

Abstract
Huntingtin-interacting protein 1 (HIP1) is an endocytic adaptor protein that plays a role in clathrin-mediated endocytosis and the ligand-induced internalization of AMPA receptors (AMPARs) (Metzler et al., 2003). In the present study, we investigated the role of HIP1 in NMDA receptor (NMDAR) function by analyzing NMDA-dependent transport and NMDA-induced excitotoxicity in neurons from HIP1−/− mice. HIP1 colocalizes with NMDARs in hippocampal and cortical neurons and affinity purifies with NMDARs by GST (glutathione S-transferase) pull down and coimmunoprecipitation. A profound decrease in NMDA-induced AMPAR internalization of 75% occurs in neurons from HIP1−/− mice compared with wild type, using a quantitative single-cell-based internalization assay. This defect in NMDA-dependent removal of surface AMPARs is in agreement with the observed defect in long-term depression induction in hippocampal brain slices of HIP1−/− mice and supports a role of HIP1 in AMPAR internalization in vivo. HIP1−/− neurons are partially protected from NMDA-induced excitotoxicity as assessed by LDH (lactate dehydrogenase) release, TUNEL (terminal deoxynucleotidyl transferase-mediated biotinylated dUTP nick end labeling) and caspase-3 activation assays, which points to a role of HIP1 in NMDA-induced cell death. Interestingly, phosphorylation of Akt and its substrate huntingtin (htt) decreases during NMDA-induced excitotoxicity by 48 and 31%, respectively. This decrease is significantly modulated by HIP1, resulting in 94 and 48% changes in P-Akt and P-htt levels in HIP1−/− neurons, respectively. In summary, we have shown that HIP1 influences important NMDAR functions and that both HIP1 and htt participate in NMDA-induced cell death. These findings may provide novel insights into the cellular mechanisms underlying enhanced NMDA-induced excitotoxicity in Huntington's disease.
Keywords: Huntington's disease, neurodegeneration, glutamate receptors, excitotoxicity, transport, synapse
Introduction
The endocytic adaptor protein huntingtin-interacting protein 1 (HIP1) has a well established role in clathrin-mediated endocytosis in vitro (Metzler et al., 2001; Mishra et al., 2001; Waelter et al., 2001; Legendre-Guillemin et al., 2002). HIP1 binds to the clathrin heavy chain and the heterotetrameric adaptor protein AP2 and triggers clathrin assembly through direct binding to the clathrin light chain (Legendre-Guillemin et al., 2005). Studies in neurons from HIP1−/− mice revealed a defect in ligand-induced and clathrin-dependent AMPA receptor (AMPAR) internalization, supporting a function of HIP1 in clathrin-mediated endocytosis in vivo (Metzler et al., 2003). AMPARs are dynamic neurotransmitter receptors, moving in and out of postsynaptic membranes through constitutive and ligand-induced internalization (Carroll et al., 1999b; Luscher et al., 1999; Lin et al., 2000; Man et al., 2000). They are concentrated in dendritic spines and responsible for fast synaptic transmission at excitatory synapses in the CNS. AMPARs colocalize with NMDA receptors (NMDARs), which play important roles in synaptic plasticity and excitotoxic cell death. Indeed, NMDAR function is enhanced in Huntington's disease (HD), a devastating neurodegenerative disorder caused by polyglutamine (polyQ) expansion in htt (Vonsattel and DiFiglia, 1998).
The earliest evidence for an involvement of NMDARs in HD came from studies by Beal et al. (1986), who replicated loss of striatal neurons in HD by injection of quinolinic acid (QA), an NMDAR agonist, into rat brain. These early observations were supported by studies in the yeast artificial chromosome (YAC) transgenic mouse model of HD expressing human full-length htt with either 46, 72, or 128 polyQ (YAC46, YAC72, and YAC128) (Hodgson et al., 1999; Slow et al., 2003; Graham et al., 2006). Injection of QA into the striatum of YAC72 and YAC128 transgenic mice led to a greater lesion volume and NMDAR-mediated excitotoxic response compared with wild-type mice. Similarly, stimulation of striatal neurons established from YAC46, YAC72, or YAC128 transgenic mice led to a significant increase in NMDA-induced excitotoxicity compared with wild-type controls (Zeron et al., 2002; Shehadeh et al., 2006). This increase in cell death is accompanied by an enhanced Ca2+ load through stimulation of NMDARs and can be blocked by administration of ifenprodil, an NMDAR subunit 2B (NR2B)-selective antagonist with high efficiency for blocking NR1/NR2B-containing receptors. This is compatible with an involvement of NR2B-containing NMDARs in the pathogenesis of HD.
The molecular mechanism responsible for enhanced NMDA-induced excitotoxicity in HD is unknown. Alterations in NMDAR trafficking to and from the cell surface with the ultimate effect of prolonging Ca2+ influx, changes in subunit composition and channel characteristics, or alterations in scaffolding and downstream signaling could all contribute to this phenomenon.
To investigate possible regulation of NMDAR function by HIP1, we analyzed NMDA-dependent excitotoxicity and NMDA-dependent transport in HIP1−/− mice. These studies reveal a direct interaction between HIP1 and NMDARs. Neurons from HIP1−/− mice exhibit a decrease in NMDA-dependent AMPAR internalization and partial protection from NMDA-induced excitotoxicity. Furthermore, alterations in phosphorylation of htt and Akt were observed after NMDA-induced excitotoxicity, which is diminished in neurons from HIP1−/− mice. Together, these findings demonstrate modulation of NMDAR function by HIP1.
Materials and Methods
Neuronal culture and mice.
Embryos were isolated from embryonic day 15.5 (E15.5)–E17.5 time-pregnant HIP1+/− females. Brain tissue was dissected, and corresponding tails were frozen and used for genotype determination as described previously (Metzler et al., 2003). Neurons were seeded onto poly-d-lysine-coated surfaces at a density of ∼1.5 × 105 cells/cm2. Cultures were maintained at 37°C under 5% CO2, and one-half of the culture media was exchanged once every 4–5 d.
All experiments were performed in accordance with protocols approved by the University of British Columbia Committee on Animal Care and the Canadian Council on Animal Care.
Antibodies.
The following antibodies (Abs) were used for expression analyses: the mouse monoclonal antibody (mAb) HIP1#9 and the rabbit polyclonal Ab (pAb) against HIP1 (HIP1FP) have been described previously (Chopra et al., 2000; Metzler et al., 2001). Abs against NR2B and postsynaptic density-95 (PSD-95) were purchased from Millipore (Billerica, MA); a goat pAb detecting NR2A was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The mouse mAb against the His6 epitope was purchased from Applied Biological Materials (Richmond, British Columbia, Canada). Total Akt and P-Akt-S437 were detected with rabbit pAbs from Cell Signaling Technology (Beverly, MA). Phosphorylation of htt on S421 was determined with a pAb, as described previously (Warby et al., 2005). Total htt was determined by probing with mAb 2166 (Millipore). Surface expression of AMPAR subunit 2 (GluR2) was determined with a mouse mAb against GluR2 (Millipore). Alexa-labeled secondary Abs were purchased from Invitrogen (Carlsbad, CA).
Immunofluorescence.
Hippocampal and cortical neurons were processed for immunofluorescence after 14–16 d in vitro (DIV) essentially as described previously (Metzler et al., 2003). For double immunofluorescence, cells were incubated with anti-HIP1 Abs in 2% normal goat serum (NGS) in PBS at 4°C overnight followed by incubation with secondary Abs for 2 h at room temperature (RT) with extensive washing in PBS and 1% BSA between each incubation. Neurons were then incubated with Alexa 488- and Alexa 568-labeled secondary Abs (Invitrogen), washed, mounted, and analyzed under a Zeiss (Oberkochen, Germany) Axioscope microscope taking pictures with a cool-snap CCD camera using MetaMorph imaging software (PerkinElmer, Woodbridge, Ontario, Canada).
Generation and purification of His6-tagged fusion proteins.
Glutathione S-transferase (GST)-HIP1-(219–616) has been described previously (Metzler et al., 2001), and GST-PSD-95-(150–253), which contains the PDZ2 domain of rat PSD-95, has been kindly provided by Dr. Alaa El-Husseini (University of British Columbia, Vancouver, British Columbia, Canada). The corresponding fragments of HIP1 and PSD-95/Discs large/zona occludens-1 domain 2 (PDZ2) were cloned into pTrc-His (Invitrogen), and the sequence of His6-tagged HIP1-(219–616) and His6-tagged PSD-95–PDZ2 was verified by DNA sequencing. Both His6-tagged fusion proteins were expressed in Escherichia coli BL21 (GE Healthcare, Piscataway, NJ) and purified from bacterial lysates on Ni–nitrilotriacetic acid–agarose (Qiagen, Mississauga, Ontario, Canada). The purified proteins were dialyzed against PBS, and 25 μg of protein was used for GST pull down.
Generation of GST fusion proteins and GST pull down.
C-terminal fragments of NR2A and NR2B were cloned into pGEX and the corresponding GST-NR2A-(1017–1464) and GST-NR2B-(1001–1484) expression constructs were verified by sequencing. Either GST alone or GST fusion proteins were expressed in E. coli BL21 after induction with 0.1 mm isopropyl-β-d-1-thiogalactopyranoside for 3 h at 37°C. Bacteria were pelleted and lysed in PBS, 0.5 mm EDTA, 1% Triton X-100, and 1 mm PMSF in the presence of a mixture of protease inhibitors (Roche Diagnostics, Indianapolis, IN). After brief sonication and incubation on ice for 20 min, bacterial lysates were centrifuged at 20,817 × g for 3 min. The protein concentration was measured in the supernatant, and samples were analyzed by SDS-PAGE and Coomassie blue staining.
Whole brain from 3-month-old mice was Dounce homogenized in buffer A (10 mm HEPES, pH 7.4, 0.83 mm benzamidine, 0.23 mm PMSF, 0.5 μg/ml aprotinin, and 0.5 μg/ml leupeptin) and centrifuged at 956 × g for 10 min at 4°C. After preclearing in the presence of 1% Triton X-100 and 1% sodium deoxycholate, the supernatant was incubated with GST-fusion proteins precoupled to glutathione-Sepharose 4B beads (GE Healthcare) overnight at 4°C. Equivalent amounts of GST fusion protein input were verified by Coomassie blue staining. After incubation, samples were washed four times in buffer A containing 1% Triton X-100. In other cases, 25 μg of recombinant HIP1-(219–616)-His6 and recombinant PSD-95-PDZ2-His6 was incubated overnight at 4°C in buffer A and 1% Triton X-100 with the various GST-fusion proteins precoupled to glutathione-Sepharose beads. After incubation, samples were washed in buffer A containing 1% Triton X-100, eluted in SDS sample buffer, and separated by SDS-PAGE.
Coimmunoprecipitation.
Forebrains from 3-month-old mice were homogenized with a Polytron at 20,000 rpm for 30 s in homogenization buffer (PBS, 1 mm EDTA, 1 mm EGTA, 1 mm PMSF, 2 μg/ml aprotinin, 2 μg/ml leupeptin, and 4 μg/ml pepstatin A) and centrifuged at 956 × g for 10 min at 4°C. The pellet was resuspended in ice-cold homogenization buffer supplemented with 0.2% SDS, 0.8% Triton X-100, and 0.5 U/ml DNase I. The homogenate was precleared with protein A/G Sepharose 4-Fast Flow beads (GE Healthcare), and 3 mg of protein was immunoprecipitated with 5 μg of HIP1- or NR2B-specific Abs or normal IgG for at least 1 h at 4°C. Subsequently, samples were pelleted and washed in ice-cold 50 mm Tris/HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, and 1% Triton X-100. Samples were resuspended in SDS sample buffer, heated to 95°C, centrifuged at 14,000 × g, and separated by SDS-PAGE.
Cell-death assays.
To measure total NMDA-induced cell death, a cytotoxicity detection kit was used that determines the amount of lactate dehydrogenase (LDH) release into the cell culture supernatant of dying cells (Roche Diagnostics). Briefly, cortical neurons at 12 DIV were stimulated with NMDA and 30 μm glycine in extracellular solution (ECS) (in mm: 14 NaCl, 0.54 KCl, 0.13 CaCl2, 0.1 MgCl2, 1 HEPES, and 2.5 glucose, pH 7.35)] in the absence of MgCl2 for 10 min at 37°C. Neurons were washed in ECS and incubated in cell-culture media for 14 h. Subsequently, LDH release was measured according to the manufacturer's instructions.
To determine apoptotic cell death after NMDAR stimulation, a combination of terminal deoxynucleotidyl transferase-mediated biotinylated dUTP nick end labeling (TUNEL) and propidium iodide (PI) staining was used as described previously (Zeron et al., 2002). Caspase-3 activation was measured 3 h after NMDA stimulation. Neurons were washed in ECS and lysed in 20 mm Tris-HCl, pH 7.5, 140 mm NaCl, 0.5 mm EDTA, 1% Triton X-100, and 4 mm Pefabloc. Samples were incubated for 10 min on ice and centrifuged for 10 min at 956 × g. The supernatant was removed, and aliquots were taken to determine protein concentration. Duplicate samples were incubated in a 96-well plate with the fluorogenic caspase-3 substrate N-acetyl-DEVD-7-amino-4-trifluoromethyl coumarin (Ac-DEVD-AFC; BIOMOL, Plymouth Meeting, PA). The amount of cleaved AFC was measured in a multiwell plate reader (Victor; PerkinElmer) equipped with fluorescence capabilities at an excitation at 400 nm and emission at 505 nm. Measurements were taken every 10 min over a time course of 1 h and compared with an AFC standard curve.
Quantification of P-Akt and P-htt.
To determine changes in phosphorylation of Akt and htt, cortical neurons at 12 DIV were harvested after NMDA stimulation in ice-cold PBS supplemented with 2.5 mm NaF, 1 μg/ml aprotinin, 1 μg/ml leupeptin, 1 mm NaVO4, 1 mm β-glycero-PO4, and 4 mm Pefabloc. The pellet was resuspended in lysis buffer [50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1% IGEPAL (Sigma, St. Louis, MO), 2.5 mm NaF, 1 μg/ml aprotinin, 1 μg/ml leupeptin, 1 mm NaVO4, 1 mm β-glycero-PO4, 4 mm Pefabloc, and 5 μm Z-VAD (EMD Biosciences, La Jolla, CA)] and incubated for 20 min on ice. After centrifugation at 20,817 × g, the protein concentration was measured in the supernatant, and 30 μg of protein was resuspended in lithium dodecyl sulfate (LDS) sample buffer (Invitrogen) and separated by 7.5% SDS-PAGE, blotted onto membranes, and probed with Abs recognizing P-Akt-S473 and total Akt, respectively.
To determine P-htt over total, 400 μg of protein was incubated with 2–4 μl of mouse mAb 2166 bound to protein A/G Sepharose 4 Fast Flow (GE Healthcare) in lysis buffer overnight at 4°C. After immunoprecipitation, samples were washed in lysis buffer, and bound protein was eluted in LDS-PAGE sample buffer. Proteins were separated by 3–8% Tris-acetate gel electrophoresis (Invitrogen), blotted onto membranes, and probed with a pAb specifically recognizing P-htt-S421; total htt was detected by probing with mAb 2166.
Colorimetric assay to determine GluR2 surface expression.
The colorimetric assay was performed with cortical neurons at 14 DIV, essentially as described previously (Metzler et al., 2003). Briefly, cortical neurons were treated with 100 μm NMDA, 10 μm glycine, and 5 μm strychnine for 10 min at 37°C, washed, and incubated for 20 min at 37°C. After fixation in 4% PFA/4% sucrose in PBS for 10 min, one-half of the neurons in each group were permeabilized with 0.2% Triton X-100 in PBS for 5 min and washed three times in PBS. Nonspecific binding was blocked in 5% NGS for 1 h at RT. Neurons were then incubated with a mAb recognizing the extracellular domain of GluR2 (1 μg/ml; Millipore) to label surface-expressed receptors under nonpermeant conditions, or the entire receptor pool was labeled under permeant conditions as described previously (Man et al., 2000). Values for each group were averaged using at least three separate plates from three independent cortical cultures.
Internalization assay of GluR2.
Striatal neurons at 11 DIV were live labeled with an mAb against GluR2 (5 μg/ml; Millipore) for 15 min at 37°C as described previously (Metzler et al., 2003). Neurons were then washed with ECS and treated for 5 min at 37°C with either ECS alone or (in μm) 50 NMDA, 10 glycine, and 5 strychnine in ECS in the absence of MgCl2. Cultures were washed and incubated for 20 min at 37°C to allow internalization. Neurons were fixed in 4% PFA/4% sucrose in PBS, washed, and incubated with goat anti-mouse Alexa 568 F(ab′) fragment-labeled secondary antibodies for 1 h at RT to visualize surface-expressed GluR2. Subsequently, neurons were permeabilized in 0.2% Triton X-100 in PBS, washed, and treated with goat anti-mouse Alexa 488-labeled secondary Abs for 1 h at RT to detect prelabeled internalized GluR2s. Neurons were washed, mounted in Mowiol, and analyzed under a Zeiss Axioscope microscope. Images were selected under differential interference contrast to avoid bleaching of red and green fluorescence intensities and acquired using a CCD camera. Exposure times for each red and green channel were kept constant in each independent experiment. Fluorescence intensities in regions of interest (ROIs) at both somatic and dendritic areas were measured for both red and green channels using MetaMorph imaging software. The signal intensities in ROIs were calculated as green-to-red signal ratios to determine the degree of internalization. Background ROIs were also measured and subtracted. Approximately 240 ROIs were measured for each sample and each condition. The values of the green-to-red ratio were averaged for each group in each experiment. Figure panels are presented without additional enhancement.
Electrophysiology.
Hippocampal brain slices (350 μm thickness) from wild-type and HIP1−/− littermates at 2–3 weeks of age were obtained in ice-cold artificial CSF (ACSF) containing (in mm) 126 NaCl, 2.5 KCl, 1 MgCl2, 1 CaCl2, 1.25 KH2PO4, 26 NaHCO3 and 20 glucose that was bubbled continuously with carbogen (95% O2/5% CO2) to adjust the pH to 7.4. For electrophysiological recordings, slices were transferred to a recording chamber perfused continuously by carbogenated ACSF containing bicuculline methiodide (10 μm) to block GABAA receptor-mediated inhibitory synaptic currents. Whole-cell recordings of CA1 neurons in brain slices were performed at RT using the “blind” method with a MultiClamp 700A amplifier. Recording pipettes were filled with solution containing (in mm) 132.5 Cs-gluconate, 17.5 CsCl, 2 MgCl2, 0.5 EGTA, 10 HEPES, 4 ATP, and 5 QX-314 [N-(2,6-dimethylphenylcarbamoylmethyl)triethylammonium bromide], with pH adjusted to 7.2 by CsOH. EPSCs were evoked by stimulating the Schaffer collateral–commissural pathway via constant-current pulses (0.05 ms) delivered through a tungsten bipolar electrode and recorded while the voltage was clamped at −60 mV. Long-term depression (LTD) of EPSCs was induced by bath application of 20 μm NMDA for 3 min.
Results
HIP1 colocalizes with NMDARs
We performed colocalization studies in hippocampal and cortical neurons between HIP1, NMDAR components, and the scaffolding protein PSD-95, which is concentrated at the postsynaptic membrane in dendritic spines. These studies revealed high expression of HIP1 in neuronal cell bodies and in dendrites, as observed previously (Fig. 1) (data not shown) (Metzler et al., 2003). HIP1 expression was diffuse along neuronal dendrites and partially concentrated at postsynaptic membranes, at which colocalization with PSD-95 was observed (Fig. 1). Moreover, partial colocalization between HIP1 and both NMDAR subunits, NR2A and NR2B, occurred at distinct puncta along neuronal dendrites (Fig. 1). The observed localization of HIP1 to dendritic spines and its colocalization with NMDAR components is compatible with a proposed role of HIP1 influencing NMDAR function.
Figure 1.

HIP1 colocalizes with NMDARs. HIP1 expression was analyzed by double immunofluorescence in primary hippocampal neurons after 14 DIV. HIP1 immunostaining was either detected with the mAb HIP1#9 (top and middle) or the pAb HIP1FP (bottom) shown in green. Expression of PSD-95, NR2A, and NR2B is shown in red. HIP1 localizes to synapses, at which it colocalizes with PSD-95. HIP1 also shows partial colocalization with NR2A (middle) and NR2B (bottom). Arrows indicate colocalization. Scale bars, 10 μm.
HIP1 interacts with NMDARs
To test for interaction between HIP1 and NMDAR components, GST pull-down assays were performed with equivalent amounts of GST alone and GST-HIP1-(219–616) fusion protein containing the central portion of HIP1 (Fig. 2A) (Metzler et al., 2003). These studies revealed interaction between HIP1 and NMDAR subunits NR2A and NR2B (Fig. 2A). Similarly, when HIP1 was immunoprecipitated from brain lysate of wild-type mice, coimmunoprecipitation of NR2B was observed. In contrast, no immunoprecipitating band was present in brain lysate from HIP1−/− mice or after immunoprecipitation with normal IgG, used here as negative controls (Fig. 2B). Vice versa, immunoprecipitation of NR2B copurifies HIP1 in brain lysate from wild-type but not HIP1−/− mice, and no immunoprecipitating bands were evident in the presence of normal IgG (Fig. 2C).
Figure 2.

HIP1 interacts with NMDARs in vitro and in brain lysate. A, Soluble proteins from brain lysate were affinity purified with either GST alone or GST-HIP1-(219–616) bound to glutathione-Sepharose beads. The input amount of GST alone and GST fusion protein was verified by Coomassie blue staining, and their sizes were compared with a molecular-weight (MW) marker. Bound proteins were eluted and analyzed by Western blot and probed with anti-NR2A and anti-NR2B Abs as indicated. B, Brains from 3-month-old wild-type and HIP1−/− mice were homogenized and solubilized in the presence of 0.2% SDS and 0.8% Triton X-100. Proteins were immunoprecipitated with mAb HIP1#9 or normal IgG, separated by Western blot, and probed with Abs against HIP1 and NR2B. The same amount of sample was loaded on gels stained for NR2B and HIP1. C, Proteins were immunoprecipitated as described in B with an Ab against NR2B and normal IgG. D, Recombinant HIP1-(219–616)-His6 and recombinant PSD-95-PDZ2-His6 were expressed and purified from bacterial lysate. Twenty-five micrograms of purified HIP1 or PDZ2 were incubated with GST alone, GST-NR2A-(1017–1464), and GST-NR2B-(1001–1484) bound to glutathione-Sepharose beads. The input amount of GST alone and GST fusion proteins was verified by Coomassie blue staining, and their sizes were compared with a molecular-weight marker. The amount of specifically bound protein was eluted, immunoblotted, and probed with mAb HIP1#9 or an anti-His Ab, demonstrating direct interaction between HIP1 and NMDARs and PDZ2 and NMDARs, respectively. IP, Immunoprecipitate.
The NMDAR subunit NR2B and the endocytic protein HIP1 both contain binding sites for the endocytic adaptor protein AP2, which is responsible for AP2-dependent internalization of NR2B in cerebellar granule neurons (Beattie et al., 2000; Ehlers, 2000; Metzler et al., 2001; Roche et al., 2001; Lee et al., 2002; Prybylowski et al., 2005). Therefore, it is possible that interaction between HIP1 and NMDAR components is indirect and is mediated through binding to AP2. To test for direct interaction between HIP1 and NMDARs, a recombinant His-tagged HIP1 fusion protein, HIP1-(219–616)-His6, was tested for interaction with equivalent amounts of GST-NR2A-(1017–1464), GST-NR2B-(1001–1484), or GST alone (Fig. 2D). Interestingly, His-tagged HIP1 was pulled down by the C-terminal portion of both NR2 subunits, demonstrating direct interaction between HIP1 and NMDAR components (Fig. 2D). Furthermore, this interaction is specific, because no binding between HIP1 and GST alone was observed. To ascertain correct folding of the C-terminal NR2 fusion proteins, we studied their interaction with PSD-95. This interaction is mediated through the PDZ2 domain in PSD-95, which was expressed and purified from bacterial lysates. As shown in Figure 2D, both C-terminal fragments of NR2A and NR2B interact with recombinant PDZ2. No interaction is observed with GST alone. A specific interaction was also observed in pull-down assays using cell lysate from COS7 cells overexpressing PSD-95, providing additional evidence for interaction of HIP1 with correctly folded NR2 subunits (data not shown). In summary, we have demonstrated interaction between HIP1 and NR2 subunits, suggesting a possible role of HIP1 in NR2A- and NR2B-dependent NMDAR functions.
NMDA-induced excitotoxicity is reduced in HIP1−/− neurons
A contributing factor to neuronal death in HD is overactivation of NMDARs and enhanced excitotoxicity in striatal and cortical neurons (Beal et al., 1986; Zeron et al., 2002; Van Raamsdonk et al., 2005). To assess a possible role of HIP1 in NMDA-induced excitotoxicity in neurons susceptible to HD, we analyzed striatal and cortical cultures. First, we established cortical neurons from all three genotypes to assess total cell death after treatment with varying doses of NMDA for 10 min. Excitotoxicity was measured by LDH release 14 h after NMDA treatment. These studies revealed a 42.8 ± 6.9% increase in LDH release in wild-type cultures after stimulation with 250 μm NMDA compared with control cultures (Fig. 3A). In heterozygous neurons, LDH release increased by 36.6 ± 4.5%. In contrast, neurons from HIP1−/− mice showed an increase in LDH release of only 14.2 ± 2.3% (p ≤ 0.0006; n = 10 for HIP1−/− compared with wild type). This result suggests protection from NMDA-induced cell death in HIP1−/− neurons.
Figure 3.

Reduced NMDA-induced excitotoxicity in HIP1−/− neurons. A, Cortical neurons at 12 DIV were treated with different doses of NMDA and 30 μm glycine for 10 min. After drug removal, neurons were cultured for 14 h, and release of LDH into the cell culture supernatant was measured. Duplicate samples for each condition and genotype in each of six independent experiments were determined. B, Striatal neurons from wild-type and HIP1−/− mice were treated with 500 μm NMDA and 30 μm glycine for 5 min at 9 DIV. After drug removal, neurons were cultured for 20 h and stained for TUNEL-positive neurons (green) followed by PI counterstaining (red). C, Cortical neurons at 12 DIV from wild-type and HIP1−/− mice were treated with 100 or 500 μm NMDA and 30 μm glycine for 5 min in the presence or absence of 50 μm MK-801 and processed as described in B. The number of TUNEL-positive neurons was determined and expressed relative to the number of total neurons. For each sample, 200 neurons were counted for each condition and genotype in each of three independent experiments. D, Cortical neurons at 12 DIV were treated with varying doses of NMDA and 30 μm glycine for 10 min. After drug removal, neurons were cultured for 3 h, and caspase-3 activation was measured fluorometrically by cleavage of Ac-DEVD-AFC. Duplicate samples for each condition and genotype in each of seven independent experiments were determined. *p < 0.05; **p < 0.005; ***p < 0.0005 compared with wild type; t test. Error bars denote ±SEM.
Neurodegeneration in HD occurs by an apoptotic mechanism, and a number of studies report activation of caspases, including caspase-3, after NMDA-induced excitotoxicity (Okamoto et al., 2002; Zeron et al., 2002, 2004). To determine apoptosis after NMDAR stimulation, we first identified apoptotic cells by TUNEL and counterstaining with PI (Fig. 3B,C) (data not shown). These studies revealed significant increases in the number of apoptotic neurons in cortical and striatal cultures from wild-type mice after NMDA treatment. In contrast, NMDA-induced cell death was reduced in neurons from HIP1−/− mice. The number of apoptotic neurons in striatal cultures from HIP1−/− mice after 500 μm NMDA stimulation was 16 ± 2.2% compared with 21.9 ± 1.5% in wild-type cultures (p ≤ 0.04; n = 9 for wild type and n = 4 for HIP1−/−) (Fig. 3B) (data not shown).
A similar reduction in NMDA-induced cell death was observed in cortical neurons from HIP1−/− mice after treatment with 500 μm NMDA (17.5 ± 1.5% for wild type and 7.7 ± 2.0% for HIP1−/−; p ≤ 0.02 with n = 3 for both genotypes). These results suggest a role for HIP1 in the execution of NMDA-induced apoptotic cell death. Moreover, the observed effect is NMDAR specific, because pretreatment of cortical neurons with dizocilpine (MK-801) abolished NMDA-induced cell death to near control levels (Fig. 3C).
To assess caspase-3 activation, cortical cultures from all three genotypes were established and stimulated with varying doses of NMDA for 10 min. Caspase-3 activation was measured 3 h after NMDA treatment using a fluorogenic caspase-3 activation assay. These studies revealed significant increases in caspase-3 activation in wild-type cultures after NMDA treatment (50–500 μm) compared with unstimulated controls (Fig. 3D). For example, after 500 μm NMDA stimulation, caspase-3 activation increased by 128 ± 9.9% in wild-type neurons. In contrast, neurons from HIP1+/− and HIP1−/− mice showed an increase in caspase-3 activation of only 84.5 ± 9.9 and 55.7 ± 10.2%, respectively (p ≤ 0.016, n = 14 for HIP1+/− compared with wild type; p ≤ 0.0004; n = 12 for HIP1−/− neurons compared with wild type). Together, these findings demonstrate that HIP1 modulates NMDAR function and participates in a pathway executing NMDA-induced cell death.
HIP1 promotes dephosphorylation of Akt during NMDA-induced excitotoxicity
NMDARs serve two opposing functions. Neuronal survival is promoted by synaptic physiological stimulation of NMDARs through activation of Akt and the Ras–extracellular signal-regulated kinase 1/2 (ERK1/2) pathways, phosphorylation of cAMP response element (CRE)-binding protein (CREB), and CRE-mediated expression of survival genes (Yano et al., 1998; Perkinton et al., 2002; Sutton and Chandler, 2002; Lee et al., 2005; Papadia et al., 2005). In contrast, overactivation of NMDARs leads to increased phosphatase activity and the reversal of these responses, thereby effectively shutting down neuronal survival (Hardingham and Bading, 2002; Hardingham et al., 2002). To test whether alterations in this shut-off pathway occur in neurons from HIP1−/− mice, we determined the level of active and phosphorylated Akt at serine 473 after NMDA-induced excitotoxicity (Fig. 4A,B). Stimulation of wild-type cortical neurons with 250 μm NMDA for 10 min led to a 48 ± 5% decrease in P-Akt-S473 with no additional reduction after administration of 500 μm NMDA. In contrast, a reduction in P-Akt-S473 level of only 3 ± 10% occurred after stimulation with 250 μm NMDA in neurons from HIP1−/− mice, and only a 16 ± 10% reduction was observed after stimulation with 500 μm NMDA. Both responses were significantly different from wild type (p ≤ 0.001 for 250 μm NMDA; p ≤ 0.015 for 500 μm NMDA; n = 11 for wild type; n = 6 for HIP1−/− neurons). A half-maximal response was observed in neurons from HIP1+/− mice, although not significantly different from wild type (p ≤ 0.06 for 250 μm NMDA; p ≤ 0.18 for 500 μm NMDA; n = 11 for wild type; n = 7 for HIP1+/−). Moreover, the ratio between P-Akt-S473 and total Akt was similar under control conditions among all three genotypes (Fig. 4C). Together, the increased level of P-Akt-S473 in neurons from HIP−/− mice is in agreement with the notion that these neurons are protected from NMDA-induced excitotoxicity, supporting a role for HIP1 in this process.
Figure 4.
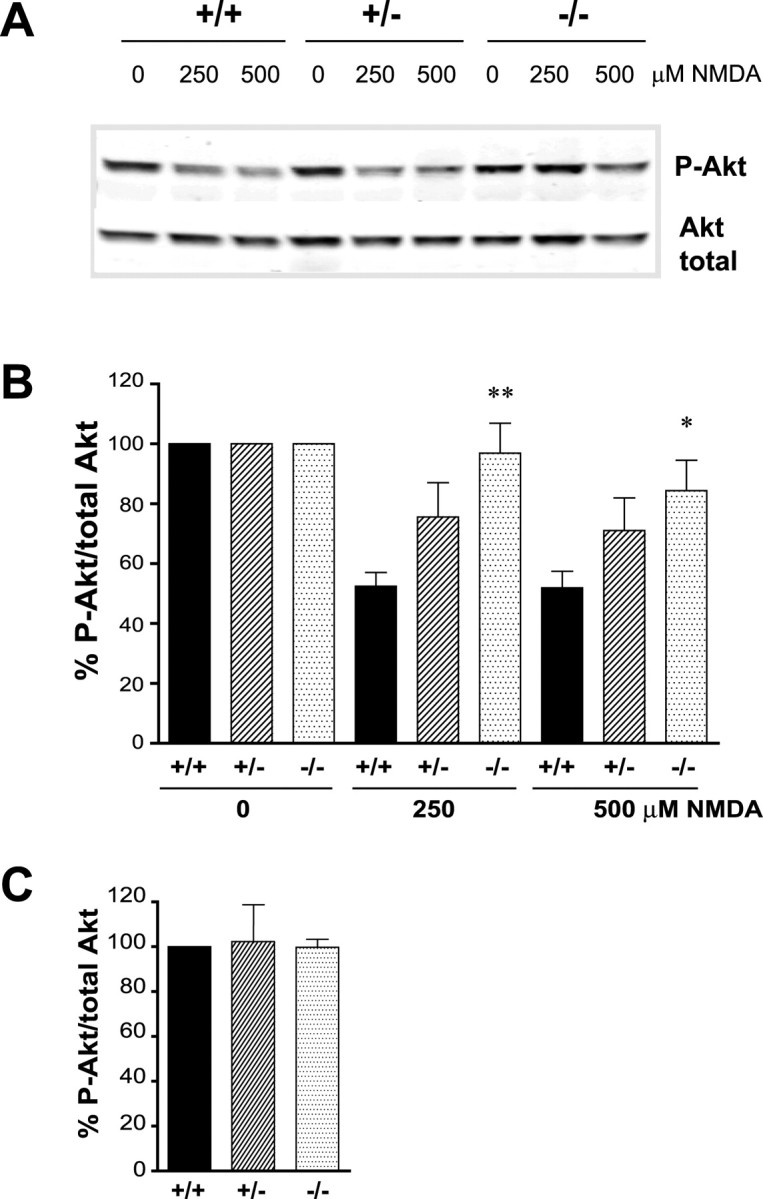
HIP1 promotes dephosphorylation of Akt during NMDA-induced excitotoxicity. A, Cortical neurons from wild-type and HIP1−/− mice were treated with varying doses of NMDA and 30 μm glycine for 10 min at 12 DIV. After drug removal, neurons were cultured for 90 min and processed for Western blotting. Membranes were probed with Abs against P-Akt-S473 and total Akt. B, The ratio between P-Akt-S473 and total Akt was determined by densitometry and expressed relative to controls. Duplicate samples for each condition and genotype in each of eight independent experiments were determined (*p < 0.05; **p < 0.005 compared with wild type; t test). C, The absolute ratio of P-Akt-S473 and total Akt was determined by densitometry under control conditions for all three genotypes and expressed relative to wild type. Error bars denote ±SEM.
HIP1 promotes dephosphorylation of htt during NMDA-induced excitotoxicity
A number of reports demonstrate a role for htt in neuronal survival that is at least in part dependent on phosphorylation of htt by Akt on serine 421 (P-htt-S421) (Humbert et al., 2002; Zuccato et al., 2003; Warby et al., 2005; Leavitt et al., 2006). Therefore, it is possible that in addition to changes in phosphorylation of Akt, changes in phosphorylation of htt are also present during NMDA-induced excitotoxicity and shut off of neuronal survival. To test for changes in phosphorylation of htt, a phospho-specific antibody recognizing phosphorylation of htt at serine 421 was used, and the level of P-htt-S421 over total was determined (Warby et al., 2005). Treatment of cortical neurons from wild-type mice with 250 and 500 μm NMDA for 10 min led to 31 ± 3 and 34 ± 3% decreases, respectively, in P-htt-S421 (Fig. 5A,B). In contrast, the level of P-htt-S421 over total in neurons from HIP1−/− mice was significantly increased after NMDA stimulation compared with wild type (48 ± 2.5%, p ≤ 0.003 for 250 μm NMDA; 76 ± 6.9%, p ≤ 0.033 for 500 μm NMDA; n = 6 for each genotype). Again, no significant difference in P-htt-S421 was observed under basal conditions comparing all three genotypes (Fig. 5C). The reduction in phosphorylation of htt on S421 during NMDA-induced excitotoxicity suggests a role of htt in the pathway executing NMDA-induced cell death. Moreover, the increased level of P-htt-S421 in neurons from HIP1−/− mice compared with wild type is in agreement with protection from NMDA-induced excitotoxicity of HIP1−/− neurons.
Figure 5.
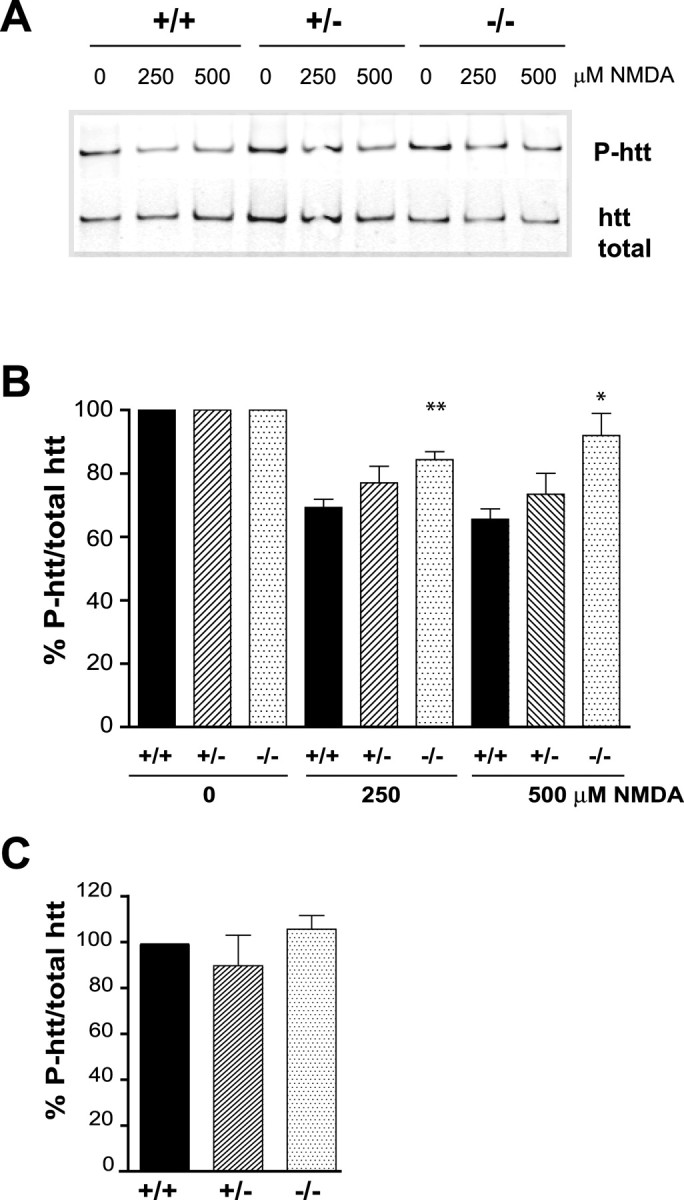
HIP1 promotes dephosphorylation of htt during NMDA-induced excitotoxicity. A, Cortical neurons from wild-type and HIP1−/− mice were treated with varying doses of NMDA and 30 μm glycine for 10 min at 12 DIV. After drug removal, neurons were cultured for 90 min and processed for immunoprecipitation of htt. Precipitated protein was separated by Western blot, and membranes were probed with Abs against P-htt-S421 and total htt (mAb 2166). B, The ratio between P-htt-S421 and total htt was determined by densitometry and expressed relative to controls. Ratios for each condition and genotype in each of six independent experiments were determined (*p < 0.05; **p < 0.005 compared with wild type; t test). C, The absolute ratio between P-htt-S421 and total htt was determined by densitometry under control conditions for all three genotypes and expressed relative to wild type (n = 4). Error bars denote ±SEM.
NMDA-induced AMPAR internalization is diminished in neurons from HIP1−/− mice
Activation of NMDARs leads to rapid removal of AMPARs from the cell surface in cell culture and in hippocampal brain slices (Carroll et al., 1999a; Beattie et al., 2000; Ehlers, 2000). This process is dependent on clathrin-mediated endocytosis (Carroll et al., 1999b; Lee et al., 2002). Moreover, evidence obtained in a recent study suggests that such facilitated AMPAR endocytosis plays an essential role in mediating NMDA-induced neuronal apoptosis that could be regulated by the endocytic protein HIP1 (Wang et al., 2004).
To investigate a possible involvement of HIP1 in this process, primary cortical neurons from all three genotypes were established, stimulated with 100 μm NMDA for 10 min, and assessed for alterations in surface expression of the AMPAR subunit GluR2 using a quantitative ELISA-based assay (Man et al., 2000). These studies revealed a significant blockade in NMDA-induced removal of GluR2 from the cell surface in neurons from HIP1−/− mice compared with wild type (Fig. 6A). As expected, stimulation of wild-type neurons with 100 μm NMDA led to a significant reduction in surface expression of GluR2 by 18.6 ± 2.5% (p ≤ 0.01 for wild type; between-treatment comparison; n = 3; t test). In contrast, surface expression of GluR2 increased by 10.2 ± 5.6% after NMDA treatment in cultures from HIP1−/− mice and is significantly different from wild type (p ≤ 0.03 between genotypes, for within-treatment comparison; n = 3; t test). A similar trend, although not significant, was observed in neurons from HIP1+/− littermates, in which NMDA treatment led to an increase in surface expression of GluR2 by 3.9 ± 5.1% (p ≤ 0.06 between genotypes, for within-treatment comparison; n = 3). The observed net increase in surface GluR2 expression in neurons from HIP1−/− mice suggests a deficit in receptor internalization, although enhanced receptor insertion cannot be ruled out.
Figure 6.
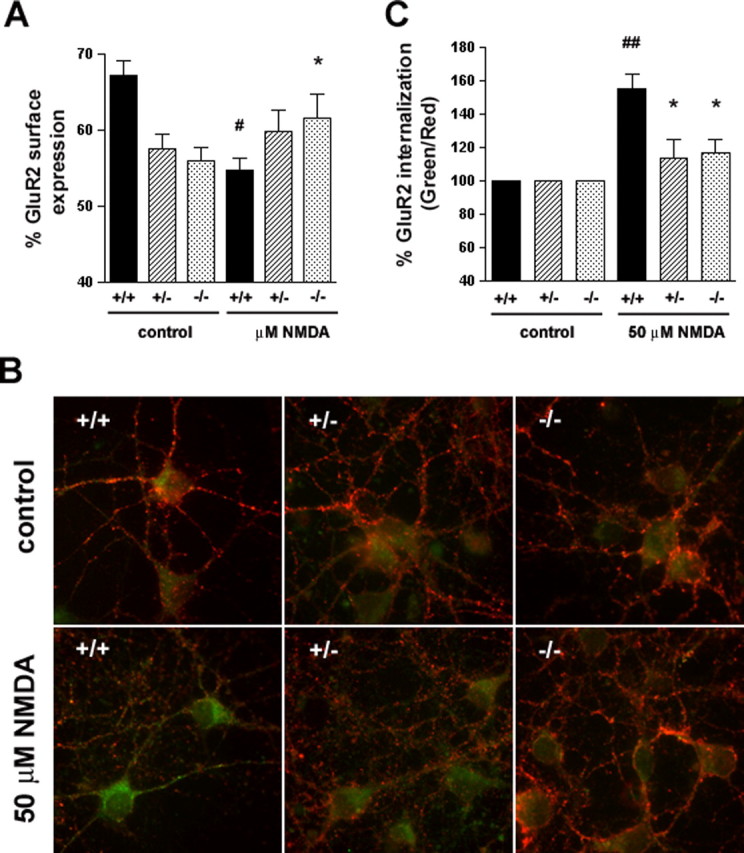
Reduced NMDA-induced AMPAR internalization in HIP1−/− neurons. A, HIP1 modulates NMDA-induced GluR2 trafficking in cortical neurons. Cortical neurons were established from wild-type (+/+), heterozygous (+/−), and homozygous (−/−) littermates. After 12–15 DIV, the percentage of surface-expressed GluR2 was determined in a quantitative colorimetric assay in control and 100 μm NMDA-stimulated cultures under nonpermeant and permeant conditions. Results were averaged for each group in each of three independent experiments (#p < 0.05 for within-genotype, between-treatment comparison; *p < 0.05 for within-treatment, between-genotype comparison; t test). B, HIP1 modulates NMDA-induced GluR2 internalization in striatal neurons. Striatal neurons were cultured from wild-type (+/+), heterozygous (+/−), or homozygous (−/−) littermates and live-labeled with an Ab recognizing the extracellular domain of GluR2, followed by treatment with either control solution (ECS alone) or 50 μm NMDA, 10 μm glycine, and 5 μm strychnine for 5 min at 37°C. Cultures were washed and incubated for 20 min at 37°C to allow internalization. Surface GluR2 was then visualized using a red secondary Ab, whereas internalized GluR2 was labeled with a green secondary Ab. C, Internalization was quantified as the green-to-red signal ratio, which represents the ratio of the internalized GluR2 to the GluR2 remaining on the cell surface after different treatments (see Materials and Methods). Green-to-red ratios were averaged for each group in each of three independent experiments (##p < 0.005 for within-genotype, between-treatment comparison; *p < 0.05 for within-treatment, between-genotype comparison; t test). Error bars denote ±SEM.
To directly address this question, a quantitative single-cell-based internalization assay was used, which determines receptor internalization. This assay is based on the live labeling of surface-expressed GluR2 and the differential detection of surface-expressed and internalized GluR2 comparing control and stimulated cultures. No detection of surface-expressed GluR2 with the green secondary Ab was observed in nonpermeabilized cells, demonstrating specific detection of internalized GluR2 by this Ab (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material) (Beattie et al., 2000; Ehlers, 2000; Lee et al., 2002). In wild-type neurons, the ratio between internalized (green) and surface-expressed GluR2 (red) increases significantly by 56% after stimulation with 50 μm NMDA for 5 min compared with untreated cultures (Fig. 6B,C) (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material) (p ≤ 0.004 between treatment; n = 6 for wild type), demonstrating removal of GluR2 from the cell surface. In contrast, a significant defect in GluR2 internalization exists in striatal neurons from HIP1+/− and HIP1−/− mice, in which only a minimal and nonsignificant increase in GluR2 internalization is observed after NMDA stimulation. Accordingly, the green/red ratio in neurons from HIP1+/− and HIP1−/− mice increases by only 14 and 17%, respectively, compared with 56% in wild-type neurons, and represents a significant difference between wild type and mutant (p ≤ 0.02 for HIP1+/−, n = 4; p ≤ 0.01, n = 6 for HIP1−/−) (Fig. 6C). This failure to reduce GluR2 surface expression after NMDA stimulation demonstrates a role of HIP1 in the NMDAR-dependent internalization of AMPARs.
NMDA treatment can also stimulate AMPAR endocytosis, thereby resulting in chemical LTD of EPSCs in brain slices acutely prepared from adult rats or mice. To assess changes in basal glutamatergic responses in brain slices of HIP1−/− mice, we performed whole-cell recordings of excitatory glutamatergic currents at CA1 pyramidal neurons after single-pulse stimulation of the Schaffer collaterals. These studies revealed no obvious changes in HIP1−/− mice compared with wild-type littermates. Sample EPSCs from neurons of HIP1−/− mice are provided in supplemental Figure 2A (available at www.jneurosci.org as supplemental material). Addition of AP5 to block NMDARs attenuated EPSCs obtained at positive holding potentials and current–voltage relationships in the absence and presence of AP5 (supplemental Fig. 2B,C, respectively, available at www.jneurosci.org as supplemental material) were similar between genotypes. Specifically, neither the NMDA component nor the remaining current, which is mediated primarily by AMPARs, is altered in HIP1−/− mice. In summary, the basal activity of AMPARs and NMDARs after single-pulse stimulation of the Schaffer's collateral is unaltered in HIP1−/− mice.
Next, to determine whether the deficit in NMDA-induced AMPAR endocytosis is also observed in hippocampal slices from HIP1−/− mice, we investigated NMDA-induced chemical LTD. As shown in Figure 7, bath application of 20 μm NMDA facilitated AMPAR endocytosis as gauged by a significant reduction in the amplitude of EPSCs in slices from wild-type mice. Consistent with a deficit in NMDA-induced AMPAR endocytosis, the NMDA-induced inhibition of EPSCs was reduced in slices from HIP1−/− mice. Thus, 55 min after NMDA stimulation, the normalized amplitude of evoked EPSCs was reduced by 71.8 ± 11% in wild-type slices and by 35.3 ± 10.8% in brain slices from HIP1−/− mice (p ≤ 0.048 between genotypes; t test). These defects in LTD induction in HIP1−/− mice and alterations in GluR2 surface expression support a role for HIP1 in NMDA-dependent AMPAR internalization in vivo.
Figure 7.

LTD is reduced in hippocampal slices of HIP1−/− mice. A, Chemically induced LTD was established in hippocampal slices of wild-type mice (+/+; n = 8) and compared with HIP1−/− littermates (−/−; n = 7). B, The normalized amplitude of evoked EPSCs 55 min after bath application of NMDA is significantly reduced in hippocampal brain slices from wild-type mice compared with HIP1−/− littermates (*p < 0.05; t test). Error bars denote ±SEM.
Discussion
In the present study, we have demonstrated an essential role for HIP1 in NMDA-dependent AMPAR internalization and the induction of NMDA-induced cell death. Furthermore, we have identified htt as a novel component of this NMDA-induced cell-death pathway. Htt is dephosphorylated at serine 421 together with dephosphorylation of Akt during this process. Together, we have demonstrated a role for HIP1 in NMDAR function and participation of both HIP1 and htt in NMDA-induced cell death.
NMDARs are well known for their important role as mediators of neuronal death in many neuropathological conditions, including HD. NMDARs are also increasingly recognized for their function in activity-dependent neuronal survival, although detailed knowledge about the mechanisms is still lacking (Ikonomidou et al., 1999; Mennerick and Zorumski, 2000). Recent evidence suggests that this dualistic nature of NMDARs is mostly dependent on their localization at synaptic and extrasynaptic sites and associated with different electrophysiological properties, altered protein composition, and initiation of distinct signaling pathways (Li et al., 1998, 2002; Tovar and Westbrook, 1999; Hardingham and Bading, 2002). For example, activation of synaptic NMDARs promotes neuronal survival through activation of phosphoinositide 3-kinase (PI3K)/Akt and Ras/ERK pathways leading to CREB activation and BDNF gene expression (Chandler et al., 2001; Perkinton et al., 2002; Lee et al., 2005; Papadia et al., 2005). In contrast, activation of extrasynaptic NMDARs is coupled to loss of mitochondrial membrane potential and inhibition of PI3K/Akt and Ras/ERK, as part of a dominant CREB shut-off pathway triggering cell death (Chalecka-Franaszek and Chuang, 1999; Hardingham et al., 2002; Hardingham, 2006). Importantly, we observe protection from NMDA-induced cell death in cortical and striatal neurons without HIP1. For example, total NMDA-induced cell death, as measured by LDH release, is reduced by 53% in HIP1−/− neurons compared with wild type after stimulation with 50 μm NMDA (Fig. 3A). At this relatively low dose of NMDA, most neurons undergo apoptotic cell death (Bonfoco et al., 1995). In fact, the number of apoptotic neurons after NMDAR stimulation is significantly reduced in striatal and cortical neurons from HIP1−/− mice compared with wild type, as assessed by TUNEL (Fig. 3B,C) (data not shown). Furthermore, a 61% reduction in apoptotic cell death occurs in cortical neurons from HIP1−/− mice as measured by caspase-3 activation (50 μm NMDA) (Fig. 3D). Together, these results demonstrate a role for HIP1 in NMDA-induced cell death that involves, at least in part, neuronal death by apoptosis.
One exciting possibility to explain this result is based on the function of HIP1 in clathrin-mediated endocytosis and the observed defect in neurotransmitter receptor transport in neurons from HIP1−/− mice (Metzler et al., 2001, 2003; Legendre-Guillemin et al., 2005). According to this hypothesis, the internalization of AMPARs in response to NMDA is needed for the execution of cell death. This view is in agreement with the observed 75% reduction in NMDA-induced AMPAR internalization in HIP1−/− neurons in cell culture and the observed defect in LTD induction in hippocampal brain slices of HIP1−/− mice (Figs. 6, 7). This idea is also supported by the recent observation that a blockade in clathrin-mediated endocytosis of AMPARs protects neurons from NMDA-induced cell death (Wang et al., 2004).
A number of receptors need to be internalized to promote apoptotic cell death (Alloway et al., 2000; Kiselev et al., 2000; Lee et al., 2006). The most recent example is the death receptor CD95, whose internalization is needed for efficient DISC (death-inducing signaling complex) assembly and activation of caspase-8 and caspase-3 (Lee et al., 2006). Furthermore, growth factor receptors such as TRKA (tyrosine receptor kinase A) and EGF (epidermal growth factor) are needed for the generation of de novo signaling complexes at the endosome to effectively promote retrograde signaling (Miaczynska et al., 2004). Indeed, localization studies between AMPAR components and endosomal markers after receptor internalization demonstrate transit of AMPAR through endosomal compartments and the possibility of AMPAR signaling from endosomes (Ehlers, 2000; Lin et al., 2000; Metzler et al., 2003). Finally, we cannot rule out the possibility that components of the endocytic machinery themselves need to be internalized to promote cell death.
One possible mechanism of how HIP1 could influence NMDA-dependent functions is based on its interaction with NMDARs. Because no changes in basal NMDAR activity were evident in hippocampal brain slice of HIP1−/− mice (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), a role of HIP1 in the coupling of NMDAR activation to signaling pathways mediating NMDA-dependent AMPAR internalization and NMDA induced cell death is possible. This is supported by our previous observation that ligand-induced rather than constitutive endocytosis is altered in HIP1−/− neurons and that HIP1 can interact with both NMDARs and AMPARs (Metzler et al., 2003). This hypothesis is in agreement with a role of AMPAR transport in the induction of cell death.
Htt plays important functions in cellular and neuronal survival and its expression is required for normal transcription of BDNF through binding of the neuron-restrictive REST (repressor element 1-silencing transcription factor) (Dragatsis et al., 2000; Metzler et al., 2000; Zuccato et al., 2003). Htt is also a substrate for a number of kinases and phosphorylation of htt at serine 421 by Akt and SGK (serum and glucocorticoid-inducible kinase) in response to IGF-1 (insulin-like growth factor-1) has been linked to protection against cell death in the presence of mutant htt (Humbert et al., 2002; Rangone et al., 2004; Warby et al., 2005).
In the present study, we show a 31% decrease in phosphorylation of htt at serine 421 in response to NMDA-induced excitotoxicity (Fig. 5). This indicates that htt itself, through changes in its phosphorylation state, participates in NMDA-induced signaling. In addition, the decrease in P-htt-S421 in response to NMDA-induced cell death suggests a beneficial role of P-htt-S421 in neuronal survival and is in agreement with the neuroprotective function of P-htt-S421 (Humbert et al., 2002; Warby et al., 2005). Dephosphorylation of htt is likely to occur as part of an NMDA-induced CREB shut off characterized by dephosphorylation of a number of proteins important for neuronal survival, such as Akt. In our studies, a 48% decrease in P-Akt-S473 is seen after NMDA-induced cell death (Fig. 4). In contrast, phosphorylation of Akt and htt are increased by 94 and 48% (250 μm NMDA), respectively, in neurons from HIP1−/− mice compared with wild type, providing some mechanism for the neuroprotection observed in HIP1−/− neurons.
Although htt is a substrate for Akt, it is less likely that dephosphorylation and inactivation of Akt leads to a decline in htt phosphorylation as a consequence of reduced kinase activity. Changes in phosphorylation of htt occur within minutes after NMDA stimulation (data not shown), and this is far more likely a direct consequence of phosphatase activation. In fact, htt has recently been identified as a substrate for protein phosphatase 2B (PP2B/calcineurin) (Luo et al., 2005; Pardo et al., 2006), and dephosphorylation of P-Akt-S473 (Sutton and Chandler, 2002), P-GSK3β-S9 (Szatmari et al., 2005), and P-CREB-S133 (Sala et al., 2000; Kopnisky et al., 2003) during NMDA-induced excitotoxicity is the result of PP1 activity.
Together, our findings demonstrate that HIP1 influences important NMDAR functions and that both HIP1 and htt participate in a pathway leading to NMDA-induced cell death. NMDA-induced excitotoxicity is enhanced in HD, and this raises the interesting question as to whether polyQ expansion in htt, which leads to altered interaction with HIP1, modulates the function of both proteins, thereby sensitizing the NMDA-dependent excitotoxic pathway in HD.
Footnotes
This work was supported by Canadian Institutes of Health Research (CIHR) Grant MT-9133 (M.R.H.). Additional support was provided by Merck-Frosst Canada (M.R.H., M.M.). Y.T.W. is a CIHR investigator. M.R.H. is a holder of a Canada Research Chair in Human Genetics. PSD-95 constructs have been kindly provided by Alaa El-Husseini. We thank Simon Warby for the P-htt-S421 antibody and Jing Wang, Yu-Zhou Yang, Anita Kwok, Kuljeet Vaid, and Lisa Bertram for technical support.
References
- Alloway PG, Howard L, Dolph PJ. The formation of stable rhodopsin-arrestin complexes induces apoptosis and photoreceptor cell degeneration. Neuron. 2000;28:129–138. doi: 10.1016/s0896-6273(00)00091-x. [DOI] [PubMed] [Google Scholar]
- Beal MF, Kowall NW, Ellison DW, Mazurek MF, Swartz KJ, Martin JB. Replication of the neurochemical characteristics of Huntington's disease by quinolinic acid. Nature. 1986;321:168–171. doi: 10.1038/321168a0. [DOI] [PubMed] [Google Scholar]
- Beattie EC, Carroll RC, Yu X, Morishita W, Yasuda H, von Zastrow M, Malenka RC. Regulation of AMPA receptor endocytosis by a signaling mechanism shared with LTD. Nat Neurosci. 2000;3:1291–1300. doi: 10.1038/81823. [DOI] [PubMed] [Google Scholar]
- Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA. Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-d-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci USA. 1995;92:7162–7166. doi: 10.1073/pnas.92.16.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll RC, Lissin DV, von Zastrow M, Nicoll RA, Malenka RC. Rapid redistribution of glutamate receptors contributes to long-term depression in hippocampal cultures. Nat Neurosci. 1999a;2:454–460. doi: 10.1038/8123. [DOI] [PubMed] [Google Scholar]
- Carroll RC, Beattie EC, Xia H, Luscher C, Altschuler Y, Nicoll RA, Malenka RC, von Zastrow M. Dynamin-dependent endocytosis of ionotropic glutamate receptors. Proc Natl Acad Sci USA. 1999b;96:14112–14117. doi: 10.1073/pnas.96.24.14112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalecka-Franaszek E, Chuang DM. Lithium activates the serine/threonine kinase Akt-1 and suppresses glutamate-induced inhibition of Akt-1 activity in neurons. Proc Natl Acad Sci USA. 1999;96:8745–8750. doi: 10.1073/pnas.96.15.8745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler LJ, Sutton G, Dorairaj NR, Norwood D. N-methyl d-aspartate receptor-mediated bidirectional control of extracellular signal-regulated kinase activity in cortical neuronal cultures. J Biol Chem. 2001;276:2627–2636. doi: 10.1074/jbc.M003390200. [DOI] [PubMed] [Google Scholar]
- Chopra VS, Metzler M, Rasper DM, Engqvist-Goldstein AE, Singaraja R, Gan L, Fichter KM, McCutcheon K, Drubin D, Nicholson DW, Hayden MR. HIP12 is a non-proapoptotic member of a gene family including HIP1, an interacting protein with huntingtin. Mamm Genome. 2000;11:1006–1015. doi: 10.1007/s003350010195. [DOI] [PubMed] [Google Scholar]
- Dragatsis I, Levine MS, Zeitlin S. Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nat Genet. 2000;26:300–306. doi: 10.1038/81593. [DOI] [PubMed] [Google Scholar]
- Ehlers MD. Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron. 2000;28:511–525. doi: 10.1016/s0896-6273(00)00129-x. [DOI] [PubMed] [Google Scholar]
- Graham RK, Slow EJ, Deng Y, Bissada N, Lu G, Pearson J, Shehadeh J, Leavitt BR, Raymond LA, Hayden MR. Levels of mutant huntingtin influence the phenotypic severity of Huntington disease in YAC128 mouse models. Neurobiol Dis. 2006;21:444–455. doi: 10.1016/j.nbd.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Hardingham GE. 2B synaptic or extrasynaptic determines signalling from the NMDA receptor. J Physiol (Lond) 2006;572:614–615. doi: 10.1113/jphysiol.2006.109603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Bading H. Coupling of extrasynaptic NMDA receptors to a CREB shut-off pathway is developmentally regulated. Biochim Biophys Acta. 2002;1600:148–153. doi: 10.1016/s1570-9639(02)00455-7. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Hodgson JG, Agopyan N, Gutekunst CA, Leavitt BR, LePiane F, Singaraja R, Smith DJ, Bissada N, McCutcheon K, Nasir J, Jamot L, Li XJ, Stevens ME, Rosemond E, Roder JC, Phillips AG, Rubin EM, Hersch SM, Hayden MR. A YAC mouse model for Huntington's disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron. 1999;23:181–192. doi: 10.1016/s0896-6273(00)80764-3. [DOI] [PubMed] [Google Scholar]
- Humbert S, Bryson EA, Cordelieres FP, Connors NC, Datta SR, Finkbeiner S, Greenberg ME, Saudou F. The IGF-1/Akt pathway is neuroprotective in Huntington's disease and involves Huntingtin phosphorylation by Akt. Dev Cell. 2002;2:831–837. doi: 10.1016/s1534-5807(02)00188-0. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Bosch F, Miksa M, Bittigau P, Vockler J, Dikranian K, Tenkova TI, Stefovska V, Turski L, Olney JW. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. 1999;283:70–74. doi: 10.1126/science.283.5398.70. [DOI] [PubMed] [Google Scholar]
- Kiselev A, Socolich M, Vinos J, Hardy RW, Zuker CS, Ranganathan R. A molecular pathway for light-dependent photoreceptor apoptosis in Drosophila. Neuron. 2000;28:139–152. doi: 10.1016/s0896-6273(00)00092-1. [DOI] [PubMed] [Google Scholar]
- Kopnisky KL, Chalecka-Franaszek E, Gonzalez-Zulueta M, Chuang DM. Chronic lithium treatment antagonizes glutamate-induced decrease of phosphorylated CREB in neurons via reducing protein phosphatase 1 and increasing MEK activities. Neuroscience. 2003;116:425–435. doi: 10.1016/s0306-4522(02)00573-0. [DOI] [PubMed] [Google Scholar]
- Leavitt BR, Raamsdonk JM, Shehadeh J, Fernandes H, Murphy Z, Graham RK, Wellington CL, Raymond LA, Hayden MR. Wild-type huntingtin protects neurons from excitotoxicity. J Neurochem. 2006;96:1121–1129. doi: 10.1111/j.1471-4159.2005.03605.x. [DOI] [PubMed] [Google Scholar]
- Lee B, Butcher GQ, Hoyt KR, Impey S, Obrietan K. Activity-dependent neuroprotection and cAMP response element-binding protein (CREB): kinase coupling, stimulus intensity, and temporal regulation of CREB phosphorylation at serine 133. J Neurosci. 2005;25:1137–1148. doi: 10.1523/JNEUROSCI.4288-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KH, Feig C, Tchikov V, Schickel R, Hallas C, Schutze S, Peter ME, Chan AC. The role of receptor internalization in CD95 signaling. EMBO J. 2006;25:1009–1023. doi: 10.1038/sj.emboj.7601016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Liu L, Wang YT, Sheng M. Clathrin adaptor AP2 and NSF interact with overlapping sites of GluR2 and play distinct roles in AMPA receptor trafficking and hippocampal LTD. Neuron. 2002;36:661–674. doi: 10.1016/s0896-6273(02)01024-3. [DOI] [PubMed] [Google Scholar]
- Legendre-Guillemin V, Metzler M, Charbonneau M, Gan L, Chopra V, Philie J, Hayden MR, McPherson PS. HIP1 and HIP12 display differential binding to F-actin, AP2, and clathrin. Identification of a novel interaction with clathrin light chain. J Biol Chem. 2002;277:19897–19904. doi: 10.1074/jbc.M112310200. [DOI] [PubMed] [Google Scholar]
- Legendre-Guillemin V, Metzler M, Lemaire JF, Philie J, Gan L, Hayden MR, McPherson PS. Huntingtin interacting protein 1 (HIP1) regulates clathrin assembly through direct binding to the regulatory region of the clathrin light chain. J Biol Chem. 2005;280:6101–6108. doi: 10.1074/jbc.M408430200. [DOI] [PubMed] [Google Scholar]
- Li B, Chen N, Luo T, Otsu Y, Murphy TH, Raymond LA. Differential regulation of synaptic and extra-synaptic NMDA receptors. Nat Neurosci. 2002;5:833–834. doi: 10.1038/nn912. [DOI] [PubMed] [Google Scholar]
- Li JH, Wang YH, Wolfe BB, Krueger KE, Corsi L, Stocca G, Vicini S. Developmental changes in localization of NMDA receptor subunits in primary cultures of cortical neurons. Eur J Neurosci. 1998;10:1704–1715. doi: 10.1046/j.1460-9568.1998.00169.x. [DOI] [PubMed] [Google Scholar]
- Lin JW, Ju W, Foster K, Lee SH, Ahmadian G, Wyszynski M, Wang YT, Sheng M. Distinct molecular mechanisms and divergent endocytotic pathways of AMPA receptor internalization. Nat Neurosci. 2000;3:1282–1290. doi: 10.1038/81814. [DOI] [PubMed] [Google Scholar]
- Luo S, Vacher C, Davies JE, Rubinsztein DC. Cdk5 phosphorylation of huntingtin reduces its cleavage by caspases: implications for mutant huntingtin toxicity. J Cell Biol. 2005;169:647–656. doi: 10.1083/jcb.200412071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscher C, Xia H, Beattie EC, Carroll RC, von Zastrow M, Malenka RC, Nicoll RA. Role of AMPA receptor cycling in synaptic transmission and plasticity. Neuron. 1999;24:649–658. doi: 10.1016/s0896-6273(00)81119-8. [DOI] [PubMed] [Google Scholar]
- Man HY, Lin JW, Ju WH, Ahmadian G, Liu L, Becker LE, Sheng M, Wang YT. Regulation of AMPA receptor-mediated synaptic transmission by clathrin-dependent receptor internalization. Neuron. 2000;25:649–662. doi: 10.1016/s0896-6273(00)81067-3. [DOI] [PubMed] [Google Scholar]
- Mennerick S, Zorumski CF. Neural activity and survival in the developing nervous system. Mol Neurobiol. 2000;22:41–54. doi: 10.1385/MN:22:1-3:041. [DOI] [PubMed] [Google Scholar]
- Metzler M, Helgason CD, Dragatsis I, Zhang T, Gan L, Pineault N, Zeitlin SO, Humphries RK, Hayden MR. Huntingtin is required for normal hematopoiesis. Hum Mol Genet. 2000;9:387–394. doi: 10.1093/hmg/9.3.387. [DOI] [PubMed] [Google Scholar]
- Metzler M, Legendre-Guillemin V, Gan L, Chopra V, Kwok A, McPherson PS, Hayden MR. HIP1 functions in clathrin-mediated endocytosis through binding to clathrin and adaptor protein 2. J Biol Chem. 2001;276:39271–39276. doi: 10.1074/jbc.C100401200. [DOI] [PubMed] [Google Scholar]
- Metzler M, Li B, Gan L, Georgiou J, Gutekunst CA, Wang Y, Torre E, Devon RS, Oh R, Legendre-Guillemin V, Rich M, Alvarez C, Gertsenstein M, McPherson PS, Nagy A, Wang YT, Roder JC, Raymond LA, Hayden MR. Disruption of the endocytic protein HIP1 results in neurological deficits and decreased AMPA receptor trafficking. EMBO J. 2003;22:3254–3266. doi: 10.1093/emboj/cdg334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miaczynska M, Pelkmans L, Zerial M. Not just a sink: endosomes in control of signal transduction. Curr Opin Cell Biol. 2004;16:400–406. doi: 10.1016/j.ceb.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Mishra SK, Agostinelli NR, Brett TJ, Mizukami I, Ross TS, Traub LM. Clathrin- and AP-2-binding sites in HIP1 uncover a general assembly role for endocytic accessory proteins. J Biol Chem. 2001;276:46230–46236. doi: 10.1074/jbc.M108177200. [DOI] [PubMed] [Google Scholar]
- Okamoto S, Li Z, Ju C, Scholzke MN, Mathews E, Cui J, Salvesen GS, Bossy-Wetzel E, Lipton SA. Dominant-interfering forms of MEF2 generated by caspase cleavage contribute to NMDA-induced neuronal apoptosis. Proc Natl Acad Sci USA. 2002;99:3974–3979. doi: 10.1073/pnas.022036399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadia S, Stevenson P, Hardingham NR, Bading H, Hardingham GE. Nuclear Ca2+ and the cAMP response element-binding protein family mediate a late phase of activity-dependent neuroprotection. J Neurosci. 2005;25:4279–4287. doi: 10.1523/JNEUROSCI.5019-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo R, Colin E, Regulier E, Aebischer P, Deglon N, Humbert S, Saudou F. Inhibition of calcineurin by FK506 protects against polyglutamine-huntingtin toxicity through an increase of huntingtin phosphorylation at S421. J Neurosci. 2006;26:1635–1645. doi: 10.1523/JNEUROSCI.3706-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkinton MS, Ip JK, Wood GL, Crossthwaite AJ, Williams RJ. Phosphatidylinositol 3-kinase is a central mediator of NMDA receptor signalling to MAP kinase (Erk1/2), Akt/PKB and CREB in striatal neurones. J Neurochem. 2002;80:239–254. doi: 10.1046/j.0022-3042.2001.00699.x. [DOI] [PubMed] [Google Scholar]
- Prybylowski K, Chang K, Sans N, Kan L, Vicini S, Wenthold RJ. The synaptic localization of NR2B-containing NMDA receptors is controlled by interactions with PDZ proteins and AP-2. Neuron. 2005;47:845–857. doi: 10.1016/j.neuron.2005.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangone H, Poizat G, Troncoso J, Ross CA, MacDonald ME, Saudou F, Humbert S. The serum- and glucocorticoid-induced kinase SGK inhibits mutant huntingtin-induced toxicity by phosphorylating serine 421 of huntingtin. Eur J Neurosci. 2004;19:273–279. doi: 10.1111/j.0953-816x.2003.03131.x. [DOI] [PubMed] [Google Scholar]
- Roche KW, Standley S, McCallum J, Dune LC, Ehlers MD, Wenthold RJ. Molecular determinants of NMDA receptor internalization. Nat Neurosci. 2001;4:794–802. doi: 10.1038/90498. [DOI] [PubMed] [Google Scholar]
- Sala C, Rudolph-Correia S, Sheng M. Developmentally regulated NMDA receptor-dependent dephosphorylation of cAMP response element-binding protein (CREB) in hippocampal neurons. J Neurosci. 2000;20:3529–3536. doi: 10.1523/JNEUROSCI.20-10-03529.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shehadeh J, Fernandes HB, Zeron Mullins MM, Graham RK, Leavitt BR, Hayden MR, Raymond LA. Striatal neuronal apoptosis is preferentially enhanced by NMDA receptor activation in YAC transgenic mouse model of Huntington disease. Neurobiol Dis. 2006;21:392–403. doi: 10.1016/j.nbd.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Slow EJ, van Raamsdonk J, Rogers D, Coleman SH, Graham RK, Deng Y, Oh R, Bissada N, Hossain SM, Yang YZ, Li XJ, Simpson EM, Gutekunst CA, Leavitt BR, Hayden MR. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet. 2003;12:1555–1567. doi: 10.1093/hmg/ddg169. [DOI] [PubMed] [Google Scholar]
- Sutton G, Chandler LJ. Activity-dependent NMDA receptor-mediated activation of protein kinase B/Akt in cortical neuronal cultures. J Neurochem. 2002;82:1097–1105. doi: 10.1046/j.1471-4159.2002.01031.x. [DOI] [PubMed] [Google Scholar]
- Szatmari E, Habas A, Yang P, Zheng JJ, Hagg T, Hetman M. A positive feedback loop between glycogen synthase kinase 3beta and protein phosphatase 1 after stimulation of NR2B NMDA receptors in forebrain neurons. J Biol Chem. 2005;280:37526–37535. doi: 10.1074/jbc.M502699200. [DOI] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL. The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J Neurosci. 1999;19:4180–4188. doi: 10.1523/JNEUROSCI.19-10-04180.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Raamsdonk JM, Murphy Z, Slow EJ, Leavitt BR, Hayden MR. Selective degeneration and nuclear localization of mutant huntingtin in the YAC128 mouse model of Huntington disease. Hum Mol Genet. 2005;14:3823–3835. doi: 10.1093/hmg/ddi407. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, DiFiglia M. Huntington disease. J Neuropathol Exp Neurol. 1998;57:369–384. doi: 10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]
- Waelter S, Scherzinger E, Hasenbank R, Nordhoff E, Lurz R, Goehler H, Gauss C, Sathasivam K, Bates GP, Lehrach H, Wanker EE. The huntingtin interacting protein HIP1 is a clathrin and alpha-adaptin-binding protein involved in receptor-mediated endocytosis. Hum Mol Genet. 2001;10:1807–1817. doi: 10.1093/hmg/10.17.1807. [DOI] [PubMed] [Google Scholar]
- Wang Y, Ju W, Liu L, Fam S, D'Souza S, Taghibiglou C, Salter M, Wang YT. alpha-Amino-3-hydroxy-5-methylisoxazole-4-propionic acid subtype glutamate receptor (AMPAR) endocytosis is essential for N-methyl-d-aspartate-induced neuronal apoptosis. J Biol Chem. 2004;279:41267–41270. doi: 10.1074/jbc.C400199200. [DOI] [PubMed] [Google Scholar]
- Warby SC, Chan EY, Metzler M, Gan L, Singaraja RR, Crocker SF, Robertson HA, Hayden MR. Huntingtin phosphorylation on serine 421 is significantly reduced in the striatum and by polyglutamine expansion in vivo. Hum Mol Genet. 2005;14:1569–1577. doi: 10.1093/hmg/ddi165. [DOI] [PubMed] [Google Scholar]
- Yano S, Tokumitsu H, Sodeling TR. Calcium promotes cell survival through CaM-K kinase activation of the protein-kinase-B pathway. Nature. 1998;396:584–587. doi: 10.1038/25147. [DOI] [PubMed] [Google Scholar]
- Zeron MM, Hansson O, Chen N, Wellington CL, Leavitt BR, Brundin P, Hayden MR, Raymond LA. Increased sensitivity to N-methyl-d-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington's disease. Neuron. 2002;33:849–860. doi: 10.1016/s0896-6273(02)00615-3. [DOI] [PubMed] [Google Scholar]
- Zeron MM, Fernandes HB, Krebs C, Shehadeh J, Wellington CL, Leavitt BR, Baimbridge KG, Hayden MR, Raymond LA. Potentiation of NMDA receptor-mediated excitotoxicity linked with intrinsic apoptotic pathway in YAC transgenic mouse model of Huntington's disease. Mol Cell Neurosci. 2004;25:469–479. doi: 10.1016/j.mcn.2003.11.014. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Tartari M, Crotti A, Goffredo D, Valenza M, Conti L, Cataudella T, Leavitt BR, Hayden MR, Timmusk T, Rigamonti D, Cattaneo E. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat Genet. 2003;35:76–83. doi: 10.1038/ng1219. [DOI] [PubMed] [Google Scholar]