networks. Science. 2005;310:113–116. doi: 10.1126/science.1116916. [DOI] [PubMed] [Google Scholar]
- Pasti L, Zonta M, Pozzan T, Vicini S, Carmignoto G. Cytosolic calcium oscillations in astrocytes may regulate exocytotic release of glutamate. J Neurosci. 2001;21:477–484. doi: 10.1523/JNEUROSCI.21-02-00477.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patwardhan RV, Nanda A. Implanted ventricular shunts in the United States: the billion-dollar-a-year cost of hydrocephalus treatment. Neurosurgery. 2005;56:139–144. 144–135. doi: 10.1227/01.neu.0000146206.40375.41. discussion. [DOI] [PubMed] [Google Scholar]
- Perez-Figares JM, Jimenez AJ, Perez-Martin M, Fernandez-Llebrez P, Cifuentes M, Riera P, Rodriguez S, Rodriguez EM. Spontaneous congenital hydrocephalus in the mutant mouse hyh. Changes in the ventricular system and the subcommissural organ. J Neuropathol Exp Neurol. 1998;57:188–202. doi: 10.1097/00005072-199802000-00010. [DOI] [PubMed] [Google Scholar]
- Perez-Figares JM, Jimenez AJ, Rodriguez EM. Subcommissural organ, cerebrospinal fluid circulation, and hydrocephalus. Microsc Res Tech. 2001;52:591–607. doi: 10.1002/1097-0029(20010301)52:5<591::AID-JEMT1043>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Pople IK, Quinn MW, Bayston R. Morbid and outcome of shunted hydrocephalus. Z Kinderchir. 1990;45(Suppl 1):29–31. doi: 10.1055/s-2008-1042631. [DOI] [PubMed] [Google Scholar]
- Porter JT, McCarthy KD. Hippocampal astrocytes in situ respond to glutamate released from synaptic terminals. J Neurosci. 1996;16:5073–5081. doi: 10.1523/JNEUROSCI.16-16-05073.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redfern CH, Coward P, Degtyarev MY, Lee EK, Kwa AT, Hennighausen L, Bujard H, Fishman GI, Conklin BR. Conditional expression and signaling of a specifically designed Gi-coupled receptor in transgenic mice. Nat Biotechnol. 1999;17:165–169. doi: 10.1038/6165. [DOI] [PubMed] [Google Scholar]
- Redfern CH, Degtyarev MY, Kwa AT, Salomonis N, Cotte N, Nanevicz T, Fidelman N, Desai K, Vranizan K, Lee EK, Coward P, Shah N, Warrington JA, Fishman GI, Bernstein D, Baker AJ, Conklin BR. Conditional expression of a Gi-coupled receptor causes ventricular conduction delay and a lethal cardiomyopathy. Proc Natl Acad Sci USA. 2000;97:4826–4831. doi: 10.1073/pnas.97.9.4826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Perez LM, Perez-Martin M, Jimenez AJ, Fernandez-Llebrez P. Immunocytochemical characterisation of the wall of the bovine lateral ventricle. Cell Tissue Res. 2003;314:325–335. doi: 10.1007/s00441-003-0794-1. [DOI] [PubMed] [Google Scholar]
- Sakakibara S, Nakamura Y, Yoshida T, Shibata S, Koike M, Takano H, Ueda S, Uchiyama Y, Noda T, Okano H. RNA-binding protein Musashi family: roles for CNS stem cells and a subpopulation of ependymal cells revealed by targeted disruption and antisense ablation. Proc Natl Acad Sci USA. 2002;99:15194–15199. doi: 10.1073/pnas.232087499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid RS, Pruitt WM, Maness PF. A MAP kinase-signaling pathway mediates neurite outgrowth on L1 and requires Src-dependent endocytosis. J Neurosci. 2000;20:4177–4188. doi: 10.1523/JNEUROSCI.20-11-04177.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott MA, Fletcher JM, Brookshire BL, Davidson KC, Landry SH, Bohan TC, Kramer LA, Brandt ME, Francis DJ. Memory functions in children with early hydrocephalus. Neuropsychology. 1998;12:578–589. doi: 10.1037//0894-4105.12.4.578. [DOI] [PubMed] [Google Scholar]
- Takahashi S, Makita Y, Okamoto N, Miyamoto A, Oki J. L1CAM mutation in a Japanese family with X-linked hydrocephalus: a study for genetic counseling. Brain Dev. 1997;19:559–562. doi: 10.1016/s0387-7604(97)00079-x. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Ohkura A, Hirohata M, Tokutomi T, Shigemori M. Ultrastructure of obstructive tissue in malfunctioning ventricular catheters without infection. Neurol Med Chir (Tokyo) 1998;38:399–404. 403–394. doi: 10.2176/nmc.38.399. discussion. [DOI] [PubMed] [Google Scholar]
- Takano T, Rutka JT, Becker LE. Overexpression of nestin and vimentin in ependymal cells in hydrocephalus. Acta Neuropathol (Berl) 1996;92:90–97. doi: 10.1007/s004010050493. [DOI] [PubMed] [Google Scholar]
- Takeuchi IK, Takeuchi YK. Congenital hydrocephalus following X-irradiation of pregnant rats on an early gestational day. Neurobehav Toxicol Teratol. 1986;8:143–150. [PubMed] [Google Scholar]
- Wagner C, Batiz LF, Rodriguez S, Jimenez AJ, Paez P, Tome M, Perez-Figares JM, Rodriguez EM. Cellular mechanisms involved in the stenosis and obliteration of the cerebral aqueduct of hyh mutant mice developing congenital hydrocephalus. J Neuropathol Exp Neurol. 2003;62:1019–1040. doi: 10.1093/jnen/62.10.1019. [DOI] [PubMed] [Google Scholar]
- Walz W. Role of astrocytes in the clearance of excess extracellular potassium. Neurochem Int. 2000;36:291–300. doi: 10.1016/s0197-0186(99)00137-0. [DOI] [PubMed] [Google Scholar]
- Yashon D, Jane JA, Sugar O. The course of severe untreated infantile hydrocephalus. Prognostic significance of the cerebral mantle. J Neurosurg. 1965;23:509–516. doi: 10.3171/jns.1965.23.5.0509. [DOI] [PubMed] [Google Scholar]