

Abstract
Kv3 (Shaw) channels regulate rapid spiking, transmitter release and dendritic integration of many central neurons. Crucial to functional diversity are the complex targeting patterns of channel proteins. However, the targeting mechanisms are not known. Here we report that the axon–dendrite targeting of Kv3.1 is controlled by a conditional interaction of a C-terminal axonal targeting motif (ATM) with the N-terminal T1 domain and adaptor protein ankyrin G. In cultured hippocampal neurons, although the two splice variants of Kv3.1, Kv3.1a and Kv3.1b, are differentially targeted to the somatodendritic and axonal membrane, respectively, the lysine-rich ATM is surprisingly common for both splice variants. The ATM not only directly binds to the T1 domain in a Zn2+-dependent manner, but also associates with the ankyrin-repeat domain of ankyrin G. However, the full-length channel proteins of Kv3.1b display stronger association to ankyrin G than those of Kv3.1a, suggesting that the unique splice domain at Kv3.1b C terminus influences ATM binding to T1 and ankyrin G. Because ankyrin G mainly resides at the axon initial segment, we propose that it may function as a barrier for axon–dendrite targeting of Kv3.1 channels. In support of this idea, disrupting ankyrin G function either by over-expressing a dominant-negative mutant or by siRNA knockdown decreases polarized axon–dendrite targeting of both Kv3.1a and Kv3.1b. We conclude that the conditional ATM masked by the T1 domain in Kv3.1a is exposed by the splice domain in Kv3.1b, and is subsequently recognized by ankyrin G to target Kv3.1b into the axon.
Keywords: Kv3 channel, neuron, axonal targeting motif, T1 domain, ankyrin G, axon initial segment
Introduction
Characterized by high activation threshold and fast deactivation kinetics, Kv3 (Shaw) voltage-gated potassium channels are required for rapid spiking of many central neurons (Rudy and McBain, 2001). Kv3.3 gene mutation is implicated in adult-onset dominant ataxia in humans (Waters et al., 2006), and Kv3 knock-out mice generally show motor and other behavioral abnormalities (Lau et al., 2000; Espinosa et al., 2001; Matsukawa et al., 2003; Espinosa et al., 2004; Joho et al., 2006). In addition to the unique channel properties, targeting into distinct subcellular domains in neurons is also crucial to the diverse functions of Kv3 channels. However, what controls Kv3 channel targeting is not known.
Kv3 channels display intriguingly complex targeting patterns in dendrites and axons, depending on the isoform and alternative splicing (Misonou and Trimmer, 2004; Lai and Jan, 2006). There are four Kv3 isoforms, Kv3.1, Kv3.2, Kv3.3, and Kv3.4. Whereas Kv3.1, Kv3.2 and Kv3.4 are targeted to the nerve terminals of central neurons to regulate action potential duration and transmitter release (Rettig et al., 1992; Weiser et al., 1994; Ishikawa et al., 2003; Lien and Jonas, 2003; Matsukawa et al., 2003; Brooke et al., 2004; Goldberg et al., 2005), Kv3.3 and Kv3.4 are localized in the somatodendritic regions of cerebellar Purkinje cells to shape the large depolarization events (Martina et al., 2003). Each Kv3 isoform gives rise to multiple products by alternative splicing, which exclusively occurs at the C-terminal region. Of the two Kv3.1 splice variants, the longer Kv3.1b is localized primarily in proximal dendrites, soma, and axons, particularly in axonal terminal fields (Weiser et al., 1995; Sekirnjak et al., 1997; Ozaita et al., 2002). The shorter Kv3.1a is less abundant than Kv3.1b, replaced gradually by Kv3.1b in adults (Perney et al., 1992; Gan and Kaczmarek, 1998).
Multiple targeting motifs have been identified for different Kv channels (Misonou and Trimmer, 2004; Lai and Jan, 2006). Axonal targeting of Kv1 involves the N-terminal T1 domain (Gu et al., 2003, 2006; Rivera et al., 2005). Dendritic targeting of Kv2.1 and Kv4.2 requires the proximal clustering and a di-leucine motif, respectively, located at the C termini (Lim et al., 2000; Rivera et al., 2003). Other trafficking signals, e.g., the YXXΦ motif (at Y458) at the C terminus of Kv1.2 (Gu et al., 2003) and the kinesin (KIF17)-binding motif of Kv4.2, also participate in channel targeting (Chu et al., 2006). In KCNQ channels, an ankyrin G-binding motif and A domain are involved in localizing the channels to the axon initial segment (AIS) and the distal axon, respectively (Chung et al., 2006; Pan et al., 2006). However, for Kv3 channels, little is known about the targeting signal and mechanism except that a PDZ-domain binding motif at the C terminus of Kv3.3 is required to target the channels to distal dendrites of pyramidal neurons of Apteronotus leptorhynchus (Deng et al., 2005).
Our results reveal a novel mechanism governing Kv3.1 axon–dendrite targeting that involves the T1 domain and ankyrin G for binding to a newly identified ATM at C termini. Although the ATM is present in both Kv3.1a and Kv3.1b, its binding to ankyrin G is more permissive in the presence of the splice domain at Kv3.1b C terminus, allowing the channel complex to cross the AIS. Because the ATM and T1 domain are highly conserved within Kv3 channels from fly to human, their interaction may represent an evolutionarily conserved mechanism by which channel targeting is regulated.
Materials and Methods
Materials.
The following reagents were used: rabbit polyclonal anti-MAP2 (Chemicon, Temecula, CA), rat monoclonal anti-HA antibody (Roche, Indianapolis, IN), mouse monoclonal anti-human CD4 antibody (Caltag, Burlingame, CA), mouse monoclonal anti-pan-Nav channel antibody (Sigma, St. Louis, MO), rabbit polyclonal anti-Kv3.1b antibody (Alomone Labs, Jerusalem, Israel), rabbit polyclonal anti-EGFP and anti-6×His antibodies (Invitrogen, Carlsbad, CA), rabbit polyclonal anti-ankyrin G (a kind gift from Dr. Vann Bennett) and anti-Tau1 (Abcam, Cambridge, MA), Cy2-, Cy5- and HRP-conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA), Kv3.1a and Kv3.1b (a kind gift from Dr. Bernardo Rudy), transferrin receptor-GFP fusion (TfR–GFP, a kind gift from Dr. Gary Banker), ankyrin G-GFP (a kind gift from Drs. Lori Isom and Vann Bennett).
cDNA constructs.
Kv3.1aHA and Kv3.1bHA were constructed by inserting an HA tag (YPYDVPDYA) into the first extracellular loop right behind T231. Kv3.1T1–TfR–GFP was constructed by inserting the PCR fragment encoding the Kv3.1 N-terminal region [amino acids (aa) 1–186] right before the N terminus of TfR–GFP between engineered NotI and XhoI sites (Gu et al., 2003). CD4–31aC (aa 442–511), CD4–31bC (aa 442–585), CD4–31sC (aa 500–585), CD4–31aC475–511 (aa 475–511) and CD4–31aC482–511 (aa 482–511) were constructed by inserting the PCR fragments right after the CD4 C terminus between engineered NotI and XhoI sites (Gu et al., 2003). With the Quickchange strategy, CD4-aC442–489, CD4–31aC442–469, CD4–31aC442–464 and CD4–31aC442–452 were constructed by engineering stop codons at these positions (490, 470, 465, 453 respectively) of CD4–31aC. CD4–31aC447–464 (aa 447–464) and CD4–31aC453–464 (aa 453–464) were constructed by inserting synthesized DNA fragments right after the CD4 C terminus between NotI and XhoI sites. Kv3.1bHA1–502, Kv3.1bHA1–489, Kv3.1bHA1–469 and Kv3.1bHA1–464 were also constructed by engineering stop codons in Kv3.1bHA. Indicated residues in the ATM were mutated to alanines in Kv3.1aHAIPR, Kv3.1aHAPP, Kv3.1bHAKKK, Kv3.1bHAKKH, CD4–31aCKKK, CD4–31aCKKH, CD4–31aCIPR and CD4–31aCPP using the Quickchange strategy. GST-31N (aa 1–186), GST-31aC (aa 442–511), GST-31bC (aa 442–585) and GST-31sC (aa 500–585) were made by inserting the corresponding PCR fragments into pGEX4T-2 between EcoRI and XhoI. GST-31aC442–489, GST-31aC442–469 and GST-31aC442–452 were constructed by engineering a stop codon at these indicated positions (490, 470, 453 respectively) in GST-31aC. His-31N (aa 1–186), His-31T1 (aa 1–110), and His-MB (aa 1–890) were constructed by inserting the corresponding PCR fragments into pRSET B between BglII and EcoRI. MB–GFP was constructed by digesting ankyrin G-GFP with ApaI and religating the fragment containing the vector. All constructs were confirmed by sequencing.
Hippocampal neuron culture.
Hippocampal neuron culture was prepared as previously described (Gu et al., 2003). In brief, 2 d after neuron plating, 1 μm cytosine arabinose (Sigma) was added to the neuronal culture medium to inhibit glial growth for the subsequent 2 d, then replaced with the normal neuronal culture medium. The culture medium was replenished twice a week by replacing a half volume. For transient transfection, neurons in culture at 5–7 DIV were incubated in Opti-MEM containing 0.8 μg of cDNA plasmid and 1.5 μl of Lipofectamine 2000 (Invitrogen) for 20 min at 37°C.
Whole-cell patch-clamp recording.
Kv3.1a, Kv3.1b, Kv3.1aHA and Kv3.1bHA were subcloned into pIRES2EGFP vector. The transfected HEK293 cells were identified by GFP fluorescence and recorded in Hank's buffer (150 mm NaCl, 4 mm KCl, 1.2 mm MgCl2, 10 mg/ml glucose, 1 mm CaCl2, 20 mm HEPES, pH 7.4). The internal solution for electrical pipettes is composed of (in mm) 122 KMeSO4, 20 NaCl, 5 Mg-ATP, 0.3 GTP and 10 HEPES, pH 7.2,. The resistance of electrodes is between 2 and 5 MΩ. Isolated cells were voltage-clamped in the whole-cell mode using an EPC9 amplifier (HEKA Elektronik, Southboro, MA), held at −80 mV, and given 250 ms voltage episodes from −50 to 40 mV with a 10-mV increment. Voltage commands were made from the Pulse+Pulse Fit program (version 8.53; HEKA Elektronik), and the currents were recorded at 5 kHz.
Antibody uptake experiments.
Living neurons transfected with CD4–31aC and point mutations were incubated with a monoclonal anti-hCD4 antibody (1:200 dilution in culture medium) for 8 min at 37°C, rinsed briefly in prewarmed PBS, and fixed with 4% formaldehyde (from 10% ultrapure EM grade and methanol free; Polysciences, Warrington, PA) and 0.4% sucrose in PBS for 20 min. Under nonpermeabilized conditions, the neurons were further incubated with secondary antibodies in PBS with 2.5% normal goat serum, followed by extensive washing. Then the neurons were permeabilized with PBS containing 0.2% Triton X100 for 15 min, and stained for the dendritic marker MAP2. Under permeabilized conditions, the neurons were permeabilized with 0.2% Triton X100 in PBS for 15 min, incubated with the rabbit polyclonal anti-MAP antibody in PBS with 0.02% Triton X100 and 2.5% normal goat serum, washed and then further incubated with secondary antibodies.
Immunofluorescence staining.
The procedure of immunocytochemistry and fluorescence microscopy were described previously (Gu et al., 2003). In brief, the neurons expressing HA-tagged channels or CD4 fusion proteins were stained under both permeabilized conditions to label total proteins and nonpermeabilized conditions to label the surface pool.
Fluorescence microscopy and quantification.
Fluorescence images were captured with a Spot CCD camera RT slider (Diagnostic Instruments, Sterling Heights, MI) in a Zeiss (Oberkochen, Germany) upright microscope, Axiophot, using Plan Apo objectives 20×/0.75 and 100×/1.4 oil, saved as 16-bit TIFF files, and analyzed with NIH Image J and SigmaPlot 8.0 for fluorescence intensity quantification. Exposure times were controlled so that the pixel intensities in dendrites and axons were below saturation, but the same exposure time was used within each group of an experiment. The quantification procedure was described previously (Gu et al., 2003). Only transfected neurons with clearly separated dendrites and axons, and isolated from other transfected cells were chosen for analysis. To distinguish axons from dendrites, axonal marker Tau1 and dendritic marker MAP2 were used for costaining. We determined the targeting ratio (Faxon/Fdendrite), the average fluorescence intensity in axons divided by the average fluorescence intensity in dendrites, by laying lines along the major processes to acquire the fluorescence intensity profiles (in arbitrary unit) of these processes excluding segments fasciculated with other neurites, regions with crossings, proximal dendritic segments within 20 μm from the soma, and fine terminal branches.
In vitro protein binding assay.
The expression of GST fusion proteins was induced with 1 mm IPTG in E. coli BL21 cells for 5 h at 37°C. The bacterial pellets were solubilized with sonication in a pull-down buffer [50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% Triton X100, and protease inhibitor cocktail (Roche)] at 4°C, and centrifuged at 50,000 × g for 30 min at 4°C. The supernatants were incubated with 50 μl of glutathione beads at 4°C either for 3 h (for mixed pull down) or overnight (for sequential pull down). After extensive washing, the beads coated with purified GST fusion proteins were further incubated with either bacterial lysate supernatant containing His-tagged fusion proteins or the supernatant from HEK293 cells expressing ankyrin G-GFP or MB–GFP. The proteins precipitated were eluted with a 2× sample buffer, and resolved in SDS-PAGE, transferred to PVDF membrane and subjected to immunoblotting with either an anti-His antibody or anti-GFP antibody. The SDS-PAGE gels with GST fusion inputs were stained with Coomassie Blue.
Pull-down assays with purified fusion proteins.
GST- and His-tagged fusion proteins were purified with glutathione and Ni2+ beads, respectively. Under normal condition, purified His-31T1 (0.5 mg) and GST fusion (0.5 mg) were incubated in the pull-down buffer (total volume 1.2 ml) at 4°C overnight, and then precipitated with glutathione beads. Under EDTA condition, the two were incubated in the pull-down buffer with 1 mm EDTA at 4°C overnight, and then precipitated with glutathione beads. Under Zn2+ re-association condition, the two were first incubated in the pull-down buffer with 1 mm EDTA and 20 mm 2-mercaptoethanol at 4°C overnight. Next the mixture was dialyzed in pull-down buffer with 100 μm ZnSO4 and 20 mm 2-mercaptoethanol, and then precipitated with glutathione beads. This procedure was adopted with minor modification from an early study (Nanao et al., 2003).
Coimmunoprecipitation.
HEK293 cells cotransfected with HA-tagged Kv3.1 channels and either ankyrin G-GFP or GFP for 48 h were solubilized in 1 ml of IP buffer [50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% Triton X100, and protease inhibitor cocktail (Roche)] for 1 h at 4°C, and then centrifuged at 50,000 × g for 20 min at 4°C. 0.9 ml of the supernatant was incubated (for 2 h, 4°C) with 1 μg of polyclonal anti-GFP antibody and 50 ml of protein A agarose beads (Roche). The beads were washed six times with 1 ml of IP buffer and then eluted with 100 μl of 2× sample buffer at 95°C for 15 min. The immunoprecipitates were resolved by SDS-PAGE, transferred to a PVDF membrane, and subjected to immunoblotting with monoclonal anti-HA antibody.
siRNA suppression of endogenous ankyrin G expression in rat hippocampal neurons.
We used vector-based siRNA strategy to suppress the endogenous ankyrin G expression in cultured rat hippocampal neurons. The procedure was described previously (Gu et al., 2006). In brief, four 21 nucleotide sequences unique to rat ankyrin G were selected from the open reading frame. For constructing each siRNA construct, two complementary strands of DNA, from 5′ to 3′, containing a BamH1 site, 21-nucleotide antisense, loop (TTGATATCCG), 21-nucleotide sense, and a HindIII site, were chemically synthesized, and inserted into the BamHI/HindIII sites of pRNAT-H1.1/neo vector (GenScript, Piscataway, NJ), which also contains a GFP gene. One probe, AnkG siR (nucleotide 294–314, CAACGTCGATGCAGCCACAAA), can efficiently suppress expression of ankyrin G in rat hippocampal neurons. AnkG siR or control siRNA was usually transfected into hippocampal neurons with Lipofectamine 2000 (Invitrogen) at 3 DIV, and neurons were fixed and stained 5 d later. GFP fluorescence signal indicated the transfected neurons. To examine the effects of suppressing ankyrin G by siRNA, we cotransfected Kv3.1aHA or Kv3.1bHA together with siRNA probe at 3 DIV, and fixed and stained the neurons 5 d later.
Results
Differential membrane targeting of Kv3.1 splice variants, Kv3.1a and Kv3.1b
To search for targeting signals within Kv3.1 channels, we first examined membrane targeting of Kv3.1 splice variants in dissociated hippocampal neurons, a widely used model system for studying neuronal polarity (Stowell and Craig, 1999; Horton and Ehlers, 2003; Kaech and Banker, 2006). HA-tagged Kv3.1a and Kv3.1b (Kv3.1aHA and Kv3.1bHA) were constructed by inserting an HA tag into the first extracellular loop after the second N-glycosylation site (Brooks et al., 2006). When expressed in HEK293 cells, Kv3.1aHA and Kv3.1bHA formed functional channels with activation potential at approximately −10 mV; the high activation threshold is a hallmark of Kv3 channels (Fig. 1A,B). Their current amplitudes were also similar to those of wild type channels (Fig. 1A,B). Next, we transfected cultured hippocampal neurons with Kv3.1aHA and Kv3.1bHA, and examined the distribution of these channels on the plasma membrane, in which they are actively involved in neuronal electrical signaling. Membrane targeting was revealed by staining extracellular localized HA epitope under nonpermeabilized conditions. Whereas Kv3.1aHA was primarily localized on the somatodendritic surface (Fig. 1C), Kv3.1bHA was preferentially targeted to the axonal membrane (Fig. 1D). The average fluorescence intensity for Kv3.1bHA on the axonal surface is approximately twofold higher than that on the dendritic surface (Fig. 1D), which is consistent with the Kv3.1b staining pattern in brain slices (Weiser et al., 1995; Sekirnjak et al., 1997; Ozaita et al., 2002; Devaux et al., 2003). Moreover, our results show that alternative splicing indeed regulates polarized targeting of Kv3.1 channels.
Figure 1.
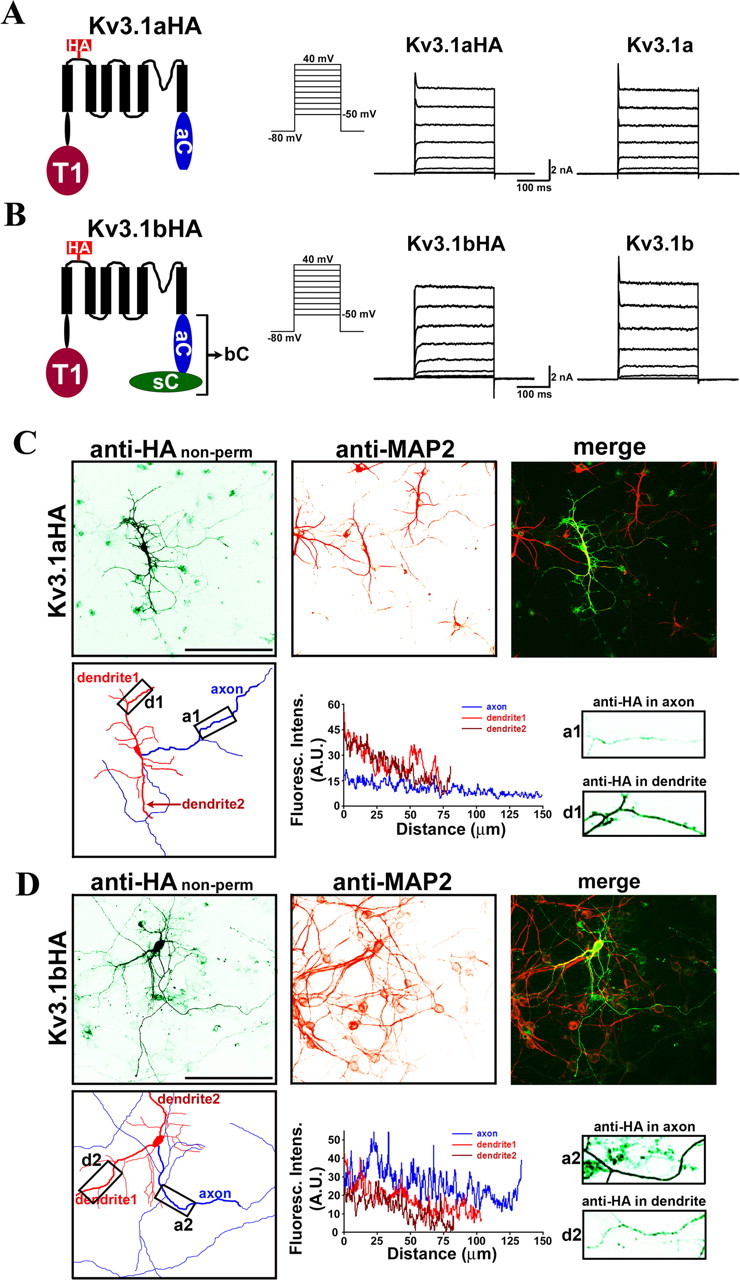
Distinct targeting patterns of Kv3.1 splice variants. HA-tagged Kv3.1a (A) and Kv3.1b (B) channels are functional and similar to the wild type in activation potential (approximately −10 mV) and current amplitude. Structural diagrams are on the left. Transmembrane segments are shown with six black bars. The HA tag (in red) is within the first extracellular loop. The T1 domain (dark red), the C-terminal region of Kv3.1a (in blue), and the splice domain in Kv3.1b (in green) are indicated. HA-tagged and wild-type channels were subcloned in pIRES2EGFP vector, expressed in HEK293 cells, and recorded by whole-cell voltage clamp. The cells expressing channel constructs were identified with GFP fluorescence, held at −80 mV, and given 250 ms voltage episodes from −50 to 40 mV with a 10 mV increment. Examples of current traces from HA-tagged and wild-type channels are given in middle and right panels, respectively. The endogenous voltage-gated outward currents only reached ∼100 pA at 40 mV and are therefore ignored (data not shown). Shown are representatives of 6–10 cells. C, When expressed in cultured hippocampal neurons, Kv3.1aHA was predominantly localized to the somatodendritic membrane. Neurons were transfected with Kv3.1aHA at 6 DIV, fixed, and stained with an anti-HA antibody (in green) under nonpermeabilized conditions 2 d later. Dendrites of the neurons were labeled by anti-MAP2 (microtubule associated protein 2) staining (in red). The camera lucida drawing of the neuron in the bottom left shows dendrites in red and axons in blue. The anti-HA fluorescence profiles along the major axon (within 200 μm from the soma) and two dendrites (including the entire dendrite, indicated by thicker lines in the drawing), and two boxed areas (in 3 times higher magnification) are shown in the bottom middle and right, respectively. Boxed areas were chosen along the major axon/dendrite processes ∼50 μm away from the soma. D, Kv3.1bHA was preferentially localized to the axonal membrane. Distance 0, Soma. Scale bars, 100 μm.
Identification of ATM in the C terminus of Kv3.1
How does alternative splicing regulate membrane protein targeting? The simplest scenario is that targeting signals are encoded within the splice domain. However, our studies have revealed a more complex mechanism.
To determine whether a targeting signal is localized in the N- or C-terminal region of Kv3.1 channels, we fused these regions to two membrane reporter proteins, dendritic transferrin receptor-GFP (TfR–GFP) (Fig. 2A) and unpolarized CD4 (Fig. 2B) (Gu et al., 2003). When expressed in neurons, TfR–GFP (Faxon/Fdendrite: 0.13 ± 0.09; n = 10) was predominantly localized to somatodendritic membranes (Fig. 2A). Whereas the N-terminal domains of Kv1 channels, including the T1 domain, targeted TfR–GFP onto axon membranes (Fig. 2A) (Faxon/Fdendrite: 1.44 ± 0.34; n = 13) (Gu et al., 2003; Rivera et al., 2005), the N-terminal domain of Kv3.1 channels did not (Fig. 2A). When expressed in neurons, CD4 (Faxon/Fdendrite: 1.0 ± 0.2; n = 20) was uniformly distributed in both dendrites and axons (Fig. 2B). Surprisingly, both CD4–31aC (the Kv3.1a C-terminal region (31aC) fused to the CD4 C terminus) (Faxon/Fdendrite: 2.95 ± 0.17; n = 25) and CD4–31bC (the Kv3.1b C-terminal region (31bC) fused to the CD4 C terminus) (Faxon/Fdendrite: 2.49 ± 0.16; n = 22) were primarily targeted to the axonal membrane (Fig. 2C–E), suggesting that at least one ATM resides in both 31aC and 31bC.
Figure 2.
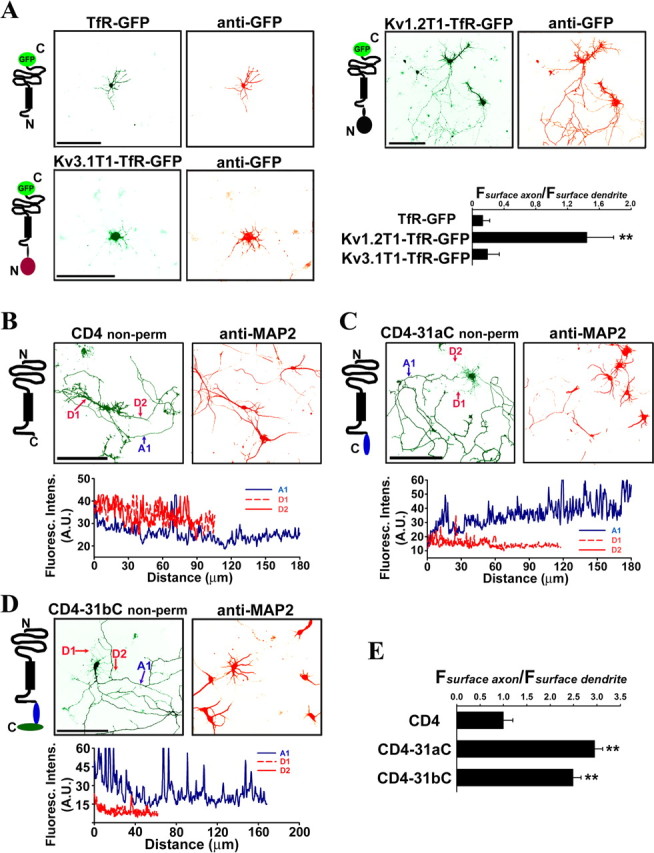
Kv3.1 C-terminal regions enrich CD4 to axonal membranes. A, The Kv3.1 T1 domain did not increase the level of GFP-tagged transferrin receptor (TfR–GFP) in axons. Diagrams of TfR–GFP fusion proteins are on the left. TfR contains an intracellular N and extracellular C terminus with a single membrane-spanning segment indicated by a black bar. GFP fluorescence indicating the total protein level (green) is shown in the middle; anti-GFP staining under nonpermeabilized condition indicating the surface level of expressed fusion proteins (red) is on the right. TfR–GFP itself was primarily localized in the somatodendritic region (top left). The Kv1.2 T1 domain increased the level of TfR–GFP on axonal membranes (top right), whereas the Kv3.1 T1 domain did not (bottom left). Kv3.1T1–TfR–GFP was mainly restricted in soma and proximal dendrites and often formed punctate clusters along dendrites. The axon–dendrite targeting ratio for a fusion construct equals to the average anti-GFP immunofluorescence intensity along axons divided by the average intensity along dendrites (Fsurface axon/Fsurface dendrite), shown at the bottom right. B, CD4 is localized to both dendrites and axons. CD4 contains an extracellular N and intracellular C terminus with a single membrane-spanning segment indicated by a black bar. The CD4-transfected neurons were stained with anti-CD4 antibodies (green, top middle) under nonpermeabilized conditions and then permeabilized and stained for the dendritic marker MAP2 (red, top right). The anti-CD4 fluorescence profiles of the major axon (A1 in blue) and two dendrites (D1 and D2 in red) are shown in the bottom. C, The Kv3.1a C terminus (31aC) polarized CD4 targeting to the axonal membrane. D, The Kv3.1b C terminus (31bC) polarized CD4 targeting to the axonal membrane. E, Summary of polarized membrane targeting of CD4 fusion proteins. The polarity ratio (Fsurface axon/Fsurface dendrite) is equal to the average anti-CD4 fluorescence intensity along axons divided by the average intensity along dendrites. Distance 0, Soma. Scale bars, 100 μm. One-way ANOVA followed by Dunnett's test. **p < 0.0001.
The C-terminal residues close to the sixth transmembrane segment are highly conserved within the Kv3 channel subfamily, whereas the residues in distal C terminus, in which alternative splicing occurs, are more divergent (Fig. 3A). To determine whether the splice domain (31sC, 80 residues) in the Kv3.1b C terminus contains targeting signals, we made a fusion construct, CD4–31sC, and found that it essentially remained unpolarized on neuronal membranes (Faxon/Fdendrite: 0.84 ± 0.06; n = 15) when expressed in hippocampal neurons (Fig. 3D). Therefore, the ATM likely resides in the 31aC region, not in the 31sC region.
Figure 3.
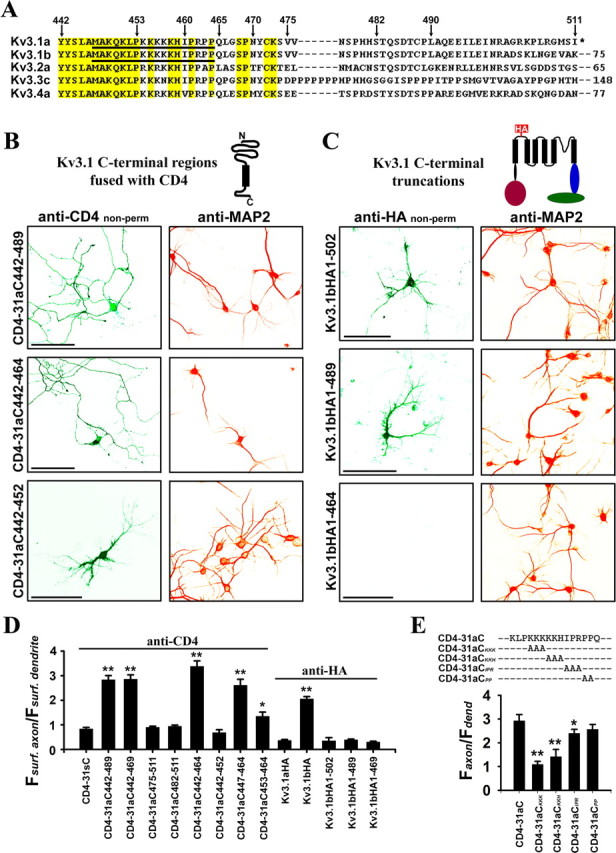
Identification of the axonal targeting motif (ATM) in a highly conserved region of Kv3.1 C terminus. A, Sequence alignment of the C-terminal regions of rat Kv3 channels. Conserved residues are highlighted in yellow. Numbers above the sequence indicate the positions of residues in Kv3.1a. Numbers after the dashed lines indicate the number of extra residues at C-terminal regions. The underlined residues are critical for axonal targeting. Asterisk indicates the end of Kv3.1a C terminus. B, Primarily localized on the axonal surface were CD4–31aC442–489 and CD4–31aC442–464, but not CD4–31aC442–452, which were constructed by fusing different regions from Kv3.1a C terminus (aa 442–489, aa 442–464, and aa 442–452, respectively) to the CD4 C terminus. C, Deleting Kv3.1bHA C-terminal region eliminated axonal membrane targeting. The transfected neurons were first stained with an anti-HA antibody (green) under nonpermeabilized conditions and then permeabilized and stained for the dendritic marker MAP2 (red). Kv3.1bHA1–502 and Kv3.1bHA1–489 became primarily localized on dendritic membranes, whereas Kv3.1bHA1–464 failed to traffic to the plasma membrane. These truncations were generated by engineering a stop codon at positions 503, 490, and 465, respectively. D, Summary of surface targeting of various constructs indicates that the ATM region (M447-Q465) is sufficient to target CD4 to the axonal membrane but fails to target Kv3.1aHA or Kv3.1bHA C-terminal deletions to the axonal membrane. CD4 (in Fig. 2E) and Kv3.1aHA were used as controls in statistics. E, The alanine scan experiment indicates that the lysine-rich motif but not the proline-rich motif is required for axonal targeting. Based on CD4–31aC, three or two consecutive residues were mutated to alanines with the Quickchange mutagenesis strategy. Antibody uptake experiments were performed on neurons expressing these constructs. Here anti-CD4 staining was done under nonpermeabilized conditions. One-way ANOVA followed by Dunnett's test. *p < 0.001; **p < 0.0001.
To identify the ATM, we made six truncations based on CD4–31aC and examined their membrane targeting patterns. Whereas the CD4 fusion constructs missing the C-terminal half of 31aC, including CD4–31aC442–489 (Faxon/Fdendrite: 2.83 ± 0.17; n = 19), CD4–31aC442–469 (Faxon/Fdendrite: 2.86 ± 0.18; n = 26), and CD4–31aC442–464 (Faxon/Fdendrite: 3.38 ± 0.22; n = 12), were still primarily localized to axonal membranes (Fig. 3B,D), a construct missing another 13 residues (aa 453–465), CD4–31aC442–452 (Faxon/Fdendrite: 0.69 ± 0.11; n = 14), and two constructs missing the N-terminal half of 31aC, CD4–31aC475–511 (Faxon/Fdendrite: 0.90 ± 0.04; n = 21) and CD4–31aC482–511 (Faxon/Fdendrite: 0.94 ± 0.05; n = 17), failed to concentrate on axonal membranes (Fig. 3B,D). Furthermore, we made two fusion constructs containing the N-terminal half of 31aC. Whereas CD4–31aC447–464 (Faxon/Fdendrite: 2.73 ± 0.23; n = 22) remained enriched on axonal membranes, CD4–31aC453–464 (Faxon/Fdendrite: 1.43 ± 0.17; n = 20) showed a significant decrease of the axon/dendrite ratio. Together, this study shows that the ATM is located within residues 447–465 in the N-terminal half of 31aC (Fig. 3A).
To examine whether the ATM functions through dendrite-specific endocytosis, we adopted a different experimental strategy, antibody uptake, which was successfully used in the studies of dendrite-specific endocytosis of VAMP2 and Nav1.2 (Garrido et al., 2001; Sampo et al., 2003). We briefly incubated the live neurons expressing Kv3.1bHA with monoclonal anti-HA antibodies added in the culture medium. The antibodies can be internalized into the neurons together with Kv3.1bHA channels if the channels undergo endocytosis. If Kv3.1b achieves axonal membrane targeting via rapid dendrite-specific endocytosis, strong anti-HA signals (most from internalized anti-HA antibodies) should be observed in dendrites under permeabilized conditions. However, the staining patterns of these neurons under both permeabilized and nonpermeabilized conditions are similar (supplemental Fig. S1, available at www.jneurosci.org as supplemental material), suggesting the ATM does not act through dendrite-specific endocytosis.
Within this ATM region, there are two interesting putative motifs. One is a lysine-rich motif (K454-K458), and the other is a proline-rich motif (P461-P464) (Fig. 3A). To determine whether the lysine- and proline-rich motifs are indispensable to the ATM, we performed an alanine scan in this region, by mutating three or two consecutive residues to alanines based on CD4–31aC, and then assayed targeting of these mutants with antibody uptake strategy (Fig. 3E). Under nonpermeabilized conditions, in contrast to CD-31aC (Faxon/Fdendrite: 2.93 ± 0.26; n = 15), the targeting patterns of KKK (Faxon/Fdendrite: 1.09 ± 0.13; n = 15) and KKH (Faxon/Fdendrite: 1.42 ± 0.30; n = 10) became unpolarized (Fig. 3E). Axonal targeting remained unchanged for PP (Faxon/Fdendrite: 2.57 ± 0.20; n = 20), and modestly decreased for IPR (Faxon/Fdendrite: 2.40 ± 0.17; n = 16) (Fig. 3E). Therefore, it is the lysine-rich motif but not the proline-rich motif that is required for axonal targeting (Fig. 3E).
Because both Kv3.1a and Kv3.1b contain this ATM, why is Kv3.1a primarily localized in the somatodendritic region (Fig. 1C)? Alternative splicing gives rise to 10 and 85 nonconserved residues at the C-terminal ends of Kv3.1a and Kv3.1b, respectively (Fig. 3A). To determine whether the last 10 residues in Kv3.1a could contain a dominant dendritic targeting signal, we made a deletion construct by introducing a stop codon at S503 of Kv3.1bHA (Kv3.1bHA1–502), which is similar to a Kv3.1aHA deletion missing the last 10 residue except for an Asp residue from Kv3.1b at the very end (Fig. 3A). In contrast to Kv3.1bHA (Faxon/Fdendrite: 2.06 ± 0.09; n = 32), Kv3.1bHA1–502 (Faxon/Fdendrite: 0.35 ± 0.13; n = 11) was primarily localized on the somatodendritic surface just like Kv3.1aHA (Faxon/Fdendrite: 0.37 ± 0.04; n = 23) (Fig. 3C,D), suggesting that the last 10 nonconserved residues at the Kv3.1a C terminus are not involved in regulating Kv3.1a targeting. To further examine whether other regions within 31aC could compromise the ATM function, we constructed more truncations of Kv3.1bHA by introducing stop codons at L490, N470 and Q465 (Fig. 3A). Whereas Kv3.1bHA1–489 (Faxon/Fdendrite: 0.39 ± 0.03; n = 16) and Kv3.1bHA1–469 (Faxon/Fdendrite: 0.30 ± 0.03; n = 10) were targeted to somatodendritic membranes similar to Kv3.1aHA, Kv3.1bHA1–464 failed to even reach the plasma membrane (Fig. 3C,D). Together, these data indicate that the ATM within residues 447–465 is the only axonal targeting signal in Kv3.1 C termini. Because axonal targeting is compromised in the presence of Kv3.1 N terminus, it is possible that the N terminus can negatively regulate the ATM function in Kv3.1a.
Direct interaction between the T1 domain and the Kv3.1 ATM
How does the Kv3.1 N terminus negatively regulate the ATM function? One possibility is that 31N directly interacts with 31aC to prevent the ATM from accessing its axonal targeting machinery, and this masking effect may be weakened in the presence of 31sC in Kv3.1b.
To examine whether this interaction occurs within Kv3.1 channels, we set out to make GST- and 6×His-tagged fusion proteins from Kv3.1 cytoplasmic regions as illustrated in Figure 4A. All these fusion proteins are soluble when expressed in E. coli BL21 cells. In the first experiment, the supernatant of bacterial lysates containing expressed GST, GST-31N [GST fused to the Kv3.1 N terminus (aa 1–186)], GST-31aC [GST fused to the Kv3.1a C terminus (aa 442–511)], GST-31bC [GST fused to the Kv3.1b C terminus (aa 442–585)] or GST-31sC [GST-fused to the Kv3.1b splice domain (aa 500–585)], was mixed in equal volume with the supernatant containing expressed His-31N [6×His tag fused to the Kv3.1 N terminus (aa 1–186)] or His-31T1 [6×His tag fused to the Kv3.1 T1 domain (aa 1–110)] for 2 h and then precipitated with glutathione agarose beads. GST-31N pulled down His-31N and His-31T1 (Fig. 4B) as expected. Interestingly, GST-31aC and GST-31bC, but not GST-31sC or GST alone, precipitated both His-31N and His-31T1 (Fig. 4B), suggesting that 31N and 31aC indeed interact.
Figure 4.
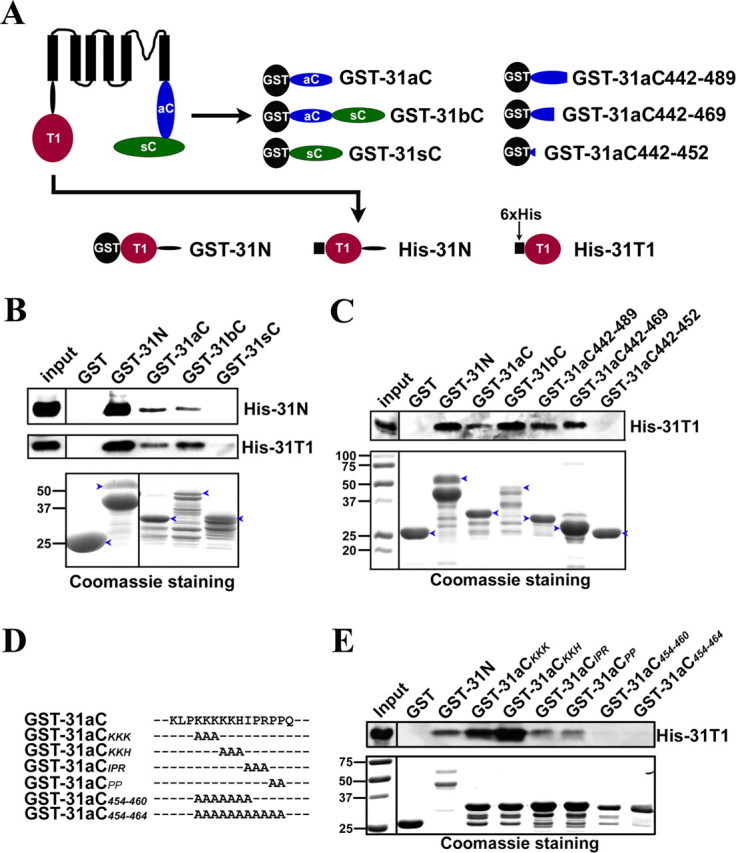
The Kv3.1 T1 domain binds to the C-terminal region. A, Diagrams of Kv3.1b and fusion constructs. Black circles, GST tag. Black squares, 6×His tag. Red circles, The T1 domain. Blue ellipses, The Kv3.1a C-terminal region (31aC). Green ellipses, The Kv3.1b splice domain (31sC). Truncations based on GST-31aC were generated with Quickchange mutagenesis. B, GST-31N, GST-31aC, and GST-31bC, but not GST and GST-31sC, pulled down both His-31N and His-31T1. The bacterial lysates containing a GST fusion protein and a His-tag fusion protein were mixed at a 1:1 ratio in volume and incubated with glutathione agarose beads for 3 h at 4°C. The precipitated proteins were eluted, resolved with SDS-PAGE, and blotted with an anti-His antibody (the top 2 panels). Four percent of the inputs of His-tagged proteins were loaded (left lanes). Fifty percent of the inputs of GST fusion proteins were loaded for Coomassie staining (bottom). C, The ATM precipitated the Kv3.1 T1 domain. GST fusion proteins were expressed in bacteria, purified with glutathione beads, and subsequently incubated on the beads with the bacterial lysates containing expressed His-31T1. The precipitants were blotted with an anti-His antibody (top). GST-31N, GST-31aC, GST-31bC, GST-31aC442–489, and GST-31aC442–469, but not GST and GST-31aC442–452, pulled down His-31T1. D, Sequence alignments of GST-31aC and point mutations in the ATM. E, Residues within the ATM are critical for binding to the T1 domain. Mutating the proline-rich motif but not the lysine-rich motif weakened its binding to the T1 domain, but the binding was completely eliminated only when both motifs were mutated. The experimental procedure here is similar to that in C. The molecular weight markers (in kilodaltons) were at the left lane in the bottom. Blue arrowheads, Full-length GST fusion proteins.
In the second experiment, to confirm the 31N/31aC interaction and identify the region in 31aC critical for the interaction, the purified GST fusion proteins, including the three truncations in 31aC, GST-31aC442–489 (GST fused to the region 442–489 in Kv3.1a C terminus), GST-31aC442–469 (GST fused to the region 442–469 in Kv3.1a C terminus) and GST-31aC442–452 (GST fused to the region 442–452 in Kv3.1a C terminus), were used to pull down His-31T1 from the supernatant of bacterial lysates (Fig. 4C). Remarkably, GST-31N, GST-31aC, GST-31bC, GST-31aC442–489, and GST-31aC442–469, but not GST and GST-31aC442–452, precipitated His-31T1 (Fig. 4C). Furthermore, residues in the ATM were mutated to alanines in the GST-31aC backbone as shown in Figure 4D. The pull down experiment showed that mutating the proline-rich motif weakened the interaction, whereas mutating the lysine-rich motif had no effect (Fig. 4E). Only after disrupting both motifs did we observe complete elimination of the interaction between GST-31aC and His-31T1 (Fig. 4E). These results suggest an interesting interaction between the Kv3.1 T1 domain and the ATM.
To determine whether the interaction is direct, we further performed pull-down experiments with purified proteins. Under the normal pull-down condition, both purified GST-31N and GST-31aC precipitated His-31T1 inefficiently. The precipitated His-31T1 was only clearly detected with Western blotting like those experiments in Figure 4, but not with Coomassie staining (data not shown). It is known that Zn2+ ions are required for Kv3 T1 domains to form a stable tetramer complex (Bixby et al., 1999; Nanao et al., 2003). After chelating Zn2+, the Kv3 T1 complex is dissembled to monomers, which can then reassemble into tetramers when Zn2+ is added back (Bixby et al., 1999; Nanao et al., 2003). We reasoned that under normal conditions, the purified His-31T1 and GST-31N likely exist as tetramers already, thus they do not interact with each other efficiently. Therefore, we adopted a different strategy, to first treat the reaction mixture with 1 mm EDTA to chelate Zn2+, next to dialyze EDTA and add Zn2+ back to the mixture, and then to perform the pull-down assay (Nanao et al., 2003) (Fig. 5A). This pull-down strategy significantly enhanced the amounts of His-31T1 precipitated by either GST-31N or GST-31aC, so that the bands of precipitated His-31T1 were clearly detected by Coomassie staining (Fig. 5B). In sharp contrast, a mutation of GST-31aC, GST-31aC454–464, in which 11 residues (454–464) in the ATM were all mutated to 11 alanines, did not pull down His-31T1 at all (Fig. 5B). We also performed the pull-down assay in the presence of 1 mm EDTA (most T1 domains in monomeric states), and obtained a result similar to the regular pull-down experiment (data not shown). Together, our studies have demonstrated a direct interaction between the T1 and the C terminus of Kv3.1a, supporting the hypothesis that the T1 domain binds to the ATM and blocks its function.
Figure 5.

Zinc regulates direct binding between purified His-31T1 and GST-31aC. GST and four other fusion proteins (His-31T1, GST-31N, GST-31aC, and GST-31aC454–464) were bacterially expressed and purified. In the normal pull-down buffer, GST-31N and GST-31aC did not efficiently precipitate His-31T1. Despite being evident in Western blotting, the precipitated His-31T1 was not detected by Coomassie staining. Therefore, we adopted a different pull-down strategy. A, Diagram of the procedure of pull-down assay. Purified His-31T1 and GST fusion proteins were first incubated overnight at 4°C in the presence of 1 mm EDTA to chelate Zn2+ to dissemble T1 tetramers into monomers. Then EDTA was dialyzed away and Zn2+ was added back to induce T1 monomers to reassemble into tetramers. B, GST-31N and GST-31aC, but not GST and GST-31aC454–464, efficiently precipitated His-31T1 in this pull-down assay. The precipitated His-31T1 was clearly detected in Coomassie staining. Identity of precipitated His-31T1 was confirmed with Western blot, in which only 10% of those in the top were loaded in each lane (bottom). For those purified fusion proteins, in addition to the main protein band, there are often a few smaller minor bands presumably attributable to protein degradation, which could occur even before the lysing and purification processes, because we have used protease inhibitors. The lower strong band in GST-31N may miss the T1-S1 linker, which does not compound our assay data. The lower bands in GST-31aC and GST-31aC454–464 are likely to be just GST. Because GST as a negative control included in our assay, these bands should not interfere with our experimental results as well. This pull-down experiment was repeated for three times. For Coomassie staining, 25% input of His-31T1 (the left lane) of the pull-down assay were loaded. Blue arrowheads, Full-length GST fusion proteins. Red arrows, His-31T1 bands. Numbers on the left, Molecular weights in kilodaltons.
Ankyrin G associates with the Kv3.1 ATM through its N-terminal ankyrin-repeat domain
We have provided evidence showing a T1-domain-mediated regulation of the Kv3.1 ATM. We next asked what is the axonal targeting machinery associated with this ATM. Ankyrin G is a good candidate for regulating Kv3.1 channel targeting, because it coimmunoprecipitated (co-IP) with Kv3.1b channels from rat brain lysates and this interaction has been suggested to play a role in targeting the channels to nodes of Ranvier in central neurons (Devaux et al., 2003). Ankyrins are adaptor proteins that link the spectrin-based membrane skeleton to integral membrane proteins, including the anion exchanger, the Na+/Ca2+ exchanger, the Na+/K+ ATPase, members of L1CAM family, and Nav and KCNQ channels (Bennett and Chen, 2001; Jenkins and Bennett, 2001; Garrido et al., 2003; Pan et al., 2006). Ankyrin G contains 24 ankyrin repeats (33-residue motifs involved in protein recognition) at its N terminus, which is also referred to as the membrane-binding (MB) domain attributable to its binding to the various membrane proteins. Following these ankyrin repeats is a spectrin-binding domain, providing a link to the actin cytoskeleton (Bennett and Chen, 2001). The binding affinity of the membrane- and spectrin-binding domains to their targets can be modulated by a C-terminal regulatory region (Bennett and Chen, 2001). Although it has been found that Kv3.1b associates with ankyrin G in rat brain lysates (Devaux et al., 2003), the ankyrin G-binding site in Kv3.1b is not known. In fact, Kv3.1b channels do not contain sequences that are homologous to the ankyrin G-binding motifs identified from other membrane proteins (Devaux et al., 2003). Moreover, although ankyrin G is localized at both nodes of Ranvier and AISs, Kv3.1b channels are essentially absent from the AIS (Devaux et al., 2003). Therefore, ankyrin G may play an important role in Kv3.1 channel targeting, but this role may be distinct from the one in clustering Nav and KCNQ channels to the AIS.
To examine the potential role of ankyrin G in Kv3.1 channel targeting, we first searched for the ankyrin G-binding site in Kv3.1b channels. Purified GST-31aC and GST-31bC, but not GST and GST-31sC, precipitated ankyrin G-GFP from transfected HEK293 cell lysates (Fig. 6A), indicating that the ankyrin G-binding site exists within the 31aC region but not the splice domain of Kv3.1b (31sC). A very faint band of ankyrin G-GFP was also observed from GST-31N precipitants (Fig. 6A), which could result from a second low-affinity binding site for ankyrin G in the N-terminal region or an indirect interaction. Next, we examined the truncations of GST-31aC. Remarkably, GST-31aC442–489 and GST-31aC442–469, but not GST-31aC442–452, pulled down ankyrin G-GFP from HEK293 cell lysates (Fig. 6B), showing that the ATM not only binds to the T1 domain, but also associates with ankyrin G.
Figure 6.

The ankyrin-repeat domain of ankyrin G binds to the Kv3.1 ATM region. A, Ankyrin G-GFP was precipitated by purified GST-31aC and GST-31bC. The diagram shows the full-length ankyrin G-GFP construct, from the N to C terminus, containing the ankyrin repeat [also called membrane-binding (MB)] domain, spectrin-binding domain, serine-rich domain, and a death domain. GST, GST-31N, GST-31aC, GST-31bC, and GST-31sC were expressed in E. coli BL21 cells and purified with glutathione agarose beads. Then the beads coated with purified GST fusion proteins were further incubated with the supernatant of cell lysates from HEK293 cells transfected with ankyrin G-GFP for 3 d. The precipitated proteins were eluted, resolved in SDS-PAGE, and blotted with polyclonal anti-GFP antibody (top). The weak band in the GST-31N precipitant may result from an indirect interaction or a minor binding site. Five percent of the inputs of ankyrin G-GFP were loaded (left lane). The Coomassie staining shows 25% input of purified GST fusion proteins (bottom). B, Ankyrin G-GFP was precipitated by purified GST-31aC442–489 and GST-31aC442–469 but not GST-31aC442–452. The same experimental procedure as A was used. The left lane of Coomassie staining panel shows molecular weight standards. C, The ankyrin repeat or MB domain of ankyrin G binds to the ATM region. The top shows the diagram of MB–GFP. The experimental procedure was the same as that in B, except that the ankyrin G truncation MB–GFP was expressed in HEK293 cells. D, The lysine-rich motif but not the proline-rich motif is required for GST-31aC binding to MB–GFP. Four point mutants of GST-31aC were purified and used to pull down MB–GFP expressed in HEK293 cells. Only GST-31aCKKK failed to precipitate MB–GFP. E, Ankyrin G-GFP preferentially binds to Kv3.1bHA but not Kv3.1aHA in coimmunoprecipitation assay. Cotransfections were performed on HEK293 cells. Two days after transfection, the supernatant of cell lysates were incubated with polyclonal anti-GFP antibody and protein A agarose beads. The immunoprecipitants were blotted with rat monoclonal anti-HA antibody (top). Five percent of the inputs were loaded (middle and bottom). Numbers on the left, Molecular weights in kilodaltons.
This ankyrin G-binding site in Kv3.1 contains a number of positively charged residues and three prolines, in contrast to the motifs identified from Nav and KCNQ channels, which contain several acidic residues (Pan et al., 2006). Because ankyrin G binds to Nav and KCNQ channels through its N-terminal MB domain, we wondered whether this MB domain also associates with the Kv3.1 ATM or the interaction is mediated through a different domain of ankyrin G. Interestingly, MB–GFP (GFP fused to the C-terminal of the MB domain of ankyrin G) expressed in HEK293 cells was precipitated by purified GST-31aC, GST-31bC, GST-31aC442–489, and GST-31aC442–469, but not by GST or GST-31aC442–452 (Fig. 6C). We further examined whether point mutations in the ATM affect the association to MB–GFP. Remarkably, GST-31aCKKK (3 residues K(454)KK in 31aC mutated to 3 Ala residues) failed to pull down MB–GFP, whereas three other mutations, GST-31aCKKH (3 residues K(457)KH in 31aC mutated to 3 Ala residues) GST-31aCIPR (3 residues I(460)PR in 31aC mutated to 3 Ala residues) and GST-31aCPP (2 residues P(463)P in 31aC mutated to 2 Ala residues) precipitated MB–GFP (Fig. 6D). Therefore, ankyrin G associates with the Kv3.1 ATM via its MB domain. Notably, the lysine-rich motif within the ATM is required for both axonal targeting and ankyrin G association, but not for binding to the T1 domain.
To examine whether and how the access of ankyrin G to the ATM in 31aC could be affected by 31T1 and 31sC in the context of channel proteins, we performed co-IP experiments using HEK293 cells that were cotransfected with ankyrin G-GFP and either Kv3.1aHA or Kv3.1bHA. Consistent with the in vivo co-IP result (Devaux et al., 2003), Kv3.1bHA was precipitated by ankyrin G-GFP (Fig. 6E). In contrast, the interaction between Kv3.1aHA and ankyrin G-GFP was much weaker (Fig. 6E). Therefore, this result that ankyrin G differentially binds to Kv3.1 splice variants, suggests that the access of ankyrin G to the ATM is regulated by the T1 domain and splice domain, which correlates well with the regulation of the ATM function.
Point mutations in the ATM alter axon–dendrite targeting of Kv3.1 channels
If the T1 domain masks the ATM function in Kv3.1a, we wondered whether disrupting the T1/ATM interaction could help Kv3.1a to traffic into axons. Because mutating the residues I(460)PR or P(463)P in the ATM to AAA or AA, weakened the GST-31aC and His-31T1 binding in the pull-down assay (Fig. 4E), we made two mutations based on Kv3.1aHA channels, Kv3.1aHAIPR and Kv3.1aHAPP, and examined their targeting patterns in neurons. Interestingly, significantly more Kv3.1aHAPP became localized in axons compared with Kv3.1aHA (Fig. 7B,E). In contrast, Kv3.1aHAIPR remained restricted on somatodendritic membranes (Fig. 7A), which is consistent with the result that axonal targeting of CD4–31aCIPR modestly decreased (Fig. 4E). Therefore, our data suggest that disrupting the T1/ATM interaction indeed can help Kv3.1a to target into axons.
Figure 7.
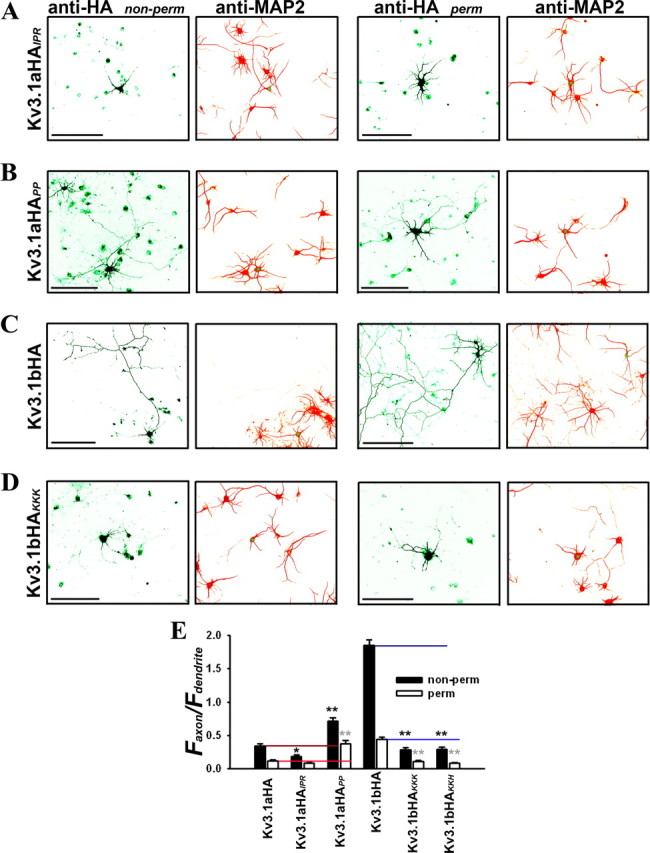
Site-directed mutagenesis in the ATM region alters axon–dendrite targeting of Kv3.1 channels. Two point mutations (Kv3.1aHAIPR and Kv3.1aHAPP) in Kv3.1aHA and two (Kv3.1bHAKKK and Kv3.1bHAKKH) in Kv3.1bHA were constructed by Quickchange strategy. Hippocampal neurons were transfected at 5 DIV, fixed, and stained 2 d later. HA staining was performed under both nonpermeabilized (non-perm; left column) and permeabilized (perm; right column) conditions. The levels of Kv3.1aHAPP (B), but not of Kv3.1aHAIPR (A), in axons significantly increased compared with Kv3.1aHA. Compared with Kv3.1bHA (C), axonal targeting of both Kv3.1bHAKKK (D) and Kv3.1bHAKKH dramatically decreased. E, Summary of polarized targeting of Kv3.1 constructs. Kv3.1aHA: non-perm Faxon/Fdendrite, 0.34 ± 0.03 (n = 45); perm Faxon/Fdendrite, 0.12 ± 0.02 (n = 32). Kv3.1aHAIPR: non-perm Faxon/Fdendrite, 0.18 ± 0.02 (n = 28); perm Faxon/Fdendrite, 0.09 ± 0.01 (n = 32). Kv3.1aHAPP: non-perm Faxon/Fdendrite, 0.72 ± 0.05 (n = 31); perm Faxon/Fdendrite, 0.37 ± 0.05 (n = 30). Kv3.1bHA: non-perm Faxon/Fdendrite, 1.85 ± 0.09 (n = 43); perm Faxon/Fdendrite, 0.45 ± 0.03 (n = 25). Kv3.1bHAKKK: non-perm Faxon/Fdendrite, 0.29 ± 0.04 (n = 20); perm Faxon/Fdendrite, 0.11 ± 0.02 (n = 31). Kv3.1bHAKKH: non-perm Faxon/Fdendrite, 0.30 ± 0.04 (n = 21); perm Faxon/Fdendrite, 0.09 ± 0.01 (n = 24). Scale bars, 100 μm. Kv3.1aHA and Kv3.1bHA are the two controls for their mutants. One-way ANOVA followed by Dunnett's test. *p < 0.001; **p < 0.0001.
Our previous experiments showed that mutating residues K(454)KK or K(457)KH to AAA primarily eliminated axonal targeting of CD4–31aC (Fig. 3E). To examine whether these residues of the ATM is essential for Kv3.1b axonal targeting, we introduced the mutations based on Kv3.1bHA, Kv3.1bHAKKK and Kv3.1bHAKKH, and determined their targeting patterns in neurons. Indeed, both mutants failed to traffic to both axonal membranes and axons (Fig. 7C–E). In addition, both mutants did not traffic to the plasma membrane well, suggesting that these residues may also be critical for the general forward trafficking of the channels. Nonetheless, these results demonstrate that the ATM we identified is indeed a key motif in Kv3 axonal targeting.
Ankyrin G differentially regulates polarized targeting of splice variants of Kv3.1 channels
Our in vitro binding assays suggest that, through association with the ATM, ankyrin G may be a critical component of the axonal targeting machinery of Kv3.1 channels. To examine how ankyrin G is involved in Kv3.1 targeting, we coexpressed ankyrin G-GFP with either Kv3.1aHA or Kv3.1bHA in cultured hippocampal neurons and examined their distribution patterns under permeabilized conditions. Kv3.1aHA (Faxon/Fdendrite: 0.10 ± 0.01; n = 22) was primarily restricted in somatodendritic regions, whereas ankyrin G-GFP was concentrated at the AIS (Fig. 8A–C). The Kv3.1aHA level decreased abruptly in the beginning part of the AIS, in which ankyrin G appeared like a barrier that prevented Kv3.1aHA from entering the axon (Fig. 8C). Conversely, Kv3.1bHA (Faxon/Fdendrite: 0.32 ± 0.03; n = 20) was distributed throughout axons as well as in soma and proximal dendrites, whereas ankyrin G-GFP was still concentrated in the proximal part of the axon (Fig. 8B,C). Interestingly, the Kv3.1bHA level increased in axons distal to the AIS (Fig. 8C), with a pattern similar to that of endogenous Kv3.1b channels in mature hippocampal neurons (28 DIV) (Fig. 8H,I) as well as in brain slices (Devaux et al., 2003). Therefore, the result that Kv3.1bHA channels can traffic through the ankyrin G-enriched AIS into distal axons is consistent with the strong association of Kv3.1bHA with ankyrin G revealed by the co-IP experiment (Fig. 6E).
Figure 8.
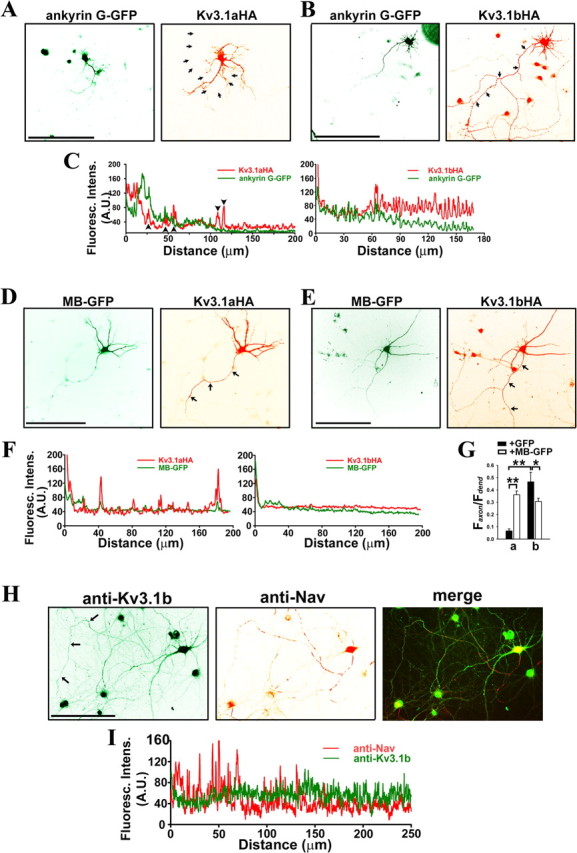
Ankyrin G differentially regulates polarized targeting of Kv3.1 splice variants. A–G, Hippocampal neurons at 7 DIV were cotransfected with ankyrin G and Kv3.1 constructs, fixed, and stained with anti-HA antibodies under permeabilized conditions. A, Kv3.1aHA (red) did not colocalize with ankyrin G-GFP (green) in axons. B, Kv3.1bHA (red) colocalized with ankyrin G-GFP (green) in proximal axons. C, The fluorescence intensity profiles along the axons in A and B indicated by arrows. Arrowheads, The fluorescence peaks attributable to crossing dendrites. D, E, The levels of Kv3.1aHA (D) and Kv3.1bHA (E) in axons markedly increased and modestly decreased, respectively, in the presence of MB–GFP (green). F, The fluorescence intensity profiles along the axons in D and E indicated by arrows. G, Summary of polarized targeting of Kv3.1aHA and Kv3.1bHA in the presence of GFP and MB–GFP. Faxon/Fdendrite is average fluorescence intensity in axons/average fluorescence intensity in dendrites. H, Endogenous Kv3.1b channels were localized to distal axons (arrows), in contrast to the localization of endogenous Nav channels in proximal axons. Hippocampal neurons at 30 DIV were costained with polyclonal anti-Kv3.1b (green) and monoclonal anti-pan-Nav channel (red) antibodies. It is important to note that Kv3.1b also localizes in soma and proximal dendrites. I, Fluorescence intensity profiles along the axon indicated by arrows in H. Distance 0, Soma. Scale bars, 100 μm. Student's t test used in the comparison of two groups. *p < 0.01; **p < 0.0001.
Ankyrin G links membrane proteins and spectrin/actin cytoskeleton via its MB domain and spectrin-binding domain, respectively. Over-expressing MB–GFP may disrupt such link and hence MB–GFP may exhibit a dominant-negative effect on the function of endogenous ankyrin G. Thus, we coexpressed MB–GFP with Kv3.1 channel constructs in cultured neurons. When expressed in neurons, MB–GFP becomes localized in both dendrites and axons, different from the AIS targeting of ankyrin G-GFP (Fig. 8A–E) (Zhang and Bennett, 1998). In the presence of MB–GFP and under permeabilized conditions, the total level of Kv3.1aHA (Faxon/Fdendrite: 0.36 ± 0.03; n = 22) in axons dramatically increased (Fig. 8D,F,G), whereas the total level of Kv3.1bHA (Faxon/Fdendrite: 0.31 ± 0.03; n = 22) in axons modestly decreased (Fig. 8E–G), when compared with those of Kv3.1aHA (Faxon/Fdendrite: 0.07 ± 0.02; n = 10) and Kv3.1bHA (Faxon/Fdendrite: 0.47 ± 0.08; n = 12) in the presence of GFP (Fig. 8G). However, over-expressing MB–GFP may cause nonspecific effects. We designed siRNA probes to suppress the expression of endogenous rat ankyrin G. One of the probes, AnkG siR (mean and SE of fluorescence intensity (A.U.) at the AIS: 72 ± 15; n = 22), when transfected into hippocampal neurons at 3 DIV, can efficiently suppress endogenous ankyrin G at 8 DIV compared with control siR (mean and SE of fluorescence intensity (A.U.) at the AIS: 198 ± 26; n = 18) (Fig. 9A). We cotransfected this probe with either Kv3.1aHA or Kv3.1bHA into hippocampal neurons, and examined channel targeting under both permeabilized and nonpermeabilized conditions. Consistent with the results of expressing MB–GFP, suppressing endogenous ankyrin G significantly decreased polarized axon–dendrite targeting of Kv3.1 channels (Fig. 9B,C). Together, our data support a pivotal role of ankyrin G in differentially targeting Kv3.1 channel splice variants.
Figure 9.
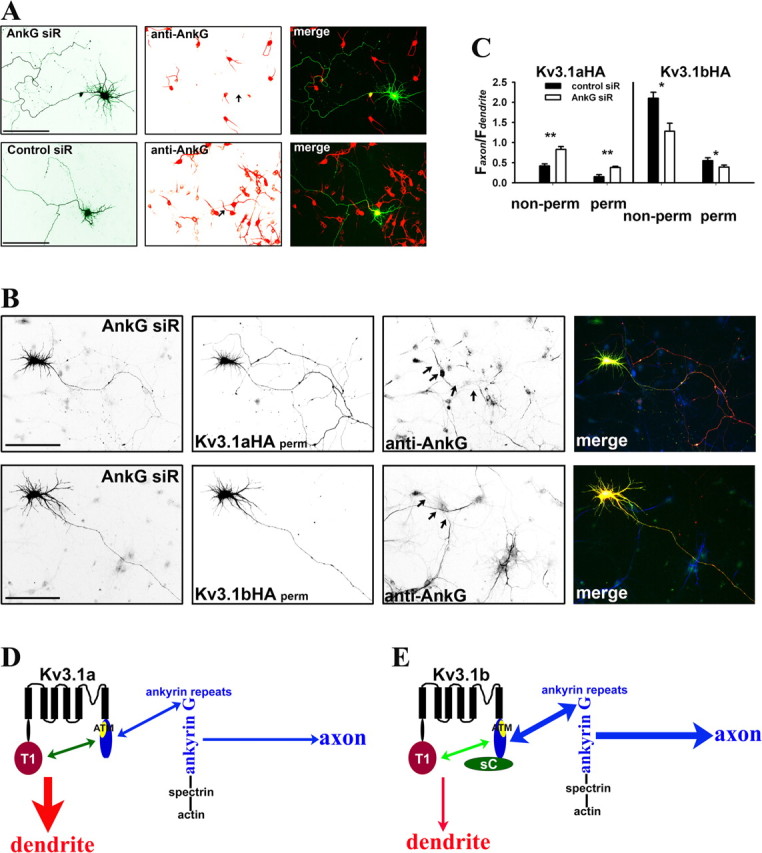
Suppressing endogenous ankyrin G by siRNA altered axon–dendrite targeting of Kv3.1 channels. A, One of four siRNA probes designed against rat ankyrin G, AnkG siR, efficiently suppressed endogenous ankyrin G levels in both soma and axon initial segments. AnkG siR and control probe (Control siR) were transfected into cultured hippocampal neurons at 3 DIV. Five days later (at 8 DIV), the neurons were fixed and stained for endogenous ankyrin G (red). Control siR did not affect ankyrin G levels in both soma and axon initial segments. B, Suppressing endogenous ankyrin G by AnkG siR increased Kv3.1aHA levels in axons and decreased the axon/dendrite ratio of Kv3.1bHA. Neurons were cotransfected with AnkG siR and Kv3.1aHA (top) or Kv3.1bHA (bottom) at 3 DIV and fixed and stained 5 d later. In merged images, AnkG siR is in green, anti-HA staining is in red, and anti-ankyrin G (anti-AnkG) staining is in blue. C, Summary of the effects of siRNA knockdown on Kv3.1 channel targeting. Under nonpermeabilized conditions for anti-HA staining (non-perm), the axon/dendrite ratio of Kv3.1aHA with AnkG siR (Faxon/Fdendrite: 0.83 ± 0.07; n = 19) significantly increased compared with that with Control siR (Faxon/Fdendrite: 0.42 ± 0.05; n = 20), whereas the axon/dendrite ratio of Kv3.1bHA with AnkG siR (Faxon/Fdendrite: 1.28 ± 0.20; n = 13) decreased compared with that with Control siR (Faxon/Fdendrite: 2.1 ± 0.15; n = 14). Under permeabilized conditions, the axon/dendrite ratio of Kv3.1aHA with AnkG siR (Faxon/Fdendrite: 0.38 ± 0.03; n = 15) also increased compared with that with Control siR (Faxon/Fdendrite: 0.15 ± 0.05; n = 15), whereas the axon/dendrite ratio of Kv3.1bHA with AnkG siR (Faxon/Fdendrite: 0.39 ± 0.05; n = 18) decreased compared with that with Control siR (Faxon/Fdendrite: 0.55 ± 0.07; n = 18). Arrows, Proximal axons. Scale bars, 100 μm. One-way ANOVA, followed by Dunnett's test for p values. **p < 0.0001. *p < 0.001. D, E, A model diagram for polarized targeting of Kv3.1 channels. In Kv3.1a (D), the 31T1/31aC interaction masks the ATM to prevent the channel from being accessed by axonal targeting machinery, including ankyrin G. In the axon initial segment, ankyrin G linking various membrane proteins to spectrin/actin cytoskeletons may behave as a trafficking checkpoint. Weak binding between Kv3.1a and ankyrin G does not favor the entry of Kv3.1a to the axon. In Kv3.1b (E), the 31sC domain alters the conformation and/or affinity of the 31T1/31aC interaction, resulting in exposed ATM, which binds to ankyrin G. Thus, Kv3.1b binds to ankyrin G with much higher affinity compared with Kv3.1a, and thereby Kv3.1b is attracted to enter the axon. For simplification, only one subunit of a Kv3.1 channel tetramer is shown here.
Discussion
In this study, we have identified a conditional ATM in Kv3.1 channels. This motif is not only shared by both Kv3.1a and Kv3.1b, but also highly conserved among all Kv3 channels. Remarkably, this ATM interacts with both the Kv3.1 T1 domain and adaptor protein ankyrin G. Based on several lines of evidence obtained in this study, we postulate a mechanism by which the two splice variants of Kv3.1 channels are differentially targeted to dendritic and axonal membranes (Fig. 9D,E).
The Kv3.1 ATM is a conditional lysine-rich targeting signal
To our surprise, the Kv3.1 ATM is located in a lysine-rich region of its C terminus. Using membrane reporter proteins, including unpolarized CD4 and dendritic TfR–GFP, we have thoroughly examined the potential roles played by cytoplasmic domains of Kv3.1 channels in polarized membrane targeting. Only one region, right after the sixth membrane-spanning segment, was sufficient to polarize CD4 targeting to the axonal membrane (Fig. 3). Analysis of this region by site-directed mutagenesis showed that the lysine-rich motif but not the proline-rich motif is required for axonal targeting (Fig. 3E). Consistent with the CD4 fusion protein studies, this lysine-rich motif is also required for axonal targeting of Kv3.1bHA (Fig. 7D,E).
The Kv3 ATM is unique to the Kv3 (Shaw) family among Kv channels. It has been shown that axonal targeting of Kv1 channels requires the microtubule plus end tracking protein EB1 and KIF3/kinesin II (Gu et al., 2006). Different targeting signals and mechanisms between Kv1 and Kv3 are actually consistent with their differences in channel activity, T1 domain structure, expression and subcellular distribution patterns, and associated neurological diseases. Moreover, in contrast to the dominant axonal/dendritic targeting signals of Kv1, Kv2, and Kv4 channels (Lim et al., 2000; Gu et al., 2003; Rivera et al., 2003), the axonal targeting signal of Kv3.1 channels is conditional. The initial puzzling result that the Kv3.1 ATM targets CD4 and Kv3.1b, but not Kv3.1a, into axons, has led us to uncover an intriguing interaction between the N- and C-terminal domains. This interaction is mediated by direct and Zn2+-dependent binding between the T1 domain and the ATM region (Figs. 4, 5). Thus, 31T1 likely masks the ATM to lessen the access of ankyrin G, a critical component of Kv3.1b axonal targeting machinery.
Direct interaction between the T1 domain and the ATM region
The T1 domains in the N termini of Kv channels are conserved and they form tetramers (Li et al., 1992; Choe, 2002; Nanao et al., 2003; Long et al., 2005) right after emerging from the ribosomal exiting tunnel once synthesis of the T1-S1 linker is completed (Kosolapov et al., 2004). It is important to note that although the T1 domains are highly conserved within the Kv3 family (> 70% identify), the Kv3 T1 domain shares only ∼40% homology with the Kv1 T1 domain and requires four Zn2+ ions to assemble into tetramers (Nanao et al., 2003). Interestingly, addition and removal of Zn2+ reversibly switches the T1 domains of both Kv3.1 and Kv4.2 channels between stable tetrameric and monomeric states (Jahng et al., 2002; Nanao et al., 2003). These differences between the Kv1 T1 and Kv3 T1 are consistent with the result that only the Kv1 T1, but not the Kv3 T1, targets TfR–GFP into axons (Fig. 2A).
The interaction between N and C termini of several Kv channels, including Shaker, Kv1.2 and Kv2.1, has been suggested before (Schulteis et al., 1996; Ju et al., 2003; Scholle et al., 2004; Long et al., 2005; Kobrinsky et al., 2006). In this study, we have provided evidence that the N/C terminal interaction of Kv3.1 channels is mediated by a direct, Zn2+-dependent binding between the T1 domain and the ATM region (Figs. 4, 5). Mutating 11 residues in the ATM to alanines completely abolished the N/C terminal interaction between purified proteins (Fig. 5B). The lack of efficient precipitation of His-31T1 by both GST-31N and GST-31aC under normal pull-down conditions is likely attributable to the high stability of T1 domain tetramers, because the 31T1/31T1 and 31T1/31aC interactions were both significantly enhanced by chelating Zn2+ with EDTA and subsequently re-addition of Zn2+ to allow tetramers to reform (Fig. 5). Nonetheless, it remains possible that there are multiple binding interfaces between 31N and 31aC. The structure, exact conformation(s) and the stoichiometry of the Kv3.1 N/C termini complex remain to be determined.
How may alternative splicing regulate polarized targeting of Kv3.1 channels?
In targeting assays, we found that CD4–31aC and CD4–31bC are both targeted to the axonal membrane, whereas the distribution pattern of CD4–31sC resembles that of wild type CD4 (Figs. 2, 3D), suggesting that the 31sC domain does not contain any axonal or dendritic targeting signals. GST-31sC does not precipitate either His-31T1 or ankyrin G-GFP in pull-down assays (Figs. 4B, 6A). However, the 31sC domain apparently enhances both axonal targeting (Fig. 1D) as well as Kv3.1b binding to ankyrin G (Fig. 6E). Therefore, the 31sC domain likely exerts its function by facilitating the access of ankyrin G to the ATM through altering the interaction between 31T1 and 31aC. Whether 31sC may alter the binding affinity or the conformation of 31T1/31aC remains to be determined. Our in vitro binding assays only show that both GST-31aC and GST-31bC precipitate His-31T1 (Fig. 4). Quantitative comparison of their binding affinities with 31T1 is difficult because of the presence of a significant amount of multiple small-size polypeptides in the GST-31bC preparation. Moreover, it cannot be ruled out that the bacterially expressed channel domains may behave differently from those expressed in vivo. Therefore, exactly how 31sC regulates 31T1/31aC and 31aC/ankyrin G binding will be determined in future studies.
Both the T1 domain and ATM region of Kv3.1 channels are highly conserved within the Kv3 subfamily from flies to humans. Kv3 channels share >70% identity in their T1 domains, whereas the Kv3 ATM regions are nearly identical except for one or two residues (Fig. 3A). Thus, the interaction between the 31T1 domain and the ATM region may be a conserved mechanism by which Kv3 channel targeting is regulated. In different Kv3 isoforms and their alternatively spliced variants, residues in their distal C termini (varying from ∼50–135 aa) may regulate the 31T1/31aC complex differently. Alternatively, they may contain an additional targeting motif, e.g., a PDZ domain-binding ligand identified in Kv3.3 for targeting to distal dendrites (Deng et al., 2005). This explains why Kv3 channels have complex targeting patterns, and why Kv3.1a is targeted to axons in many neurons in vivo (Ozaita et al., 2002), but to dendrites in cultured hippocampal neurons. For the latter, a missing regulatory component might be the reason. Dendritic targeting of Kv3.1a or other Kv3.1b truncations, although not the primary focus of this study, may result from the following two possibilities. First, 31N might contain a dendritic targeting signal that was not revealed by the dendritic reporter TfR–GFP. Second, dendritic targeting may result from default forward trafficking. Indeed both Kv3.1a and Kv3.1b channels traffic to the plasma membrane efficiently in both HEK293 cells and neurons.
The potential role of ankyrin G in differential targeting of Kv3.1 splice variants
The presence of multiple basic residues in the ankyrin G-binding motif of Kv3.1 is consistent with the idea that the role of ankyrin G in targeting Kv3.1 is different from those in clustering Nav and KCNQ channels in the AIS. The conserved ankyrin G-binding motifs identified from Nav1.2 and KCNQ channels contain multiple acidic residues (Bennett and Chen, 2001; Jenkins and Bennett, 2001; Garrido et al., 2003; Pan et al., 2006). Disrupting these motifs eliminates clustering of Nav1.2 and KCNQ channels in the AIS (Garrido et al., 2003; Pan et al., 2006). In contrast, ankyrin G does not cluster Kv3.1 channels at the AIS.
Ankyrin G may function as a barrier or check point to exclude Kv3.1a from entering axons, because disrupting ankyrin G function by either MB–GFP expression (Fig. 8D,F,G) or siRNA knockdown (Fig. 9B,C) enhanced the level of Kv3.1aHA in axons. In addition, ankyrin G may attract Kv3.1b into axons or prevent axonal Kv3.1b from retreating back to the soma, because axonal targeting of Kv3.1bHA modestly decreased in the presence of MB–GFP (Fig. 8E–G) or AnkG siR (Fig. 9B,C). These distinct effects of ankyrin G on the two Kv3.1 splice variants may result from their different binding affinities; a stronger interaction between ankyrin G and Kv3.1b has been detected (Fig. 6E). However, this raises an interesting question. How does Kv3.1b dissociate from ankyrin G after it passes the barrier? Because protein phosphorylation has been indicated in regulating the interaction between L1-CAM and ankyrin (Whittard et al., 2006), it is possible that phosphorylation of the Kv3.1b C terminus might be involved in regulating Kv3.1b/ankyrin G interaction during barrier crossing. Released Kv3.1b channel proteins may cross the AIS barrier through intracellular transport mediated by motor proteins and/or passive diffusion. Finally, it is important to note that our studies do not exclude the involvement of other proteins in the targeting process, because precise targeting of neuronal membrane proteins likely requires multiple steps (Boiko et al., 2007). Our results suggest that ankyrin G plays a key role in controlling axon–dendrite targeting of Kv3.1 channels, and lay the foundation for additional studies aimed at a complete understanding of Kv3 channel targeting and function in neurons.
Footnotes
This work was supported by a Career Transition Fellowship Award (C.G.) from the National Multiple Sclerosis Society (Grant TA3012A1) and an Ohio State University startup fund (C.G.). Additional support was provided by National Institutes of Health Grant P30-NS045758. We thank Dr. B. Rudy for Kv3.1a and Kv3.1b cDNA constructs; Drs. V. Bennett and L. Isom for ankyrin G-GFP and polyclonal anti-ankyrin G antibody; Dr. G. Banker for transferrin receptor-GFP; Dr. S. Qin for technical assistance; and Drs. L. Y. Jan, V. Bennett, A. P. Young, and J. Oberdick for valuable comments on this manuscript.
References
- Bennett V, Chen L. Ankyrins and cellular targeting of diverse membrane proteins to physiological sites. Curr Opin Cell Biol. 2001;13:61–67. doi: 10.1016/s0955-0674(00)00175-7. [DOI] [PubMed] [Google Scholar]
- Bixby KA, Nanao MH, Shen NV, Kreusch A, Bellamy H, Pfaffinger PJ, Choe S. Zn2+-binding and molecular determinants of tetramerization in voltage-gated K+ channels. Nat Struct Biol. 1999;6:38–43. doi: 10.1038/4911. [DOI] [PubMed] [Google Scholar]
- Boiko T, Vakulenko M, Ewers H, Yap CC, Norden C, Winckler B. Ankyrin-dependent and -independent mechanisms orchestrate axonal compartmentalization of L1 family members neurofascin and L1/neuron-glia cell adhesion molecule. J Neurosci. 2007;27:590–603. doi: 10.1523/JNEUROSCI.4302-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooke RE, Moores TS, Morris NP, Parson SH, Deuchars J. Kv3 voltage-gated potassium channels regulate neurotransmitter release from mouse motor nerve terminals. Eur J Neurosci. 2004;20:3313–3321. doi: 10.1111/j.1460-9568.2004.03730.x. [DOI] [PubMed] [Google Scholar]
- Brooks NL, Corey MJ, Schwalbe RA. Characterization of N-glycosylation consensus sequences in the Kv3.1 channel. FEBS J. 2006;273:3287–3300. doi: 10.1111/j.1742-4658.2006.05339.x. [DOI] [PubMed] [Google Scholar]
- Choe S. Potassium channel structures. Nat Rev Neurosci. 2002;3:115–121. doi: 10.1038/nrn727. [DOI] [PubMed] [Google Scholar]
- Chu PJ, Rivera JF, Arnold DB. A role for KIF17 in transport of Kv4.2. J Biol Chem. 2006;281:365–373. doi: 10.1074/jbc.M508897200. [DOI] [PubMed] [Google Scholar]
- Chung HJ, Jan YN, Jan LY. Polarized axonal surface expression of neuronal KCNQ channels is mediated by multiple signals in the KCNQ2 and KCNQ3 C-terminal domains. Proc Natl Acad Sci USA. 2006;103:8870–8875. doi: 10.1073/pnas.0603376103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Q, Rashid AJ, Fernandez FR, Turner RW, Maler L, Dunn RJ. A C-terminal domain directs Kv3.3 channels to dendrites. J Neurosci. 2005;25:11531–11541. doi: 10.1523/JNEUROSCI.3672-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaux J, Alcaraz G, Grinspan J, Bennett V, Joho R, Crest M, Scherer SS. Kv3.1b is a novel component of CNS nodes. J Neurosci. 2003;23:4509–4518. doi: 10.1523/JNEUROSCI.23-11-04509.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa F, McMahon A, Chan E, Wang S, Ho CS, Heintz N, Joho RH. Alcohol hypersensitivity, increased locomotion, and spontaneous myoclonus in mice lacking the potassium channels Kv3.1 and Kv3.3. J Neurosci. 2001;21:6657–6665. doi: 10.1523/JNEUROSCI.21-17-06657.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa F, Marks G, Heintz N, Joho RH. Increased motor drive and sleep loss in mice lacking Kv3-type potassium channels. Genes Brain Behav. 2004;3:90–100. doi: 10.1046/j.1601-183x.2003.00054.x. [DOI] [PubMed] [Google Scholar]
- Gan L, Kaczmarek LK. When, where, and how much? Expression of the Kv3.1 potassium channel in high-frequency firing neurons. J Neurobiol. 1998;37:69–79. doi: 10.1002/(sici)1097-4695(199810)37:1<69::aid-neu6>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Garrido JJ, Fernandes F, Giraud P, Mouret I, Pasqualini E, Fache MP, Jullien F, Dargent B. Identification of an axonal determinant in the C-terminus of the sodium channel Na(v)1.2. EMBO J. 2001;20:5950–5961. doi: 10.1093/emboj/20.21.5950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrido JJ, Giraud P, Carlier E, Fernandes F, Moussif A, Fache MP, Debanne D, Dargent B. A targeting motif involved in sodium channel clustering at the axonal initial segment. Science. 2003;300:2091–2094. doi: 10.1126/science.1085167. [DOI] [PubMed] [Google Scholar]
- Goldberg EM, Watanabe S, Chang SY, Joho RH, Huang ZJ, Leonard CS, Rudy B. Specific functions of synaptically localized potassium channels in synaptic transmission at the neocortical GABAergic fast-spiking cell synapse. J Neurosci. 2005;25:5230–5235. doi: 10.1523/JNEUROSCI.0722-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu C, Jan YN, Jan LY. A conserved domain in axonal targeting of Kv1 (Shaker) voltage-gated potassium channels. Science. 2003;301:646–649. doi: 10.1126/science.1086998. [DOI] [PubMed] [Google Scholar]
- Gu C, Zhou W, Puthenveedu MA, Xu M, Jan YN, Jan LY. The microtubule plus-end tracking protein EB1 is required for Kv1 voltage-gated K+ channel axonal targeting. Neuron. 2006;52:803–816. doi: 10.1016/j.neuron.2006.10.022. [DOI] [PubMed] [Google Scholar]
- Horton AC, Ehlers MD. Neuronal polarity and trafficking. Neuron. 2003;40:277–295. doi: 10.1016/s0896-6273(03)00629-9. [DOI] [PubMed] [Google Scholar]
- Ishikawa T, Nakamura Y, Saitoh N, Li WB, Iwasaki S, Takahashi T. Distinct roles of Kv1 and Kv3 potassium channels at the calyx of Held presynaptic terminal. J Neurosci. 2003;23:10445–10453. doi: 10.1523/JNEUROSCI.23-32-10445.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahng AW, Strang C, Kaiser D, Pollard T, Pfaffinger P, Choe S. Zinc mediates assembly of the T1 domain of the voltage-gated K channel 4.2. J Biol Chem. 2002;277:47885–47890. doi: 10.1074/jbc.M208416200. [DOI] [PubMed] [Google Scholar]
- Jenkins SM, Bennett V. Ankyrin-G coordinates assembly of the spectrin-based membrane skeleton, voltage-gated sodium channels, and L1 CAMs at Purkinje neuron initial segments. J Cell Biol. 2001;155:739–746. doi: 10.1083/jcb.200109026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joho RH, Street C, Matsushita S, Knopfel T. Behavioral motor dysfunction in Kv3-type potassium channel-deficient mice. Genes Brain Behav. 2006;5:472–482. doi: 10.1111/j.1601-183X.2005.00184.x. [DOI] [PubMed] [Google Scholar]
- Ju M, Stevens L, Leadbitter E, Wray D. The Roles of N- and C-terminal determinants in the activation of the Kv2.1 potassium channel. J Biol Chem. 2003;278:12769–12778. doi: 10.1074/jbc.M212973200. [DOI] [PubMed] [Google Scholar]
- Kaech S, Banker G. Culturing hippocampal neurons. Nat Protoc. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- Kobrinsky E, Stevens L, Kazmi Y, Wray D, Soldatov NM. Molecular rearrangements of the Kv2.1 potassium channel termini associated with voltage gating. J Biol Chem. 2006;281:19233–19240. doi: 10.1074/jbc.M601231200. [DOI] [PubMed] [Google Scholar]
- Kosolapov A, Tu L, Wang J, Deutsch C. Structure acquisition of the T1 domain of Kv1.3 during biogenesis. Neuron. 2004;44:295–307. doi: 10.1016/j.neuron.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Lai HC, Jan LY. The distribution and targeting of neuronal voltage-gated ion channels. Nat Rev Neurosci. 2006;7:548–562. doi: 10.1038/nrn1938. [DOI] [PubMed] [Google Scholar]
- Lau D, Vega-Saenz de Miera EC, Contreras D, Ozaita A, Harvey M, Chow A, Noebels JL, Paylor R, Morgan JI, Leonard CS, Rudy B. Impaired fast-spiking, suppressed cortical inhibition, and increased susceptibility to seizures in mice lacking Kv3.2 K+ channel proteins. J Neurosci. 2000;20:9071–9085. doi: 10.1523/JNEUROSCI.20-24-09071.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Jan YN, Jan LY. Specification of subunit assembly by the hydrophilic amino-terminal domain of the Shaker potassium channel. Science. 1992;257:1225–1230. doi: 10.1126/science.1519059. [DOI] [PubMed] [Google Scholar]
- Lien CC, Jonas P. Kv3 potassium conductance is necessary and kinetically optimized for high-frequency action potential generation in hippocampal interneurons. J Neurosci. 2003;23:2058–2068. doi: 10.1523/JNEUROSCI.23-06-02058.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim ST, Antonucci DE, Scannevin RH, Trimmer JS. A novel targeting signal for proximal clustering of the Kv2.1 K+ channel in hippocampal neurons. Neuron. 2000;25:385–397. doi: 10.1016/s0896-6273(00)80902-2. [DOI] [PubMed] [Google Scholar]
- Long SB, Campbell EB, Mackinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005;309:897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- Martina M, Yao GL, Bean BP. Properties and functional role of voltage-dependent potassium channels in dendrites of rat cerebellar Purkinje neurons. J Neurosci. 2003;23:5698–5707. doi: 10.1523/JNEUROSCI.23-13-05698.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsukawa H, Wolf AM, Matsushita S, Joho RH, Knopfel T. Motor dysfunction and altered synaptic transmission at the parallel fiber-Purkinje cell synapse in mice lacking potassium channels Kv3.1 and Kv3.3. J Neurosci. 2003;23:7677–7684. doi: 10.1523/JNEUROSCI.23-20-07677.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misonou H, Trimmer JS. Determinants of voltage-gated potassium channel surface expression and localization in Mammalian neurons. Crit Rev Biochem Mol Biol. 2004;39:125–145. doi: 10.1080/10409230490475417. [DOI] [PubMed] [Google Scholar]
- Nanao MH, Zhou W, Pfaffinger PJ, Choe S. Determining the basis of channel-tetramerization specificity by x-ray crystallography and a sequence-comparison algorithm: Family Values (FamVal) Proc Natl Acad Sci USA. 2003;100:8670–8675. doi: 10.1073/pnas.1432840100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozaita A, Martone ME, Ellisman MH, Rudy B. Differential subcellular localization of the two alternatively spliced isoforms of the Kv3.1 potassium channel subunit in brain. J Neurophysiol. 2002;88:394–408. doi: 10.1152/jn.2002.88.1.394. [DOI] [PubMed] [Google Scholar]
- Pan Z, Kao T, Horvath Z, Lemos J, Sul JY, Cranstoun SD, Bennett V, Scherer SS, Cooper EC. A common ankyrin-G-based mechanism retains KCNQ and NaV channels at electrically active domains of the axon. J Neurosci. 2006;26:2599–2613. doi: 10.1523/JNEUROSCI.4314-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perney TM, Marshall J, Martin KA, Hockfield S, Kaczmarek LK. Expression of the mRNAs for the Kv3.1 potassium channel gene in the adult and developing rat brain. J Neurophysiol. 1992;68:756–766. doi: 10.1152/jn.1992.68.3.756. [DOI] [PubMed] [Google Scholar]
- Rettig J, Wunder F, Stocker M, Lichtinghagen R, Mastiaux F, Beckh S, Kues W, Pedarzani P, Schroter KH, Ruppersberg JP, et al. Characterization of a Shaw-related potassium channel family in rat brain. EMBO J. 1992;11:2473–2486. doi: 10.1002/j.1460-2075.1992.tb05312.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera JF, Ahmad S, Quick MW, Liman ER, Arnold DB. An evolutionarily conserved dileucine motif in Shal K+ channels mediates dendritic targeting. Nat Neurosci. 2003;6:243–250. doi: 10.1038/nn1020. [DOI] [PubMed] [Google Scholar]
- Rivera JF, Chu PJ, Arnold DB. The T1 domain of Kv1.3 mediates intracellular targeting to axons. Eur J Neurosci. 2005;22:1853–1862. doi: 10.1111/j.1460-9568.2005.04384.x. [DOI] [PubMed] [Google Scholar]
- Rudy B, McBain CJ. Kv3 channels: voltage-gated K+ channels designed for high-frequency repetitive firing. Trends Neurosci. 2001;24:517–526. doi: 10.1016/s0166-2236(00)01892-0. [DOI] [PubMed] [Google Scholar]
- Sampo B, Kaech S, Kunz S, Banker G. Two distinct mechanisms target membrane proteins to the axonal surface. Neuron. 2003;37:611–624. doi: 10.1016/s0896-6273(03)00058-8. [DOI] [PubMed] [Google Scholar]
- Scholle A, Zimmer T, Koopmann R, Engeland B, Pongs O, Benndorf K. Effects of Kv1.2 intracellular regions on activation of Kv2.1 channels. Biophys J. 2004;87:873–882. doi: 10.1529/biophysj.104.040550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulteis CT, Nagaya N, Papazian DM. Intersubunit interaction between amino- and carboxyl-terminal cysteine residues in tetrameric shaker K+ channels. Biochemistry. 1996;35:12133–12140. doi: 10.1021/bi961083s. [DOI] [PubMed] [Google Scholar]
- Sekirnjak C, Martone ME, Weiser M, Deerinck T, Bueno E, Rudy B, Ellisman M. Subcellular localization of the K+ channel subunit Kv3.1b in selected rat CNS neurons. Brain Res. 1997;766:173–187. doi: 10.1016/s0006-8993(97)00527-1. [DOI] [PubMed] [Google Scholar]
- Stowell JN, Craig AM. Axon/dendrite targeting of metabotropic glutamate receptors by their cytoplasmic carboxy-terminal domains. Neuron. 1999;22:525–536. doi: 10.1016/s0896-6273(00)80707-2. [DOI] [PubMed] [Google Scholar]
- Waters MF, Minassian NA, Stevanin G, Figueroa KP, Bannister JP, Nolte D, Mock AF, Evidente VG, Fee DB, Muller U, et al. Mutations in voltage-gated potassium channel KCNC3 cause degenerative and developmental central nervous system phenotypes. Nat Genet. 2006;38:447–451. doi: 10.1038/ng1758. [DOI] [PubMed] [Google Scholar]
- Weiser M, Vega-Saenz de Miera E, Kentros C, Moreno H, Franzen L, Hillman D, Baker H, Rudy B. Differential expression of Shaw-related K+ channels in the rat central nervous system. J Neurosci. 1994;14:949–972. doi: 10.1523/JNEUROSCI.14-03-00949.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiser M, Bueno E, Sekirnjak C, Martone ME, Baker H, Hillman D, Chen S, Thornhill W, Ellisman M, Rudy B. The potassium channel subunit KV3.1b is localized to somatic and axonal membranes of specific populations of CNS neurons. J Neurosci. 1995;15:4298–4314. doi: 10.1523/JNEUROSCI.15-06-04298.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittard JD, Sakurai T, Cassella MR, Gazdoiu M, Felsenfeld DP. MAP kinase pathway-dependent phosphorylation of the L1-CAM ankyrin binding site regulates neuronal growth. Mol Biol Cell. 2006;17:2696–2706. doi: 10.1091/mbc.E06-01-0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Bennett V. Restriction of 480/270-kD ankyrin G to axon proximal segments requires multiple ankyrin G-specific domains. J Cell Biol. 1998;142:1571–1581. doi: 10.1083/jcb.142.6.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]