

Abstract
Parkinson's disease is a neurodegenerative disorder characterized by the preferential loss of midbrain dopaminergic neurons in the substantia nigra (SN). One of the earliest detectable biochemical alterations that occurs in the Parkinsonian brain is a marked reduction in SN levels of total glutathione (glutathione plus glutathione disulfide), occurring before losses in mitochondrial complex I (CI) activity, striatal dopamine levels, or midbrain dopaminergic neurodegeneration associated with the disease. Previous in vitro data from our laboratory has suggested that prolonged depletion of dopaminergic glutathione results in selective impairment of mitochondrial complex I activity through a reversible thiol oxidation event. To address the effects of depletion in dopaminergic glutathione levels in vivo on the nigrostriatal system, we created genetically engineered transgenic mouse lines in which expression of γ-glutamyl cysteine ligase, the rate-limiting enzyme in de novo glutathione synthesis, can be inducibly downregulated in catecholaminergic neurons, including those of the SN. A novel method for isolation of purified dopaminergic striatal synaptosomes was used to study the impact of dopaminergic glutathione depletion on mitochondrial events demonstrated previously to occur in vitro as a consequence of this alteration. Dopaminergic glutathione depletion was found to result in a selective reversible thiol-oxidation-dependent mitochondrial complex I inhibition, followed by an age-related nigrostriatal neurodegeneration. This suggests that depletion in glutathione within dopaminergic SN neurons has a direct impact on mitochondrial complex I activity via increased nitric oxide-related thiol oxidation and age-related dopaminergic SN cell loss.
Keywords: glutathione, Parkinson's disease, mitochondrial complex I, dopaminergic neuron, S-nitrosation, substantia nigra
Introduction
Parkinson's disease (PD) is a neurodegenerative disorder characterized by the preferential loss of dopaminergic (DAergic) neurons in the substantia nigra (SN) as well as other brain regions. The pathogenesis of PD is not fully understood, but several lines of evidence suggest it may involve both increased oxidative stress and mitochondrial dysfunction. One of the earliest detectable biochemical alterations reported in parkinsonian SN is a marked reduction in total glutathione [glutathione (GSH) plus glutathione disulfide (GSSG)] levels, occurring before selective decreases in mitochondrial complex I activity, striatal dopamine loss, and dopaminergic neurodegeneration (Sian et al., 1994). Although glutathione is not the only antioxidant reported to be altered in PD, the magnitude of depletion parallels severity of the disease. Glutathione levels are not reduced in other areas in the parkinsonian brain or in other diseases affecting the SN, such as multiple system atrophy or progressive supranuclear palsy (Perry and Yong, 1986).
Glutathione plays an important role in brain by removing oxidants formed during metabolic processes, such as oxygen utilization by the mitochondria. It is one of the most abundant soluble antioxidant molecules present in brain, comprising cysteine, glycine, and glutamate. GSH is formed by a two-step reaction involving enzymes γ-glutamyl cysteine ligase (GCL) and glutathione synthetase. Brain GSH appears to arise primarily through de novo synthesis from its constituent amino acids via these enzymes (Meister, 1988). GCL is the rate-limiting enzyme in this process and composed of a heavy catalytic subunit (GCLC, 73 kDa) and a light modulatory subunit (GCLM, 27.7 kDa). The modulatory subunit increases the affinity for the substrate glutamate and decreases sensitivity to feedback inhibition by glutathione (Huang et al., 1993). Previous in vitro studies from our laboratory demonstrated that acute reduction in both cellular and mitochondrial glutathione levels in dopaminergic PC12 cells resulted in mitochondrial dysfunction (Jha et al., 2000). Recent studies from our laboratory and others demonstrated that both acute and chronic in vitro dopaminergic glutathione depletion results in reversible, selective inhibition of complex I activity via nitric oxide (NO)-mediated thiol oxidation (Hsu et al., 2005; Chinta et al., 2006; Burwell et al., 2006). Recently, GCLC knock-out mice were generated and found to be embryonic lethal when homozygous, demonstrating that this gene is essential for embryonic development (Dalton et al., 2004). This model, although helpful in determining the essential nature of GCL, is neither inducible nor cell specific. Recent in vitro studies with cortical neurons showed that short hairpin RNA-mediated knockdown of GCLC subunit lead to the time-dependent apoptotic death, and this effect was synergistic with nitric oxide-mediated toxicity (Diaz-Hernandez et al., 2005). However, there is little information on the direct effects of dopaminergic glutathione depletion in vivo on the nigrostriatal system. To address this, we have created a transgenic mouse line in which expression of antisense message against GCLC is inducible in dopaminergic SN neurons in adult animals using inducible reverse tetracycline expression system. Results from our studies suggest that dopaminergic GSH depletion results in increases in dopaminergic S-NO levels and mitochondrial S-nitrosation correlating with selective, thiol reversible inhibition of mitochondrial complex I and age-related dopaminergic SN neurodegeneration.
Materials and Methods
Generation of inducible antisenseGCL transgenic mouse lines.
Generation of transgenic mice was performed using standard protocols (Hogan et al., 1986). Constructs for generation of the animals included the following: (1) a regulatory transgene consisting of a rat tyrosine hydroxylase (TH) promoter constitutively expressing a reverse tetracycline repressor-transcription activator gene (pTH-rTetR-VP16) that produces reverse tetracycline transcription activator (rTta) protein and (2) a response transgene containing tetracycline operator sequence from which antisense GCLC message can be inducibly expressed via doxycycline (dox) (Tet-O-antisenseGCL). The antisense GCL response transgene also contains luciferase cDNA, which was inserted on the other side of the bidirectional cytomegalovirus (CMV) promoter to act as a cellular marker for inducible gene expression. These constructs were linearized and coinjected into the pronuclei of eggs from C57BL/6 mice. Eggs were implanted into pseudopregnant females to produce double transgenic offspring. Transgenic founders containing both transgenes were identified by PCR analysis of genomic DNA isolated from mouse tail using specific primer pairs (pTH-rtTA, 5′ TGT CCC ACA GAA TAC CAG CCA G-3′ and 5′CCT TCG ATT CCG ACC TCA TTA AG-3′; antisenseGCL, 5′ CCA CAA ACA CAA CTC CTC C-3′ and 5′ GAC CAA CGC CTT GAT TGA C-3′). Genomic DNA from tails of pTH-rTta-antisense GCL founder animals was digested with XbaI enzyme, separated on a 0.8% agarose gel, and transferred to positively charged nylon membrane (Roche, Indianapolis, IN) for Southern blot analyses. The membrane was hybridized with a 32P-labeled 1.6 kb XbaI–HindIII luciferase cDNA probe, and the presence of the 4.2 kb XbaI luciferase-containing transgene band was assessed.
Dox administration.
Three- and 12-month-old antisenseGCL transgenics and their wild-type C57BL/6 littermates were used for all subsequent studies. Double transgenics and nontransgenic littermates from each line were fed either dox-containing (2 mg/g; Purina Rodent Chow; Research Diets, New Brunswick, NJ) or normal feed for a 3 week period. For dox-withdrawal studies, the dox-fed mice were switched back to a normal no-dox diet for 3 weeks before the animals were killed. All protocols were in accordance with the National Institutes of Health guidelines for use of live animals and were previously approved by the Institutional Animal Care and Use Committee of the Buck Institute.
Reverse transcription-PCR analysis for verification of constitutive expression of rTta and dox-dependent antisenseGCL transgene induction.
Total RNA was isolated from dissected SN tissues from either dox-fed or no-dox-fed transgenic animals or wild-type littermates using TRIzol reagent, followed by reverse transcription (RT) using Super-Script reverse transcriptase (Invitrogen, Carlsbad, CA). From this reaction, 2 μl was used as a template for RT-PCR amplification with TaqDNA polymerase. PCR analyses were performed as described above.
Immunohistochemistry.
dox-fed versus no-dox-fed antisenseGCL and wild-type mouse brains were fixed and processed for immunohistochemistry as described previously (Viswanath et al., 2001). Primary antibodies used were as follows: rabbit polyclonal tyrosine hydroxylase antibody (1:500; Chemicon, Temecula, CA) and mouse monoclonal antibody to firefly luciferase (1:500; Novus Biologicals, Littleton, CO). Immunofluorescent reactivity was detected using standard protocols. For detection of S-nitrosated proteins by immunohistochemistry, antisenseGCL transgenic mice with or without dox were anesthetized and perfused transcardially with freshly prepared N-ethylmaleimide/diethylenetriaminepentaacetic acid (at 10 and 2 mm, respectively) in PBS to alkylate in situ the free thiols and complex any free transition metals. The mouse brains were then fixed and processed for immunocytochemistry (ICC) as described above. ICC was performed on nigrostriatal sections from dox- versus no-dox-fed transgenics and wild-type littermates using monoclonal nitrosocysteine antibody (1:1000; A.G. Scientific, San Diego, CA).
Luciferase assay.
Striatal tissue was dissected from the brains of antisenseGCL transgenic animals (n = 4) fed either dox-containing or regular chow. Tissues were homogenized in 1× lysis buffer according to the specifications of the manufacturer using a sonicator. Cellular homogenates were then centrifuged for 15 min at 10,000 × g at 4°C. Twenty microliters of the supernatant was used for measurement of luciferase activity in a luminometer using a luciferase reporter gene assay kit (Promega, Madison, WI). All measurements were performed in triplicate. Luciferase activity was normalized to total protein concentration.
Isolation of primary mesencephalic cultures and 1-methyl-4-phenylpyridinium treatment.
Ventral mesencephalon was dissected from embryonic gestation day 14 of wild-type and antisenseGCL transgenic mice. Neurons were dissociated mechanically and then digested enzymatically for 30 min at 37°C in papain solution (Worthington Biochemical, Lakewood, NJ). The suspension was centrifuged, and cells were placed at a density of 3 × 106 live cells/ml (Smith and Friedmann, 2004). All cultures were incubated at 37°C at 5% CO2. After 4 d in vitro, one-half of the media was removed and replaced with either an equal volume of media containing dox at 20 μg/ml or regular media for 24 h. Afterward, a portion of the cell samples were exposed to 5 μm 1-methyl-4-phenylpyridinium (MPP+) (Sigma-Aldrich, St. Louis, MO) for 6 h.
ICC and SN TH-positive cell counts in mesencephalic cultures.
Primary mesencephalic cultures were fixed with 4% paraformaldehyde (PFA) and incubated with blocking buffer. ICC was performed using standard protocols. Total numbers of TH-positive (TH+) neurons were counted in at least three separate wells after 24 h of dox treatment with or without MPP+, and the percentage of TH+ neurons in treated versus untreated cells was calculated. Experiments were repeated with cultures isolated from three independent dissections.
Magnetic bead purification of dopaminergic versus GABAergic striatal synaptosomes.
DAergic and GABAergic striatal (ST) synaptosomes were isolated using a modified immunomagnetic protocol (Docherty et al., 1991). Briefly, synaptosomes were prepared from dissected striatal tissue (n = 5 animals in each group) (Budd and Nicholls, 1995). From a single striatum, we were able to isolate 20–25 μg of DA synaptosomal protein. Dopaminergic synaptosomes were then purified using a novel magnetic bead approach using antibody against the cell surface dopamine transporter (DAT) protein. Isolated striatal synaptosomes were incubated with primary DAT antibody (Chemicon), followed by incubation with rat anti-mouse IgG microbeads (Miltenyi Biotec, Auburn, CA). Labeled beads were then separated using a high-gradient magnetic-positive separation column placed in a strong magnetic field according to the instructions of the manufacturer. The magnetically labeled DAT+ cells are retained in the column, whereas nonlabeled non-DAT+ synaptosomes (GABA+) pass through. When the column is removed from the magnetic field, the magnetically retained DAT+ synaptosomes are eluted.
Western blot analysis of DAergic versus GABAergic synaptosomal fractions.
Striatal synaptosomal magnetic bead column fractions were collected, and protein content was measured using the Bradford method (Bradford, 1976). Equal amounts of protein from each sample were fractionated on a 10% SDS-PAGE gel and electrophoretically transferred to polyvinylidene difluoride membranes. Membranes were incubated with primary antibodies [TH antibody, 1:500; GABA, 1:500; and synaptosome-associated protein of 25 kDa (SNAP-25), 1:1000] overnight at 4°C. Immunoblotting was performed using horseradish peroxidase-conjugated secondary antibody and an ECL kit (GE Healthcare, Little Chalfont, UK). Levels of TH and GABA were normalized versus SNAP-25 (a synaptosomal protein) to assess the degree of DA versus GABA-specific ST synaptosomal enrichment in each fraction.
Total glutathione levels in DAergic versus GABAergic ST synaptosomes with or without dox induction.
Total glutathione levels (GSH plus GSSG) was measured in ST DAergic versus GABAergic synaptosomes from dox-fed and no-dox-fed transgenic mice and after dox withdrawal using HPLC, according to the method of Fariss and Reed (1987). For total glutathione measurements, the DA synaptosomes were isolated in medium containing 0.5% 5-sulfosalicylic acid to prevent the oxidation of glutathione. Briefly, DA synaptosomes isolated by magnetic bead method were washed in 1× PBS, acidified in 10% perchloric acid/2 mm EDTA, supplemented with an internal standard, γ-glutamylglutamic acid (Bachem, Torrance, CA), and derivatized with 2,4-fluorodinitrobenzene. The samples were precipitated and sonicated twice in this solution to ensure complete GSH recovery. The samples were further processed and analyzed for their glutathione content using PerkinElmer (Wellesley, MA) HPLC system, equipped with LC-890 UV spectrophotometric detector and an autoinjector (Choi et al., 2000). Glutathione concentrations were normalized by protein assay using BCA protein assay kit, per the recommendations of the manufacturer (Pierce, Rockford, IL).
Measurement of mitochondrial complex I, II–III, and IV activities in DAergic versus GABAergic ST synaptosomes with or without dox induction.
Isolated ST synaptosomes were found to be electrophysiologically viable for up to 6 h after magnetic bead separation. Isolated synaptosomes were resuspended in isolation buffer (215 mm mannitol, 75 mm sucrose, 0.1% BSA, and 20 mm HEPES, pH 7.2) before measurement of complex I activity. Complex I activities were assayed in isolated DAergic versus GABAergic ST synaptosomal fractions from dox-induced versus no-dox transgenics and after dox withdrawal as rotenone-sensitive Nicotinamide adenine dinucleotide (NADH) dehydrogenase activity by measuring 2,6-dichlorophenolindophenol (DCPIP) reduction in synaptosomal extracts after addition of 200 μm NADH, 200 μm decylubiquinone, 2 mm KCN, and 50 μm DCPIP in the presence and absence of 2 μm rotenone at 600 nm at 30°C (Trounce et al., 1996). This was done in the presence and absence of 4 mm dithiothreitol (DTT) to test whether inhibition was attributable to a reversible thiol oxidation event as observed in our in vitro studies (Jha et al., 2000; Hsu et al., 2005; Chinta et al., 2006). Complex II–III activity was assayed as described previously (Hsu et al., 2005). Synaptosomal preparations from dox- and no-dox-fed groups were added to buffer containing 50 mm Tris–SO4, pH 7.4, 100 mm EDTA, 2 mm succinate, and 1% maltoside. The assay was initiated with 100 mm cytochrome c, and absorbance was measured at 550 nm at 30°C in the presence and absence of 4 mm antimycin A. Complex IV activity was assayed as cytochrome c oxidase activity by observing the rapid (1–2 s) oxidation of freshly reduced 40 μm ferrocytochrome c in a 10 mm K-PO4 buffer, pH 7.2, containing 100 mm KCl and 0.025% maltoside at 30°C at 550 nm averaged over two concentrations per sample (SpectraMax 340PC384 microplate spectrophotometer; Molecular Devices, Sunnyvale, CA). Values for all assays were normalized per protein using Bio-Rad (Hercules, CA) reagent.
Measurement of NO levels after glutathione depletion.
NO levels were measured using the fluorescent probe 4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate (DAF-FM diacetate) (Invitrogen). After glutathione depletion, dox-treated and control synaptosomes were loaded with 10 μm DAF-FM and incubated at 37°C for 30 min. Synaptosomes were then washed, aliquoted into 96-well plates, and allowed to rest for 30 min to maximize deacetylation of the dye by intracellular esterases. Fluorescence was determined on a Molecular Devices Gemini EM microplate reader at excitation/emission of 496/524 nm. Samples were normalized per total cellular protein.
Immunoprecipitation of mitochondrial complex I.
Mitochondria were prepared by the method of Trounce et al. (1996). Briefly, freshly dissected mouse SN tissues were washed and homogenized in ice-cold isolation buffer (in mm: 320 sucrose, 5 N-[tris(hydroxymethyl)methyl]-2-aminoethanesulfonic acid (Tes), and 1 EGTA, pH 7.2). The homogenate was centrifuged at 1000 × g for 5 min at 4°C, followed by centrifugation of the supernatant at 8500 × g for 10 min at 4°C. The mitochondrially enriched pellet was resuspended in isolation buffer and layered on top of 6% (w/v) Ficoll solution (F-4375; Sigma-Aldrich) and centrifuged at 75,000 × g for 30 min at 4°C to remove myelin, which forms a layer at the top. The pellet was resuspended in reconstitution buffer (250 mm sucrose and 10 mm Tes, pH 7.2). Approximately 2 mg of mitochondria were washed with 20 mm Tris-HCl, pH 7.5, and 1 mm EDTA, and suspended in the same buffer containing protease inhibitor mixture. This suspension was solubilized by adding n-dodecyl-β-d-maltoside to a final concentration of 1% at 5 mg/ml protein concentration and incubated for 30 min on ice. Insoluble material from this suspension was removed by centrifugation at 55,000 × g for 30 min at 4°C, and soluble material was taken for immunoprecipitation of mitochondrial complex I using MS101 Complex I Immunocapture kit from MitoSciences (Eugene, OR) (Schilling et al., 2005). Immunoprecipitated complex I was incubated with radiolabeled 14C-iodoacetamide (14C-IAA) (25 μCi from GE Healthcare) for 30 min at room temperature, shaking at 1300 rpm before passing through Micro Bio-Spin 6 chromatography columns to remove excess unbound 14C-IAA. The samples were run on 10% SDS-PAGE gel, followed by air drying under vacuum for 3 h at 85°C before exposing to radioactive film for 7 d at −80°C. Syproruby staining was used to normalize protein levels in SN glutathione-depleted versus control samples.
Measurement of mitochondrial S-nitrosothiol levels isolated from ST DAergic synaptosomes.
The method used was according to Cook et al. (1996). In brief, both DAergic and GABAergic ST synaptosomes from dox- versus no-dox-fed transgenics and after dox withdrawal were incubated with maltoside for 10 min on ice and centrifuged at 10,000 × g for 10 min. The supernatant containing mitochondria from the ST DAergic synaptosomes were then mixed with neutral Griess reagent [57 mm sulfanilamide and 1.2 mm N-(1-naphthyl) ethylenediamine in PBS; Sigma-Aldrich] followed by addition of HgCl2 to a final concentration of 100 μm. The mixture was gently shaken and allowed to stand for 20 min at room temperature in the dark before its absorbance at 496 nm was measured.
Evaluation of S-nitrosation of mitochondrial SN DA proteins via immunogold electron microscopy.
The SN region from coronal brain sections was immediately fixed in 2% PFA and 0.2% glutaraldehyde in 0.1 m sodium phosphate buffer, pH 7.4, for 2 h at room temperature. Fifty-nanometer-thick sections were cut at room temperature by using an Ultracut S ultramicrotome (Leica, Nussloch, Germany). The sections were collected on carbon-coated Formvar grids using a mixture of 1.8% methylcellulose and 2.3 m sucrose and incubated with primary nitrosocysteine antibody (1:250; A.G. Scientific) and 12 nm of protein A gold. After immunogold labeling, the sections were fixed with 1% glutaraldehyde, counterstained with uranyl acetate, and embedded in methylcellulose–uranyl acetate (Greco et al., 2006). The specificity of the labeling was verified in control experiments in which sections were treated with 3.5 mm p-hydroxymercuricbenzoate for 30 min (three 10-min treatments). The sections were viewed in a Jeol (Peabody, MA) 1200CX electron microscope.
Measurement of striatal dopamine levels.
Three- and 12-month-old antiGCL transgenic mice and their wild-type littermates were fed dox or no-dox containing chow for 3 weeks as described above. Mice were killed, and both striata were rapidly dissected on a chilled glass plate and were frozen at −70°C. The samples were subsequently thawed in 0.4 ml of chilled 0.1 m perchloric acid and sonicated. Aliquots were taken for protein quantification whereas other aliquots were centrifuged, and dopamine levels were measured in supernatants by HPLC with electrochemical detection as described previously (Kaur et al., 2003). The concentration of dopamine was expressed as nanograms per milligram protein.
Stereological counts of SN TH+ neurons in vivo.
For stereology studies, cryostat-cut sections (40 μm) were taken through the entire midbrain of dox and no-dox animals. TH-positive neurons were immunolabeled by incubating the tissue sections successively with a rabbit polyclonal anti-TH antibody (1:500 dilution; Chemicon), followed by biotinylated goat anti-rabbit IgG (1:1000; Vector Laboratories, Burlingame, CA) following the staining procedure outlined by the manufacturer (Vector Laboratories) in combination with 3,3′-diaminobenzidine reagent. Slides were coded, and SN cell counts were performed using the unbiased dissector method (McCormack et al., 2002). TH-positive cells were counted from a total of 8–10 sections in each field per brain (i.e., every fourth section) at a magnification of 100× in a 0.2 × 0.2 mm area using the optical fractionator approach on a Neurolucida stereo investigator (MicroBrightField, Colchester, VT). Nissl staining was performed on alternate sections to assess the specificity of dopaminergic cell loss in the SN.
Results
Generation of dox-inducible dopaminergic antisenseGCL transgenics
To study the effects of glutathione depletion within midbrain dopaminergic neurons in adult animals, transgenic mice were created using the reverse dox-inducible Tet-on system (Clontech, Mountain View, CA). Constructs for this purpose consisted of a regulatory transgene in which expression of the dox-dependent rTta is under the control of the tyrosine hydroxylase promoter (pTH-rTta) and a response transgene containing antisenseGCL mouse heavy-chain cDNA under the control of a Tet operator site linked to a minimal CMV promoter (Tet-O-antisenseGCL). Luciferase cDNA was inserted into the response transgene construct on the other side of the bidirectional CMV promotor to act as a cellular marker for inducible gene expression (Fig. 1A). PCR analysis of tail genomic DNA isolated from founder animals (rTta-antisenseGCL) confirmed the presence of both the regulatory and response transgenes in several of the founder animals (rTta and luciferase) (Fig. 1B). Integration of the antisenseGCL transgene in these founder lines was further verified via Southern blot analysis (Fig. 1C).
Figure 1.

A, Schematic of pTH-rTta/antisenseGCL-luciferase constructs used for creation of dox-inducible antisenseGCL transgenics. When dox is administered in the culture media of cells or the feed of animals containing these constructs, this allows the rTta protein to bind Tet-O sequence and induce expression of transgenes from the bidirectional minimal pCMV. pTH, Tyrosine hydroxylase promoter; rtetR, reverse tetracycline repressor; VP16, herpes simplex virus transcription activator; dox, doxycycline; rTta, reverse tetracycline transcription activator protein; luc, luciferase; pCMV, minimal cytomegaloviral promotor; Tet-O, tetracycline operator sequence; AntiGCL, antisenseGCL catalytic subunit cDNA. B, Representative PCR analysis of genomic DNA isolated from double pTH-rTta/antiGCL transgenic mice to verify the presence of rTta (551 bp, lanes 1–4), antisenseGCL/luc (337 bp, lanes 7–10) transgenes versus nontransgenic littermates (lanes 5, 6). C, Southern blot analysis for the presence of the 4.2 kb XbaI antisenseGCL/luc genomic DNA band (M, MW markers; lanes 1–3, antisenseGCL genomic DNA; W, nontransgenic littermate DNA).
Verification of inducible antisenseGCL transgene expression
To verify the expression of transgenes in this newly created line, antisenseGCL transgenics and their wild-type littermates were fed either dox-containing or regular feed for a period of 3 weeks, and the nigrostriata were dissected out. Total RNA was isolated from these tissues, and RT-PCR analyses was performed to evaluate the expression of the transgenic mRNAs. rTta mRNA was found to be constitutively expressed in both no-dox and dox-fed transgenics but was absent in wild-type animals (Fig. 2A). Luciferase marker mRNA expression, conversely, was induced only in dox-fed antisenseGCL transgenics.
Figure 2.
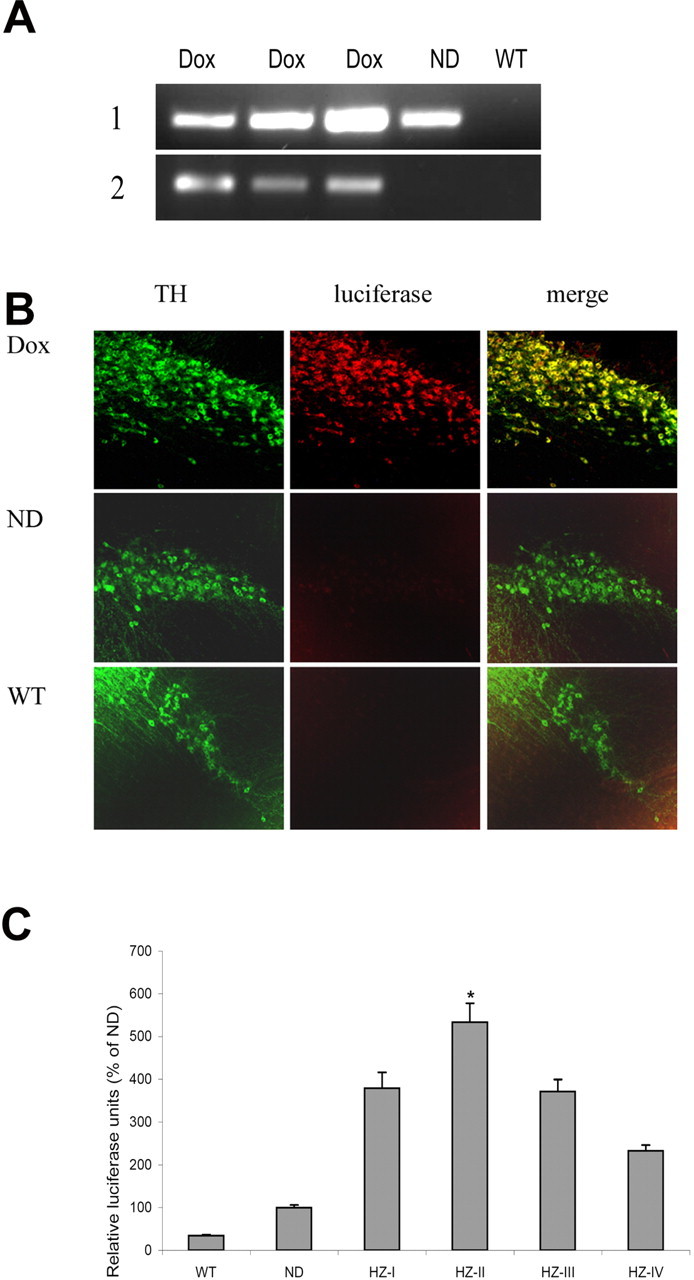
A, Representative RT-PCR analyses of total RNA isolated from the SN of antiGCL transgenics versus wild-type littermates to assess dox-inducible expression of transgenic mRNAs. 1, Constitutive expression of 551 bp rTta mRNA in dox- and no-dox-fed transgenics but not in wild-type littermates (Dox, antisenseGCL + dox; ND, antisenseGCL − dox; WT, nontransgenic littermate). 2, Inducible expression of 337 bp luciferase mRNA in dox-fed but not no-dox-fed antisenseGCL transgenics (Dox, antisenseGCL + dox; ND, antisenseGCL − dox; WT, nontransgenic littermate + dox). B, ICC analysis of luciferase marker expression in TH+ SN neurons (green, TH; red, luciferase; yellow, merged; Dox, antisenseGCL + dox; ND, antisenseGCL − dox; WT, nontransgenic littermate). Original magnification, 20×. C, Measurement of transgenic SN luciferase activity in various dox-fed antisenseGCL transgenic founder lines. WT, Nontransgenic littermate; ND, no-dox-fed antisenseGCL transgenic; HZ1-HZIV, dox-fed antisenseGCL transgenic founder lines. Note that the HZ-II line, which gave the highest transgene induction versus ND, was used for all subsequent studies. *p < 0.05 versus WT control.
Transgene expression is detected in TH-positive neurons in the SN
To further verify the tissue-specific inducible expression of the antisenseGCL transgene within SN TH-positive neurons, dual immunofluorescence for luciferase and TH was performed on SN sections from dox- versus no-dox-fed antisenseGCL transgenics and their wild-type littermates. Double labeling of TH-expressing cells with a luciferase antibody demonstrated that the luciferase marker protein was only expressed in the dox-fed antisenseGCL transgenic line and expression was within dopaminergic SN TH-positive neurons. Induction of luciferase protein was not observed either in no-dox-fed antisenseGCL transgenics or in wild-type SN TH+ neurons, indicating appropriate regulation of transgene expression (Fig. 2B).
Analyses of transgene-derived luciferase expression in antisenseGCL transgenic lines
To validate the levels of transgene expression in various founder lines, dox-inducible alterations in the luciferase activity reporter molecule was measured. Luciferase activity levels were found to be induced in all of the dox-fed antisenseGCL transgenic lines to varying degrees compared with no-dox fed transgenic and wild-type littermate controls (Fig. 2B). Maximal induction (fivefold) was observed in the HZII line; this line was chosen for all subsequent experiments.
Effect of glutathione depletion on viability of dopaminergic neurons within primary mesencephalic cultures
We first examined the effect of dopaminergic glutathione depletion on dopaminergic cell viability in primary mesencephalic cultures isolated from antiGCL transgenics. Primary mesencephalic cultures were isolated from antisenseGCL transgenics, and, after 4 d in vitro, the cultures were exposed to media containing either 20 or 0 μg/ml dox for 24 h. Dox treatment significantly reduced (26 ± 4% vs ND) the viability of TH+ neurons in the antisenseGCL cultures, whereas no change was observed in either wild-type mesencephalic cultures treated with dox compared with no-dox controls nor were numbers of other cell types in the culture altered (data not shown). In conjunction with dopaminergic cell loss, shortening of neuritic processes were also observed in the remaining surviving TH+ neurons in the dox-treated cultures (Fig. 3A,B). Toxicity of glutathione depletion was further enhanced after exposure of cultures to the Parkinsonian-inducing agent MPP+ (Fig. 3A,B).
Figure 3.
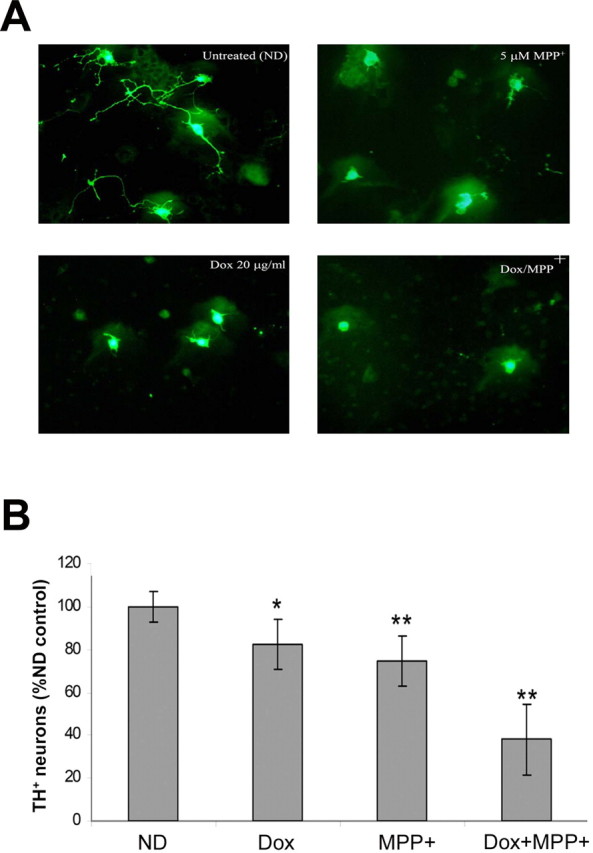
Decreased survival of DA neurons in antisenseGCL primary mesencephalic cultures treated with dox with or without MPP+. After 4 d in vitro, cultures were induced with 20 μg/ml dox for 24 h and then coexposed to 5 μm MPP+ for an additional 6 h. Values represent the mean ± SEM from n = 4 individual experiments. A, Representative photomicrographs illustrating morphology of DA neurons after glutathione depletion with or without MPP+. B, Quantitation of DA cell counts after TH ICC. *p < 0.05 versus ND control; **p < 0.01 versus ND control. Values are represented as percentage of no-dox (ND) control.
Measurement of glutathione levels in ST DAergic versus GABAergic synaptosomes isolated from transgenics using a novel magnetic bead enrichment technique
Dopaminergic neurons constitute only 1–5% of the entire SN neuronal population. Therefore, detecting cytoplasmic alterations in GSH as well as other biochemical parameters specifically within these cells in our transgenic lines would likely be extremely difficult using whole SN homogenates. Because axons originating from dopaminergic SN neurons innervate the striatum, it is possible to use striatal synaptosomes for such measurements. However, the axons from SN dopaminergic neurons still correspond to only ∼20% of total ST nerve terminals. To achieve meaningful data as to the impact of dox feeding on dopaminergic glutathione levels and other parameters in our inducible antiGCL transgenics, we developed a novel technique for isolating dopaminergic striatal synaptosomes based on modification of a published magnetic bead column purification method (Docherty et al., 1991) (Fig. 4A). This method involved using antibody against the dopaminergic cell surface membrane protein DAT to selectively bind DA synaptosomes to magnetic beads bound to secondary antibody. ST DAergic-enriched synaptosomes can be subsequently eluted from the column by removal of the magnetic field (Fig. 4A, fraction E). Synaptosomal fractions in the column flow-though wash (W) are GABAergic enriched and therefore useful as negative controls for subsequent biochemical assays. Synaptosomal proteins were normalized using synaptosomal-specific antibody against SNAP-25 and DAergic versus GABAergic enrichment in elute versus flow-through verified by Western blot analysis (Fig. 4A). Measurement of total glutathione levels from dox-fed antisenseGCL ST DA synaptosomes isolated by this method displayed a significant (33.05 ± 4.1%, 9.45 nmol/mg) decrease compared with no dox-fed controls (14.11 nmol/mg). This decrease is further enhanced in 12-month-old dox-fed mice (39.6 ± 6.4%; 8.52 nmol/mg). In contrast, no difference in glutathione levels was observed in GABAergic ST synaptosomal preparations from dox- versus no-dox-fed transgenic animals (21.16 nmol/mg) (Fig. 4B). Dopaminergic glutathione levels returned to normal after removal of dox from the diet for 3 weeks in young animals.
Figure 4.

A, Quantitation and representative Western blot analyses of TH versus GABA levels in various fractions from magnetic bead enrichment of DA versus GABAergic ST synaptosomes (normalized per SNAP-25). C, Whole synaptosomal fraction; U, unbound excess; FT, column flow-through; W, wash (GABA+); E, eluate (DA+). Values are reported as percentage densitometric intensity versus C. B, Total glutathione levels in isolated GABA versus DA synaptosomes from dox-fed (Dox) versus no-dox-fed (ND) young and old antiGCL transgenics. GABA, GABA synaptosomal fraction (from W above) and antiGCL dopaminergic synaptosomal fractions (from E above) from antisenseGCL transgenic mice. *p < 0.05 versus ND DA levels; **p < 0.01 versus ND DA levels; ***p > 0.05 compared with ND DA levels. Values are represented as percentage of no-dox (ND) control. DW represents dox withdrawal for 3 weeks from diet.
Decreased dopaminergic glutathione levels in vivo results in selective inhibition of mitochondrial complex I activity in isolated striatal dopaminergic synaptosomes that are DTT reversible
Previous in vitro studies have demonstrated that dopaminergic glutathione depletion selectively inhibits mitochondrial complex I activity. To test whether this occurs in vivo after dopaminergic GSH depletion in the SN of adult animals, we compared the activities of complex I versus complex II–III and IV in isolated DAergic synaptosomes from dox-treated versus untreated antisenseGCL mice. Lowering of GSH levels in these DA synaptosomes resulted in a significant decrease in rotenone-inhibited CI activity (26% vs no dox; 24.42 ± 3.8 nm NADH reduced per minute per milligram of protein vs 33.1 ± 4.1 nm NADH reduced per minute milligram of protein), whereas no significant changes were detected in complex II–III (no-dox values are 14.8 ± 1.73 vs 14.1 ± 2.3 nmol · min−1 · mg−1 protein of dox-treated group) and complex IV activities (no-dox values are 190 nm ferrocytochrome c reduced per minute per milligram of protein vs 181 nm dox treated). The addition of the thiol-reducing agent DTT was found to restore complex I activity to levels higher than those found in no-dox controls, indicating that the inhibition of enzyme activity is a reversible thiol-oxidation-dependent event. The increased activity is likely attributable to the fact that, during synaptosomal isolation, a small percentage of thiol groups in complex I isolated from the control no-dox group are susceptible to mild oxidative stress, undergoes reversible oxidation, and are reduced back to normal after DTT treatment. CI activity was also found to restore to the levels in no-dox controls after dox withdrawal from the diet for 3 weeks (Fig. 5). Previous in vitro studies have demonstrated that addition of doxycycline to human osteosarcoma cells interferes with mitochondrial protein translation including CI, thereby affecting enzyme activity (Yadava et al., 2004). However, in our studies, in addition to lack of effect on CI activity in dox-treated wild-type mice, we also observed no change in CI subunit protein levels as a consequence of dox treatment (3 week period) as assessed via immunoprecipitation/gel electrophoretic analyses (data not shown).
Figure 5.

Effect of GSH depletion on mitochondrial complex I activity. Measurement of complex I activity in striatal dopaminergic synaptosomes isolated from 3-month-old mice. Rotenone-sensitive NADH dehydrogenase activity measured in dopaminergic synaptosomal preparations from uninduced (ND), induced (Dox), and dox withdrawal transgenic antisenseGCL animals. n = 5 animals per group and a total of 3 separate experiments; values are expressed as mean ± SD. *p < 0.01 compared with ND; **p > 0.05 compared with ND. 100% complex I activity is at 33 nm NADH reduced per minute per milligram of protein for the ND values for DA synaptosomes. DW, Dox withdrawal.
Inducible dopaminergic SN glutathione depletion in adult mice in vivo results in direct thiol oxidation of mitochondrial complex I subunits
Previous in vitro studies including those from our own laboratory (Clementi et al., 1998; Chinta and Andersen, 2006) have demonstrated that NO can result in inhibition of mitochondrial complex I activity. So, we first assessed the levels of NO after glutathione depletion in synaptosomes isolated from our dopaminergic glutathione-depleted transgenic mice. dox treatment significantly elevated NO levels in DA synaptosomes (22% of no-dox control), whereas there is no change in GABA synaptosomes (Fig. 6A). We next assessed whether increased NO levels have any effect on the thiol redox status of mitochondrial complex I. To determine whether increased dopaminergic NO levels in vivo results in direct thiol oxidation of mitochondrial complex I subunits as demonstrated after its depletion in vitro, we immunoprecipitated complex I from the SN of glutathione depleted versus controls and subjected the isolated protein to incubation with 14C-IAA. Incubation of isolated mitochondrial complex I protein from the SN of glutathione-depleted mice resulted in a decrease in labeling in complex I subunits in the dox-fed samples, suggesting increased thiol oxidation of these subunits (Fig. 6B).
Figure 6.
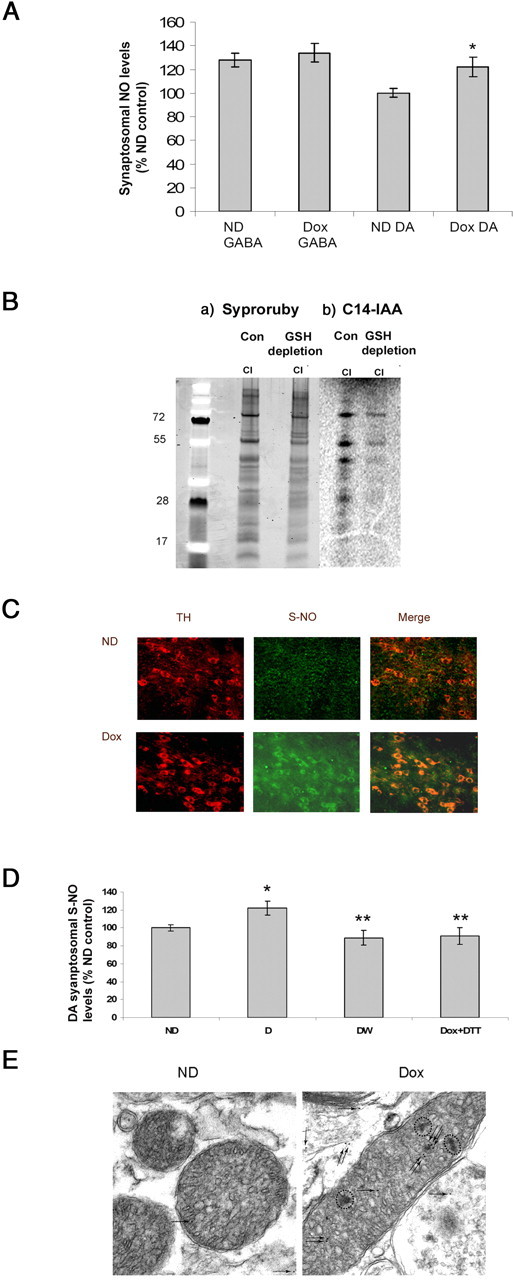
Increased sulfhydryl group modification of dopaminergic mitochondrial proteins in the SN after glutathione depletion. A, Nitric oxide levels assayed via DAF-FM fluorescence in synaptosomes from dox-treated antiGCL transgenics. *p < 0.01 compared with control. B, Decreased sulfhydryl group staining of mitochondrial complex I subunits after glutathione depletion. a, SDS-PAGE analysis of immunoprecipitated complex I subunits stained with syproruby. b, 14C-IAA-labeled complex I subunits from control and glutathione-depleted mice separated on SDS-PAGE, followed by exposure to radioactive film. C, Increased S-NO levels after glutathione depletion in TH neurons of antisenseGCL mice. Double immunostaining of tyrosine hydroxylase (red) and S-NO (green) in SN regions from dox- and no-dox-fed mice antiGSH mice. D, Measurement of mitochondrial S-NO levels in DAergic synaptosomes after 3 weeks of GSH depletion by the Greiss-based colorimetric assay. *p < 0.05 compared with control; **p < 0.05 compared with ND control. E, High-resolution immunoelectron microscopy of SN region from dox- and no-dox-fed antisenseGCL. Immunoreactivity for S-nitrosocysteine-containing proteins was visualized by 10 nm protein A gold particles (arrows). Increased immunoreactivity for S-nitrosated proteins with abnormal spots (dotted circles) noticed in glutathione-depleted mitochondria. ND, No-dox-fed mice.
Mitochondrial dopaminergic SN S-nitrosation after GSH depletion
In previous in vitro studies from our laboratory, we demonstrated that dopaminergic glutathione depletion resulted in reversible inhibition of mitochondrial complex I activity via a reversible thiol oxidation NO-mediated event (Hsu et al., 2005; Chinta et al., 2006). Brookes and colleagues have demonstrated recently that S-nitrosation of complex I results in its enzymatic inhibition both in situ and recently in vivo in an acute cardiac ischemia reperfusion model (Burwell et al., 2006; Tompkins et al., 2006; Nadtochiy et al., 2007). We first assessed S-NO protein levels within SN dopaminergic neurons by immunohistochemistry using monoclonal antibody directed against S-nitrosocysteine. Immunohistochemistry demonstrated a selective increase in S-NO-protein levels within DAergic SN neurons after glutathione depletion (Fig. 6C). To assess whether dopaminergic glutathione depletion results in increased S-nitrosation in DAergic SN mitochondria, we measured the levels of S-nitrosothiols after glutathione depletion in mitochondria isolated from ST DAergic synaptosomes. As demonstrated in the Figure 6D, there is significant increase in S-NO-protein content in mitochondria isolated from glutathione depleted ST DAergic synaptosomes compared with no dox controls. Furthermore, S-NO protein content was restored by DTT treatment and dox withdrawal, confirming reversible sulfhydryl modification of CI subunits. Finally, immunogold EM labeling for detection of subcellular localization of S-NO-protein revealed distinct S-NO protein labeling within dopaminergic glutathione-depleted mitochondria. This was accompanied by an abnormal mitochondrial morphology within glutathione-depleted DAergic SN neurons (Fig. 6E).
Striatal dopamine levels
We next assessed the impact of dopaminergic glutathione reduction in adult animals on striatal dopamine content in young and old mice transgenic lines. At 3 months of age, dopaminergic SN depletion via dox feeding has no significant impact on ST dopamine levels. However by 12 months of age, dopaminergic SN glutathione depletion was found to result in a significant decrease in ST dopamine levels compared with no-dox-fed antiGCL transgenics (Fig. 7A) (601 ± 16 vs 786 ± 12 pmol DA/mg protein). dox feeding alone was found to have no effect on ST dopamine levels in wild-type littermate controls (data not shown).
Figure 7.

Effect of age-related glutathione alterations on ST DA content and on SN TH+ cell numbers in dox- versus no-dox-fed young and old mice. A, Striatal dopamine levels were measured in transgenic antisenseGCL versus wild-type mice via reverse-phase HPLC (n = 6 mice per group) in young (3 months) and old (12 months) mice. Values represented as picomoles of DA per milligram of protein. *p < 0.05, dox antisenseGCL versus wild-type or no-dox-fed mice. B, Cell counts of SN TH+ cell numbers after GSH depletion demonstrate a significant loss of DA SN neurons in old mice (12 months). Cells were counted from a total of 15–20 sections in each field per brain at a magnification of 100× using the optical fractionator approach. n = 5 animals per group. *p < 0.01, dox antisenseGCL versus WT or ND antisenseGCL.
Stereological SN TH+ cell counts
We next assessed the effect of reductions in dopaminergic SN glutathione content on DAergic SN cell viability. Stereological SN TH+ cell counts were performed after a 3 week period of dox feeding in young and old mice. TH immunohistochemistry revealed that dox feeding had no significant impact on DA neurons in young mice. However, glutathione depletion resulted in a significant loss (20.2 ± 6.2%) in TH+ neurons in the SN of the old mice, confirming increased susceptibility to midbrain dopaminergic glutathione depletion with age (Fig. 7B). dox feeding has no effect on TH+ neurons in wild-type mice (data not shown).
Discussion
Glutathione plays an important protective role in the brain removing toxic oxidant species (Bharath et al., 2002). Total SN glutathione (GSH plus GSSG) levels have been reported to be selectively depleted in the early stages of PD (Bannon et al., 1984; Sian et al., 1994). In PD patients, glutathione loss has been found to occur within the soma and dendrites of dopaminergic SN neurons (Pearce et al., 1997). Glutathione levels are not reduced in other areas of the parkinsonian brain or in other diseases affecting the basal ganglia, such as multiple system atrophy or progressive supranuclear palsy (Perry and Yong, 1986; Fitzmaurice et al., 2003). Although glutathione is not the only antioxidant compound whose levels are altered in PD, it is the earliest reported biochemical change that occurs, and its depletion appears to correlate with disease progression (Jenner, 1993). Because glutathione is one of the prevalent thiol antioxidant compounds in the CNS (Cooper et al., 1980), its depletion in the SN has been hypothesized to be an important biochemical event associated with the development of subsequent PD pathology. Whether its loss may be directly involved in subsequent dopaminergic SN neurodegeneration, however, is unclear. Previous studies using chemical agents such as the GCL inhibitor buthionine sulfoxamine to acutely deplete glutathione in vivo have produced global glutathione depletion, including in the brain (Jain et al., 1991; Heales et al., 1995; Martinez et al., 1995; Meister, 1995; Andersen et al., 1996). Because this agent does not easily cross the blood–brain barrier, most of these studies have been performed in neonatal rather than adult animals, making it difficult to separate the effects of glutathione depletion on normal developmental processes in the brain from effects in the adult animal. Creation of a transgenic model in which dopaminergic glutathione levels can be reduced in the adult animal has allowed us to demonstrate that in vivo reductions in the range associated with early PD can result in selective inhibition of mitochondrial complex I activity within DAergic SN neurons in association with their subsequent age-related neurodegeneration. Neurodegeneration occurs even in the presence of astrocytes that normally supply cysteine and glycine to neurons in the form of cysteinylglycine (Dringen et al., 1999; Dringen, 2000; Dringen and Hirrlinger, 2003); inhibition of de novo synthesis in our model prevents the use of these GSH precursors for GSH production.
We reported previously that both acute and chronic depletion of glutathione within dopaminergic cell lines in vitro results in reversible inhibition of mitochondrial complex I activity. In more recent studies from our laboratory and others, both acute and chronic in vitro glutathione depletion was found to result in reversible inhibition of mitochondrial complex I activity via S-nitrosation of mitochondrial complex I subunits (Hsu et al., 2005; Burwell et al., 2006; Chinta et al., 2006). S-nitrosation of complex I subunits in these studies was found to involve elevations in NO and peroxynitrite (ONOO−) production as a consequence of glutathione depletion and to be reversible by either increasing cellular GSH levels or DTT administration. Others have demonstrated that long-term exposure of complex I itself or mitochondria or mitochondrial membranes to NO or NO donors results in selective inhibition of complex I activity via S-nitrosation of critical thiols in the enzyme complex, which was reversible on addition of GSH or other thiol-reducing agents (Bolanos et al., 1996; Clementi et al., 1998; Burwell et al., 2006; Dahm et al., 2006). The inability to demonstrate direct S-nitrosation of dopaminergic complex I subunits after glutathione depletion in vivo in our model is likely attributable in part to technical limitations of available methodologies and also as a consequence of the subtle effects of pathophysiological elevations in reactive nitrogen species elicited by lowered glutathione. However, detection of S-nitrosation has been made possible only with use of a large amount of starting material and making acute bolus additions of exogenous NO-producing compounds (Burwell et al., 2006; Dahm et al., 2006). Attempts to use available S-nitrosylation antibodies for either immunoprecipitation or Western blot analysis have also thus far proven unsuccessful (Gow et al., 2002). However, based on direct evidence for S-nitrosation of complex I after depletion of glutathione in our in vitro dopaminergic cell model that results in reduced cell viability (Hsu et al., 2005, Chinta et al., 2006) coupled with reversibility of complex I inhibition in vivo by DTT and the presence of increased S-nitrosylated CI subunit levels in the DAergic SN neurons after glutathione depletion, we propose that the observed DAergic SN neurodegeneration in our antiGCL transgenic mouse model is most likely attributable to selective decreases in complex I activity elicited via the S-nitrosation of complex I subunits. This is particularly intriguing in the context of mass spectrometric data generated recently in our laboratory, demonstrating an increase in the oxidation of sulfhydryl groups within immunoprecipitated mitochondrial complex I subunits isolated from human postmortem parkinsonian SN tissues versus age-matched controls (Schilling et al., 2005; Andersen, 2006).
Previous in vitro studies have demonstrated the interactive nature of various antioxidants during oxidative stress conditions associated with Parkinson's disease. Decrease in total glutathione in mesencephalic cells via treatment with chemical agents such as buthionine sulfoximine potentiated both malonate-induced neurotoxicity and generation of ROS. Ascorbate completely ameliorated the increase in free radicals and toxicity in GSH-depleted cultures, indicating that, during oxidative stress, ascorbate can act cooperatively with GSH to remove reactive oxygen species and control the intracellular GSSG/GSH redox status. These studies exemplify the importance of glutathione and other antioxidants in the protection of dopaminergic neurons during oxidative stress conditions (Ehrhart and Zeevalk, 2001, 2003) such as those that arise in Parkinson's disease.
Recent studies from our laboratory and others have demonstrated that the GSH precursors glutamyl cysteine ethyl ester and glutathione ethyl ester both significantly elevate intracellular glutathione levels in neuronal cells and provide significant protection of dopamine cells against oxidative stress or mitochondrial impairment both in vitro and in vivo (Chinta et al., 2006; Zeevalk et al., 2007). A pilot study in which a small group of untreated PD patients were given daily intravenous infusions of glutathione over the period of 1 month reportedly resulted in a significant improvement in disability (Sechi et al., 1996). Whether such treatment was affective in alternating brain levels of glutathione or having lasting effects is unclear, but our data suggests that maintaining glutathione levels is critical for protecting dopaminergic SN neurons from neurodegeneration and that its replacement may be a possible therapeutic avenue for PD and other related disorders.
Footnotes
This work was supported by National Institutes of Health Grant AG12141 (J.K.A.). S.J.C. is a recipient of a postdoctoral fellowship from American Parkinson's Disease Association. We thank Daniel Crippen for technical assistance with EM studies.
References
- Andersen JK. Translational and post-translational modifications to mitochondrial complex I in the post-mortem Parkinsonian midbrain. post-genomic symposium; May; Beijing, China. 2006. Paper presented at. [Google Scholar]
- Andersen JK, Mo JQ, Hom DG, Lee FY, Harnish P, Hamill RW, McNeill TH. Effect of buthionine sulfoximine, a synthesis inhibitor of the antioxidant glutathione, on the murine nigrostriatal neurons. J Neurochem. 1996;67:2164–2171. doi: 10.1046/j.1471-4159.1996.67052164.x. [DOI] [PubMed] [Google Scholar]
- Bannon MJ, Goedert M, Williams B. The possible relation of glutathione, melanin and 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine (MPTP) to Parkinson's disease. Biochem Pharmacol. 1984;33:2697–2698. doi: 10.1016/0006-2952(84)90682-8. [DOI] [PubMed] [Google Scholar]
- Bharath S, Hsu M, Kaur D, Rajagopalan S, Andersen JK. Glutathione, iron and Parkinson's disease. Biochem Pharmacol. 2002;64:1037–1048. doi: 10.1016/s0006-2952(02)01174-7. [DOI] [PubMed] [Google Scholar]
- Bolanos JP, Heales SJ, Peuchen S, Barker JE, Land JM, Clark JB. Nitric oxide-mediated mitochondrial damage: a potential neuroprotective role for glutathione. Free Radic Biol Med. 1996;21:995–1001. doi: 10.1016/s0891-5849(96)00240-7. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Budd DC, Nicholls DG. Protein kinase C-mediated suppression of the presynaptic adenosine A1 receptor by a facilitatory metabotropic glutamate receptor. J Neurochem. 1995;65:615–621. doi: 10.1046/j.1471-4159.1995.65020615.x. [DOI] [PubMed] [Google Scholar]
- Burwell LS, Nadtochiy SM, Tompkins AJ, Young S, Brookes PS. Direct evidence for S-nitrosation of mitochondrial complex I. Biochem J. 2006;394:627–634. doi: 10.1042/BJ20051435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinta SJ, Andersen JK. Reversible inhibition of mitochondrial complex I activity following chronic dopaminergic glutathione depletion in vitro: implications for Parkinson's disease. Free Radic Biol Med. 2006;41:1442–1448. doi: 10.1016/j.freeradbiomed.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Chinta SJ, Rajagopalan S, Butterfield DA, Andersen JK. In vitro and in vivo neuroprotection by gamma-glutamylcysteine ethyl ester against MPTP: relevance to the role of glutathione in Parkinson's disease. Neurosci Lett. 2006;402:137–141. doi: 10.1016/j.neulet.2006.03.056. [DOI] [PubMed] [Google Scholar]
- Choi J, Liu RM, Kundu RK, Sangiorgi F, Wu W, Maxson R, Forman HJ. Molecular mechanism of decreased glutathione content in human immunodeficiency virus type 1 Tat-transgenic mice. J Biol Chem. 2000;275:3693–3698. doi: 10.1074/jbc.275.5.3693. [DOI] [PubMed] [Google Scholar]
- Clementi E, Brown GC, Feelisch M, Moncada S. Persistent inhibition of cell respiration by nitric oxide: crucial role of S-nitrosylation of mitochondrial complex I and protective action of glutathione. Proc Natl Acad Sci USA. 1998;95:7631–7636. doi: 10.1073/pnas.95.13.7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook JA, Kim SY, Teague D, Krishna MC, Pacelli R, Mitchell JB, Vodovotz Y, Nims RW, Christodoulou D, Miles AM, Grisham MB, Wink DA. Convenient colorimetric and fluorometric assays for S-nitrosothiols. Anal Biochem. 1996;238:150–158. doi: 10.1006/abio.1996.0268. [DOI] [PubMed] [Google Scholar]
- Cooper AJ, Pulsinelli WA, Duffy TE. Glutathione and ascorbate during ischemia and postischemic reperfusion in rat brain. J Neurochem. 1980;35:1242–1245. doi: 10.1111/j.1471-4159.1980.tb07882.x. [DOI] [PubMed] [Google Scholar]
- Dahm CC, Moore K, Murphy MP. Persistent S-nitrosation of complex I and other mitochondrial membrane proteins by S-nitrosothiols but not nitric oxide or peroxynitrite: implications for the interaction of nitric oxide with mitochondria. J Biol Chem. 2006;281:10056–10065. doi: 10.1074/jbc.M512203200. [DOI] [PubMed] [Google Scholar]
- Dalton TP, Chen Y, Schneider SN, Nebert DW, Shertzer HG. Genetically altered mice to evaluate glutathione homeostasis in health and disease. Free Radic Biol Med. 2004;37:1511–1526. doi: 10.1016/j.freeradbiomed.2004.06.040. [DOI] [PubMed] [Google Scholar]
- Diaz-Hernandez JI, Almeida A, Delgado-Esteban M, Fernandez E, Bolanos JP. Knockdown of glutamate-cysteine ligase by small hairpin RNA reveals that both catalytic and modulatory subunits are essential for the survival of primary neurons. J Biol Chem. 2005;280:38992–39001. doi: 10.1074/jbc.M507065200. [DOI] [PubMed] [Google Scholar]
- Docherty M, Bradford HF, Cash CD, Ehret M, Maitre M, Joh TH. Isolation of monoaminergic synaptosomes from rat brain by immunomagnetophoresis. J Neurochem. 1991;56:1569–1580. doi: 10.1111/j.1471-4159.1991.tb02053.x. [DOI] [PubMed] [Google Scholar]
- Dringen R. Metabolism and functions of glutathione in brain. Prog Neurobiol. 2000;62:649–671. doi: 10.1016/s0301-0082(99)00060-x. [DOI] [PubMed] [Google Scholar]
- Dringen R, Hirrlinger J. Glutathione pathways in the brain. Biol Chem. 2003;384:505–516. doi: 10.1515/BC.2003.059. [DOI] [PubMed] [Google Scholar]
- Dringen R, Pfeiffer B, Hamprecht B. Synthesis of the antioxidant glutathione in neurons: supply by astrocytes of CysGly as precursor for neuronal glutathione. J Neurosci. 1999;19:562–569. doi: 10.1523/JNEUROSCI.19-02-00562.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrhart J, Zeevalk GD. Hydrogen peroxide removal and glutathione mixed disulfide formation during metabolic inhibition in mesencephalic cultures. J Neurochem. 2001;77:1496–1507. doi: 10.1046/j.1471-4159.2001.00355.x. [DOI] [PubMed] [Google Scholar]
- Ehrhart J, Zeevalk GD. Cooperative interaction between ascorbate and glutathione during mitochondrial impairment in mesencephalic cultures. J Neurochem. 2003;86:1487–1497. doi: 10.1046/j.1471-4159.2003.01954.x. [DOI] [PubMed] [Google Scholar]
- Fariss MW, Reed DJ. High-performance liquid chromatography of thiols and disulfides: dinitrophenol derivatives. Methods Enzymol. 1987;143:101–109. doi: 10.1016/0076-6879(87)43018-8. [DOI] [PubMed] [Google Scholar]
- Fitzmaurice PS, Ang L, Guttman M, Rajput AH, Furukawa Y, Kish SJ. Nigral glutathione deficiency is not specific for idiopathic Parkinson's disease. Mov Disord. 2003;18:969–976. doi: 10.1002/mds.10486. [DOI] [PubMed] [Google Scholar]
- Gow AJ, Chen Q, Hess DT, Day BJ, Ischiropoulos H, Stamler JS. Basal and stimulated protein S-nitrosylation in multiple cell types and tissues. J Biol Chem. 2002;277:9637–9640. doi: 10.1074/jbc.C100746200. [DOI] [PubMed] [Google Scholar]
- Greco TM, Hodara R, Parastatidis I, Heijnen HF, Dennehy MK, Liebler DC, Ischiropoulos H. Identification of S-nitrosylation motifs by site-specific mapping of the S-nitrosocysteine proteome in human vascular smooth muscle cells. Proc Natl Acad Sci USA. 2006;103:7420–7425. doi: 10.1073/pnas.0600729103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heales SJ, Davies SE, Bates TE, Clark JB. Depletion of brain glutathione is accompanied by impaired mitochondrial function and decreased N-acetyl aspartate concentration. Neurochem Res. 1995;20:31–38. doi: 10.1007/BF00995149. [DOI] [PubMed] [Google Scholar]
- Hogan B, Costantini F, Lacy E. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1986. Manipulating the mouse embryo. [Google Scholar]
- Hsu M, Srinivas B, Kumar J, Subramanian R, Andersen J. Glutathione depletion resulting in selective mitochondrial complex I inhibition in dopaminergic cells is via an NO-mediated pathway not involving peroxynitrite: implications for Parkinson's disease. J Neurochem. 2005;92:1091–1103. doi: 10.1111/j.1471-4159.2004.02929.x. [DOI] [PubMed] [Google Scholar]
- Huang CS, Chang LS, Anderson ME, Meister A. Catalytic and regulatory properties of the heavy subunit of rat kidney gamma-glutamylcysteine synthetase. J Biol Chem. 1993;268:19675–19680. [PubMed] [Google Scholar]
- Jain A, Martensson J, Stole E, Auld PA, Meister A. Glutathione deficiency leads to mitochondrial damage in brain. Proc Natl Acad Sci USA. 1991;88:1913–1917. doi: 10.1073/pnas.88.5.1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenner P. Altered mitochondrial function, iron metabolism and glutathione levels in Parkinson's disease. Acta Neurol Scand Suppl. 1993;146:6–13. [PubMed] [Google Scholar]
- Jha N, Jurma O, Lalli G, Liu Y, Pettus EH, Greenamyre JT, Liu RM, Forman HJ, Andersen JK. Glutathione depletion in PC12 results in selective inhibition of mitochondrial complex I activity. Implications for Parkinson's disease. J Biol Chem. 2000;275:26096–26101. doi: 10.1074/jbc.M000120200. [DOI] [PubMed] [Google Scholar]
- Kaur D, Yantiri F, Rajagopalan S, Kumar J, Mo JQ, Boonplueang R, Viswanath V, Jacobs R, Yang L, Beal MF, DiMonte D, Volitaskis I, Ellerby L, Cherny RA, Bush AI, Andersen JK. Genetic or pharmacological iron chelation prevents MPTP-induced neurotoxicity in vivo: a novel therapy for Parkinson's disease. Neuron. 2003;37:899–909. doi: 10.1016/s0896-6273(03)00126-0. [DOI] [PubMed] [Google Scholar]
- Martinez M, Ferrandiz ML, Diez A, Miquel J. Depletion of cytosolic GSH decreases the ATP levels and viability of synaptosomes from aged mice but not from young mice. Mech Ageing Dev. 1995;84:77–81. doi: 10.1016/0047-6374(95)01644-f. [DOI] [PubMed] [Google Scholar]
- McCormack AL, Thiruchelvam M, Manning-Bog AB, Thiffault C, Langston JW, Cory-Slechta DA, Di Monte DA. Environmental risk factors and Parkinson's disease: selective degeneration of nigral dopaminergic neurons caused by the herbicide paraquat. Neurobiol Dis. 2002;10:119–127. doi: 10.1006/nbdi.2002.0507. [DOI] [PubMed] [Google Scholar]
- Meister A. Glutathione metabolism and its selective modification. J Biol Chem. 1988;263:17205–17208. [PubMed] [Google Scholar]
- Meister A. Glutathione biosynthesis and its inhibition. Methods Enzymol. 1995;252:26–30. doi: 10.1016/0076-6879(95)52005-8. [DOI] [PubMed] [Google Scholar]
- Nadtochiy SM, Burwell LS, Brookes PS. Cardioprotection and mitochondrial S-nitrosation: effects of S-nitroso-2-mercaptopropionyl glycine (SNO-MPG) in cardiac ischemia-reperfusion injury. J Mol Cell Cardiol. 2007;42:812–825. doi: 10.1016/j.yjmcc.2007.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce RK, Owen A, Daniel S, Jenner P, Marsden CD. Alterations in the distribution of glutathione in the substantia nigra in Parkinson's disease. J Neural Transm. 1997;104:661–677. doi: 10.1007/BF01291884. [DOI] [PubMed] [Google Scholar]
- Perry TL, Yong VW. Idiopathic Parkinson's disease, progressive supranuclear palsy and glutathione metabolism in the substantia nigra of patients. Neurosci Lett. 1986;67:269–274. doi: 10.1016/0304-3940(86)90320-4. [DOI] [PubMed] [Google Scholar]
- Schilling B, Bharath MMS, Row RH, Murray J, Cusack MP, Capaldi RA, Freed CR, Prasad KN, Andersen JK, Gibson BW. Rapid purification and mass spectrometric characterization of mitochondrial NADH dehydrogenase (Complex I) from rodent brain and a dopaminergic neuronal cell line. Mol Cell Proteomics. 2005;4:84–96. doi: 10.1074/mcp.M400143-MCP200. [DOI] [PubMed] [Google Scholar]
- Sechi G, Deledda MG, Bua G, Satta WM, Deiana GA, Pes GM, Rosati G. Reduced intravenous glutathione in the treatment of early Parkinson's disease. Prog Neuropsychopharmacol Biol Psychiatry. 1996;20:1159–1170. doi: 10.1016/s0278-5846(96)00103-0. [DOI] [PubMed] [Google Scholar]
- Sian J, Dexter DT, Lees AJ, Daniel S, Agid Y, Javoy-Agid F, Jenner P, Marsden CD. Alterations in glutathione levels in Parkinson's disease and other neurodegenerative disorders affecting basal ganglia. Ann Neurol. 1994;36:348–355. doi: 10.1002/ana.410360305. [DOI] [PubMed] [Google Scholar]
- Smith DW, Friedmann T. Discrepant effects of culture conditions on survival and function of dopaminergic neurons. NeuroReport. 2004;15:1025–1028. doi: 10.1097/00001756-200404290-00018. [DOI] [PubMed] [Google Scholar]
- Tompkins AJ, Burwell LS, Digerness SB, Zaragoza C, Holman WL, Brookes PS. Mitochondrial dysfunction in cardiac ischemia-reperfusion injury: ROS from complex I, without inhibition. Biochim Biophys Acta. 2006;1762:223–231. doi: 10.1016/j.bbadis.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Trounce IA, Kim YL, Jun AS, Wallace DC. Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. Methods Enzymol. 1996;264:484–509. doi: 10.1016/s0076-6879(96)64044-0. [DOI] [PubMed] [Google Scholar]
- Viswanath V, Wu Y, Boonplueang R, Chen S, Stevenson FF, Yantiri F, Yang L, Beal MF, Andersen JK. Caspase-9 activation results in downstream caspase-8 activation and bid cleavage in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinson's disease. J Neurosci. 2001;21:9519–9528. doi: 10.1523/JNEUROSCI.21-24-09519.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadava N, Houchens T, Potluri P, Scheffler IE. Development and characterization of a conditional mitochondrial complex I assembly system. J Biol Chem. 2004;279:12406–12413. doi: 10.1074/jbc.M313588200. [DOI] [PubMed] [Google Scholar]
- Zeevalk GD, Manzino L, Sonsalla PK, Bernard LP. Characterization of intracellular elevation of glutathione (GSH) with glutathione monoethyl ester and GSH in brain and neuronal cultures: relevance to Parkinson's disease. Exp Neurol. 2007;203:512–520. doi: 10.1016/j.expneurol.2006.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]