

Abstract
Mutations in the parkin gene are a major cause of autosomal recessive Parkinson's disease. Here we show that the E3 ubiquitin ligase parkin activates signaling through the IκB kinase (IKK)/nuclear factor κB (NF-κB) pathway. Our analysis revealed that activation of this signaling cascade is causally linked to the neuroprotective potential of parkin. Inhibition of NF-κB activation by an IκB super-repressor or a kinase-inactive IKKβ interferes with the neuroprotective activity of parkin. Furthermore, pathogenic parkin mutants with an impaired neuroprotective capacity show a reduced ability to stimulate NF-κB-dependent transcription. Finally, we present evidence that parkin interacts with and promotes degradation-independent ubiquitylation of IKKγ/NEMO (NF-κB essential modifier) and TRAF2 [TNF (tumor necrosis factor) receptor-associated factor 2], two critical components of the NF-κB pathway. Thus, our results support a direct link between the neuroprotective activity of parkin and ubiquitin signaling in the IKK/NF-κB pathway.
Keywords: IKK, NF-κB, parkin, Parkinson's disease, TRAF, ubiquitin
Introduction
Among the genes that are responsible for monogenic familial variants of Parkinson's disease (PD), the parkin gene (PARK2) seems to play a prominent role accounting for the majority of autosomal recessive PD cases (Mata et al., 2004; West and Maidment, 2004). Parkin, a 465 amino acid E3 ubiquitin ligase, is characterized by an ubiquitin-like (UBL) domain at the N terminus and a RING box close to the C terminus (Kitada et al., 1998). A consistent observation in cell culture and animal models is the neuroprotective capacity of parkin. Parkin has been shown to protect cultured cells against cell death induced by kainate, proteasomal inhibition, α-synuclein, ceramide, manganese, dopamine, and unfolded protein stress (Imai et al., 2000; Petrucelli et al., 2002; Darios et al., 2003; Staropoli et al., 2003; Higashi et al., 2004; Jiang et al., 2004; Muqit et al., 2004). In Drosophila, overexpression of parkin can suppress loss of dopaminergic neurons induced by α-synuclein or Pael-R (parkin-associated endothelin-like receptor) (Yang et al., 2003; Haywood and Staveley, 2004). Furthermore, lentiviral delivery of parkin prevents dopaminergic degeneration caused by mutant α-synuclein in a rat model and protects mouse skeletal muscle cells against mitochondrial toxins (Lo Bianco et al., 2004; Rosen et al., 2006).
To gain insight into the mechanism underlying the broad neuroprotective capacity of parkin, we investigated a possible role of parkin in stress response pathways. This analysis revealed that parkin specifically activates the nuclear factor-κB (NF-κB) pathway and that this pathway is essential for the neuroprotective action of parkin. NF-κB transcription factors regulate various biological processes, including apoptosis, differentiation, and immunity. Although almost ubiquitously expressed, NF-κB proteins are inhibited by their association with IκBs (Hayden and Ghosh, 2004). Many stimuli activate NF-κB through distinct upstream pathways that converge at the IκB kinase (IKK) complex, consisting of two catalytic (IKKα and IKKβ) and one regulatory [IKKγ/NEMO (NF-κB essential modifier)] subunit. Classical IKK/NF-κB signaling involves IKKβ/γ-mediated phosphorylation of IκBs, which targets the inhibitors for ubiquitylation and subsequent proteasomal degradation. Recent research has established a major role for ubiquitylation in the regulation of IKK/NF-κB signaling pathways (for review, see Ravid and Hochstrasser, 2004; Chen, 2005; Haglund and Dikic, 2005; Krappmann and Scheidereit, 2005). Besides the well known role of lysine 48 (K48)-linked polyubiquitylation in the regulation of IκB degradation and precursor proteolysis, activation of the IKK complex and other upstream regulators, such as TRAF2 [tumor necrosis factor (TNF) receptor-associated factor] and TRAF6, is dependent on the assembly of lysine 63 (K63)-linked ubiquitin chains.
NF-κB is present throughout the nervous system, and there is accumulating evidence that the NF-κB signaling pathway plays a central role in neuronal integrity and synaptic plasticity (for review, see Karin and Lin, 2002). Importantly, recent cell culture and animal models established a role of NF-κB in preventing neuronal death induced by oxidative stress, excitotoxicity, ischemia, or glucose deprivation (for review, see Mattson et al., 2000; Kaltschmidt et al., 2005). In this study, we identified parkin as a novel activator of IKK/NF-κB signaling, suggesting that parkin mediates its neuroprotective effect by modulating ubiquitin signaling in the IKK/NF-κB pathway.
Materials and Methods
DNA constructs.
The following constructs were described previously: wild-type (wt) human parkin, full-length parkin (M80T), ΔN parkin (Δ1–79), and the pathogenic parkin mutants R42P and G430D (Winklhofer et al., 2003; Henn et al., 2005), FLAG–IKKβ K/A, FLAG–TRAF2, and FLAG–TRAF6 (Krappmann et al., 2000), FLAG–IKKγ (Tegethoff et al., 2003), FLAG–TRAF2ΔN (Rothe et al., 1995), hemagglutinin (HA)–wt–ubiquitin and FLAG-IκBΔN (Krappmann et al., 1996), HA–K48-only and HA–K63-only ubiquitin (Evans et al., 2004), heat shock element (HSE)–luciferase (Luc) (Winklhofer et al., 2001), and NF-κB–Luc (Krappmann et al., 2001). The phosphorylated forms of enhanced yellow fluorescent protein (EYFP) and enhanced green fluorescent protein were purchased from Clontech (Mountain View, CA).
Antibodies and reagents.
The following antibodies were used: anti-parkin hP1 polyclonal antibody (pAb) (Winklhofer et al., 2003), anti-parkin pAb (catalog #2132; Cell Signaling Technology, Danvers, MA), anti-parkin PRK28 and PRK8 monoclonal antibody (mAb) (Pawlyk et al., 2003), anti-HA mAb (12CA5; Hiss Diagnostics, Freiburg, Germany), anti-HA pAb (H 6908; Sigma, Taufkirchen, Germany), anti-ACTIVE caspase-3 pAb (Promega, Mannheim, Germany), cyanine 3 (Cy3)-conjugated anti-rabbit IgG antibody (Dianova, Hamburg, Germany), FITC-conjugated anti-mouse IgG antibody (Sigma), anti-FLAG M2 mAb (Sigma), anti-α-tubulin mAb (N356; Amersham Biosciences, Freiburg, Germany), anti-β-actin mAb (AC-74; Sigma), anti-TRAF2 mAb (33a1293; Abcam, Cambridge, UK), anti-TRAF2 pAb (C-20; Santa Cruz Biotechnology, Heidelberg, Germany), anti-IKKγ mAb (B-3; Santa Cruz Biotechnology), anti-IKKγ pAb (FL-419; Santa Cruz Biotechnology), anti-p65 pAb (C-20; Santa Cruz Biotechnology), and horseradish peroxidase (HRP)-conjugated anti-mouse and anti-rabbit IgG antibody (Amersham Biosciences). Glutamate, rotenone, and phorbol 12-myristate 13-acetate (PMA) were purchased from Sigma, kainic acid was from Calbiochem (Darmstadt, Germany), recombinant human TNFα was from Biomol (Hamburg, Germany), and complete protease inhibitor cocktail was from Roche (Mannheim, Germany).
Cell culture and transfection.
SH-SY5Y and HEK293T (CRL-1573; American Type Culture Collection, Manassas, VA) cells were cultivated and transfected as described previously (Henn et al., 2005). Fibroblasts from a PARK2 patient were described previously (Nakaso et al., 2006).
Preparation and cultivation of primary rat cortical neurons.
Cortices were removed from the brains of embryonic day 18 rats, and cells were dissociated by mild trypsination and trituration as described previously (Mattson et al., 1993). The dissociated neurons were then seeded onto polyethyleneimine-coated 60 mm culture dishes and grown in Neurobasal medium (Invitrogen, Karlsruhe, Germany) supplemented with 5 mm HEPES, 1.2 mm glutamine, B27 supplement (20 ml/L; Invitrogen), and gentamicin (0.1 mg/ml). All experimental treatments were performed on 8- to 10-d-old cultures, at which time they contained <5% astrocytes as determined previously by GFAP immunocytochemistry. To induce glutamate excitotoxicity, neurons were exposed to 5 and 10 μm glutamate in EBSS medium (6800 mg/L NaCl, 400 mg/L KCl, 264 mg/L CaCl2 · 2H2O, 200 mg/L MgCl2 · 7H2O, 2200 mg/L NaHCO3, and 140 mg/L NaH2PO4 · H2O, pH 7.2) with 10 mm glucose. After the indicated time, cells were harvested for RNA extraction as described below.
Western blot analysis and metabolic labeling.
Protein analysis by Western blotting and metabolic labeling of cellular proteins were performed as described previously (Henn et al., 2005). For the detection, we used the enhanced chemiluminescence detection system (Amersham Biosciences) or the Immobilon Western chemiluminescent HRP substrate (Millipore, Schwalbbach, Germany).
Immunoprecipitation.
Cells were washed twice with cold PBS, scraped off the plate, pelleted by centrifugation, and lysed in cold lysis buffer A (0.1% Triton X-100 in PBS, supplemented with protease inhibitor cocktail). Cell lysates were cleared by centrifugation (16,000 × g, 20 min, 4°C), and equal amounts of protein of the detergent-soluble fraction were incubated overnight at 4°C with the antibody indicated. The antigen–antibody complexes were captured by the addition of immobilized protein A or G (Pierce, Bonn, Germany) and then washed three times with lysis buffer. Proteins present in the immunoprecipitates were released from the protein A/G-beads by the addition of Laemmli sample buffer containing 1% SDS and heating at 100°C for 5 min.
Immunofluorescence and confocal microscopy.
SH-SY5Y cells were grown on glass coverslips, transfected, fixed 16 h after transfection in 3% paraformaldehyde and 3% sucrose in PBS for 10 min at room temperature, and permeabilized with 0.1% Triton X-100. Fixed cells were incubated with primary antibody (diluted in 1% BSA and 10% goat serum) for 1 h at room temperature. For double staining, antibody incubations were performed sequentially. After washes with PBS, the coverslips were incubated with Cy3- or FITC-conjugated secondary antibodies. Finally, cells were embedded in Mowiol mounting medium (Calbiochem) supplemented with 4′,6′-diamidino-2-phenylindole (DAPI). Images were obtained on a Leica (Wetzlar, Germany) DM IRE2 confocal microscope and evaluated with the Leica confocal software version 2.6.1.
Apoptosis assay.
SH-SY5Y cells were grown on glass coverslips. At 24 h after transfection, cells were incubated with kainate (500 μm) or rotenone (10 μm) for 3 h. The cells were then fixed with 3% paraformaldehyde for 20 min, permeabilized with 0.2% Triton X-100 for 10 min at room temperature, and blocked with 5% horse serum, 0.1% Tween 20 in PBS for 1 h at room temperature. Fixed cells were incubated with anti-ACTIVE caspase-3 antibody overnight at 4°C, followed by an incubation with Cy3-conjugated secondary antibody for 1 h at room temperature. After extensive washing, cells were mounted onto glass slides and examined by fluorescence microscopy using a Zeiss (Jena, Germany) Axioscope2 plus microscope. To detect cells undergoing apoptosis, the number of activated caspase-3-positive cells of at least 300 transfected cells was determined. Quantifications were based on at least three independent experiments.
Luciferase assay.
Luciferase reporter plasmid (HSE–Luc or NF-κB–Luc) and wt parkin or the indicated parkin mutants were cotransfected into HEK293T or SH-SY5Y cells. At 24 h after transfection, cells were heat shocked (HSE–Luc) at 42°C for 15 min and further incubated at 37°C for 16 h, or incubated with the agents indicated (NF-κB–Luc) at 37°C for 3 h and harvested after additional 8–16 h. Luciferase activity of cell lysates was determined luminometrically by the dual-luciferase assay system (Promega) as specified by the manufacturer. Each transfection was performed in triplicate, and all experiments were repeated at least three times.
RNA interference.
Human parkin was targeted with Stealth small interfering RNA (siRNA) duplex 1 synthesized by Invitrogen (sense 5′-UUCGCAGGUGACUUUCCUCUGGUCA-3′); Stealth siRNA negative control duplex was used as a control. siRNA duplexes were transfected into HEK293T cells using Lipofectamine 2000 (Invitrogen) according to the protocol of the manufacturer.
Ubiquitylation assay.
Full-length parkin, HA-tagged ubiquitin, or the indicated ubiquitin mutants and either FLAG-tagged IKKγ or TRAF2 were cotransfected into HEK293T cells. One day after transfection, protein extracts were prepared in denaturing lysis buffer (50 mm Tris/HCl, pH 7.4, 5 mm EDTA, 1% SDS, 15 U/ml DNase I, and protease inhibitor cocktail) and boiled for 5 min. Protein extracts were diluted 1:10 with nondenaturing lysis buffer (50 mm Tris/HCl, pH 7.4, 300 mm NaCl, 5 mm EDTA, 1% Triton X-100, and protease inhibitor cocktail), and immunoprecipitation was performed as described above. Proteins present in the immunoprecipitates were analyzed by Western blotting using an anti-HA antibody.
Real-time PCR.
SH-SY5Y cells were incubated with 1 μm rotenone or 50 μm kainate for 3 h. Cells were harvested 5, 8, 12, and 24 h after drug treatment, respectively. Primary rat cortical neurons were incubated with 5 or 10 μm glutamate for 6 h and then harvested. Total cellular RNA was isolated and treated with DNase I according to the instructions of the manufacturer (RNeasy mini kit; Qiagen, Hilden, Germany). cDNA was synthesized from 1 μg of total RNA using iScript cDNA Synthesis kit (Bio-Rad, Munich, Germany). Each quantitative PCR (20 μl) included 2 μl of cDNA solution, 2× TaqMan Universal PCR Master Mix, and TaqMan Gene Expression Assay (parkin; Hs01038827-m1; Applied Biosystems, Foster City, CA). For rat neurons, quantitative PCR was performed with 2× Power SYBR Green PCR Master Mix (Applied Biosystems) and 1 μm of each primer pair (rat parkin forward primer, 5′-AGA CAA GCA ACC CTC ACC TT-3′; rat parkin reverse primer, 5′-TGG CAC TCT CCA CTC ATC C-3′; rat β-actin forward primer, 5′- CTC TCT TCC AGC CTT CCT TC-3′; rat β-actin reverse primer, 5′-GGT CTT TAC GGA TGT CAA CG-3′). Quantification was performed with 7500 Fast Real Time System (Applied Biosystems). Triplicates were performed with each primer set for each RNA sample. RNA expression was normalized with respect to β-actin (P/N 4326315 E; Applied Biosystems). Relative expression was calculated for each gene using the ΔΔCT method.
Electrophoretic mobility shift assays.
To prepare nuclear extracts, cells were scraped into ice-cold PBS and pelleted by brief centrifugation. Cell pellets were resuspended in three packed cell volumes of hypotonic cell lysis buffer (20 mm HEPES, pH 7.9, 10 mm KCl, 5 mm MgCl2, 0.5 mm EDTA, 0.1% Triton X-100, 20% glycerol, and 2 mm DTT). After 10 min incubation on ice, the nuclei were pelleted by centrifugation for 5 min at 1000 rpm at 4°C. The nuclei were resuspended in three pellet volumes of high-salt buffer (20 mm HEPES, pH 7.9, 10 mm KCl, 5 mm MgCl2, 0.5 mm EDTA, 0.1% Triton X-100, 20% glycerol, 2 mm DTT, and 300 mm NaCl) and incubated for 15 min at 4°C. After centrifugation (5 min at 1000 rpm, 4°C), protein concentration of the supernatants (nuclear extracts) was determined by the Bradford assay. Binding reactions were performed in a final volume of 20 μl consisting of 10 mm HEPES, pH 7.9, 50 mm NaCl, 5 mm MgCl2, 2 mm DTT, 0.1 mm EDTA, 5% glycerol, 10 μg of BSA, 2 μg of poly(dI-dC), 5 μg (cell culture) or 10 μg (fibroblasts) nuclear extract, and 0.15 ng (15,000 cpm) of 32P-labeled, double-stranded NF-κB consensus oligonucleotide (5′-AGT TGA GGG TTT CCC AGG C-3′; Promega) or OCT1 consensus oligonucleotide (5′-TGT CGS ATG CAA ATC ACT AGA A-3′; Promega). After binding on ice for 30 min, mixtures were loaded onto nondenaturing 4% polyacrylamide gels in 0.5× TBE (45 mm Tris borate and 1 mm EDTA). Gels were electrophoresed at 4°C for 4 h at 160 V, dried, and exposed for autoradiography at −80°C.
Statistical analysis.
Data were expressed as means ± SE. All transfections were performed in triplicates and repeated at least three times. Statistical analysis among groups was performed using Student's t test. p values are as follows: *p < 0.01, **p < 0.001, and *** p < 0.0001.
Results
Parkin is a stress-inducible protein with neuroprotective capacity
Parkin has been shown to protect neurons against diverse cellular insults in different model systems, indicating that it might play a central role in maintaining neuronal integrity. To confirm and extend these studies, we analyzed the effect of parkin on cell viability under stress conditions that play a prominent pathophysiological role in PD, namely inhibition of complex I of the mitochondrial electron transport chain and excitotoxicity. To identify cells undergoing apoptosis, human SH-SY5Y neuroblastoma cells transiently transfected with wild-type parkin were analyzed by indirect immunofluorescence using an antibody specific for activated caspase-3. A protective effect of parkin was observed both in cells treated with the complex I inhibitor rotenone and in those treated with the excitotoxin kainate (Fig. 1A), whereas the pathogenic parkin mutants R42P and G430D showed a reduced neuroprotective capacity (Fig. 1B). Interestingly, a smaller parkin species lacking the N-terminal UBL domain (ΔN parkin) was also impaired in protecting cells from stress-induced cell death (Fig. 1B). We have shown previously that an internal translation initiation site in the human parkin gene is responsible for the generation of ΔN parkin (Henn et al., 2005). Of note, ΔN parkin is also found in human brain (Shimura et al., 1999; Schlossmacher et al., 2002; Staropoli et al., 2003). The protective activity of parkin was observed not only at early time points after stress treatment (determined by caspase-3 assay) but also after 24 h (determined by Hoechst 33342 staining; data not shown).
Figure 1.

Parkin is a stress-inducible protein with neuroprotective capacity. A, B, Parkin has a neuroprotective capacity in cultured cells. A, SH-SY5Y cells were cotransfected with EYFP and wt parkin. At 24 h after transfection, cells were incubated with kainate (500 μm) or rotenone (10 μm) at 37°C for 3 h, fixed, permeabilized, and analyzed by indirect immunofluorescence. Activation of caspase-3 was detected using the anti-ACTIVE caspase-3 pAb. Shown is the percentage of apoptotic cells among the transfected cells. B, SH-SY5Y cells were cotransfected with EYFP and either wt parkin or the parkin mutants indicated. At 24 h after transfection, cells were incubated with kainate (500 μm) at 37°C for 3 h and analyzed by indirect immunofluorescence as described in A. *p < 0.01, **p < 0.001 compared with cells expressing wt parkin. As a control for parkin expression, an aliquot of the cell lysates was immunoblotted with the anti-parkin pAb hP1 (bottom). C–F, Endogenous parkin is upregulated after cellular stress. SH-SY5Y cells were incubated with 50 μm kainate (C) or 1 μm rotenone (D) for 3 h. Cells were harvested 5, 8, 12, and 24 h after the drug treatment, respectively. Total cellular RNA was isolated and subjected to quantitative RT-PCR using parkin-specific primers. The amount of RNA of each sample was normalized with respect to the endogenous housekeeping control gene β-actin. Shown is the fold increase in the amount of parkin-specific mRNA compared with the untreated control. E, Primary cortical neurons derived from embryonic rat brain were incubated with glutamate (5 or 10 μm) for 6 h and then analyzed as described in C and D. F, SH-SY5Y cells were treated with rotenone (1 μm, 3 h) or mock treated, and expression of endogenous parkin was analyzed by Western blotting using the PRK8 mAb.
To increase evidence for a functional role of parkin in cellular stress management, we analyzed whether transcription/translation of endogenous parkin might be regulated by stress. Quantitative reverse transcription (RT)-PCR analysis revealed that parkin-specific mRNA levels were significantly increased in response to kainate or rotenone treatment with a maximal increase 5–8 h after drug treatment (Fig. 1C,D). Stress inducibility of parkin was also observed in primary cortical neurons derived from embryonic rat brain (Fig. 1E). Importantly, the stress-induced increase in parkin mRNA translates into elevated parkin protein levels (Fig. 1F).
Parkin stimulates NF-κB-dependent transcription
The spectrum of stressors parkin can cope with is remarkably wide, suggesting an influence on key signaling pathways. To address the question of which mechanism might integrate all protective effects described, we tested whether parkin has an impact on general stress response pathways. A major anti-stress pathway involves the upregulation of molecular chaperones in response to proteotoxic stress via activation of heat shock transcription factors (HSF), which bind to HSEs in the regulatory region of genes coding for diverse chaperones. Another stress response pathway is associated with the activation of the NF-κB transcription factor, which induces the expression of antiapoptotic genes after binding to specific NF-κB-responsive elements in their enhancer regions. To test for a possible role of parkin in the activation of these pathways, SH-SY5Y cells and human embryonic kidney (HEK293T) cells were transiently transfected with a plasmid coding for human parkin and a reporter plasmid expressing firefly luciferase under the control of an inducible promotor regulated by either the heat shock transcription factor 1 (HSE–Luc) or NF-κB (NF-κB–Luc). As a positive control, cells were subjected to a moderate heat shock or treated with PMA, a classical activator of the NF-κB pathway. Although parkin showed no effect on the HSF-inducible pathway, luciferase activity was significantly increased by parkin in cells harboring the NF-κB-responsive reporter (Fig. 2A,B). An increase in luciferase-specific transcripts was confirmed by quantitative RT-PCR, indicating that differences in the activity of luciferase observed in the presence of parkin were not attributable to alterations of luciferase at the protein level, such as folding or stability (data not shown).
Figure 2.

Parkin stimulates NF-κB-dependent transcription. A, HEK293T cells were cotransfected with a reporter plasmid encoding luciferase under control of an HSF1-inducible promoter (HSE–Luc) and wt parkin. At 24 h after transfection, the cells were subjected to a heat shock (42°C for 15 min), returned to 37°C, and harvested after an additional 16 h. Shown is the fold induction of luciferase activity compared with the EYFP control. B, HEK293T cells were cotransfected with a reporter plasmid encoding luciferase under control of an NF-κB-responsive promoter (NF-κB–Luc) and wt parkin. At 24 h after transfection, the cells were incubated with PMA (10 ng/ml) at 37°C for 3 h and further incubated at 37°C for 16 h. C–E, Parkin sensitizes cells to NF-κB-activating stimuli. C, D, HEK293T cells were cotransfected with NF-κB–Luc and wt parkin. At 24 h after transfection, the cells were incubated with TNFα (25 ng/ml; C) or PMA (5 ng/ml; D) at 37°C and harvested after 7 h. E, HEK293T cells transfected with either wt parkin or vector were incubated with TNFα for 30 min. Nuclear extracts were prepared and probed for NF-κB binding activity by an electrophoretic mobility shift assay. The higher-molecular-weight complex contains p65 as determined by supershift experiments (data not shown).
With regard to the neuroprotective potential of parkin, we reasoned that parkin might increase or even potentiate the activation of NF-κB in response to stress. To address this possibility experimentally, we treated cells with TNFα or PMA under conditions that only moderately activated NF-κB. When parkin was expressed in TNFα- or PMA-treated cells, a supra-additive increase in luciferase activity was observed (Fig. 2C,D). To verify this observation with an independent approach, we analyzed NF-κB activation by monitoring its DNA binding activity. Electrophoretic mobility shift assays indicated that, in the presence of parkin, NF-κB binding activity was significantly increased after stimulation of cells with TNFα. Activation of NF-κB was even observed with low amounts of TNFα, which did not induce NF-κB binding activity in the absence of parkin (Fig. 2E). Thus, parkin sensitizes cells to NF-κB-activating stimuli.
Pathogenic mutations impair the NF-κB-activating potential of parkin
To investigate whether the neuroprotective potential of parkin is linked to the activation of the NF-κB pathway, we included a variety of pathogenic parkin mutants in our analysis. Several studies indicated that different mechanisms account for the loss-of-function phenotype of parkin mutants, such as parkin misfolding and aggregation or a decrease in the stability of full-length parkin (Winklhofer et al., 2003; Henn et al., 2005; Sriram et al., 2005; Wang et al., 2005). The pathogenic parkin mutants R42P and G430D as well as ΔN parkin, which are characterized by a reduced neuroprotective capacity (Fig. 1B), also showed a decreased potential to activate NF-κB-dependent transcription (Fig. 3). Although the decreased stability of full-length R42P parkin may account for the reduced NF-κB activation, steady-state levels of the G430D mutant were not decreased. Remarkably, ΔN parkin was characterized by the lowest potential to activate NF-κB. Conversely, full-length parkin devoid of the internal initiation site showed the highest potential to induce NF-κB-dependent transcription (Fig. 3). This suggests that the N-terminal UBL domain of parkin plays an important role in mediating activation of NF-κB-dependent transcription and neuroprotection.
Figure 3.

Pathogenic parkin mutations decrease the NF-κB-activating potential of parkin. HEK293T cells cotransfected with the NF-κB reporter plasmid and either wt parkin or the parkin mutants indicated were harvested 24 h after transfection. Shown is the fold induction of luciferase activity compared with the EYFP control. *p < 0.01, **p < 0.001, ***p < 0.0001 compared with cells expressing wt parkin. As a control for parkin expression, an aliquot of the cell lysates was immunoblotted with the anti-parkin pAb hP1 (bottom panels). α-Tubulin was used as a loading control.
Activation of NF-κB is crucial for the neuroprotective capacity of parkin
To confirm that the effect of parkin on NF-κB-dependent transcription involves activation of NF-κB and results from enhanced IKK signaling, we made use of the NF-κB super-repressor IκBΔN. IκBΔN lacks a N-terminal domain (amino acids 71–317) that contains the IKK phospho-acceptor sites (Krappmann et al., 1996). When IκBΔN was coexpressed, parkin could no longer stimulate NF-κB-dependent transcription (Fig. 4A), indicating that indeed activation of NF-κB is responsible for the parkin-induced effect. Importantly, this effect was independent of the cell line used (data not shown). To further explore at which site parkin might activate the NF-κB signaling cascade, we tested an inhibitor upstream of IκB. A kinase-inactive IKKβ mutant (IKKβ K/A) carrying two mutations in the activation loop of IKKβ blocks activation of the NF-κB pathway at the level of the IKK signalosome (Delhase et al., 1999). Similar to IκBΔN, the kinase-inactive IKKβ K/A mutant suppressed the effect of parkin on NF-κB-dependent transcription (Fig. 4B), indicating that parkin may act on the IKK complex and/or on upstream signaling components.
Figure 4.

The effect of parkin on NF-κB-dependent transcription can be antagonized by dominant-negative IκB or kinase-inactive IKKβ. A, HEK293T cells were cotransfected with NF-κB reporter plasmid, wt parkin, and the NF-κB super-repressor IκBΔN. At 24 h after transfection, cell lysates were analyzed for luciferase activity. Shown is the fold induction of luciferase activity compared with the EYFP control. As a control for protein expression, aliquots of the cell lysates were immunoblotted with the anti-parkin pAb hP1 and anti-FLAG mAb (bottom panels). α-Tubulin was used as a loading control. B, HEK293T cells were cotransfected with the NF-κB reporter plasmid, wt parkin, and the kinase-inactive IKKβ mutant IKKβ K/A. Luciferase assay and immunoblotting were performed as described in A.
To test whether there is a causal relationship between the neuroprotective potential of parkin and its ability to activate NF-κB signaling, we analyzed the neuroprotective capacity of parkin under conditions that specifically interfere with the activation of NF-κB, i.e., in the presence of IκBΔN. Remarkably, expression of the NF-κB super-repressor IκBΔN completely abrogated the antiapoptotic effect of parkin in kainate-treated cells (Figs. 5A, 6). Similarly, the kinase-inactive IKKβ K/A mutant suppressed cell survival induced by parkin (Fig. 5B), demonstrating that the neuroprotective potential of parkin is critically dependent on IKK/NF-κB signaling.
Figure 5.
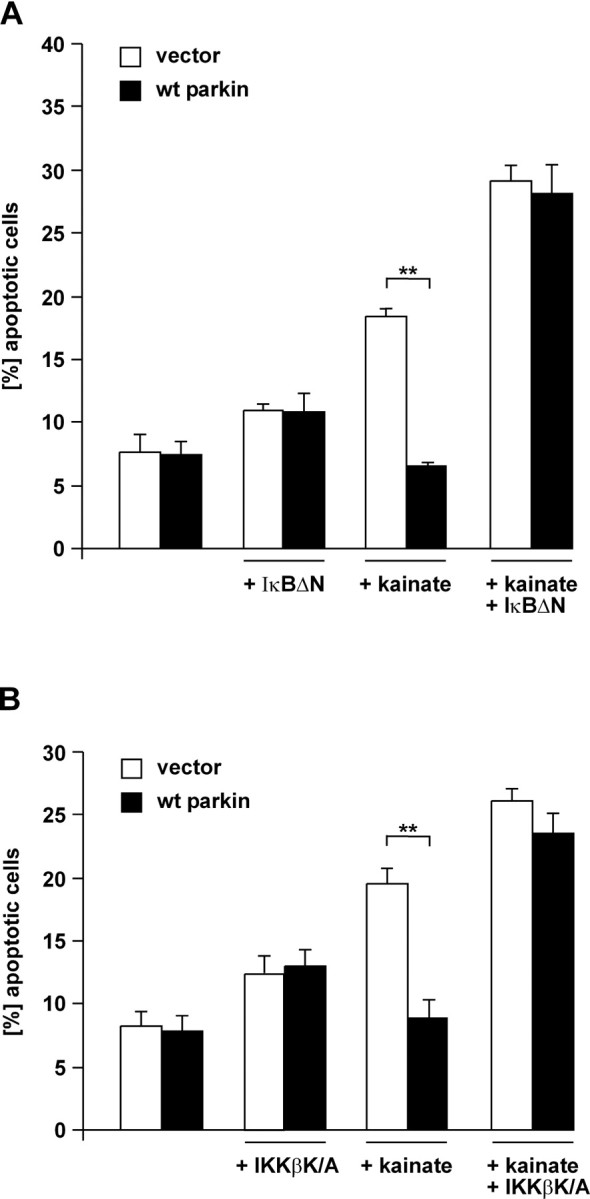
Activation of NF-κB is essential for the neuroprotective effect of parkin. A, SH-SY5Y cells were cotransfected with EYFP, wt parkin, and IκBΔN. At 24 h after transfection, cells were incubated with kainate (500 μm) at 37°C for 3 h, fixed, permeabilized, and analyzed by indirect immunofluorescence. Activation of caspase-3 was detected by using the anti-ACTIVE caspase-3 pAb. Shown is the percentage of apoptotic cells among the transfected cells. **p < 0.001 compared with EYFP-expressing cells. B, SH-SY5Y cells were cotransfected with EYFP, wt parkin, and the kinase-inactive IKKβ mutant IKKβ K/A and analyzed as described in A. **p < 0.001 compared with EYFP-expressing cells.
Figure 6.
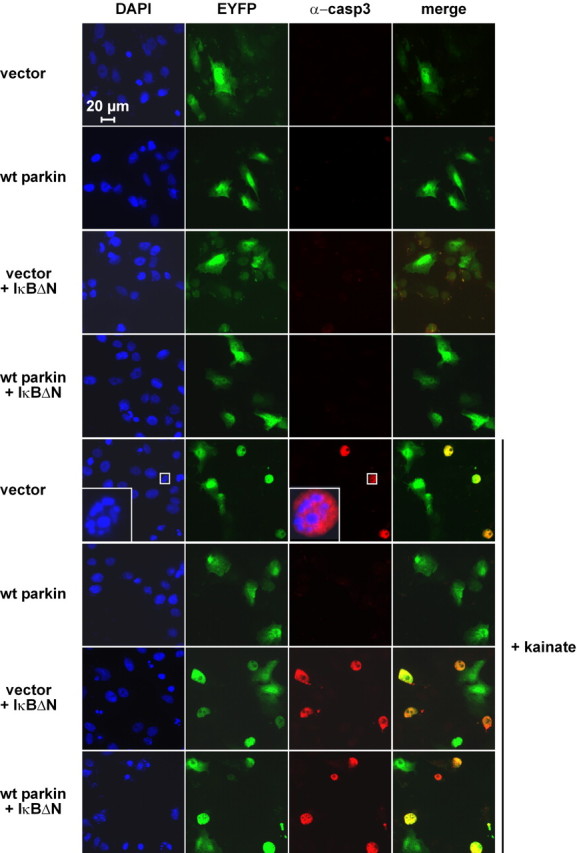
In the presence of the NF-κB super-repressor IκBΔN, parkin shows no neuroprotective activity. Immunofluorescence analysis of the experiment described in Figure 5A. Activation of caspase-3 was detected by using the anti-ACTIVE caspase-3 pAb (red), and nuclei were stained with DAPI (blue). Scale bar, 20 μm
Parkin promotes ubiquitylation of IKKγ and TRAF2
Activation of the NF-κB pathway involves ubiquitylation at different levels of the signaling cascade. To determine which protein(s) in the signaling cascade might be a target for parkin-mediated ubiquitylation, we performed ubiquitylation assays in HEK293T cells coexpressing full-length parkin, HA-tagged ubiquitin, and FLAG-tagged TRAF2, TRAF6, or IKKγ. Immunoprecipitation under denaturing conditions was performed with an anti-FLAG antibody, and precipitated proteins were subjected to a Western blot analysis using an anti-HA antibody. We observed that full-length parkin can increase the amount of ubiquitylated IKKγ and TRAF2, whereas TRAF6 ubiquitylation was not increased in the presence of parkin (Fig. 7A–C). ΔN parkin and R42P parkin, which showed a significantly lower activity in stimulating NF-κB-dependent transcription as well as protecting cells from toxic insults, were also compromised in mediating ubiquitylation of IKKγ and TRAF2. Notably, parkin also promotes ubiquitylation of TRAF2ΔN, which lacks the N-terminal RING domain essential for its E3 ligase activity, indicating that the increased ubiquitylation is not attributable to autoubiquitylation mediated by TRAF2 itself (Fig. 7B).
Figure 7.

Parkin promotes ubiquitylation of IKKγ and TRAF2. A–C, Full-length (fl) parkin, ΔN parkin, or R42P, HA-tagged ubiquitin, and either FLAG-tagged TRAF6 (A), TRAF2 (B), or IKKγ (C) were cotransfected into HEK293T cells. At 24 h after transfection, cells were harvested, lysed, and cleared by centrifugation. Equal amounts of protein in the supernatants were subjected to an immunoprecipitation (IP) under denaturing conditions using an anti-FLAG mAb. The immunoprecipitates were resolved by SDS-PAGE and immunoblotted (IB) with an anti-HA mAb (top panels). Aliquots of the supernatant before the immunoprecipitation were immunoblotted with antibodies against parkin (hP1), TRAF6 (mAb), TRAF2 (mAb), and IKKγ (mAb), respectively (input controls; bottom panels). Molecular size markers are indicated as bars at the left side of the panels and represent 148, 98, and 64 kDa (A, B) or 98, 64, and 50 kDa (C). v, Vector control; b, buffer control (no lysate). D, Parkin has no effect on the stability of IKKγ and TRAF2. HEK293T cells were cotransfected with either IKKγ or TRAF2 and wt parkin. One day after transfection, cells were metabolically labeled with [35S]methionine for 30 min and either analyzed directly or incubated in fresh medium for an additional 6, 12, and 24 h (chase) before the immunoprecipitation of IKKγ or TRAF2. E, Parkin preferentially promotes K63-linked polyubiquitylation of IKKγ. Full-length parkin, FLAG-tagged IKKγ, and HA-tagged wild-type ubiquitin, K48-only, or K63-only ubiquitin mutant were cotransfected into HEK293T cells. At 24 h after transfection, cells were harvested and analyzed as described in A.
To assess whether parkin-mediated ubiquitylation targets IKKγ and TRAF2 for proteasomal degradation, we analyzed the stability of both proteins in the presence of parkin. Pulse-chase experiments revealed that coexpression of parkin has no effect on the stability of IKKγ and TRAF2 over a period of 24 h (Fig. 7D), indicating that parkin does not induce proteasomal degradation of these proteins but rather promotes nondegradative ubiquitylation. To provide experimental evidence for this assumption, we made use of ubiquitin mutants lacking all but one lysine (K48-only and K63-only, HA-tagged). These mutants either promote ubiquitin chain assembly via lysine 48, targeting the ubiquitylated protein for proteasomal degradation, or via lysine 63, which serves nonproteolytic regulatory functions (for review, see Pickart and Fushman, 2004). Ubiquitylation assays performed as described above revealed that, in the presence of K48-only ubiquitin, parkin-mediated ubiquitylation of IKKγ is significantly decreased, whereas K63-only ubiquitin can maintain IKKγ ubiquitylation (Fig. 7E). This indicates that parkin preferentially promotes K63-linked ubiquitylation.
Parkin is found in a complex together with IKKγ and TRAF2
The data presented above point to IKKγ and TRAF2 as potential targets of parkin. To increase evidence for a physiological relevance of the observed effects, we examined a potential interaction of these proteins. First, we performed coimmunoprecipitation experiments with lysates prepared from transiently transfected HEK293T cells using a polyclonal anti-IKKγ or anti-TRAF2 antibody. Coprecipitating proteins were subjected to SDS-PAGE, and immunoblotting with a monoclonal anti-parkin antibody revealed that IKKγ as well as TRAF2 were present in a complex together with parkin, indicating that both proteins can directly or indirectly interact with parkin (Fig. 8A). To provide evidence that the observed interaction is not attributable to the overexpression of the individual proteins, we performed a coimmunoprecipitation analysis with endogenous proteins (Fig. 8B). Finally, confocal microscopy was used to examine the subcellular localization of parkin, IKKγ, and TRAF2. When expressed in SH-SY5Y cells individually, parkin, TRAF2, and IKKγ were distributed throughout the cytosol. Coexpression of parkin and TRAF2 or IKKγ, however, induced the accumulation of TRAF2 and IKKγ in the perinuclear region, in which they colocalized with parkin (Fig. 9).
Figure 8.

Parkin is found in a complex together with IKKγ and TRAF2. A, HEK293T cells were cotransfected with full-length (fl) parkin and either IKKγ (top left panel) or TRAF2 (top right panel). One day after transfection, cells were harvested, lysed, and cleared by centrifugation. Equal protein amounts of the supernatant were incubated with a pAb against IKKγ or TRAF2 overnight at 4°C. Proteins present in the immunoprecipitates (IP) were resolved by SDS-PAGE and immunoblotted (IB) with the anti-parkin mAb PRK28. Aliquots of the supernatant were immunoblotted with antibodies against parkin (pAb hP1), IKKγ (mAb), and TRAF2 (mAb), respectively (input controls; bottom panels). B, Untransfected SH-SY5Y cells were lysed as described in A and incubated with the polyclonal anti-parkin antibody hP1 or with a polyclonal anti-HA antibody (both antibodies cross-linked to protein A-agarose) overnight at 4°C (* indicates buffer instead of cell lysate). Proteins present in the immunoprecipitates were resolved by SDS-PAGE and immunoblotted with monoclonal antibodies against IKKγ (left) or TRAF2 (right). For input controls, a parallel sample was analyzed by immunoprecipitation, followed by immunoblotting for the respective proteins. v, Vector control; b, buffer control (no lysate).
Figure 9.

IKKγ and TRAF2 colocalize with parkin. SH-SY5Y cells attached to coverslips were transfected with wt parkin and FLAG-tagged IKKγ or TRAF2. One day after transfection, cells were fixed, permeabilized, and stained with the following antibodies: anti-parkin hP1 pAb (red) and anti-FLAG mAb (green). DAPI was added to the mounting medium to label nuclei. Red arrows indicate the regions in which intensity profiles (bottom panels) along a line were determined using Leica confocal software version 2.6.1. Scale bars, 10 μm
Loss of endogenous parkin increases cell death and compromises NF-κB activation
To increases evidence for a role of endogenous parkin in protecting cells from stress-induced cell death, we downregulated parkin expression by a RNA interference approach. HEK293T cells were transfected with parkin-specific siRNA duplexes and, 48 h later, were exposed to rotenone. After an additional 24 h, cell viability was determined by trypan blue exclusion. Downregulation of parkin (Fig. 10A) significantly reduced cell viability compared with control siRNA-treated cells (Fig. 10B). To address the question of whether the increase in cell death observed in parkin knockdown cells is accompanied by an alteration in NF-κB signaling, we performed NF-κB reporter assays in siRNA-treated cells. Indeed, NF-κB-dependent transcription was reduced in parkin knockdown cells after stress treatment (Fig. 10C).
Figure 10.

Loss of endogenous parkin increases cell death and compromises NF-κB activation in response to stress. A, B, HEK293T cells were transfected with parkin-specific or control siRNA duplexes. A, At 48 h later, parkin mRNA levels were analyzed by RT-PCR (described in Fig. 1C), and endogenous parkin protein was analyzed by Western blotting (bottom) as described in Figure 1F. B, Parallel cultures were exposed to rotenone (0.05 μm, 3 h), and, after additional 24 h, cell viability was determined by trypan blue exclusion. C, HEK293T cells cotransfected with parkin-specific siRNA and the NF-κB reporter plasmid were incubated with PMA (10 and 20 ng/ml, 3 h). At 5 h later, cells were harvested and analyzed for luciferase activity. D, Fibroblasts from a PARK2 patient or control fibroblasts were exposed to PMA for 30 min. Nuclear extracts were prepared, and NF-κB binding activity was determined by electrophoretic mobility shift assays as described in Figure 2E. Bottom left, Supershift induced by a pAb against p65 in nuclear extracts from PMA-treated control fibroblasts. Bottom right, As a control, OCT1 binding activity was determined in nuclear extracts from untreated and PMA-treated PARK2 and control fibroblasts.
Finally, we analyzed fibroblasts obtained from a patient with compound heterozygous deletions in the parkin gene (deleted exon 3–4/deleted exon 3–5). Parkin protein is not detectable in this patient (Nakaso et al., 2006). Consistent with our results from cell culture experiments, activation of the NF-κB pathway in response to stress was significantly compromised in the patient's fibroblasts compared with age-matched control fibroblasts (Fig. 10D).
Discussion
A milestone in PD research was the discovery of genes that are responsible for familial variants of this disease. Sporadic and familial variants of PD share important pathological features, most notably the demise of dopaminergic neurons in the substantia nigra. Consequently, insights into the function of PD-associated genes may facilitate a better understanding of the pathomechanism not only of hereditary PD but also of sporadic PD. In this study, we focused on the physiological role of parkin and showed that activation of NF-κB signaling underlies the neuroprotective potential of parkin.
Parkin can protect neuronal cells against a remarkably wide array of intrinsic and extrinsic stressors. Most studies reporting on the protective potential of parkin are consistent concerning the nature of the stressors tested, and discrepancies can be explained by the fact that high-level stress conditions inactivate parkin because of its tendency to misfold (Winklhofer et al., 2003; LaVoie et al., 2005; Wang et al., 2005; Winklhofer and Tatzelt, 2006). In our study, we therefore chose conditions of moderate stress, which more likely occur under physiological conditions. We first evaluated the cytoprotective activity of parkin for two stressors relevant to PD, inhibition of complex I of the electron transport chain (induced by rotenone) and excitotoxicity (induced by kainate or glutamate). In support of a role of parkin in coping with cellular stress, we found that both stressors significantly increased the amount of parkin-specific mRNA in cultured neuroblastoma cells as well as primary neurons, resulting in an increased expression of parkin protein. These observations are in line with a recent publication showing activation of the parkin promoter after hydrogen peroxide treatment by a luciferase reporter assay (Tan et al., 2005).
Because of the broad cytoprotective activity of parkin, we reasoned that it may have an impact on a key survival pathway. Reporter assays specific for stress-inducible pathways revealed that parkin stimulates NF-κB-dependent transcription. In the course of this study, three lines of evidence have converged to suggest that activation of the NF-κB signaling cascade is essential for the neuroprotective activity of parkin. First, there is a correlation between the neuroprotective activity of parkin and its ability to activate NF-κB. Pathogenic parkin mutants impaired in their neuroprotective capacity are also compromised in their ability to efficiently stimulate NF-κB-dependent transcription. In this context, it should be pointed out that pathogenic parkin mutants do not show a complete loss of function, at least during overexpression in cultured cells. However, subtle differences in the activity of parkin may be pathophysiologically relevant over decades, especially under conditions of increased cellular stress. Second, when the NF-κB pathway is blocked, parkin loses its protective activity. In the presence of the NF-κB super-repressor IκBΔN or the kinase-inactive IKKβ mutant IKKβ K/A, parkin no longer protects cells from kainate-induced toxicity. Finally, downregulation of endogenous parkin by an RNA interference approach results in an increase in cell death along with a decrease in NF-κB signaling in response to stress. Notably, a smaller parkin species, which occurs in human brain attributable to the presence of an internal initiation site and lacks the N-terminal UBL (ΔN parkin), is significantly impaired in activating the NF-κB pathway and thus in protecting cells from toxic insults. This observation is consistent with the finding that a mutation in the authentic initiation codon of parkin is pathogenic (Rawal et al., 2003; Mata et al., 2004). In this case, the second initiation codon is presumably used, giving rise to N-terminally truncated, functionally less active parkin. The internal initiation site at codon 80 is only present in human parkin; thus, two parkin species with different functional activities coexist in neuronal cells, which might explain why humans are particularly vulnerable to the inactivation of full-length parkin.
Seminal research in the last decade has revealed that ubiquitin is a highly versatile protein and that ubiquitylation can serve a variety of nonproteolytic functions, such as regulation of intracellular trafficking, gene transcription, DNA repair, and cell signaling. At the same time, a considerable variability of the ubiquitylation process emerged, spanning mono-ubiquitylation, multi-ubiquitylation, and polyubiquitylation as well as the formation of different polyubiquitin chains involving distinct lysine residues of ubiquitin.
The NF-κB pathway is a paradigm for the role of ubiquitylation in mediating degradation-dependent and degradation-independent functions. Conventional roles of ubiquitin in the NF-κB pathway include targeting of IκB for degradation as well as inducing the proteasomal processing of the NF-κB precursors p105 and p100. Unconventional degradation-independent polyubiquitin chain attachment is essential for the activation of the IKK signalosome and involves ubiquitylation of TRAF2, TRAF6, receptor-interacting protein, and IKKγ/NEMO (Chen, 2005; Krappmann and Scheidereit, 2005).
In an attempt to establish a possible role of the E3 ubiquitin ligase parkin in the NF-κB pathway, we analyzed whether parkin has an impact on the ubiquitylation of specific signaling proteins. The NF-κB super-repressor IκBΔN as well as the kinase-inactive IKKβ mutant IKKβ K/A interfered with the potential of parkin to activate NF-κB-dependent transcription, indicating that parkin may act on the level of the IKK complex or upstream in the signaling cascade. Indeed, we observed that parkin can promote the ubiquitylation of IKKγ and TRAF2 in a degradation-independent manner. It is not clear at the present time whether parkin induces the ubiquitylation of IKKγ and TRAF2 directly or indirectly, but also ubiquitylation of TRAF2ΔN lacking the N-terminal RING domain is increased in the presence of parkin. This indicates that the E3 ligase activity of TRAF2 is dispensable for the observed effects. Coimmunoprecipitation experiments revealed that IKKγ as well as TRAF2 can form a complex together with parkin. Importantly, it has been described recently that parkin can interact with the E2 heterodimer Ubc13/Uev1a (Doss-Pepe et al., 2005; Matsuda et al., 2006), which is the cognate E2 complex required for TRAF2- and TRAF6-mediated NF-κB activation and involves K63-linked polyubiquitylation (Deng et al., 2000; Andersen et al., 2005).
What might be the role of parkin, particularly in dopaminergic neurons that are exposed to oxidative and excitotoxic stress even under physiological conditions? By modulating the NF-κB pathway, parkin may initiate a neuroprotective program under low-level and moderate stress (Fig. 11). In line with this scenario, parkin induces a supra-additive stress response under conditions that only weakly stimulate the NF-κB pathway, indicating a sensitizing effect. Moreover, parkin is upregulated in response to cellular stress, bringing up the question of whether parkin itself is regulated by NF-κB. Several stress-responsive binding elements are located in the promoter region of parkin, but we could not identify a NF-κB-responsive element. In our experimental model, overexpression of parkin is sufficient to activate the NF-κB signaling cascade, but the E3 ligase activity of parkin is regulated rather than constitutive under physiological conditions. In this context, it is interesting to note that the phosphorylation status of parkin has an impact on its activity (Yamamoto et al., 2005).
Figure 11.
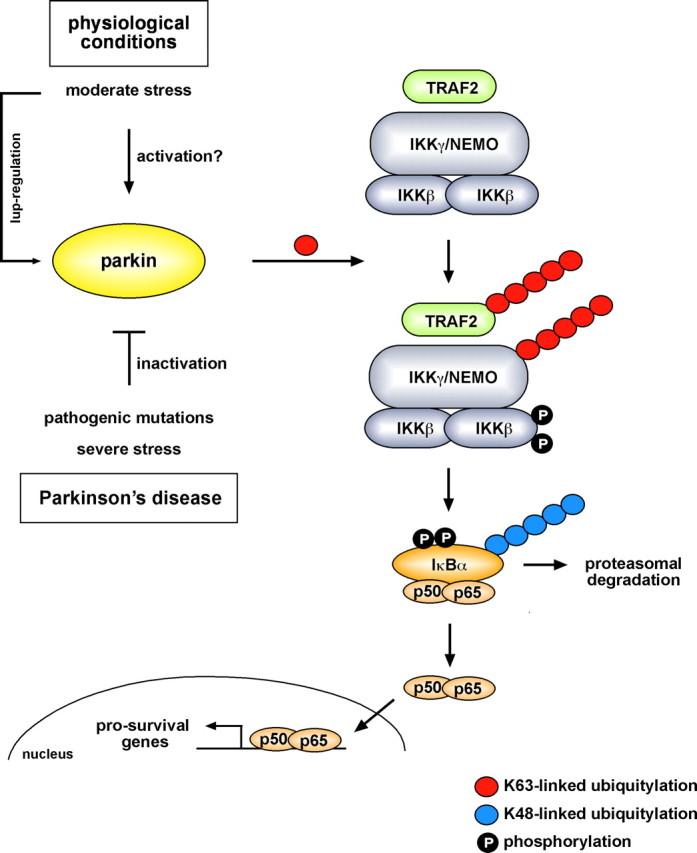
Model of the functional role of parkin. Under physiological conditions, parkin promotes regulatory ubiquitylation of TRAF2 and IKKγ during stress, leading to the activation of NF-κB. As a consequence, transcription of prosurvival genes is upregulated. The activity of parkin can be modulated by stress. Under moderate stress, parkin is upregulated. For an immediate response, it is conceivable that parkin activity can be regulated by posttranslational modifications. In contrast, severe proteotoxic stress, for example induced by oxidized dopamine, causes misfolding and thus inactivation of parkin. Mutations in the parkin gene linked to familial PD also interfere with the capacity of parkin to stimulate the IKK/NF-κB signaling pathway.
What might the implications of these findings be for PD? First, this study links the function of parkin to IKK/NF-κB signaling, suggesting that a dysregulation of this neuroprotective pathway plays a pathophysiological role in PD. Second, it might pave the way for new pharmacological interventions. To explore new targets for a specific neuroprotective or therapeutic approach, it will be important to elucidate the regulation of parkin activity.
Footnotes
This work was supported by Deutsche Forschungsgemeinschaft Grant TA 167/3. We are grateful to Christian Haass for helpful discussions. We thank Virginia M. Lee for providing the PRK28 and PRK8 antibody; Paul C. Evans for the ubiquitin plasmids; Martina Bauer for help with RT-PCR experiments; Ana Luisa Cardoso, Tatiana Simon-Ebert, and Melinda Kiss for excellent technical assistance; and Lindsay A. Smale for critically reading this manuscript.
References
- Andersen PL, Zhou H, Pastushok L, Moraes T, McKenna S, Ziola B, Ellison MJ, Dixit VM, Xiao W. Distinct regulation of Ubc13 functions by the two ubiquitin-conjugating enzyme variants Mms2 and Uev1A. J Cell Biol. 2005;170:745–755. doi: 10.1083/jcb.200502113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZJ. Ubiquitin signalling in the NF-kappaB pathway. Nat Cell Biol. 2005;7:758–765. doi: 10.1038/ncb0805-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darios F, Corti O, Lucking CB, Hampe C, Muriel MP, Abbas N, Gu WJ, Hirsch EC, Rooney T, Ruberg M, Brice A. Parkin prevents mitochondrial swelling and cytochrome c release in mitochondria-dependent cell death. Hum Mol Genet. 2003;12:517–526. doi: 10.1093/hmg/ddg044. [DOI] [PubMed] [Google Scholar]
- Delhase M, Hayakawa M, Chen Y, Karin M. Positive and negative regulation of IkappaB kinase activity through IKKbeta subunit phosphorylation. Science. 1999;284:309–313. doi: 10.1126/science.284.5412.309. [DOI] [PubMed] [Google Scholar]
- Deng L, Wang C, Spencer E, Yang L, Braun A, You J, Slaughter C, Pickart C, Chen ZJ. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell. 2000;103:351–361. doi: 10.1016/s0092-8674(00)00126-4. [DOI] [PubMed] [Google Scholar]
- Doss-Pepe EW, Chen L, Madura K. Alpha-synuclein and parkin contribute to the assembly of ubiquitin lysine 63-linked multiubiquitin chains. J Biol Chem. 2005;280:16619–16624. doi: 10.1074/jbc.M413591200. [DOI] [PubMed] [Google Scholar]
- Evans PC, Ovaa H, Hamon M, Kilshaw PJ, Hamm S, Bauer S, Ploegh HL, Smith TS. Zinc-finger protein A20, a regulator of inflammation and cell survival, has de-ubiquitinating activity. Biochem J. 2004;378:727–734. doi: 10.1042/BJ20031377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haglund K, Dikic I. Ubiquitylation and cell signaling. EMBO J. 2005;24:3353–3359. doi: 10.1038/sj.emboj.7600808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- Haywood AF, Staveley BE. Parkin counteracts symptoms in a Drosophila model of Parkinson's disease. BMC Neurosci. 2004;5:14. doi: 10.1186/1471-2202-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henn IH, Gostner JM, Tatzelt J, Winklhofer KF. Pathogenic mutations inactivate parkin by distinct mechanisms. J Neurochem. 2005;92:114–122. doi: 10.1111/j.1471-4159.2004.02854.x. [DOI] [PubMed] [Google Scholar]
- Higashi Y, Asanuma M, Miyazaki I, Hattori N, Mizuno Y, Ogawa N. Parkin attenuates manganese-induced dopaminergic cell death. J Neurochem. 2004;89:1490–1497. doi: 10.1111/j.1471-4159.2004.02445.x. [DOI] [PubMed] [Google Scholar]
- Imai Y, Soda M, Takahashi R. Parkin suppresses unfolded protein stress-induced cell death through its E3 ubiquitin-protein ligase activity. J Biol Chem. 2000;275:35661–35664. doi: 10.1074/jbc.C000447200. [DOI] [PubMed] [Google Scholar]
- Jiang H, Ren Y, Zhao J, Feng J. Parkin protects human dopaminergic neuroblastoma cells against dopamine-induced apoptosis. Hum Mol Genet. 2004;13:1745–1754. doi: 10.1093/hmg/ddh180. [DOI] [PubMed] [Google Scholar]
- Kaltschmidt B, Widera D, Kaltschmidt C. Signaling via NF-kappaB in the nervous system. Biochim Biophys Acta. 2005;1745:287–299. doi: 10.1016/j.bbamcr.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. 2002;3:221–227. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- Krappmann D, Scheidereit C. A pervasive role of ubiquitin conjugation in activation and termination of IkappaB kinase pathways. EMBO Rep. 2005;6:321–326. doi: 10.1038/sj.embor.7400380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krappmann D, Wulczyn FG, Scheidereit C. Different mechanisms control signal-induced degradation and basal turnover of the NF-kappaB inhibitor IkappaB alpha in vivo. EMBO J. 1996;15:6716–6726. [PMC free article] [PubMed] [Google Scholar]
- Krappmann D, Hatada EN, Tegethoff S, Li J, Klippel A, Giese K, Baeuerle PA, Scheidereit C. The I kappa B kinase (IKK) complex is tripartite and contains IKK gamma but not IKAP as a regular component. J Biol Chem. 2000;275:29779–29787. doi: 10.1074/jbc.M003902200. [DOI] [PubMed] [Google Scholar]
- Krappmann D, Patke A, Heissmeyer V, Scheidereit C. B-cell receptor- and phorbol ester-induced NF-kappaB and c-Jun N-terminal kinase activation in B cells requires novel protein kinase C's. Mol Cell Biol. 2001;21:6640–6650. doi: 10.1128/MCB.21.19.6640-6650.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaVoie MJ, Ostaszewski BL, Weihofen A, Schlossmacher MG, Selkoe DJ. Dopamine covalently modifies and functionally inactivates parkin. Nat Med. 2005;11:1214–1221. doi: 10.1038/nm1314. [DOI] [PubMed] [Google Scholar]
- Lo Bianco C, Schneider BL, Bauer M, Sajadi A, Brice A, Iwatsubo T, Aebischer P. Lentiviral vector delivery of parkin prevents dopaminergic degeneration in an alpha-synuclein rat model of Parkinson's disease. Proc Natl Acad Sci USA. 2004;101:17510–17515. doi: 10.1073/pnas.0405313101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata IF, Lockhart PJ, Farrer MJ. Parkin genetics: one model for Parkinson's disease. Hum Mol Genet. 2004;13:R127–R133. doi: 10.1093/hmg/ddh089. [DOI] [PubMed] [Google Scholar]
- Matsuda N, Kitami T, Suzuki T, Mizuno Y, Hattori N, Tanaka K. Diverse effects of pathogenic mutations of Parkin that catalyze multiple monoubiquitylation in vitro. J Biol Chem. 2006;281:3204–3209. doi: 10.1074/jbc.M510393200. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Cheng B, Culwell AR, Esch FS, Lieberburg I, Rydel RE. Evidence for excitoprotective and intraneuronal calcium-regulating roles for secreted forms of the beta-amyloid precursor protein. Neuron. 1993;10:243–254. doi: 10.1016/0896-6273(93)90315-i. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Culmsee C, Yu Z, Camandola S. Roles of nuclear factor kappaB in neuronal survival and plasticity. J Neurochem. 2000;74:443–456. doi: 10.1046/j.1471-4159.2000.740443.x. [DOI] [PubMed] [Google Scholar]
- Muqit MM, Davidson SM, Payne Smith MD, MacCormac LP, Kahns S, Jensen PH, Wood NW, Latchman DS. Parkin is recruited into aggresomes in a stress-specific manner: over-expression of parkin reduces aggresome formation but can be dissociated from parkin's effect on neuronal survival. Hum Mol Genet. 2004;13:117–135. doi: 10.1093/hmg/ddh012. [DOI] [PubMed] [Google Scholar]
- Nakaso K, Adachi Y, Yasui K, Sakuma K, Nakashima K. Detection of compound heterozygous deletions in the parkin gene of fibroblasts in patients with autosomal recessive hereditary parkinsonism (PARK2) Neurosci Lett. 2006;400:44–47. doi: 10.1016/j.neulet.2006.02.035. [DOI] [PubMed] [Google Scholar]
- Pawlyk AC, Giasson BI, Sampathu DM, Perez FA, Lim KL, Dawson VL, Dawson TM, Palmiter RD, Trojanowski JQ, Lee VM. Novel monoclonal antibodies demonstrate biochemical variation of brain parkin with age. J Biol Chem. 2003;278:48120–48128. doi: 10.1074/jbc.M306889200. [DOI] [PubMed] [Google Scholar]
- Petrucelli L, O'Farrell C, Lockhart PJ, Baptista M, Kehoe K, Vink L, Choi P, Wolozin B, Farrer M, Hardy J, Cookson MR. Parkin protects against the toxicity associated with mutant alpha-synuclein: proteasome dysfunction selectively affects catecholaminergic neurons. Neuron. 2002;36:1007–1019. doi: 10.1016/s0896-6273(02)01125-x. [DOI] [PubMed] [Google Scholar]
- Pickart CM, Fushman D. Polyubiquitin chains: polymeric protein signals. Curr Opin Chem Biol. 2004;8:610–616. doi: 10.1016/j.cbpa.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Ravid T, Hochstrasser M. NF-kappaB signaling: flipping the switch with polyubiquitin chains. Curr Biol. 2004;14:R898–R900. doi: 10.1016/j.cub.2004.09.074. [DOI] [PubMed] [Google Scholar]
- Rawal N, Periquet M, Lohmann E, Lucking CB, Teive HA, Ambrosio G, Raskin S, Lincoln S, Hattori N, Guimaraes J, Horstink MW, Dos Santos Bele W, Brousolle E, Destee A, Mizuno Y, Farrer M, Deleuze JF, De Michele G, Agid Y, et al. New parkin mutations and atypical phenotypes in families with autosomal recessive parkinsonism. Neurology. 2003;60:1378–1381. doi: 10.1212/01.wnl.0000056167.89221.be. [DOI] [PubMed] [Google Scholar]
- Rosen KM, Veereshwarayya V, Moussa CE, Fu Q, Goldberg MS, Schlossmacher MG, Shen J, Querfurth HW. Parkin protects against mitochondrial toxins and beta-amyloid accumulation in skeletal muscle cells. J Biol Chem. 2006;281:12809–12816. doi: 10.1074/jbc.M512649200. [DOI] [PubMed] [Google Scholar]
- Rothe M, Sarma V, Dixit VM, Goeddel DV. TRAF2-mediated activation of NF-kappa B by TNF receptor 2 and CD40. Science. 1995;269:1424–1427. doi: 10.1126/science.7544915. [DOI] [PubMed] [Google Scholar]
- Schlossmacher MG, Frosch MP, Gai WP, Medina M, Sharma N, Forno L, Ochiishi T, Shimura H, Sharon R, Hattori N, Langston JW, Mizuno Y, Hyman BT, Selkoe DJ, Kosik KS. Parkin localizes to the Lewy bodies of Parkinson disease and dementia with Lewy bodies. Am J Pathol. 2002;160:1655–1667. doi: 10.1016/S0002-9440(10)61113-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimura H, Hattori N, Kubo S, Yoshikawa M, Kitada T, Matsumine H, Asakawa S, Minoshima S, Yamamura Y, Shimizu N, Mizuno Y. Immunohistochemical and subcellular localization of Parkin protein: absence of protein in autosomal recessive juvenile parkinsonism patients. Ann Neurol. 1999;45:668–672. doi: 10.1002/1531-8249(199905)45:5<668::aid-ana19>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Sriram SR, Li X, Ko HS, Chung KK, Wong E, Lim KL, Dawson VL, Dawson TM. Familial-associated mutations differentially disrupt the solubility, localization, binding and ubiquitination properties of parkin. Hum Mol Genet. 2005;14:2571–2586. doi: 10.1093/hmg/ddi292. [DOI] [PubMed] [Google Scholar]
- Staropoli JF, McDermott C, Martinat C, Schulman B, Demireva E, Abeliovich A. Parkin is a component of an SCF-like ubiquitin ligase complex and protects postmitotic neurons from kainate excitotoxicity. Neuron. 2003;37:735–749. doi: 10.1016/s0896-6273(03)00084-9. [DOI] [PubMed] [Google Scholar]
- Tan EK, Puong KY, Chan DK, Yew K, Fook-Chong S, Shen H, Ng PW, Woo J, Yuen Y, Pavanni R, Wong MC, Puvan K, Zhao Y. Impaired transcriptional upregulation of Parkin promoter variant under oxidative stress and proteasomal inhibition: clinical association. Hum Genet. 2005;118:484–488. doi: 10.1007/s00439-005-0038-4. [DOI] [PubMed] [Google Scholar]
- Tegethoff S, Behlke J, Scheidereit C. Tetrameric oligomerization of IkappaB kinase gamma (IKKgamma) is obligatory for IKK complex activity and NF-kappaB activation. Mol Cell Biol. 2003;23:2029–2041. doi: 10.1128/MCB.23.6.2029-2041.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Ko HS, Thomas B, Tsang F, Chew KC, Tay SP, Ho MW, Lim TM, Soong TW, Pletnikova O, Troncoso J, Dawson VL, Dawson TM, Lim KL. Stress-induced alterations in parkin solubility promote parkin aggregation and compromise parkin's protective function. Hum Mol Genet. 2005;14:3885–3897. doi: 10.1093/hmg/ddi413. [DOI] [PubMed] [Google Scholar]
- West AB, Maidment NT. Genetics of parkin-linked disease. Hum Genet. 2004;114:327–336. doi: 10.1007/s00439-003-1074-6. [DOI] [PubMed] [Google Scholar]
- Winklhofer KF, Tatzelt J. The role of chaperones in Parkinson's disease and prion diseases. In: Gaestel M, editor. Molecular chaperones in health and diseases. Berlin: Springer; 2006. pp. 221–258. [DOI] [PubMed] [Google Scholar]
- Winklhofer KF, Reintjes A, Hoener MC, Voellmy R, Tatzelt J. Geldanamycin restores a defective heat shock response in vivo. J Biol Chem. 2001;276:45160–45167. doi: 10.1074/jbc.M104873200. [DOI] [PubMed] [Google Scholar]
- Winklhofer KF, Henn IH, Kay-Jackson P, Heller U, Tatzelt J. Inactivation of parkin by oxidative stress and C-terminal truncations; a protective role of molecular chaperones. J Biol Chem. 2003;278:47199–47208. doi: 10.1074/jbc.M306769200. [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Friedlein A, Imai Y, Takahashi R, Kahle PJ, Haass C. Parkin phosphorylation and modulation of its E3 ubiquitin ligase activity. J Biol Chem. 2005;280:3390–3399. doi: 10.1074/jbc.M407724200. [DOI] [PubMed] [Google Scholar]
- Yang Y, Nishimura I, Imai Y, Takahashi R, Lu B. Parkin suppresses dopaminergic neuron-selective neurotoxicity induced by Pael-R in Drosophila. Neuron. 2003;37:911–924. doi: 10.1016/s0896-6273(03)00143-0. [DOI] [PubMed] [Google Scholar]