

Abstract
Niemann–Pick disease type C (NPC) is an inherited lipid storage disorder caused by mutations in NPC1 or NPC2 genes. Loss of function of either protein results in the endosomal accumulation of cholesterol and other lipids, progressive neurodegeneration, and robust glial cell activation. Here, we report that cultured human NPC fibroblasts secrete interferon-β, interleukin-6 (IL-6), and IL-8, and contain increased levels of signal transducers and activators of transcription (STATs). These cells also contained increased levels of Toll-like receptor 4 (TLR4) that accumulated in cholesterol-enriched endosomes/lysosomes, and small interfering RNA knockdown of this receptor reduced cytokine secretion. In the NPC1−/− mouse brain, glial cells expressed TLR4 and IL-6, whereas both glial and neuronal cells expressed STATs. Genetic deletion of TLR4 in NPC1−/− mice reduced IL-6 secretion by cultured fibroblasts but failed to alter STAT levels or glial cell activation in the brain. In contrast, genetic deletion of IL-6 normalized STAT levels and suppressed glial cell activation. These findings indicate that constitutive cytokine secretion leads to activation of STATs in NPC fibroblasts and that this secretion is partly caused by an endosomal accumulation of TLR4. These results also suggest that similar signaling events may underlie glial cell activation in the NPC1−/− mouse brain.
Keywords: Niemann–Pick, cholesterol, Toll-like receptor, IL-6, STAT, glia
Introduction
Niemann–Pick disease type C (NPC) is an inherited lipid storage disorder caused by mutations in NPC1 or NPC2 genes (Carstea et al., 1997; Naureckiene et al., 2000). NPC1 is a membrane protein that contains a sterol-sensing domain and resides primarily in late endosomes, whereas NPC2 is a soluble protein that contains an MD-2-related lipid recognition domain and resides primarily in lysosomes. The loss of function of either protein results in aberrant endosomal membrane flow that leads to an accumulation of cholesterol and other lipids in the endosomal/lysosomal compartment. The main clinical feature of NPC is progressive neurological deterioration that is eventually fatal. NPC is typically a childhood disease, but its manifestations can be variable, and depending on the onset, it is classified as infantile, juvenile, and adult forms (Patterson et al., 2001).
BALB/c NPC mice contain a retroposon insertion in NPC1 (Loftus et al., 1997). NPC1−/− mice show symptoms similar to those of human patients and survive for <3 months. Brain tissues from these mice show lipid accumulation, progressive neuronal cell loss, predominantly affecting Purkinje cells, and robust activation of astroglial and microglial cells (Higashi et al., 1993; German et al., 2002; Walkley and Suzuki, 2004). Recently, administration of the neurosteroid allopregnanolone has been shown to suppress Purkinje cell loss and glial cell activation, and increase the lifespan of these mice (Griffin et al., 2004; Ahmad et al., 2005).
The prevailing hypothesis for glial cell activation in the NPC1−/− brain is that its occurrence is secondary to neuronal damage, resulting from activation of their phagocytic function to eliminate degenerating neurons. However, histochemical studies have shown that, in the NPC1−/− mouse brain, glial cell activation preceded neuronal cell loss (Baudry et al., 2003) and that, in the monkey brain, NPC1 was predominantly localized in perisynaptic astrocytes (Patel et al., 1999). These findings raise the possibility that glial cell activation is a cell-autonomous event and is causally related to neurodegeneration, although the biochemical basis for the activation remains unknown. A recent study using chimeric mice that have both NPC1−/− and NPC1+/+ Purkinje cells revealed survival of NPC1+/+ Purkinje cells surrounded by infiltrating glial cells (Ko et al., 2005). These findings exclude the possibility that any cytokines or toxic substances secreted by glial cells are the primary cause of neuronal death, but do not exclude the possibility that such factors may trigger and/or accelerate degeneration of NPC1−/− cells.
The involvement of cytokines in the brain pathology of NPC1−/− mice has been suggested by several studies showing expression of tumor necrosis factor α (TNFα) mRNA by neurons and astrocytes (Wu et al., 2005), expression of interleukin-1β (IL-1β) by astrocytes (Baudry et al., 2003), and deregulation of nuclear factor-κB (NFκB) activity in neurons and astrocytes (Bi et al., 2005).
To gain additional insight into NPC pathogenesis, we screened for cytokines that were secreted by cultured human NPC fibroblasts and found increased secretion of interferon-β (IFN-β), IL-6, and IL-8. The purpose of the current study was to reveal the biochemical basis for the secretion and their role in NPC pathogenesis.
Materials and Methods
Cell culture.
Human skin fibroblasts were from control subjects (H1, H2, H5, H11, H17, H34), NPC1 patients (UCH, YON, KUR, MUR, OHS, SAS, END, KAI) (Yamamoto et al., 2000), an NPC2 patient (81027) (Millat et al., 2001), and patients with Niemann–Pick disease type A (NPA) (GM0112) or B (NPB) (GM0252). All cells were passaged 5–15 times. Human fibroblasts, HepG2 and HEK293 cells were maintained in DMEM/10% bovine calf serum (BCS) at 37°C in a humidified atmosphere with 5% CO2. HEK293 cells that stably expressed human Toll-like receptor 4 (TLR4) were obtained from InvivoGen (San Diego, CA). CHO cells were maintained in the same manner except for the use of Ham's F12 instead of DMEM. CHO/NPC1(−) cells that do not express any NPC1 protein and “knock-in” cells that express human NPC1 have been described previously (Sugimoto et al., 2001). For preparation of conditioned medium, cells were seeded on six-well dishes (104/well), and 24 h later, the medium was changed to serum-free DMEM. After a 24 h incubation, the medium was collected and filtered through 0.22 μm pore filters. To stimulate H11 cells or HepG2 cells, conditioned medium was concentrated 100× (v/v) by using Ultrafree-15 Biomax-30 (Millipore, Bedford, MA), and applied at 1/10 (v/v). For the assessment of IL-6 secretion by H34 cells, cells were cultured for 24 h, and then incubated for 12 h in serum-free DMEM containing lipopolysaccharide (LPS) (from Escherichia coli; Sigma, St. Louis, MO), polyinosinic:polycytidylic acid [poly(I:C)] (Sigma), macrophage-activating lipopeptide-2 (MALP-2) (from Mycoplasma fermentans; Alexis Biochemicals, San Diego, CA), or recombinant TNFα (Peprotech, Rocky Hill, NJ).
Peritoneal exudates and skin fibroblasts were isolated from 6-week-old mice. Briefly, animals were killed with diethyl ether, and 5 ml of ice-cold PBS was injected to the peritoneal cavity. Cells were recovered by gentle massage of the peritoneum and suspended in DMEM/10% BCS. To enrich for macrophages, cells were plated on serum-coated dishes for 15 min, and nonadherent cells were removed by washing with PBS. Cells that adhered to the dishes were maintained in DMEM/10% BCS. Fibroblasts that migrated out of a piece of skin in DMEM/10% BCS were maintained in the same medium.
Expression plasmids.
pEFBOS expression plasmids of human TLR4 and MD-2 were a kind gift from Dr. M. Kimoto (Saga Medical University, Saga, Japan). The entire coding sequence of TLR4 was subcloned into pEGFP-N1 in-frame with green fluorescent protein (GFP) to generate a TLR4-GFP fusion protein. Cells were transfected by using FuGENE Transfection Reagent (Roche, Indianapolis, IN).
Small interfering RNA.
Predesigned, double-stranded small interfering RNA (siRNA) against human TLR4 (sense sequence, 5′-GGUCUGGCUGGUUUAGAAG-3′) and control siRNA were purchased from Ambion (Austin, TX). They were introduced to human fibroblasts by electroporation using a Human Dermal Fibroblast Nucleofector kit (Amaxa, Gaithersburg, MD), according to the manufacturer's recommendations. At 48 h after electroporation, cells were lysed for Western blotting, or further incubated for 24 h in serum-free DMEM for preparation of conditioned medium.
Western blotting.
Triton X-100 (1%) extracts from cells and tissues were used unless otherwise indicated. Cells were cultured for 48 h. Cell pellets were resuspended in buffer (50 mm Tris-Cl, pH 7.4, 150 mm NaCl, 30 mm Na4P2O7, 50 mm NaF, 2 mm Na orthovanadate) supplemented with a protease inhibitor mixture (Boehringer, Bagnolet, France) and lysed by sonication. The lysates were mixed with Triton X-100 (1% v/v) and incubated on ice for 30 min, and insoluble materials were removed by brief centrifugation. To prepare 1% Triton X-100 insoluble fractions, the lysates were centrifuged at 100,000 × g for 30 min, and the pellet was suspended in the same buffer containing 1% Triton X-100 and 0.1% SDS. Protein concentrations were measured using the BCA protein assay kit (Bio-Rad, Hercules, CA). Brains were removed from 6-week-old mice that were killed with diethyl ether. Tissues were homogenized with Polytron in 10× (w/v) of the same buffer, and homogenates were processed as described above. SDS-PAGE, Western transfer, and immunoblotting were performed as described previously (Sugimoto et al., 2001), and bound antibodies were detected using an ECL detection kit (Amersham Biosciences, Piscataway, NJ). The following antibodies were used: myxovirus resistance A (MxA) (mouse monoclonal; kindly provided by Dr. K. Nagata, Tsukuba University, Ibaraki, Japan), signal transducers and activators of transcription 1∼6 (STAT-1∼STAT-6), tyrosine-701-phosphorylated STAT-1, IFN-stimulated gene factor 3γ (ISGF3γ) (mouse monoclonal; Transduction Laboratories, Lexington, KY), GFAP (rabbit polyclonal; Santa Cruz Biotechnology, Santa Cruz, CA), TLR4 (goat polyclonal; Santa Cruz Biotechnology) and β-actin (mouse monoclonal; Calbiochem, La Jolla, CA). Blot densitometric analysis was performed by using the NIH Image J software.
Cytochemistry.
Cells cultured on coverslips for 24 h were fixed with 3.7% paraformaldehyde/PBS and permeabilized with 0.5% Triton X-100. After blocking in BlockAce (Dainippon, Osaka, Japan), they were incubated with a primary antibody (STAT-1, 1:100; MxA, 1:200) overnight at 4°C. Bound antibodies were visualized with an Alexa 488-conjugated secondary antibody, and images were obtained using a Bio-Rad MRC 1024 confocal microscope. Procedures for cellular uptake of Alexa 546-conjugated cholera toxin B subunit and the filipin staining of fixed cells have been described previously (Sugimoto et al., 2001).
Histochemistry.
Mice were anesthetized with sodium pentobarbital (0.1 ml/100 g body weight) and perfused with 0.01% heparin/saline followed by 4% paraformaldehyde/0.1 m phosphate buffer, pH 7.4. Brains were embedded in OCT compound, and 10 μm sections were cut using a cryostat. Sections were incubated in 0.3% H2O2, blocked with BlockeAce, and incubated with primary antibodies at 4°C overnight. We used rabbit polyclonal antibodies against TLR4, GFAP, STAT-1, -3, and -6, proliferating cell nuclear antigen (PCNA), goat polyclonal antibodies against IL-6, GFAP (all at 1:100; Santa Cruz Biotechnology), and rat antibody against MAC1 (1:200; Chemicon, Temecula, CA). Bound antibodies were detected by HRP-conjugated secondary antibodies and visualized with DAB, and the sections were counterstained with hematoxylin. For immunofluorescence costaining, sections were incubated with antibodies against TLR4, IL-6, STATs together with antibodies against GFAP or MAC1. Bound antibodies were visualized with Alexa 488- or 568-conjugated secondary antibodies and imaged using a confocal microscope.
Luciferase reporter gene assay.
HepG2 cells were cultured for 24 h and transfected with a luciferase reporter gene construct (pISRE-Luc, pSTAT3-Luc, or pGAS-Luc; Clontech, Cambridge, UK) together with pEGFP (Clontech) for normalization of transfection efficiency. After incubation in DMEM/0.5% BCS for 24 h, cells were further incubated for 6 h in the same medium containing cytokines or conditioned medium. Luciferase activities in cell lysates were determined using Picagene kits (Toyo Ink, Tokyo, Japan), and GFP fluorescence was determined using a fluorophotometer (Perspective Biosystems, Framingham, MA). Human IFN-α and IFN-β were from PBL Biomedical Laboratories (Piscataway, NJ): IFN-γ, IL-6, and IL-8 were from Peprotech. Where indicated, conditioned medium was incubated at 37°C for 1 h with neutralizing antibodies against IFN-α, IFN-β, IFN-γ (PBL Biomedical Laboratories), IL-6, or IL-6 receptor (R&D Systems, Minneapolis, MN).
For NFκB reporter gene assays in HEK293 cells, cells were transfected with pNFκB-luc (Clontech) together with pEFBOS/TLR4 and/or MD-2. After incubation in DMEM/10% BCS for 24 h, cells were stimulated for 6 h with LPS or TNFα.
ELISA.
Cytokines concentrations in conditioned media were determined by an ELISA assay (Biosource, Camarillo, CA) according to the manufacturer's instructions.
Generation of NPC mice that lack TLR4 or IL-6.
BALB/c NPC mice and C57BL IL-6 knock-out mice were obtained from the The Jackson Laboratory (Bar Harbor, ME). C57BL TLR4 knock-out mice were a kind gift from Dr. S. Akira (Osaka University, Osaka, Japan). BALB/c NPC mice were backcrossed for four generations into the C57BL strain. Resultant NPC1+/− mice were crossed with TLR4−/− mice. Mice heterozygous for both NPC1 and TLR4 were intercrossed to generate NPC1−/− mice with TLR4 genotypes of TLR4 +/+, +/−, and −/−. NPC1−/− mice with different IL-6 genotypes were generated in the same way. The genotype of each mouse was determined by genomic PCR. All experiments were performed according to Tottori University Animal Care and Use Committee guidelines.
Statistical analysis.
Two-way ANOVA and Student's t test were used with p < 0.05 regarded as statistically significant.
Results
Increased secretion of IFN-β, IL-6, and IL-8 by NPC cells
Concentrations of 22 cytokines and soluble IL-6 receptor in serum-free conditioned medium (CM) were determined by ELISA using cells from two control subjects (H11 and H34), one patient (UCH) with NPC1 mutations, and another (81027) with NPC2 mutations (Table 1). Both UCH and 81027 had an infantile form of NPC (Yamamoto et al., 2000; Millat et al., 2001). The secreted cytokine profiles were remarkably similar between UCH and 81027 cells. Most significantly, secretion of IL-6 and IL-8 was markedly increased. Of the three IFNs, only IFN-β was detectable. Other detectable cytokines included IL-17, TNFα, transforming growth factor β (TGFβ), and three types of colony-stimulating factors (CSFs), whose concentrations were unaltered or marginally increased in NPC cells.
Table 1.
Cytokine concentrations in conditioned medium of cultured human skin fibroblasts
| Control(H11) | Control(H34) | NPC1(UCH) | NPC2(81027) | |
|---|---|---|---|---|
| IFN-β | ND | ND | 83 | 63 |
| IL-6 | 1.4 | 17 | 2860 | 2040 |
| IL-8 | ND | ND | 1560 | 2320 |
| IL-17 | 27 | 55 | 21 | 52 |
| TNFα | 0.49 | 0.49 | 0.59 | 0.66 |
| TGFβ | ND | 0.08 | 0.04 | 0.15 |
| G-CSF | 15 | 14 | 16 | 36 |
| GM-CSF | 2.8 | ND | 4.4 | 7.5 |
| M-CSF | 121 | 212 | 411 | 977 |
Medium was collected from cells cultured in serum-free DMEM for 24 h and filtered through 0.22 mm pore filters. Concentrations of each cytokine were determined by ELISA. Units are picograms/milliliter. Shown are the mean values obtained in a single determination performed in duplicate. Similar results were obtained in another independent experiment. The following cytokines were at undetectable levels in all of the four cell lines: IFN-α, IFN-γ, IL-1α, IL-1β, IL-2, IL-4, IL-5, IL-10, IL-11, IL-12, IL-13, IL-18, and leptin. Soluble IL-6 receptor was also undetectable. G-CSF, Granulocyte colony-stimulating factor; GM-CSF, granulocyte–macrophage colony-stimulating factor; M-CSF, macrophage colony-stimulating factor; ND, not detectable.
To confirm these results, we determined the concentrations of IL-6 and IL-8 in CM of cells from seven other patients with NPC1 mutations. Of these subjects, SAS and END had a juvenile form, KAI had an adult form, and all others had an infantile form (Yamamoto et al., 2000). IL-6/IL-8 levels were increased except for KAI (Fig. 1A). Their levels were marginally increased in cells from patients with NPA and NPB, but not in cells from four other control subjects, or from an NPC1 heterozygote. We also measured concentrations of IL-6 in CM of primary cultured peritoneal exudates enriched for macrophages and skin fibroblasts obtained from NPC mice. In both types of cell preparations, there was a clear increase in NPC1−/− cells compared with the levels in heterozygous and wild-type (wt) cells (Fig. 1B). Concentrations of IFN-β and IL-8 were not determined in the mouse samples because an ELISA was not available. Thus, the increased secretion of IL-6 appeared to be a common feature of NPC cells.
Figure 1.

Secretion of IL-6 and IL-8 by NPC cells. A, Secretion by human skin fibroblasts. Conditioned medium was prepared as described in Materials and Methods. Each bar represents the mean value of duplicates, obtained in a single experiment. Similar results were reproduced in another independent experiment. B, Secretion by primary cultured mouse cells obtained from 6-week-old littermates. Peritoneal exudates were enriched for macrophages as described in Materials and Methods. Each bar represents the mean ± SEM of three determinations, each performed in duplicate.
Activation of STATs by IFN-β and IL-6 in NPC cell CM
Both IFN-β and IL-6 activate STATs, which have six subtypes. The three response elements, IFN-stimulated response element (ISRE), STAT3, and γ-activated site (GAS) are the targets of STAT-1/2 heterodimer, STAT-3 homodimer, and STAT-1 homodimer, respectively (Ihle, 1996). By using reporter gene assays in HepG2 cells, we examined whether (1) IFN-β and IL-6 secreted by NPC cells were biologically active, (2) there were any other cytokines that could activate STATs, and (3) there were any substances that could induce secretion of IFN-β or IL-6.
In control experiments, recombinant IFN-α activated ISRE, IFN-β and IFN-γ activated ISRE and GAS, IL-6 activated STAT3 and GAS, and IL-8 had no effect, as expected from their specificity (Schindler, 1999) (Fig. 2A). None of the other cytokines detected in CM (IL-17, TNFα, TGFβ, and three CSFs) at 10 ng/ml activated any of these response elements (data not shown). UCH CM, but not H34 CM, activated all of the three response elements (Fig. 2B). For absorption experiments, UCH CM was incubated with neutralizing antibodies for 1 h before application. An antibody against IFN-β abolished its effect on ISRE, whereas those against IL-6 or IL-6 receptor abolished its effects on STAT3 and GAS. Antibodies against IFN-α or IFN-γ failed to neutralize the activities (Fig. 2C). These findings indicated that both IFN-β and IL-6 in UCH CM were biologically active, and that it contained no other cytokine that could activate STATs. The same results were reproduced when antibodies against IFN-β or IL-6 were precipitated with protein A-Sepharose after the 1 h incubation (data not shown), suggesting that UCH CM contained no substance that could induce secretion of IFN-β or IL-6.
Figure 2.
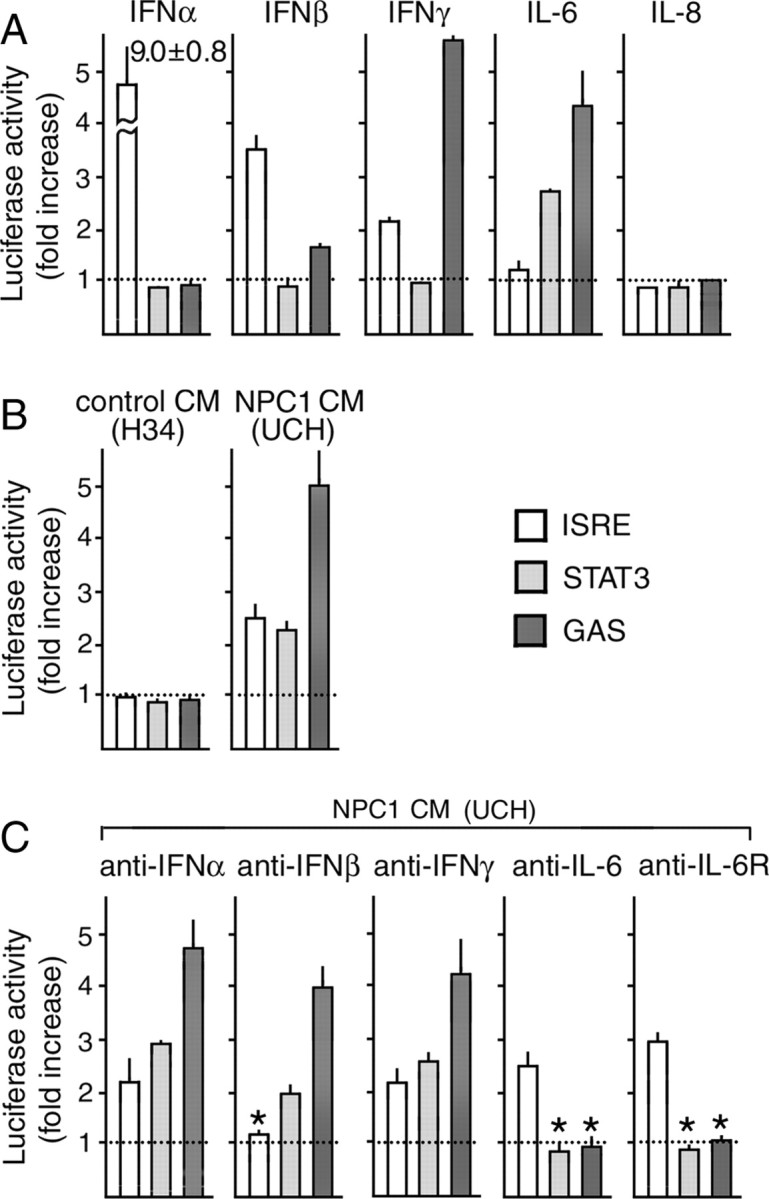
Activation of STATs by NPC cell CM assessed by reporter gene assays in HepG2 cells. Cells were transfected with a reporter construct containing each response element. A, B, At 24 h after transfection, they were stimulated for 6 h with recombinant cytokines (A) or CM of H34 or UCH cells (B). The concentration of each cytokine was 10 ng/ml. In C, UCH CM was incubated with neutralizing antibodies at 37°C for 1 h before application. The concentration of each antibody was 2 μg/ml. Each bar represents the mean ± SEM of three determinations, each performed in triplicate. *p < 0.01, significantly different from the values without neutralization.
As an alternative method to access the activity of the cytokines, we examined whether UCH CM could induce expression of MxA in control cells. MxA is one of the representative ISRE-driven genes (Der et al., 1998), encoding a dynamin family GTPase required for lipid transport in the smooth endoplasmic reticulum (Accola et al., 2002). Application of UCH CM induced expression of MxA in H11 cells (Fig. 3A). This activity was not contained in CM of H34 cells but was present in CM of another NPC1 cell line (YON) as well as 81027 cells (Fig. 3B). Consistent with the role of ISRE in MxA expression, a neutralizing antibody against IFN-β partially absorbed the activity of UCH CM and this effect was not observed with any of the antibodies against IFN-α, IFN-γ, IL-6, and IL-6 receptor (Fig. 3C).
Figure 3.

Activity of NPC cell CM to induce expression of MxA. In A–D, MxA protein in H11 cells was detected by anti-MxA Western blotting. NPC cells were from patients with NPC1 mutations (UCH and YON) and NPC2 mutations (81027). A, Time-dependent effects of UCH CM. H11 cells were incubated with CM from H34 or UCH cells for the time indicated. B, Effects of YON and 81027 CM. CM was applied to H11 cells for 12 h. C, Effects of neutralizing antibodies. UCH CM was incubated with each neutralizing antibody (2 μg/ml) at 37°C for 1 h before application to H11 cells. D, Time-dependent induction of MxA expression in Transwell cultures. H11 cells were seeded in the lower chamber, and H34 or UCH cells were seeded in the upper chamber. E, Expression of MxA by NPC cells. H11, UCH, and 81027 cells were subjected to anti-MxA Western blotting or immunofluorescence. The images were obtained with confocal microscopy. Shown are the representative results, which were reproduced at least twice.
To examine whether the similar activity was present in ordinary serum-containing medium, we used Transwell cultures in which cells were separated by a 0.22 μm pore filter. MxA was expressed by H11 cells when they were cocultured with UCH cells (Fig. 3D). We then examined whether NPC cells expressed MxA and found a dramatic increase of this protein both in UCH and 81027 cells. Anti-MxA immunofluorescence revealed its presence in the endoplasmic reticulum where it is normally localized (Accola et al., 2002), consistent with increased expression of the protein (Fig. 3E). These findings suggested that NPC cells cultured in normal serum-containing medium also secreted IFN-β and that this cytokine could act in both autocrine and paracrine manner.
Increases of STATs in NPC cells
Chronic secretion of inflammatory cytokines occurs in various disease states such as rheumatoid arthritis. This leads to increased protein levels of STATs in target cells and tissues, which serve as a marker of inflammation (Ivashkiv and Hu, 2003; Walker et al., 2006). Therefore, we examined the levels of STATs in NPC cells.
Western blotting showed that both UCH and 81027 cells contained increased levels of STAT-1 to -5 compared with H11 cells. Similarly, ISGF3γ, a protein that forms a complex with STAT-1 and -2 (Martinez-Moczygemba et al., 1997), was detectable in NPC cells, but not in H11 cells (Fig. 4A). In UCH cells, some of the STAT-1 protein was in its active state, because it was localized in the nucleus (Fig. 4B) and some of the protein was tyrosine phosphorylated (Fig. 4C).
Figure 4.
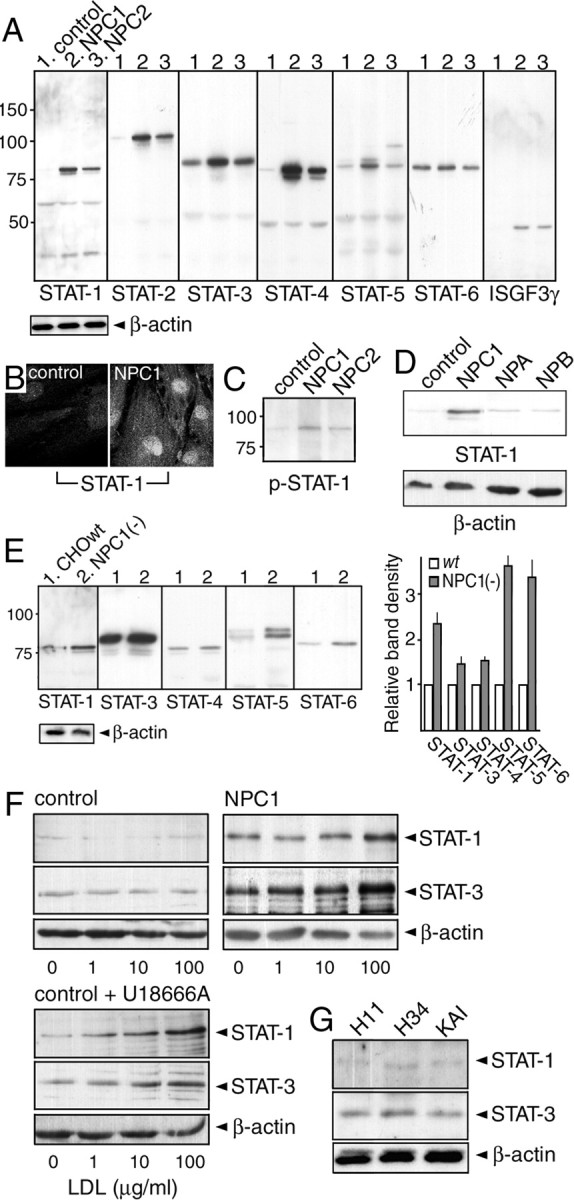
Levels of STATs in NPC cells. A, C, D, Levels of STATs and ISGF3γ in human fibroblasts. Proteins in 1% Triton X-100 extracts were detected by Western blotting with indicated antibodies. Cells were from a control subject (H11) and patients with NPC1 mutations (UCH), NPC2 mutations (81027), NPA (GM0112), and NPB (GM0252). A total of 10 μg of protein was loaded in each lane. Molecular weights are given on the left (in kilodaltons). B, STAT-1 immunofluorescence observed by confocal microscopy. E, Levels of STATs in NPC1-deficient CHO cells [NPC1(−) cells]. Signal intensity of the bands was normalized to that of β-actin, and the intensity in NPC1(−) cells was expressed as relative to that in wt cells. Each bar represents the mean ± SEM of three determinations. F, Effects of U18666A and LDL loading on STAT-1/STAT-3 levels. H34 and UCH cells were cultured in lipoprotein-deficient serum for 3 d, and then loaded with increasing concentrations of LDL for 24 h (top panels). In a separate set of H34 cells, they were loaded with LDL in the presence of 2 μg/ml U18666A (bottom panels). G, Levels of STAT-1/STAT-3 in KAI cells. Shown are the representative results, which were reproduced at least twice.
To see whether the increase of STATs was specific for NPC, we examined their levels in NPA and NPB cells, and found no increase of STAT-1 (Fig. 4D) or other STATs (data not shown). To determine whether the increase was a common feature of NPC cells, levels in NPC1-deficient CHO cells were examined (Sugimoto et al., 2001). Similar to human NPC cells, NPC1-deficient CHO cells expressed increased levels of STAT-1, -3, -4, and -5. In these cells, the level of STAT-6 was also increased. The increase of STAT-3 and -4 was less clear than in human cells, but densitometric blot analysis showed a ∼50% increase in signal intensity (Fig. 4E). This increase was not observed in “knock-in” cells that expressed human NPC1 (data not shown). No STAT-2 protein was detectable in CHO cell extracts, most likely because the antibody did not recognize the hamster protein. These findings suggested that the increase of STATs was a common and specific feature of NPC cells.
We also examined whether a similar increase of STATs was induced in control cells treated with 3β-[2-(diethylamino)ethoxy]-androst-5-en-17-one monohydrochloride (U18666A), which induces an NPC phenotype (Ko et al., 2001). Low-density lipoprotein (LDL) loading of cholesterol-depleted UCH cells caused a dose-dependent increase of STAT-1 and -3, but not in H11 cells. When H11 cells were loaded with LDL in the presence of U18666A, however, STAT levels increased in a dose-dependent manner (Fig. 4F). Thus, the increase of STATs could be induced in normal cells by U18666A treatment.
This observed increase of STATs in NPC cells was most likely attributable to autocrine effects of IFN-β and IL-6. In support of this notion, we found no increase in the levels of STAT-1 and STAT-3 in KAI cells (Fig. 4G).
Endosomal accumulation of TLR4 in NPC cells
We next investigated the biochemical basis for increased secretion of cytokines by NPC cells. Results from reporter gene assays in HepG2 cells (Fig. 2) and MxA induction experiments in H11 cells (Fig. 3) indicated that there was no activity in NPC cell CM that could induce secretion of IFN-β or IL-6, suggesting that their secretion was caused by some other intracellular event. NPC cells are characterized by aberrant endosomal membrane flow and it is possible that the activity of any signaling molecule in the endosomal compartment is altered.
We hypothesized that their secretion was attributable to activation of TLR4. TLRs are a family of receptor proteins that play a crucial role in innate immune responses. There are currently 11 known members of TLRs. TLR4 is selectively activated by bacterial LPS, and its activation by LPS requires an accessory protein MD-2 (Akashi et al., 2000). Our hypothesis was based on following observations. First, TLR4 activates transcription factors interferon regulatory factor-3 (IRF-3) and NFκB, which leads to production of IFN-β, IL-6, and IL-8 (Akira and Takeda, 2004), the same set of cytokines with elevated secretion in NPC cells. Second, TLR4 is localized both on the cell surface and endosome. It can be active on the endosome and its proper sorting to the lysosome is required to turn off its activity (Blander and Medzhitov, 2004; Doyle et al., 2004; Guillot et al., 2004; Husebye et al., 2006). Third, TLR4 is localized in raft microdomains and its activity can be modulated by the levels of cellular cholesterol (Triantafilou et al., 2002; Olsson and Sundler, 2006). To test our hypothesis, we examined whether (1) there were any alterations in the protein level and/or intracellular localization of TLR4, (2) siRNA against TLR4 could suppress cytokine secretion, and (3) U18666A could alter cellular responses to LPS.
Western blotting showed that UCH, YON, and 81027 cells contained markedly increased levels of TLR4. TLR4 was also detectable in 1% Triton X-100-insoluble fractions of NPC cells, suggesting its association with raft microdomains. As above, the cells from an adult-onset patient KAI proved to be the exception, because TLR4 was only marginally increased in cell extracts and was not detectable in 1% Triton X-100-insoluble fractions (Fig. 5A).
Figure 5.
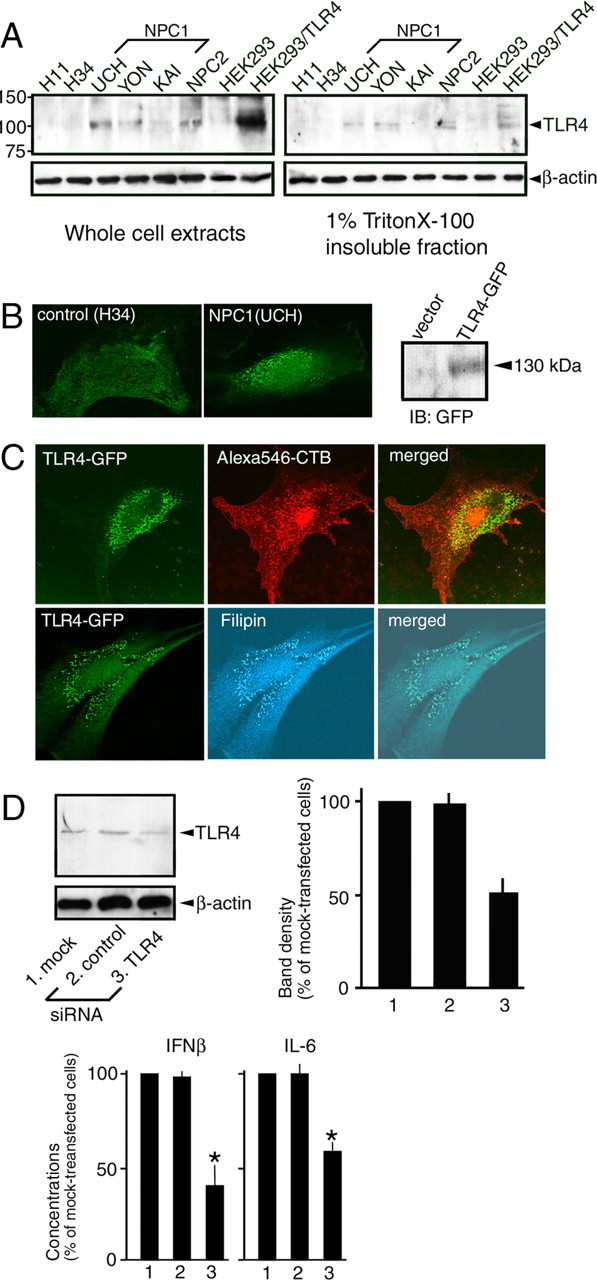
Levels of TLR4 and intracellular localization of TLR4-GFP in human NPC fibroblasts. A, Levels of TLR4. Whole-cell extracts were prepared in 1% Triton X-100/0.2% SDS. The 1% Triton X-100-insoluble fractions were prepared as described in Materials and Methods. Extracts from native HEK293 cells and HEK293 cells expressing TLR4 were included. Molecular weights are given on the left (in kilodaltons). B, C, Intracellular localization of TLR4-GFP. In B, TLR4-GFP was expressed in H34 or UCH cells. Shown on the right is an anti-GFP Western blot of H34 cell extracts transfected with vector or TLR4-GFP construct. In C, 24 h after transfection with TLR4-GFP construct, UCH cells were exposed to Alexa 546-conjugated cholera toxin B subunit (CTB) before fixation (top panels) or fixed and stained with filipin (bottom panels). Images were obtained by confocal microscopy. In A–C, shown are the representative results, which were reproduced at least twice. D, Effects of TLR4 siRNA. siRNA was introduced to UCH cells by electroporation, and at 48 h after electroporation, whole-cell extracts were processed for Western blotting or the cells were further incubated for 24 h in serum-free DMEM for preparation of CM. Signal intensity of the bands was normalized to that of β-actin, and the intensity in siRNA-transfected cells was expressed as relative to that in mock-transfected cells. Each bar represents the mean ± SEM of three determinations. Because absolute values varied between experiments, concentrations of IFN-β and IL-6 were also expressed as relative to the values of mock-transfected cells. Each bar represents the mean ± SEM of three determinations, each performed in duplicate. *p < 0.01, significantly different from the values of mock-transfected cells.
Because endogenous TLR4 could not be detected by immunocytochemistry, we determined the intracellular localization of a TLR4-GFP fusion protein, which has been shown to behave similar to the endogenous protein (Latz et al., 2002). In H34 cells, TLR4-GFP was located both on the cell surface and in tiny vesicles that were diffusely distributed throughout the cytoplasm. In contrast, in UCH cells, it was predominantly found in perinuclear vesicles (Fig. 5B). In NPC cells, ganglioside GM1 accumulates in the early endosome, whereas cholesterol accumulates in the late endosome and lysosome (Sugimoto et al., 2001). Intracellular vesicles that contained TLR4-GFP did not incorporate Alexa 546-conjugated cholera toxin B subunit, but were stained with filipin, suggesting the accumulation of TLR4-GFP in cholesterol-enriched late endosomes/lysosomes (Fig. 5C).
siRNA was introduced into UCH cells by electroporation. TLR4 siRNA decreased the level of this protein in cell extracts, and also caused significant reductions in IFN-β and IL-6 in CM (Fig. 5D). Similar results were obtained with 81027 cells (data not shown).
The response of H34 cells to LPS was assessed by quantification of secreted IL-6. LPS caused dose-dependent secretion of IL-6 by H34 cells. When cells were pretreated with U18666A, the amounts of secreted IL-6 were significantly increased. In these experiments, we also examined the cellular response to poly(I:C), MALP-2, and TNFα, all of which can activate NFκB. Poly(I:C) is a selective ligand for TLR3 (Akira and Takeda, 2004). Similar to TLR4, TLR3 activates both IRF-3 and NFκB and is located in endosomal compartments (de Bouteiller et al., 2005). However, unlike TLR4, it has never been demonstrated that this receptor is associated with raft microdomains or that the levels of cellular cholesterol influence its function. Similar to LPS, MALP-2 is a lipoprotein; however, it is a selective ligand for TLR2 and -6 (Akira and Takeda, 2004), whereas TNFα is not a ligand for TLRs. U18666A caused no significant changes in the cellular responses to these ligands (Fig. 6A).
Figure 6.

Effects of U18666A on cellular responses to LPS. A, IL-6 secretion by H34 control cells. Cells were incubated with or without U18666A (2 μg/ml) for 16 h and further incubated for 12 h in serum-free DMEM containing LPS, poly(I:C), MALP-2, or TNFα. Concentrations of IL-6 in the medium were determined by ELISA. Each point represents the mean ± SEM of three determinations each performed in duplicate. B, NFκB reporter gene assay. HEK293 cells were transfected with NFκB luciferase construct together with expression plasmids for TLR4 and/or MD-2 as indicated. At 24 h after transfection, they were stimulated with LPS or TNFα for 6 h in the absence or presence of U18666A (2 μg/ml). Each bar represents the mean ± SEM of three determinations, each performed in triplicate. *p < 0.01, significantly different from the values in the absence of U18666A.
LPS has multiple effects and some of them are independent from TLR4. To confirm the effect of U18666A on TLR4, we used an NFκB reporter gene assay in HEK293 cells, which do not express TLR4 or MD-2 (Fig. 6B). When cells were transfected with TLR4 and MD-2 constructs, LPS caused a dose-dependent activation of the reporter, and its effect was significantly enhanced in the presence of U18666A. Unexpectedly, we found that U18666A alone caused activation of the reporter in the presence of TLR4/MD-2, and was effective only when both TLR4 and MD-2 were expressed. The effect of TNFα was not affected by U18666A, confirming the specificity of this effect for TLR4/MD-2. Using this reporter gene assay, we detected no effect of UCH CM regardless of expression of TLR4/MD-2 (data not shown), consistent with the lack of activity in NPC cell CM to induce cytokine secretion.
Expression of STATs, TLR4, and IL-6 in the NPC mouse brain
The above findings in cultured NPC fibroblasts suggested that secretion of cytokines leads to activation of STATs and that this secretion is attributable, at least in part, to the endosomal accumulation of TLR4. To explore the role of these molecules in NPC pathogenesis, we first examined the protein levels and cellular localization of STATs in the NPC mouse brain, using tissues from 6-week-old wt and NPC1−/− littermates.
Western blotting of cerebral and cerebellar extracts revealed a clear increase of STAT-3 and -6 and a marginal increase of STAT-1, -4, and -5 in NPC1−/− mice compared with wt mice (Fig. 7). The profile of the increased STATs was different from that observed in cultured human fibroblasts because STAT-3 was increased most prominently. STAT-2 was undetectable, most likely because the antibody did not recognize the mouse protein. We then examined the cellular origins of STAT-1, -3, and -6 by immunohistochemistry of cerebellar sections.
Figure 7.

Levels of STATs in the NPC mouse brain. Brain extracts were obtained from 6-week-old NPC1+/+ (wt) and NPC1−/− littermates and analyzed by Western blotting. A total of 10 μg of protein was loaded in each lane. Molecular weights are given on the right (in kilodaltons). Shown are the representative results, which were reproduced at least twice.
In the following analysis, activated astroglial and microglial cells were detected with antibodies against GFAP and MAC1, respectively. Glial cell activation in the NPC mouse brain is a progressive process, and in the cerebellar cortex of 6-week-old mice, both GFAP-positive and MAC1-positive cells can be seen in every layer. There is, however, a regional difference in their abundance: GFAP-positive astroglial cells are most abundant in the granule cell layer, whereas MAC1-positive microglial cells are most abundant in the white matter. These glial cells contained undetectable levels of immunoreactivity for PCNA (data not shown), suggesting that they are not actively proliferating.
In wt sections, STAT-1 immunoreactivity was observed in Purkinje and granule cell layers. Immunoreactivity in these regions was increased and signal was also observed in the molecular cell layer of the NPC1−/− cerebellum (Fig. 8A). Double immunofluorescence analysis did not reveal any colocalization of STAT-1 with either GFAP or MAC1 (data not shown). In wt sections, STAT-3 immunoreactivity was observed in molecular and Purkinje cell layers. Immunoreactivity was increased in NPC1−/− cerebellum, and strong signal was also observed in the granule cell layer (Fig. 8B). STAT-3 immunoreactivity colocalized with both GFAP and MAC1 as shown in the representative images of the granule cell layer and the white matter, respectively (Fig. 8C). STAT-6 immunoreactivity was not observed in neuronal or glial cells (data not shown) but was confined to vascular endothelial cells, and it appeared to be increased in NPC1−/− sections (Fig. 8D). These findings suggested that, in the NPC1−/− mouse cerebellum, STAT-1 was expressed by neuronal cells, STAT-3 by both neuronal and glial cells, and STAT-6 by endothelial cells. Similar results were obtained using immunohistochemical analysis of cerebral sections (data not shown).
Figure 8.
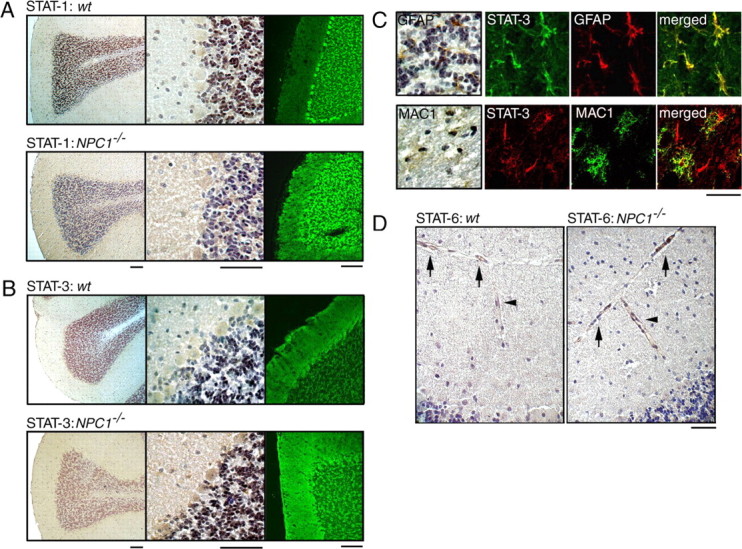
Cellular localizations of STATs in the NPC mouse cerebellum. Brain sections were obtained from 6-week-old littermates. A, B, STAT-1/STAT-3 immunostaining. Cerebellar sections were stained with indicated antibodies, and bound antibodies were detected by HRP-conjugated secondary antibody and visualized with DAB or by Alexa 488-conjugated secondary antibody followed by confocal imaging. Scale bars, 100 μm. C, Double immunofluorescence of NPC1−/− sections with STAT-3 and GFAP or MAC1. Images are from the granule cell layer (GFAP) or the white matter (MAC1). Bound antibodies were detected by Alexa 488- or Alexa 568-conjugated secondary antibodies. In the left panels, bound antibodies were detected by HRP-conjugated secondary antibody and visualized with DAB. Scale bar, 20 μm. D, STAT-6 immunostaining of blood vessels in the subarachnoid space (arrows) and the molecular cell layer (arrowheads). Scale bar, 100 μm. Shown are the representative results, which were reproduced at least twice.
Next, we examined cellular localization of TLR4 and IL-6. We focused on IL-6 in the subsequent histochemical analysis and genetic deletion experiments because of the prominent increase of STAT-3 and its expression by both neuronal and glial cells. TLR4 immunoreactivity was barely detectable in wt cerebellar sections, whereas clear signal was observed in cells localized in the granule cell layer and the white matter of NPC1−/− sections (Fig. 9A). Double immunofluorescence labeling showed colocalization of TLR4 with both GFAP and MAC1 as shown in the representative images of the granule cell layer and the white matter, respectively (Fig. 9B). Similarly, IL-6 immunoreactivity was barely detectable in wt sections (data not shown), but in NPC1−/− sections, a clear colocalization with GFAP and MAC1 was observed (Fig. 9C). These findings suggested that, in the NPC1−/− mouse cerebellum, TLR4 and IL-6 are expressed by glial cells, but not by neuronal cells.
Figure 9.
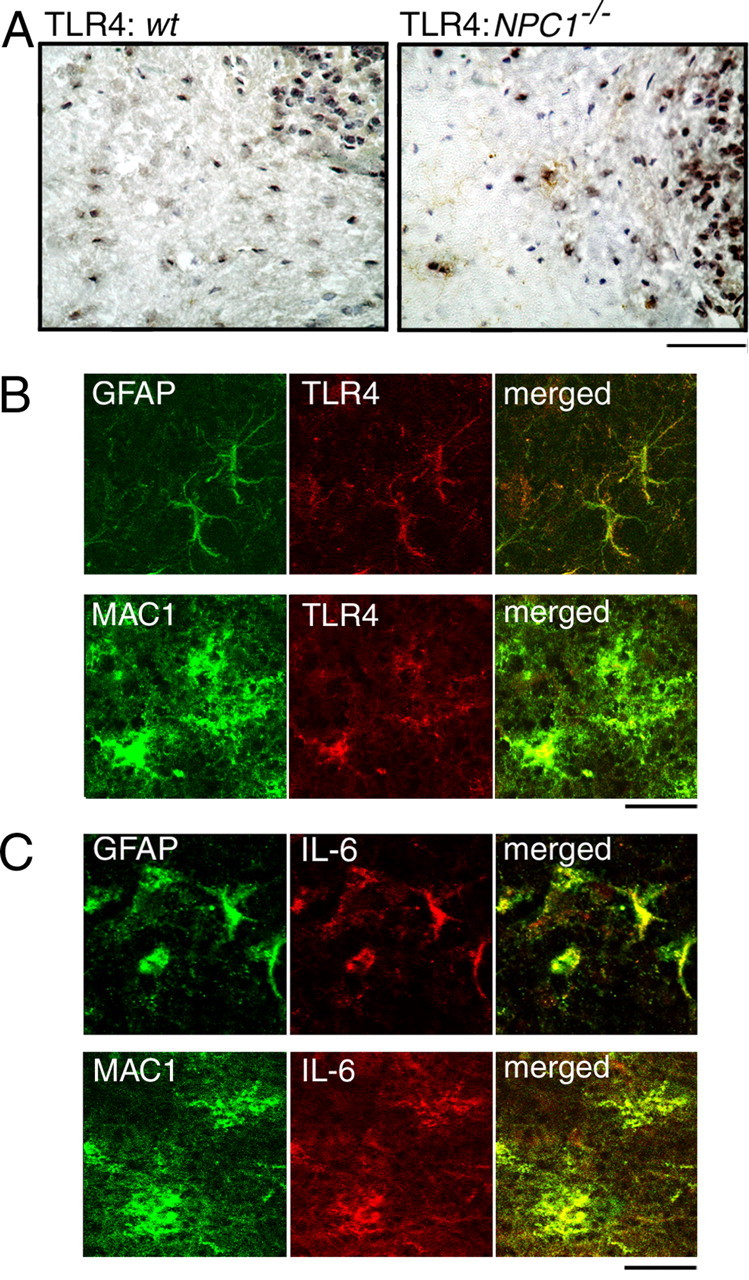
Cellular localizations of TLR4 and IL-6 in the NPC mouse cerebellum. Brain sections were obtained from 6-week-old littermates. A, TLR4 immunostaining. Scale bar, 100 μm. B, C, Double immunofluorescence of NPC1−/− sections with TLR4 or IL-6 and GFAP or MAC1. Images are from the granule cell layer (GFAP) or the white matter (MAC1). Scale bars, 20 μm. Shown are the representative results, which were reproduced at least twice.
Genetic deletion of TLR4 reduced cellular IL-6 secretion
To confirm the role of TLR4 in IL-6 secretion, we generated double gene knock-out mice that lacked NPC1 and TLR4. IL-6 secretion was examined in fibroblasts obtained from 6-week-old littermates with different TLR4 genotypes (Fig. 10, left panel). As expected, NPC1−/−/TLR4+/+ fibroblasts secreted high levels of IL-6. Its concentration was marginally reduced in NPC1−/−/TLR4+/− cells and was reduced to ∼30% in NPC1−/−/TLR4−/− cells. These results confirmed the role of TLR4 in IL-6 secretion by NPC fibroblasts, but also suggested the presence of other factors responsible for the residual secretion of IL-6.
Figure 10.

Effects of genetic deletion of TLR4 or IL-6 on IL-6 secretion by primary cultured skin fibroblasts. Cells were obtained from 6-week-old littermates with individual genotypes. Each bar represents the mean ± SEM of three determinations, each performed in duplicate. *p < 0.01, significantly different from the values of NPC1−/−/TLR4+/+ cells (left) or NPC1−/−/IL-6+/+ cells (right).
Despite this reduction of IL-6 secretion by fibroblasts, Western blotting and immunohistochemistry revealed that both STAT levels and the number of activated glial cells were indistinguishable between NPC1−/−/TLR4+/+ and NPC1−/−/TLR4−/− mice (data not shown). In addition, the lifespan of the NPC1−/−/TLR4−/− mice was not significantly different from that of NPC1−/−/TLR4+/+ mice (data not shown). Assuming that IL-6 expression by glial cells is also decreased in the double gene knock-out mice, these negative findings argue against a role of TLR4 and IL-6 in NPC pathogenesis. However, it is possible that residual IL-6, which might also be secreted by glial cells, is sufficient to induce pathological changes. To address this question, we generated double gene knock-out mice that lacked NPC1 and IL-6.
Genetic deletion of IL-6 normalized STAT levels, suppressed glial cell activation, and prolonged the life span of NPC mice.
IL-6 secretion was again assessed in fibroblasts with different IL-6 genotypes. As expected, NPC1−/−/IL-6+/+ cells secreted a high concentration of IL-6. Its concentration in NPC1−/−/IL-6+/− cells was about one-half of that found in NPC1−/−/IL-6+/+ cells, and it was undetectable in NPC1−/−/IL-6−/− cells (Fig. 10, right panel).
Genetic deletion of IL-6 caused obvious alterations in STAT levels and brain pathology. Western blotting of brain extracts from 6-week-old littermates showed that NPC1−/−/IL-6+/+ mice had increased amounts of STATs, compared with NPC1+/+/IL-6+/+ mice. The levels detected in NPC1−/−/IL-6+/− mice were comparable with those found in NPC1−/−/IL-6+/+ mice, whereas the levels in NPC1−/−/IL-6−/− mice were significantly decreased. The exception was STAT-6 that did not change significantly between IL-6 genotypes (Fig. 11A). In agreement with the Western blot analysis, STAT-3 immunoreactivity in Purkinje and molecular cell layers was decreased in NPC1−/−/IL-6−/− mice compared with NPC1−/−/IL-6+/+ or NPC1−/−/IL-6+/− mice (Fig. 11B).
Figure 11.

Effects of genetic deletion of IL-6 on STAT levels and glial cell activation in the NPC mouse brain. Brains were obtained from 6-week-old littermates with individual genotypes. A, Levels of STATs and GFAP. Western blotting of 1% Triton X-100 extracts was performed as described in Figure 7. B, STAT-3 immunostaining. Cerebellar sections were stained with anti-STAT-3 followed by HRP-conjugated secondary antibody. C, GFAP immunostaining. Sections were stained with anti-GFAP followed by Alexa 488-conjugated secondary antibody. M, P, and G denote molecular, Purkinje, and granule cell layers, respectively. D, MAC1 immunostaining. Cerebellar sections were stained with anti-MAC1 followed by HRP-conjugated secondary antibody. Shown are the representative images of the white matter. In A–D, shown are the representative results, which were reproduced at least twice. Scale bars, 50 μm.
We found that the number of activated glial cells was markedly decreased in NPC1−/−/IL-6−/− mice. The decrease of GFAP-positive astroglial cells was most apparent in the granule cell layer and the thalamic nuclei in sections from 6-week-old littermates (Fig. 11C). A similar decrease was also seen in sections from 3- and 9-week-old mice (data not shown). Accordingly, Western blotting demonstrated reduced levels of GFAP in the NPC1−/−/IL-6−/− brain (Fig. 11A). Similar to astroglial cells, the number of MAC1-positive microglial cells was decreased in NPC1−/−/IL-6−/− mice (Fig. 11D). Despite this reduction of activated glial cells, we could not see appreciable differences in the number of Purkinje cells between different genotypes (data not shown). The lifespan of NPC1−/−/IL-6−/− mice, but not that of NPC1−/−/IL-6+/− mice, was modestly increased compared with NPC1−/−/IL-6+/+ mice (Table 2).
Table 2.
Effects of genetic deletion of IL-6 on the lifespan of NPC1−/− mice
| Background | IL-6 | Male | Female | Sum |
|---|---|---|---|---|
| BALB/c | +/+ | 68 ± 4.1 (25) | 72 ± 4.2 (25) | 70 ± 4.5 (50) |
| BALB/c × C57BL | +/+ | 70 ± 2.6 (7) | 70 ± 7.2 (7) | 70 ± 5.2 (14) |
| +/− | 69 ± 1.1 (13) | 73 ± 5.6 (11) | 71 ± 4.3 (24) | |
| −/− | 77 ± 5.7* (6) | 82 ± 6.7* (6) | 80 ± 6.7* (12) |
Values represent the mean ± SD. The number of animals in each group is given in parentheses.
*p < 0.01, significantly different from the values of IL-6+/+ animals.
Discussion
The main cellular phenotype of NPC is aberrant endosomal membrane trafficking, which leads to an accumulation of cholesterol and other lipids in the endosomal/lysosomal compartment. The fundamental question in NPC pathogenesis is how this impairment of membrane flow causes pathological changes including neuronal cell loss and glial cell activation. In the current study, we presented evidence that TLR4, IL-6, and STATs constitute a potential link between impaired membrane flow and glial cell activation.
Our findings in cultured NPC fibroblasts indicate that secretion of the cytokines is attributable, at least in part, to the endosomal accumulation of TLR4. TLR4 is peculiar among the TLRs in that it can be located in endosomal compartments (Guillot et al., 2004; Husebye et al., 2006) and associates with cholesterol-enriched raft microdomains (Triantafilou et al., 2002; Olsson and Sundler, 2006). These features of TLR4 may underlie its accumulation, because NPC cells accumulate raft microdomains in their endosomes (Lusa et al., 2001). In the NFκB reporter gene assay using HEK293 cells, U18666A alone could activate NFκB in cells expressing TLR4 and MD-2, and NPC cell CM contained no activity to stimulate this receptor. These findings suggest that, in NPC cells, activation of TLR4 is caused by the endosomal accumulation in itself, and hence is a direct consequence of impaired membrane flow. This is consistent with previous findings that TLR4 could be activated by antibody-mediated aggregation in the absence of any ligand (Visintin et al., 2003) and that its proper sorting from the endosome to the lysosome was required to turn off its activity (Husebye et al., 2006). Because TLR4 triggers multiple signaling events that activate intracellular membrane trafficking (Blander and Medzhitov, 2004; Doyle et al., 2004), its activation in NPC cells may represent one of the adaptive responses of the cells to resume membrane trafficking.
Genetic deletion of TLR4 in NPC mice reduced IL-6 secretion by primary cultured fibroblasts, confirming the role of TLR4 in IL-6 secretion by these cells. Immunohistochemical analysis revealed expression of TLR4, IL-6, and STAT-3 by glial cells in the NPC mouse cerebellum, suggesting that TLR4 and downstream signaling events are also activated in glial cells in vivo. Direct evidence for the role of TLR4 in IL-6 expression by glial cells, however, is lacking. Furthermore, the negative effects of genetic deletion of TLR4 on STAT levels and glial cell activation indicate that, even if this receptor is responsible for IL-6 expression, the contribution is only partial and there must be other factors responsible for the expression. Additional analysis will be required to identify these factors and to verify the role of TLR4 in IL-6 expression by glial cells, such as other members of the TLR family, which have been shown to be expressed by glial cells (Kielian, 2006).
Selective expression of IL-6 and STATs by specific cell types in the NPC1−/− mouse cerebellum and the effects of genetic deletion of IL-6 indicate that this cytokine plays multiple roles in NPC pathogenesis, acting in both autocrine and paracrine manner. The autocrine effect is evident by expression of IL-6 and STAT-3 by glial cells and suppression of their activation in the absence of IL-6. Because this cytokine has been shown to be involved in glial cell activation that occurs secondarily to neuronal damage (Penkowa et al., 2001; Cardenas and Bolin, 2003), our findings do not exclude the occurrence of glial cell activation triggered by neuronal damage in the NPC1−/− brain. However, given the expression of TLR4, our data suggest that glial cell activation in the NPC1−/− mouse brain may occur in a cell-autonomous manner. The paracrine effect is evident by increased expression of STAT-1 and -3 by Purkinje cells and its normalization in the absence of IL-6. This is the first experimental demonstration that Purkinje cells in the NPC1−/− mouse cerebellum are under the influence of a substance secreted by glial cells. Despite its drastic effect on glial cell activation, however, genetic deletion of IL-6 in NPC1−/− mice failed to cause appreciable changes in Purkinje cell survival, and caused only a modest increase (∼11%) of their lifespan. These findings suggest a limited role of any substances secreted by glial cells in neurodegeneration, providing additional evidence for the notion that Purkinje cell death is a cell-autonomous event in these animals (Ko et al., 2005).
The roles of the other two cytokines, IFN-β and IL-8, in NPC pathogenesis remain to be investigated. The potential role of IFN-β, at least at the cellular level, is underscored by its activity to induce expression of MxA. Because MxA is a dynamin family GTPase involved in lipid transport (Accola et al., 2002), its increased expression may also represent one of the adaptive responses of the cells to resume membrane trafficking. Because IL-8 acts as a chemokine for inflammatory cells, it may also be implicated in glial cell activation in the NPC brain.
Our observations in double gene knock-out mice suggest that a benefit of an antiinflammatory therapy for NPC in this animal model is, at best, limited. This is consistent with the failure of minocycline, a drug that reduces IL-6 secretion (Zanjani et al., 2006), to delay the onset of neurological symptoms in NPC1−/− mice (Erickson and Bernard, 2002). A potential benefit of such a therapy against the human disease, however, remains to be determined. This notion is supported by our findings on KAI cells, which expressed marginally increased levels of TLR4 and did not secrete IL-6. Most of the NPC patients die within the second decade of life. The patient KAI was atypical in that he manifested an adult-onset phenotype, with neurological symptoms at the age of 25 and eventual demise at the age of 42 (Yamamoto et al., 2000). The cause–effect relationship between the lack of IL-6 secretion and the mild clinical phenotype warrants additional examination.
Finally, TLR4 has been implicated in glial cell activation that occurs in other, more common neurodegenerative diseases including Alzheimer's disease (Kakimura et al., 2002). NPC and Alzheimer's disease share several pathological features including the formation of neurofibrillary tangles (Suzuki et al., 1995) and the accumulation of β-amyloid (Saito et al., 2002), suggesting the presence of common biochemical mechanisms involved in the pathogenesis of these two diseases. Activation of TLR4 and downstream signaling events may be one of the common biochemical mechanisms involved in glial cell activation that occurs in the brains of NPC and Alzheimer's disease.
Footnotes
This work was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan. We thank K. Nagata for anti-MxA antibody, M. Kimoto for TLR4 and MD-2 expression plasmids, and S. Akira for TLR4 knock-out mice.
References
- Accola MA, Huang B, Masri AA, McNiven MA. The antiviral dynamin family member, MxA, tublates lipids and localizes to the smooth endoplasmic reticulum. J Biol Chem. 2002;277:21829–21835. doi: 10.1074/jbc.M201641200. [DOI] [PubMed] [Google Scholar]
- Ahmad I, Lope-Piedrafita S, Bi X, Hicks C, Yao Y, Yu C, Chaitkin E, Howison CM, Weberg L, Trouard TP, Erickson RP. Allopregnanolone treatment, both as a single injection or repetitively, delays demyelination and enhances survival of Niemann-Pick C mice. J Neurosci Res. 2005;82:811–821. doi: 10.1002/jnr.20685. [DOI] [PubMed] [Google Scholar]
- Akashi S, Shimazu R, Ogata H, Nagai Y, Takeda K, Kimoto M, Miyake K. Cutting edge: cell surface expression and lipopolysaccharide signaling via the Toll-like receptor 4-MD-2 complex on mouse peritoneal macrophages. J Immunol. 2000;164:3471–3475. doi: 10.4049/jimmunol.164.7.3471. [DOI] [PubMed] [Google Scholar]
- Akira S, Takeda K. Toll-like receptor signaling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- Baudry M, Yao Y, Simmons D, Liu J, Bi X. Postnatal development of inflammation in a murine model of Niemann-Pick type C disease: immunohistochemical observations of microglia and astroglia. Exp Neurol. 2003;184:887–903. doi: 10.1016/S0014-4886(03)00345-5. [DOI] [PubMed] [Google Scholar]
- Bi X, Liu J, Yao Y, Baudry M, Lynch G. Deregulation of the phosphatidylinositol-3 kinase signaling cascade is associated with neurodegeneration in npc1−/− mouse brain. Am J Pathol. 2005;167:1081–1092. doi: 10.1016/S0002-9440(10)61197-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blander JM, Medzhitov R. Regulation of phagosome maturation by signals from Toll-like receptors. Science. 2004;304:1014–1018. doi: 10.1126/science.1096158. [DOI] [PubMed] [Google Scholar]
- Cardenas H, Bolin LM. Compromised reactive microgliosis in MPTP-lesioned IL-6 KO mice. Brain Res. 2003;985:89–97. doi: 10.1016/s0006-8993(03)03172-x. [DOI] [PubMed] [Google Scholar]
- Carstea ED, Morris JA, Coleman KG, Loftus SK, Zhang D, Cummings C, Gu J, Rosenfeld MA, Pavan WJ, Krizman DB, Nagle J, Polymeropoulos MH, Sturley SL, Ioannou YA, Higgins ME, Comly M, Cooney A, Brown A, Kaneski CR, Blanchette-Mackie EJ, et al. Niemann-pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science. 1997;277:228–231. doi: 10.1126/science.277.5323.228. [DOI] [PubMed] [Google Scholar]
- de Bouteiller O, Merck E, Hasan UA, Hubac S, Benguigui B, Trinchieri G, Bates EE, Caux C. Recognition of double-stranded RNA by human Toll-like receptor 3 and downstream receptor signaling requires multimerization and an acidic pH. J Biol Chem. 2005;280:38133–38145. doi: 10.1074/jbc.M507163200. [DOI] [PubMed] [Google Scholar]
- Der SD, Zhou A, Williams BR, Silverman RH. Identification of genes differentially regulated by interferon α, β, or γ using oligonucleotide arrays. Proc Natl Acad Sci USA. 1998;95:15623–15628. doi: 10.1073/pnas.95.26.15623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle SE, O'Connell RM, Miranda GA, Vaidya SA, Chow EK, Liu PT, Suzuki S, Suzuki N, Modlin RL, Yeh WC, Lane TF, Cheng G. Toll-like receptors induce a phagocytic gene program through p38. J Exp Med. 2004;199:81–90. doi: 10.1084/jem.20031237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson RP, Bernard O. Studies on neuronal death in the mouse model of Niemann-Pick C disease. J Neurosci Res. 2002;68:738–744. doi: 10.1002/jnr.10257. [DOI] [PubMed] [Google Scholar]
- German DC, Liang CL, Song T, Yazdani U, Xie C, Dietschy JM. Neurodegeneration in the Niemann-Pick C mouse: glial involvement. Neuroscience. 2002;109:437–450. doi: 10.1016/s0306-4522(01)00517-6. [DOI] [PubMed] [Google Scholar]
- Griffin LD, Gong W, Verot L, Mellon SH. Niemann-Pick type C disease involves disrupted neurosteroidogenesis and responds to allopregnanolone. Nat Med. 2004;10:704–711. doi: 10.1038/nm1073. [DOI] [PubMed] [Google Scholar]
- Guillot L, Medjane S, Le-Barillec K, Balloy V, Danel C, Chignard M, Si-Tahar M. Response of human pulmonary epithelial cells to lipopolysaccharide involves Toll-like receptor 4 (TLR4)-dependent signaling pathways: evidence for an intracellular compartmentalization of TLR4. J Biol Chem. 2004;279:2712–2718. doi: 10.1074/jbc.M305790200. [DOI] [PubMed] [Google Scholar]
- Higashi Y, Murayama S, Pentchev PG, Suzuki K. Cerebellar degeneration in the Niemann-Pick type C mouse. Acta Neuropathol (Berl) 1993;85:175–184. doi: 10.1007/BF00227765. [DOI] [PubMed] [Google Scholar]
- Husebye H, Halaas O, Stenmark H, Tunheim G, Sandanger O, Bogen B, Brech A, Latz E, Espevik T. Endocytic pathways regulate Toll-like receptor 4 signaling and link innate and adaptive immunity. EMBO J. 2006;25:683–692. doi: 10.1038/sj.emboj.7600991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihle JN. STATs: signal transducers and activators of transcription. Cell. 1996;84:331–334. doi: 10.1016/s0092-8674(00)81277-5. [DOI] [PubMed] [Google Scholar]
- Ivashkiv LB, Hu X. The JAK/STAT pathway in rheumatoid arthritis: pathogenic or protective? Arthritis Rheum. 2003;48:2092–2096. doi: 10.1002/art.11095. [DOI] [PubMed] [Google Scholar]
- Kakimura J, Kitamura Y, Takata K, Umeki M, Suzuki S, Shibagaki K, Taniguchi T, Nomura Y, Gebicke-Haerter PJ, Smith MA, Perry G, Shimohama S. Microglial activation and amyloid-β clearance induced by exogenous heat-shock proteins. FASEB J. 2002;16:601–603. doi: 10.1096/fj.01-0530fje. [DOI] [PubMed] [Google Scholar]
- Kielian T. Toll-like receptors in central nervous system glial inflammation and homeostasis. J Neurosci Res. 2006;83:711–730. doi: 10.1002/jnr.20767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko DC, Gordon MD, Jin JY, Scott MP. Dynamic movements of organelles containing Niemann-Pick C1 protein: NPC1 involvement in late endocytic events. Mol Biol Cell. 2001;12:601–614. doi: 10.1091/mbc.12.3.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko DC, Milenkovic L, Beier SM, Manuel H, Buchanan J, Scott MP. Cell-autonomous death of cerebellar purkinje neurons with autophagy in Niemann-Pick type C disease. PLoS Genet. 2005;1:81–95. doi: 10.1371/journal.pgen.0010007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latz E, Visintin A, Lien E, Fitzgerald KA, Monks BG, Kurt-Jones EA, Golenbock DT, Espevik T. Lipopolysaccharide rapidly traffics to and from the Golgi apparatus with the toll-like receptor 4-MD-2-CD14 complex in a process that is distinct from the initiation of signal transduction. J Biol Chem. 2002;277:47834–47843. doi: 10.1074/jbc.M207873200. [DOI] [PubMed] [Google Scholar]
- Loftus SK, Morris JA, Carstea ED, Gu JZ, Cummings C, Brown A, Ellison J, Ohno K, Rosenfeld MA, Tagle DA, Pentchev PG, Pavan WJ. Murine model of Niemann-Pick C disease: mutation in a cholesterol homeostasis gene. Science. 1997;277:232–235. doi: 10.1126/science.277.5323.232. [DOI] [PubMed] [Google Scholar]
- Lusa S, Blom TS, Eskelinen EL, Kuismanen E, Mansson JE, Ikonen E. Depletion of rafts in late endocytic membranes is controlled by NPC1-dependent recycling of cholesterol to the plasma membrane. J Cell Sci. 2001;114:1893–1900. doi: 10.1242/jcs.114.10.1893. [DOI] [PubMed] [Google Scholar]
- Martinez-Moczygemba M, Gutch MJ, French DL, Reich NC. Distinct STAT structure promotes interaction of STAT2 with the p48 subunit of the interferon-α stimulated transcription factor ISGF3γ. J Biol Chem. 1997;272:20070–20076. doi: 10.1074/jbc.272.32.20070. [DOI] [PubMed] [Google Scholar]
- Millat G, Chikh K, Naureckiene S, Sleat DE, Fensom AH, Higaki K, Elleder M, Lobel P, Vanier MT. Niemann-Pick disease type C: spectrum of HE1 mutations and genotype/phenotype correlations in the NPC2 group. Am J Hum Genet. 2001;69:1013–1021. doi: 10.1086/324068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naureckiene S, Sleat DE, Lackland H, Fensom A, Vanier MT, Wattiaux R, Jadot M, Lobel P. Identification of HE1 as the second gene of Niemann-Pick C disease. Science. 2000;290:2298–2301. doi: 10.1126/science.290.5500.2298. [DOI] [PubMed] [Google Scholar]
- Olsson S, Sundler R. The role of lipid rafts in LPS-induced signaling in a macrophage cell line. Mol Immunol. 2006;43:607–612. doi: 10.1016/j.molimm.2005.04.011. [DOI] [PubMed] [Google Scholar]
- Patel SC, Suresh S, Kumar U, Hu CY, Cooney A, Blanchette-Mackie EJ, Neufeld EB, Patel RC, Brady RO, Patel YC, Pentchev PG, Ong WY. Localization of Niemann-Pick C1 protein in astrocytes: implications for neuronal degeneration in Niemann-Pick type C disease. Proc Natl Acad Sci USA. 1999;96:1657–1662. doi: 10.1073/pnas.96.4.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson MC, Vanier MT, Suzuki K, Morris JA, Carstea ED, Neufeld EB, Blanchette-Mackie EJ, Pentchev PG. Niemann-Pick disease type C: a lipid trafficking disorder. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001. pp. 3611–3633. [Google Scholar]
- Penkowa M, Molinero A, Carrasco J, Hidalgo J. Interleukin-6 deficiency reduces the brain inflammatory response and increases oxidative stress and neurodegeneration after kainic acid-induced seizures. Neuroscience. 2001;102:805–818. doi: 10.1016/s0306-4522(00)00515-7. [DOI] [PubMed] [Google Scholar]
- Saito Y, Suzuki K, Nanba E, Yamamoto T, Ohno K, Murayama S. Niemann-Pick type C disease: accelerated neurofibrillary tangle formation and amyloid β deposition associated with apolipoprotein E ε4 homozygosity. Ann Neurol. 2002;52:351–355. doi: 10.1002/ana.10266. [DOI] [PubMed] [Google Scholar]
- Schindler C. Cytokines and JAK/STAT signaling. Exp Cell Res. 1999;253:7–14. doi: 10.1006/excr.1999.4670. [DOI] [PubMed] [Google Scholar]
- Sugimoto Y, Ninomiya H, Ohsaki Y, Higaki K, Davies JP, Ioannou YA, Ohno K. Accumulation of cholera toxin and GM1 ganglioside in the early endosome of Niemann-Pick C1-deficient cells. Proc Natl Acad Sci USA. 2001;98:12391–12396. doi: 10.1073/pnas.221181998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Parker CC, Pentchev PG, Katz D, Ghetti B, D'Agostino AN, Carstea ED. Neurofibrillary tangles in Niemann-Pick disease type C. Acta Neuropathol (Berl) 1995;89:227–238. doi: 10.1007/BF00309338. [DOI] [PubMed] [Google Scholar]
- Triantafilou M, Miyake K, Golenbock DT, Triantafilou K. Mediators of innate immune recognition of bacteria concentrate in lipid rafts and facilitate lipopolysaccharide-induced cell activation. J Cell Sci. 2002;115:2603–2611. doi: 10.1242/jcs.115.12.2603. [DOI] [PubMed] [Google Scholar]
- Visintin A, Latz E, Monks BG, Espevik T, Golenbock DT. Lysines 128 and 132 enable lipopolysaccharide binding to MD-2, leading to Toll-like receptor-4 aggregation and signal transduction. J Biol Chem. 2003;278:48313–48320. doi: 10.1074/jbc.M306802200. [DOI] [PubMed] [Google Scholar]
- Walker JG, Ahern MJ, Coleman M, Weedon H, Papangelis V, Beroukas D, Roberts-Thomson PJ, Smith MD. Expression of Jak3, STAT1, STAT4 and STAT6 in inflammatory arthritis: unique Jak3 and STAT4 expression in dendritic cells in seropositive rheumatoid arthritis. Ann Rheum Dis. 2006;65:149–156. doi: 10.1136/ard.2005.037929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkley SU, Suzuki K. Consequences of NPC1 and NPC2 loss of function in mammalian neurons. Biochim Biophys Acta. 2004;1685:48–62. doi: 10.1016/j.bbalip.2004.08.011. [DOI] [PubMed] [Google Scholar]
- Wu YP, Mizukami H, Matsuda J, Saito Y, Proia RL, Suzuki K. Apoptosis accompanied by up-regulation of TNF-α death pathway genes in the brain of Niemann-Pick type C disease. Mol Genet Metab. 2005;84:9–17. doi: 10.1016/j.ymgme.2004.08.017. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Ninomiya H, Matusmoto M, Ohta Y, Nanba E, Tsutsumi Y, Yamakawa K, Millat G, Vanier MT, Pentchev PG, Ohno K. Genotype-phenotype relationship of Niemann-Pick disease type C: a possible correlation between clinical onset and levels of NPC1 protein in isolated skin fibroblasts. J Med Genet. 2000;37:707–712. doi: 10.1136/jmg.37.9.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanjani TM, Sabetkasaei M, Mosaffa N, Manaheji H, Labibi F, Farokhi B. Suppression of interleukin-6 by minocycline in a rat model of neuropathic pain. Eur J Pharmacol. 2006;538:66–72. doi: 10.1016/j.ejphar.2006.03.063. [DOI] [PubMed] [Google Scholar]