

Abstract
Cerebral amyloid angiopathy (CAA) is the accumulation of amyloid-β peptide (Aβ) in the vessel wall of arteries in the brain. Because CAA is commonly associated with Alzheimer's disease (AD), characterized by parenchymal deposition of the same peptide in the form of senile plaques, there is considerable interest in the relationship of the two deposits in generating human disease. The study of CAA is of particular importance for immunotherapeutic approaches to AD, because reports of anti-Aβ immunotherapy in mice and humans have suggested that, whereas CAA appeared resistant to clearance, its response to this treatment promoted potential adverse effects, including meningoencephalitis. We used multiphoton microscopy and longitudinal imaging to monitor CAA in a mouse model of amyloid deposition to evaluate the effects of anti-Aβ passive immunotherapy. We found detectable clearance of CAA deposits within 1 week after a single administration of antibody directly to the brain, an effect that was short-lived. Chronic administration of antibody over 2 weeks led to more robust clearance without evidence of hemorrhage or other destructive changes. We found that the progressive clearance of Aβ from vessels follows distinct kinetics from what has been previously reported for clearance of plaques (parenchymal deposits of Aβ). This quantitative in vivo imaging approach directly demonstrates that CAA in a transgenic mouse model can be cleared with an optimized immunotherapy.
Keywords: immunotherapy, CAA, neurodegeneration, in vivo imaging, Alzheimer's disease, amyloid
Introduction
The deposition of the β-amyloid peptide (Aβ) in the wall of cortical and leptomeningeal vessels [cerebral amyloid angiopathy (CAA)] is a common condition in the elderly that can lead to loss of vascular smooth muscle cells, weakening of the vessel wall, and increased risk of hemorrhagic and ischemic stroke (Zhang-Nunes et al., 2006). CAA is a frequent concomitant of Alzheimer's disease (AD), a finding that suggests that both entities may share a common pathogenic mechanism (Yamada, 2002).
Immunization with Aβ or treatment with anti-Aβ antibodies has proven to be an effective approach to clear or prevent plaque deposition, to improve cognitive performance in transgenic mouse models and other aspects of neuropathology such as neuritic dystrophy and synaptic density (Schenk et al., 1999; Bacskai et al., 2001; Kotilinek et al., 2002; Nicoll et al., 2003; Ferrer et al., 2004; Buttini et al., 2005). CAA, in contrast, appears to show little or no clearance in response to anti-Aβ immunotherapy (Bacskai et al., 2001; Pfeifer et al., 2002; Ferrer et al., 2004). Indeed, studies in some transgenic mouse lines have found treatment with anti-Aβ antibodies that bind to vascular amyloid to increase the incidence of CAA-related hemorrhage (Pfeifer et al., 2002; Wilcock et al., 2004; Racke et al., 2005), with one report even showing an association between microhemorrhages and an increase of vascular amyloid along parenchymal vessels as a result of immunotherapy (Wilcock et al., 2004).
Another recognized complication of immunotherapy possibly related to CAA is the meningoencephalitis that led to discontinuation of the clinical trial of active immunization with Aβ(1–42) (Nicoll et al., 2003; Orgogozo et al., 2003; Ferrer et al., 2004). This complication of therapy has clinical and pathological similarities to a spontaneously occurring syndrome of CAA-related vascular inflammation (Eng et al., 2004; Scolding et al., 2005).
The surface leptomeningeal vessels of the transgenic mouse brain provide an ideal system to examine the progression of CAA, because vessel segments of up to several hundred micrometers in length can be observed. We recently demonstrated that CAA developed in Tg2576 mice in a predictable manner (Domnitz et al., 2005) and have gone on to develop a quantitative approach to measure the kinetics of CAA progression in transgenic mouse models of amyloid angiopathy using serial in vivo imaging through cranial windows with multiphoton microscopy (Robbins et al., 2006). This approach allows direct imaging of identified amyloid deposits over time within living animals. With this method, we determined that CAA progresses with linear kinetics in Tg2576 mice; this system now provides us with a unique method to asses the effect of anti-Alzheimer's disease therapeutics on the rate of CAA progression. We have addressed whether the in vivo interaction of an anti-Aβ antibody influences the progression of vessel-associated amyloid. We found that single exposure to anti-Aβ antibody resulted in temporary mild regression of CAA in leptomeningeal vessels, whereas chronic exposure to antibody treatment leads to a more robust and sustained effect on CAA regression. This is the first demonstration of clearance of CAA with any therapeutic intervention.
Materials and Methods
Animals and antibody delivery.
A total of 20 Tg2576 mice (Hsiao et al., 1996), 10–13 months of age, had permanent cranial windows placed for serial in vivo imaging by multiphoton microscopy following a protocol described previously (Lombardo et al., 2003; Robbins et al., 2006). Mice were anesthetized with isoflurane, and 7 mm craniotomies were performed. The dura was carefully retracted to the midline with fine forceps. For experiments on animals receiving a single antibody application (“single dose”), 20 μl of 1 mg/ml of either an antibody against the N-terminal of Aβ (10D5) or a control antibody directed to an unrelated epitope (anti-human tau 16B5) was then applied to the surface of the brain immediately before attachment of a glass coverslip with dental cement that allowed visualization of the underlying brain surface. For animals receiving chronically administered antibody (“continuous dose”), an osmotic pump (Alzet; Durect Corporation, Cupertino, CA) was implanted subcutaneously in the interscapular region. The pump administered antibody at a rate of 0.5 ng/h during 14 d through a catheter placed along the surface of the brain at the edge of the imaging window. Indocyanine green (Akorn, Buffalo Grove, IL), a nontoxic dye that has excitation–emission spectrum in the near infrared range was included in the antibody solution to verify proper solution delivery. All animal studies were conducted under approval of the Massachusetts General Hospital Animal Care and Use Committee and in compliance with National Institutes of Health guidelines.
Imaging methods.
Mice were imaged immediately after surgery (day 0) while still under anesthesia, and then allowed to recover. To analyze changes in CAA progression, animals were anesthetized and reimaged on days 7 and 14 after surgery. To visualize Aβ, the day before each imaging session animals received an intraperitoneal injection of Methoxy-X04, a fluorescent dye that crosses the blood–brain barrier and selectively binds aggregated Aβ in blood vessels and plaques (Klunk et al., 2002). To facilitate image alignment from session to session, angiograms were performed by injecting Texas Red (0.05 ml; 35 mg/ml; 70,000 molecular weight; Invitrogen, Eugene, OR) into a lateral tail vein. Imaging was performed using a Bio-Rad 1024 MP microscope (Bio-Rad, Hercules, CA) with a Ti:Sapphire laser (Mai Tai; Spectra Physics, Mountain View, CA) at 800 nm for excitation. External photodetectors (Hamamatsu Photonics, Hamamatsu City, Japan) collected the blue fluorescent signal from bound Methoxy-X04. Multiple microscopic fields (615.8 × 615.8 μm) along the course of leptomeningeal cerebral arteries were imaged at 20× magnification [UMPlanFl; Olympus, Tokyo, Japan; numerical aperture (NA), 0.95] to a depth of ∼200 μm in 5 μm steps. Maximum intensity projections of z-series were obtained using ImageJ.
Postmortem analysis.
After the last imaging session, while still anesthetized with isoflurane, animals were euthanized by CO2 inhalation. After removal from the skull, brains were fixed in paraformaldehyde, rinsed in TBS for 10 min, immersed in 0.001% thioflavine S (ThioS) for 20 min and washed in TBS for 10 min. Brains were imaged using multiphoton microscopy as described above. Z-series were taken using an Olympus 2× objective (XL Fluor; NA, 0.14) to a depth of 4 mm with 100 μm z-steps in a 6158 × 6158 μm field. Four fields were required to image each brain dorsal surface, with photomerges performed using Adobe Photoshop. Reconstruction of Z stacks allowed definition of the area limited by the cranial window. The number of ThioS-positive plaques was counted, and the mean number of plaques inside and outside the window was calculated for each group under study.
A set of brains was used to obtain coronal sections of 30 μm thickness using a sliding microtome. Prussian blue staining for iron deposition, reflecting previous hemorrhage, was performed according to methods described previously (Wilcock et al., 2004).
Reagents.
Anti-Aβ antibody 10D5 and anti-human tau antibody 16B5 were a gift from Elan Pharmaceuticals (South San Francisco, CA); Methoxy-X04 was a gift from Dr. William Klunk (University of Pittsburgh, Pittsburgh, PA).
Texas Red dextran (70 kDa molecular weight) was purchased from Invitrogen. All other reagents, including thioflavin S, were obtained from Sigma (St. Louis, MO).
Serial imaging analyses.
Quantitative analysis of images was performed using ImageJ, Adobe Photoshop 7.0, and AutoDeblur 9.3 software packages as described previously (Robbins et al., 2006). CAA was visualized as Methoxy-X04-labeled deposits circumferential to the vessel wall. CAA-affected vessels were outlined and isolated from the background field. The CAA burden was calculated as the percentage of the vessel area affected by CAA. The vessel area from the initial imaging session was used for calculation of CAA burden because of the propensity for vasodilatation in subsequent weeks. To assess changes in CAA progression in serially imaged vessel segments, images from consecutive imaging sessions were aligned with minimal warping. Color overlays of the aligned images showed distinct areas of either clearance or new Aβ deposition. Additionally, the length of “gaps” (areas free of CAA) in micrometers in serially imaged vessel segments were measured in ImageJ at each time point. The length of gaps was measured, and the mean change in length was calculated for each group under study.
Statistical analysis.
To assess the nature of disease progression, we considered various mixed-effects models of CAA burden as a function of time as in Robbins et al., (2006). We adjusted for correlation within mice and within vessel segments through inclusion of random effects for mice and segments in the linear model. In this modeling process, variability was assumed to originate on the basis of mouse-to-mouse variation (internal variation) as well as to be influenced by location in the vascular tree (intra-animal variation). In practice, this amounted to allowing for mouse-specific and vessel segment-specific random intercepts in the linear model. To assess the effect of treatment, while allowing for the observed transient clearance of vascular Aβ, we fit a broken-line regression model that allowed for fixed treatment-specific slopes (i.e., progression rates) that could be different after day 7 versus before day 7.
To analyze the lengths of gaps between bands of CAA, we used linear normal mixed-effects models for the mean change at one time point as a function of length at the previous time points and treatment. We used Poisson regression to analyze the number of plaques, with adjustment for treatment and location (inside vs outside window). We used generalized estimating equations methodology to adjust for correlation.
Results
In vivo imaging of CAA in Tg2576 mice
Cranial windows were placed over the exposed brains of 10- to 13-month-old mice, an age range in which CAA deposition follows a linear model (Robbins et al., 2006). Mice were randomly assigned to groups exposed to a single subdural dose of an antibody against the N terminus of Aβ (10D5) or a control isotype-matched antibody directed against human tau (16B5). We chose 10D5 as the treatment antibody because of its proven efficacy on clearance of parenchymal Aβ deposits in both in vivo and ex vivo assays (Bard et al., 2000; Bacskai et al., 2001, 2003; Brendza et al., 2005; Hartman et al., 2005).
Mice were imaged with multiphoton laser-scanning microscopy on the initial day of the surgery (day 0), and the same brain volumes with identified leptomeningeal vessels were imaged again 7 and 14 d later. Fluorescent dye angiograms were used to identify stable fiduciary markers, a mapping system that allowed us to follow changes in Aβ deposition on individual leptomeningeal vessel segments over weekly imaging sessions. Quantitative imaging analyses allowed us to determine changes in the rate of CAA burden in living Tg2576 mice, measured as the percentage of vessel area involved, after antibody exposure. Through our direct visualization of the leptomeningeal vasculature with fluorescent angiograms, we did not observe any evidence of hemorrhage. This was confirmed by Prussian blue staining in postmortem tissue (data not shown).
Modest clearance of CAA by a single application of anti-Aβ antibody
Because direct application of anti-Aβ antibody to the surface of the brain had been shown to be rapidly effective at clearance of plaques in transgenic mice (Bacskai et al., 2001), we began by assessing this method of passive immunotherapy as a means of modulating CAA. The effects of a single application of 16B5 (control antibody) or 10D5 (anti-Aβ antibody) are shown in Figure 1 A1–B3. As expected, vessels in control-treated animals showed increases in CAA burden. Amyloid deposition not only did not increase in 10D5-treated vessel segments, but a modest regression was observed. Overlays of serial images from 10D5-treated vessel segments were used to highlight the changes in Aβ deposits along the vessel wall (Fig. 1 C1–C4) demonstrating that even a single application of 10D5 results in clearance of vascular Aβ by day 7. Quantitation of CAA burden at the three time points revealed a continuous progression in control-treated animals (Fig. 2 A) compared with a decline at day 7 in 10D5-treated animals (Fig. 2 B). The progression rate in the animals treated with control antibody was statistically indistinguishable from that which we had previously observed in Tg2576 mice (Robbins et al., 2006), indicating that the presence of immunoglobulin over the surface of the brain did not alter the intrinsic properties of the process. During the second week (days 7–14), both groups showed progression of CAA burden. This transient clearance of vascular Aβ is in contrast to the effects of antibody on parenchymal plaques, which remain cleared for up to 30 d after a single topical application of antibody (Bacskai et al., 2001; Lombardo et al., 2003). We confirmed the effectiveness of single-dose immunotherapy on plaques by finding a mean 60% decrease in the number of thioflavin S-positive plaques in the cortical area below the cranial window (i.e., the area of antibody exposure) relative to the number of plaques outside the window (p < 0.0001) (Table 1, Fig. 3).
Figure 1.

Serial in vivo imaging reveals progression of CAA on individual segments of leptomeningeal vessels and clearance after treatment with anti-Aβ antibody. Using multiphoton microscopy of individual segments, CAA-laden leptomeningeal vessels were visualized through cranial windows on day 0 (A1, B1), day 7 (A2, B2), and day 14 (A3, B3) after a single subdural antibody application of 16B5 (A1–A3) or 10D5 (B1–B3) antibodies. Z-stack images were taken as described previously, with Aβ deposits identified by fluorescence from systemically administered Methoxy-XO4 (gray pseudocolor). A1–A3, In 16B5-treated animals, an increase on CAA burden is observed through the three imaging sessions. B1–B3, In contrast, serial imaging of a 10D5-treated mouse revealed a modest reduction on CAA burden on day 7, with subsequent increase in vessel Aβ deposition by day 14 (B3); two areas of CAA showing these changes are indicated by arrows. C1, Amyloid deposits in a representative vessel from an animal treated with 10D5 are color-coded for changes in Aβ deposition at each time point and overlaid into a single image. C2, Deposits of CAA that were cleared between days 0 and 7. C3, Deposits of CAA that were unaltered during the same time interval. C4, Deposits of CAA that appeared during this interval (progression).
Figure 2.
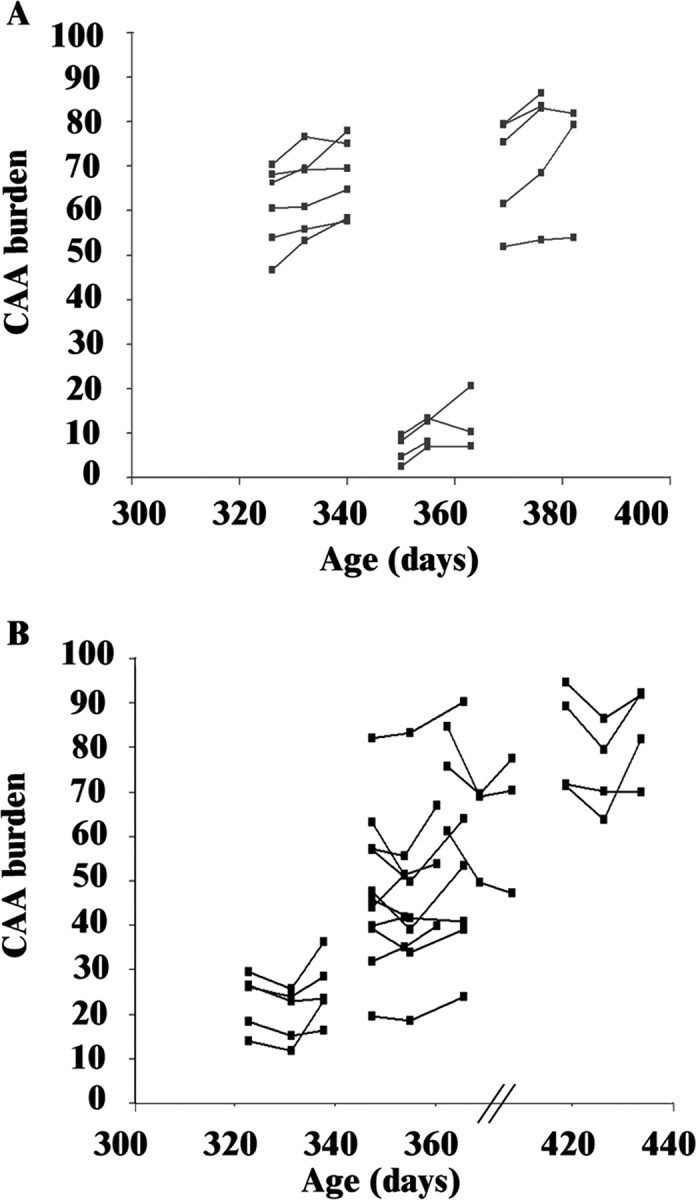
Single exposure to anti-Aβ antibody treatment decreases the progression of CAA. Measurements of CAA burden as percentage of available vessel area are plotted as a function of age in days, with treatment initiated at the time of the first data point for each animal. The variation in absolute CAA burden as a function of animal age and vessel segment involvement at that age account for the variation in position along the axes; the mixed-effects linear regression model compensates for this biological variation (Robbins et al., 2006). A, Animals treated with a single dose of 16B5 (n = 16 vessel segments from 3 mice) show progression of CAA burden over time, independent of the absolute level of disease at the time of the first observation. B, Mice treated with a single dose of 10D5 (n = 19 vessel segments from 4 mice) show overall clearance of Aβ at the second time point (day 7) but progression of disease during the interval from days 7 to 14.
Table 1.
Clearance of dense-core amyloid-β deposits after single topical application of anti-Aβ antibody
| Treatment | 16B5 | 10D5 | p |
|---|---|---|---|
| Plaque burden inside window | 6.0 (3.2–11.2)a | 3.2 (1.7–5.7)a | <0.0001 |
| Plaque burden outside window | 6.5 (1.7–5.7)a | 8.5 (5.0–14.3)a | NS |
Plaque burden indicates the mean number of ThioS-positive plaques inside and outside the window calculated for each group under study.
a95% confidence interval.
Figure 3.

Representative images of ThioS-positive plaque clearance after single topical application of anti-Aβ antibody. Images of a half-hemisphere of a brain of a mouse treated with 10D5 (left) and a mouse treated with 16B5 (right) are shown. The area of the cranial window is indicated by the yellow circle. Thio-S-positive plaques are shown in red.
To perform statistical analysis on these observations, we examined the slopes of the lines fit to the observations. Our previous studies of the kinetics of CAA progression in Tg2576 indicated that the leptomeningeal CAA burden progresses at a rate of 0.35% of available vessel area per day in the absence of antibody (Robbins et al., 2006). Using the same mixed-effects linear regression model, we found that single exposure to anti-Aβ antibody resulted in a regression of CAA deposits (negative slope) during the first week after dosing (−0.49% vessel surface area per day vs +0.5% per day progression with control antibody treatment; p < 0.0001) (Table 2). The rate of progression in animals treated with control antibody was not significantly different from the rate previously observed in animals not exposed to antibody in the subdural space (Robbins et al., 2006). Interestingly, the progression rate during the second week after anti-Aβ antibody treatment occurred at a rate greater than control antibody-treated mice (+0.84% per day vs +0.34% per day; p = 0.028) (Table 2), suggesting that deposition of amyloid in the vessel wall may accelerate after the short-lived clearance.
Table 2.
Decrease in the rate of CAA progression after treatment with anti-Aβ antibody
| 16B5 | 10D5 | p | |
|---|---|---|---|
| Single dose | |||
| Slope (days 0–7) | 0.5 ± 0.20 | −0.49 ± 0.15 | <0.0001 |
| Slope (days 7–14) | 0.34 ± 0.20 | 0.84 ± 0.10 | 0.028 |
| Continued dose | |||
| Slope (days 0–7) | 0.35 ± 0.17 | −0.6 ± 0.10 | <0.0001 |
| Slope (days 7–14) | 0.6 ± 0.13 | −0.23 ± 0.13 | <0.0001 |
The rate of progression of CAA as measured by the slope of the line determined by a mixed-effects linear regression model of quantitative measures obtained from in vivo imaging sessions after single or continued subdural antibody application of 16B5 or 10D5 antibodies is shown. Values are expressed as mean ± SE. Negative slopes (shown in italics) reflect clearance of CAA burden, whereas positive slopes reflect progression of disease.
Pattern of CAA clearance after single exposure to anti-Aβ antibody
Because our assay is based on quantitative imaging, we sought to determine whether the vascular Aβ deposits cleared by this paradigm followed a preferred pattern. We were particularly interested in whether the decrease in CAA burden occurred from random focal clearance of deposits or whether it was determined by the location and/or age of the deposits. To assess this issue, we measured the length of vessel wall free of amyloid between confluent deposits of CAA (gaps) and followed the changes in this length across three weekly imaging sessions. There was a significantly greater increase in the length of gaps between regions of confluent deposits during the first week after antibody dosing with anti-Aβ antibodies than with control antibodies (4.3 vs −2.1 μm; p < 0.0001), indicating that the confluent deposits of CAA were being cleared from their ends (Fig. 4). Combined with our previous observations that CAA grows predominantly by propagation of existing deposits (Robbins et al., 2006), these data suggested that the most recently deposited Aβ was the most readily cleared.
Figure 4.

Pattern of CAA clearance after single exposure to anti-Aβ antibody. The average change in the length of gaps (areas free of CAA) between deposits of Aβ measured at days 7 and 14 after single exposure to either 10D5 (black bars) or a 16B5 (white bars) is shown. A positive result indicates an increase in gap size, which corresponds to clearance of CAA. There was an increase in gap size only during the first week of treatment with anti-Aβ antibody (n = 29 gaps from 4 mice). The decrease in gap size during the subsequent week and during both weeks after treatment with control antibody 16B5 (n = 22 gaps from 3 mice) reflects the increase of CAA burden through propagation of deposits, as previously shown (Robbins et al., 2006). Error bars represent mean ± SD.
Greater clearance of CAA by chronic exposure to anti-Aβ antibody
The ability of topical application of a single dose of anti-Aβ antibodies to produce short-lived clearance of CAA whereas similar dosing approaches resulted in more persistent clearance of plaques in the same animals led us to consider whether only a portion of vascular amyloid was susceptible to clearance through antibody-based therapies. For this reason, we examined whether greater clearance of CAA could be achieved by chronic exposure to anti-Aβ antibody. Serial imaging was performed at days 0, 7, and 14 relative to initiation of continuous delivery of 10D5 and 16B5 antibodies by an osmotic pump. Whereas CAA progressed in the face of treatment with control antibody, we found that continuous delivery of anti-Aβ antibody resulted in significant reduction of CAA burden, which continued over 14 d. The rate of CAA progression in 16B5-treated animals was similar to previous observations (Robbins et al., 2006), although there was an acceleration in disease progression over time (+0.35% per day vs +0.6% per day; p < 0.046) (Fig. 5 A, Table 2). In contrast, with continued administration of anti-Aβ antibody, there was clearance of Aβ from vessel walls during both time periods examined Fig. 5 B, 6; Table 2).
Figure 5.
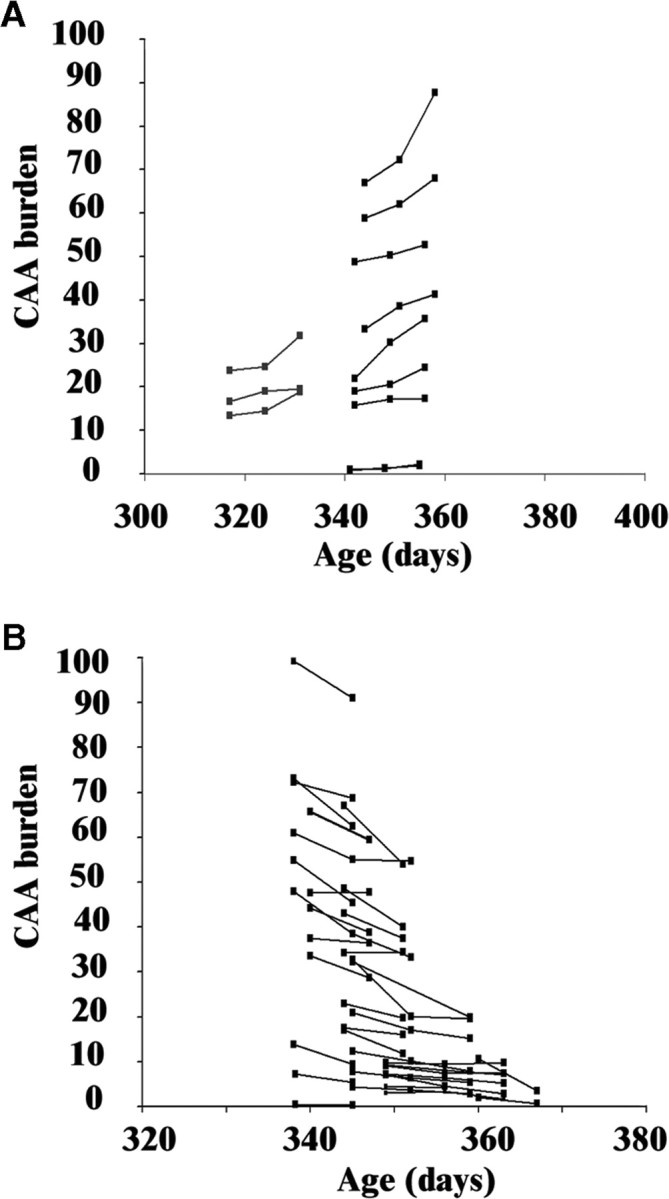
Chronic exposure to anti-Aβ antibody results in greater clearance of CAA. Measurements of CAA burden are plotted as in Figure 2. A, Animals treated with continuous infusion of 16B5 (n = 12 vessel segments from 4 mice) show progression of CAA burden over time, independent of the absolute level of disease at the time of the first observation. B, Mice treated with continuous infusion of 10D5 (n = 36 vessel segments from 8 mice) show overall clearance of Aβ at both the second time point (day 7) and third (day 14) imaging sessions. Experimental animals had either three or two imaging sessions.
Figure 6.

Serial in vivo imaging reveals greater clearance of CAA on individual segments of leptomeningeal vessels after continuous treatment with anti-Aβ antibody. Serial imaging of individual segments of CAA-laden leptomeningeal vessels was performed at day 0 (A1, B1), day 7 (A2, B2), and day 14 (A3, B3) relative to initiation of continuous delivery of 16B5 (A1–A3) or 10D5 (B1–B3) antibodies by an osmotic pump. Z-stack images were taken as described previously, with Aβ deposits identified by fluorescence from systemically administered Methoxy-XO4 (red pseudocolor). Angiograms were performed to identify fiduciary markers (blue pseudocolor). A1–A3, In 16B5-treated animals, an increase on CAA burden is observed through the three imaging sessions. B1–B3, In contrast, serial imaging of a 10D5-treated mouse revealed a significant reduction on CAA burden on day 7, which persisted through day 14 (B3). Scale bar, 100 μm.
Discussion
Although CAA is a well recognized pathological event, with >80% prevalence in AD, it has been difficult to study independently because of the lack of a specific biomarker in humans, and limited results in animal models. The recent characterization of the kinetics of CAA deposition in Tg2576 provides us with a tool well suited to study not only the underlying pathophysiological processes of CAA but also the consequences of therapies on this devastating vascular pathology (Domnitz et al., 2005; Robbins et al., 2006). The current study drew on our previous experience in the quantification of CAA progression and allowed us to examine the effects of anti-Aβ antibody treatment on affected blood vessels in an animal model of amyloid deposition.
Previous studies of the response of CAA to immunotherapy have suggested that the vascular deposits might at best be unresponsive to this treatment, and at worst lead to treatment-associated complications such as hemorrhage or inflammation (Bacskai et al., 2001; Pfeifer et al., 2002; Nicoll et al., 2003; Ferrer et al., 2004; Wilcock et al., 2004; Racke et al., 2005). However, it is important to note that there are several ways in which our study differs from these previous reports, including age of the animals, severity of the pathology, and the method of antibody administration.
Results presented here indicate that antibody treatment results in clearance of preexisting vascular amyloid in leptomeningeal vessels, just as had been previously described for plaques. The effect of a single dose of antibody on CAA clearance was modest, a result that is consistent with previous studies that reported no observable change on CAA burden in heavily affected vessels by antibody treatment (Bacskai et al., 2001; Pfeifer et al., 2002). To be able to detect this effect, our experimental design required examining mice at ages when CAA progressed at a predictably linear rate, serial observation of individual vascular segments at high spatial resolution, and a validated approach to quantification.
Our results demonstrate that, whereas plaques remain cleared after a single dose of antibody, the effect on CAA was short-lived. The fact that the effect of this paradigm of antibody treatment on CAA clearance is different from the striking effect on plaque burden may be attributable to factors intrinsic to vessel-associated Aβ deposits, including differential stability or rates of replenishment [as influenced by pathways of efflux of Aβ from the brain (Weller et al., 2004)]. Within the brain parenchyma, there are potential contributions to clearance from microglia and astrocytes (cell types that are not present in the subarachnoid space associated with leptomeningeal vessels). It is unlikely that local antibody concentration explains the relative resistance of CAA compared with plaques, because the vasculature is likely to be exposed to higher concentrations of antibody based on this method of delivery.
The progression rate during the second week after single dose treatment was at a rate greater than control antibody-treated mice. This result raises the possibility that CAA deposits may be more rapidly replenished in the presence of high levels of available Aβ when the clearance-promoting effects of anti-Aβ antibody are diminishing. Future investigations will be required to determine whether this represents reaccumulation of CAA deposits in the regions of previous clearance; if this were to be true, it would suggest that the presence of amyloid deposition had resulted in a structural change in the vessel wall that rendered it proamyloidogenic.
With continuous infusion of anti-Aβ antibody, the clearance of CAA was more robust, and continued throughout the duration of the treatment. This indicated that the results from single-dose treatments did not have limited efficacy because only a portion of the CAA was susceptible to clearance. Experiments with longer perfusion times would be required to determine whether all the vessel-associated Aβ deposits would be cleared by this approach.
In contrast to previous studies that reported an increase in CAA-associated microhemorrhages in APP and PDAPP transgenic mice that received passive anti-Aβ immunotherapy, (Pfeifer et al., 2002; Racke et al., 2005), Prussian blue staining to label hemosiderin in histological brain sections from our experimental animals did not detect hemorrhages. The efficacy of the staining was confirmed by inclusion of a known positive control (data not shown). Nor did we see hemorrhage during our in vivo imaging sessions with fluorescent angiography. Our results are also in contrast with a previous report by Wilcock et al., (2004), which found immunotherapy-related increase in vascular amyloid and microhemorrhage. A variety of possible explanations exist for these differences in results, including distinct pathologic mechanisms between parenchymal and leptomeningeal vessels as well as differences in experimental methods. All of the previous studies of immunotherapy-associated hemorrhagic outcomes have been reported on aged transgenic mice (20 months and older), whereas we studied significantly younger mice. In addition, the possibility that age plays a determinant factor on immunotherapy-associated microhemorrhages is underscored by two recent reports examining the effect of active immunization in transgenic mice. Whereas active Aβ vaccination resulted in microhemorrhages in 20-month-old double transgenic APP-PS1 mice (Wilcock et al., 2007), an immunization protocol in 10-month-old J20 APP mice, resulted in no instances of microhemorrhages (Seabrook et al., 2006). Other studies examining the effect of anti-Aβ in two different transgenic mouse models at a younger age (6 and 10 months) have not detected such events (Pfeifer et al., 2002; Chauhan and Siegel, 2003). Our findings are in agreement with these observations.
Although a study by Racke et al., (2005) in aged PDAPP mice found a relationship between CAA-associated microhemorrhage and the epitope targeted by the Aβ antibody being used, in the present study we also used an antibody directed against the N-terminal of Aβ and found no evidence of hemorrhagic events in younger Tg2576 mice. This supports our conclusion that mouse age and consequently the presence of advance degree of CAA may be a determinant factor in the increased incidence of microhemorrhage reported in anti-Aβ-treated mice.
In addition, we used direct administration of anti-Aβ antibody on CAA-affected leptomeningeal vessels, instead of systemic passive immunotherapy, which relies on the use of large quantities of antibody for long periods of time. Direct antibody administration limits the exposure of the cerebral vasculature to continuous high levels of luminal circulating anti-Aβ, which may increase the risk of adverse antibody-related vascular effects. This possibility is supported by the work of Chauhan and Siegel, (2003, 2005), which used intracerebral passive immunization in Tg2576 and TgCRND8 mice and found no evidence of adverse vascular effects.
Finally, our experimental approach used lower levels of antibody (total dose and duration of treatment) to produce significant clearance of CAA; these parameters also avoid the exposure of the cerebral vasculature to sustained high levels of antibody and may also influence the risk of hemorrhage. Together, our findings suggest that there may be a safe therapeutic window for passive anti-Aβ treatment and raises the possibility that methods of delivery using direct antibody administration to the CNS may constitute a more attractive mode of immunotherapy that can result in the clearance of plaques and CAA without eliciting adverse effects in the vasculature. This possibility may be further explored by future experiments using other methods of direct antibody administration to the CNS or antibody production within the CNS, such as the use of viral-mediated antibody expression in transgenic models of amyloid deposition.
Together, the data presented here suggest that passive immunotherapy may be tailored to result in clearance of CAA without resulting in deleterious vascular effects, provided that there is not an advanced degree of vascular involvement. In fact, the potential clinical impact of this finding is underlined by recent advances in the development of neuroimaging techniques that offer the promise of an early biomarker for Aβ deposition. An early detection of Aβ deposition would result in therapeutic strategies being directed toward patients in the early stages of the disease, a time when therapeutics have a higher possibility of modifying its progression.
It will be important in future studies to assess vascular function and determine whether application of an anti-Aβ antibody could actually influence the structural vascular pathology associated with CAA such as loss of smooth muscle cells, changes in blood flow, vasodilatation, and finally hemorrhagic outcomes.
Despite the large amount of encouraging evidence indicating that immune-based therapies ameliorate Aβ pathology, the finding that active immunization against the Aβ peptide elicits a possible autoimmune meningoencephalitis in a subset of human patients has served as a caution about potential adverse effects of this therapeutic strategy. A better understanding of the similarities and differences between vascular and parenchymal Aβ deposits in the context of immunotherapy will be needed to ensure the safety of this therapeutic approach. Our current results indicate that immunotherapy can be tailored not only for the treatment of AD, but also for CAA, leading to rationally designed immunotherapeutic approaches.
Footnotes
This work was supported by National Institutes of Health Grants AG020570, EB00768, and AG21084; Caja Madrid fellowship (M.G.-A.); Harvard Center for Neurodegeneration and Repair (R.A.B.); and a Howard Hughes Medical Institute Medical Trainee Research Fellowship (S.X.Z.-N.).
References
- Bacskai BJ, Kajdasz ST, Christie RH, Carter C, Games D, Seubert P, Schenk D, Hyman BT. Imaging of amyloid-beta deposits in brains of living mice permits direct observation of clearance of plaques with immunotherapy. Nat Med. 2001;7:369–372. doi: 10.1038/85525. [DOI] [PubMed] [Google Scholar]
- Bacskai BJ, Hickey GA, Skoch J, Kajdasz ST, Wang Y, Huang GF, Mathis CA, Klunk WE, Hyman BT. Four-dimensional multiphoton imaging of brain entry, amyloid binding, and clearance of an amyloid-beta ligand in transgenic mice. Proc Natl Acad Sci USA. 2003;100:12462–12467. doi: 10.1073/pnas.2034101100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- Brendza RP, Bacskai BJ, Cirrito JR, Simmons KA, Skoch JM, Klunk WE, Mathis CA, Bales KR, Paul SM, Hyman BT, Holtzman DM. Anti-Abeta antibody treatment promotes the rapid recovery of amyloid-associated neuritic dystrophy in PDAPP transgenic mice. J Clin Invest. 2005;115:428–433. doi: 10.1172/JCI23269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttini M, Masliah E, Barbour R, Grajeda H, Motter R, Johnson-Wood K, Khan K, Seubert P, Freedman S, Schenk D, Games D. β-Amyloid immunotherapy prevents synaptic degeneration in a mouse model of Alzheimer's disease. J Neurosci. 2005;25:9096–9101. doi: 10.1523/JNEUROSCI.1697-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan NB, Siegel GJ. Intracerebroventricular passive immunization with anti-Abeta antibody in Tg2576. J Neurosci Res. 2003;74:142–147. doi: 10.1002/jnr.10721. [DOI] [PubMed] [Google Scholar]
- Chauhan NB, Siegel GJ. Efficacy of anti-Abeta antibody isotypes used for intracerebroventricular immunization in TgCRND8. Neurosci Lett. 2005;375:143–147. doi: 10.1016/j.neulet.2004.10.090. [DOI] [PubMed] [Google Scholar]
- Domnitz SB, Robbins EM, Hoang AW, Garcia-Alloza M, Hyman BT, Rebeck GW, Greenberg SM, Bacskai BJ, Frosch MP. Progression of cerebral amyloid angiopathy in transgenic mouse models of Alzheimer disease. J Neuropathol Exp Neurol. 2005;64:588–594. doi: 10.1097/01.jnen.0000171644.00180.fc. [DOI] [PubMed] [Google Scholar]
- Eng JA, Frosch MP, Choi K, Rebeck GW, Greenberg SM. Clinical manifestations of cerebral amyloid angiopathy-related inflammation. Ann Neurol. 2004;55:250–256. doi: 10.1002/ana.10810. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Boada Rovira M, Sanchez Guerra ML, Rey MJ, Costa-Jussa F. Neuropathology and pathogenesis of encephalitis following amyloid-beta immunization in Alzheimer's disease. Brain Pathol. 2004;14:11–20. doi: 10.1111/j.1750-3639.2004.tb00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman RE, Izumi Y, Bales KR, Paul SM, Wozniak DF, Holtzman DM. Treatment with an amyloid-β antibody ameliorates plaque load, learning deficits, and hippocampal long-term potentiation in a mouse model of Alzheimer's disease. J Neurosci. 2005;25:6213–6220. doi: 10.1523/JNEUROSCI.0664-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Klunk WE, Bacskai BJ, Mathis CA, Kajdasz ST, McLellan ME, Frosch MP, Debnath ML, Holt DP, Wang Y, Hyman BT. Imaging Abeta plaques in living transgenic mice with multiphoton microscopy and methoxy-X04, a systemically administered Congo red derivative. J Neuropathol Exp Neurol. 2002;61:797–805. doi: 10.1093/jnen/61.9.797. [DOI] [PubMed] [Google Scholar]
- Kotilinek LA, Bacskai B, Westerman M, Kawarabayashi T, Younkin L, Hyman BT, Younkin S, Ashe KH. Reversible memory loss in a mouse transgenic model of Alzheimer's disease. J Neurosci. 2002;22:6331–6335. doi: 10.1523/JNEUROSCI.22-15-06331.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardo JA, Stern EA, McLellan ME, Kajdasz ST, Hickey GA, Bacskai BJ, Hyman BT. Amyloid-β antibody treatment leads to rapid normalization of plaque-induced neuritic alterations. J Neurosci. 2003;23:10879–10883. doi: 10.1523/JNEUROSCI.23-34-10879.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med. 2003;9:448–452. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, Jouanny P, Dubois B, Eisner L, Flitman S, Michel BF, Boada M, Frank A, Hock C. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology. 2003;61:46–54. doi: 10.1212/01.wnl.0000073623.84147.a8. [DOI] [PubMed] [Google Scholar]
- Pfeifer M, Boncristiano S, Bondolfi L, Stalder A, Deller T, Staufenbiel M, Mathews PM, Jucker M. Cerebral hemorrhage after passive anti-Abeta immunotherapy. Science. 2002;298:1379. doi: 10.1126/science.1078259. [DOI] [PubMed] [Google Scholar]
- Racke MM, Boone LI, Hepburn DL, Parsadainian M, Bryan MT, Ness DK, Piroozi KS, Jordan WH, Brown DD, Hoffman WP, Holtzman DM, Bales KR, Gitter BD, May PC, Paul SM, DeMattos RB. Exacerbation of cerebral amyloid angiopathy-associated microhemorrhage in amyloid precursor protein transgenic mice by immunotherapy is dependent on antibody recognition of deposited forms of amyloid β. J Neurosci. 2005;25:629–636. doi: 10.1523/JNEUROSCI.4337-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins EM, Betensky RA, Domnitz SB, Purcell SM, Garcia-Alloza M, Greenberg C, Rebeck GW, Hyman BT, Greenberg SM, Frosch MP, Bacskai BJ. Kinetics of cerebral amyloid angiopathy progression in a transgenic mouse model of Alzheimer disease. J Neurosci. 2006;26:365–371. doi: 10.1523/JNEUROSCI.3854-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- Scolding NJ, Joseph F, Kirby PA, Mazanti I, Gray F, Mikol J, Ellison D, Hilton DA, Williams TL, MacKenzie JM, Xuereb JH, Love S. Abeta-related angiitis: primary angiitis of the central nervous system associated with cerebral amyloid angiopathy. Brain. 2005;128:500–515. doi: 10.1093/brain/awh379. [DOI] [PubMed] [Google Scholar]
- Seabrook TJ, Jiang L, Thomas K, Lemere CA. Boosting with intranasal dendrimeric Abeta1–15 but not Abeta1–15 peptide leads to an effective immune response following a single injection of Abeta1–40/42 in APP-tg mice. J Neuroinflammation. 2006;3:14. doi: 10.1186/1742-2094-3-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller RO, Cohen NR, Nicoll JA. Cerebrovascular disease and the pathophysiology of Alzheimer's disease. Implications for therapy. Panminerva Med. 2004;46:239–251. [PubMed] [Google Scholar]
- Wilcock DM, Rojiani A, Rosenthal A, Levkowitz G, Subbarao S, Alamed J, Wilson D, Wilson N, Freeman MJ, Gordon MN, Morgan D. Passive amyloid immunotherapy clears amyloid and transiently activates microglia in a transgenic mouse model of amyloid deposition. J Neurosci. 2004;24:6144–6151. doi: 10.1523/JNEUROSCI.1090-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, Jantzen PT, Li Q, Morgan D, Gordon MN. Amyloid-beta vaccination, but not nitro-nonsteroidal anti-inflammatory drug treatment, increases vascular amyloid and microhemorrhage while both reduce parenchymal amyloid. Neuroscience. 2007;144:950–960. doi: 10.1016/j.neuroscience.2006.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M. Risk factors for cerebral amyloid angiopathy in the elderly. Ann NY Acad Sci. 2002;977:37–44. doi: 10.1111/j.1749-6632.2002.tb04797.x. [DOI] [PubMed] [Google Scholar]
- Zhang-Nunes SX, Maat-Schieman ML, van Duinen SG, Roos RA, Frosch MP, Greenberg SM. The cerebral beta-amyloid angiopathies: hereditary and sporadic. Brain Pathol. 2006;16:30–39. doi: 10.1111/j.1750-3639.2006.tb00559.x. [DOI] [PMC free article] [PubMed] [Google Scholar]