

Abstract
Neurotransmitters have emerged as important players in the control of programmed cell death in the cerebral cortex. We report that genetic depletion of serotonin, dopamine, and norepinephrine in mice lacking the vesicular monoamine transporter (VMAT2 KO mice) causes an increase in cell death in the superficial layers of the cingulate and retrosplenial cortices during early postnatal life (postnatal days 0–4). Electron microscopy and terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling indicated that this represents a form of apoptosis. Caspase-3 and -9 are over activated in the VMAT2 KO cortex and Bcl-XL is downregulated, whereas the apoptosis-inducing factor caspase-8 and FasL/FasR pathway are not involved. Partial inhibition of serotonin or/and catecholamine synthesis by pharmacological treatments or genetic reduction of serotonin neuron number in mice lacking the transcription factor Pet-1 (pheochromocytoma 12 E26 transformation-specific) did not modify the cell death ratios in the cerebral cortex. However, when monoamine oxidase type A was invalidated in the VMAT2 KO background (VMAT2-MAOA DKO mice), increases in 5-HT levels coincided with a reduction of cell death and a normalization of Bcl-XL expression. trkB signaling is not implicated in the anti-apoptotic effects of MAOA inhibition because BDNF mRNA levels were unchanged in VMAT2-MAOA DKO mice and because the massive cell death in the cerebral cortex of trkB KO mice is also reverted by genetic invalidation of the MAOA gene. Finally the broad 5-HT2 receptor agonist (−)-2,5-dimethoxy-4-iodoamphetamine hydrochloride prevented the increase in cell death of VMAT2 KO mice. Altogether, these results suggest that high levels of serotonin, acting through 5-HT2 receptors, have neuroprotective action on cortical neurons by controlling Bcl-XL mRNA levels and that this action is independent of trkB signaling.
Keywords: neurotrophin, serotonin, dopamine, cingulate cortex, monoamine oxidase A, apoptosis
Introduction
The development of the cerebral cortex is a sequential process that includes programmed cell death (PCD). Approximately half of the neurons produced during corticogenesis are thought to die (Ferrer et al., 1992; Blaschke et al., 1996). Two consecutive waves of PCD affect the cortical neurons at different periods of their development. The first wave consists of cell death of the proliferating precursors in the ventricular and subventricular zones, which appears to be closely linked to cell cycle regulation (Thomaidou et al., 1997). The second wave affects postmitotic neurons at later stages and may be involved in matching the size of neuronal populations to that of their targets during the formation and the maintenance of synapses (Ferrer et al., 1992). At both periods, cell death is apoptotic and the molecular machinery relies on the mitochondrial pathways of intracellular signal transduction (Dikranian et al., 2001).
Postmitotic developmental cell death is not homogeneously distributed in the different cortical areas and layers. In rodents, superficial layers of the frontal and limbic cortices are much more affected than other cortical areas (Finlay and Slattery, 1983; Ferrer et al., 1990; Verney et al., 2000). Molecules that convey trophic support to the cortical neurons could be particularly important in the control of the survival of postmitotic neurons (Catapano et al., 2001). Genetic studies showed that invalidation of trkB, the high-affinity receptor of brain-derived neurotrophic factor (BDNF) and neurotrophin-4 (NT4), substantially increased cell death in the cerebral cortex (Alcantara et al., 1997). More recently, a role of glutamatergic and GABAergic neurotransmission was shown in the control of PCD in the cerebral cortex whereby inhibition of the NMDA receptors or excessive activation of the GABAA receptors greatly enhanced cell death in the cerebral cortex, particularly in layers II and V (Ikonomidou et al., 1999, 2001; Pohl et al., 1999). Interestingly, this effect was observed only during a limited period of development, the first postnatal week.
In the present study, we evaluated the impact of serotoninergic and catecholaminergic neurotransmission on PCD in the developing cortex. Monoamine neurotransmitter systems differentiate early during development and play important roles in controlling neuronal proliferation, migration, differentiation, and cell survival (Lauder, 1993; Vitalis and Parnavelas, 2003). For instance, serotonin enhances the survival of cortical glutamatergic neurons in vitro (Dooley et al., 1997; Lavdas et al., 1997). Genetic approaches now allow to tease out the developmental function of amines in vivo (Gaspar et al., 2003). Using such models combined with pharmacological approaches, we show here that a decreased production of serotonin or catecholamines has no clear effect on cortical PCD, whereas altered storage of monoamines increases cell death in selected cortical areas. This effect can be rescued by the genetic ablation of monoamine oxidase type A (MAOA) or by the administration of serotonin receptor type 2 agonists during the perinatal period. Thus, although serotonin does not appear to directly control developmental cell death in the cerebral cortex, it can display neuroprotective properties through a mechanism that could involve upregulation of the anti-apoptotic factor Bcl-XL via 5-HT2 activation, independently of trkB signaling.
Materials and Methods
Animals.
Animal procedures were conducted in strict compliance with approved institutional protocols and in accordance with the provisions for animal care and use described in the European Communities council directive of November 24, 1986 (86/609/EEC). Vesicular monoamine transporter knock-out (VMAT2 KO) mice were generated with a targeted mutation in the first protein-coding exon of the VMAT2 gene (Fon et al., 1997). trkB knock-out mice were generated with a targeted mutation in the catalytic domain (Klein et al., 1993). MAOA knock-out mice displayed a deletion of exons 2 and 3 in the gene encoding MAOA (Cases et al., 1995). Pet-1 [pheochromocytoma 12 E26 transformation-specific (ETS)] null mice were generated with a targeted mutation of all coding regions (Hendricks et al., 2003). Mouse lines were maintained on a mixed C57BL/6/129Sv genetic background enriched in C3H/HeJ. For VMAT2, MAOA, trkB, and Pet-1 genotyping, we used the same primers as described previously, respectively, by Fon et al. (1997), Vitalis et al. (2002b), and Hendricks et al. (2003). Some pharmacological experiments were made on Swiss mice. The day of birth was counted as postnatal day 0 (P0).
Pharmacological treatments.
dl-P-Chlorophenylalanine methyl ester hydrochloride (PCPA) (300 mg/kg; Sigma/RBI, Natick, MA), α-methyl-paratyrosine (αMT) (300 mg/kg; Interchim, Montlucon, France), or sterile physiological saline was administered subcutaneously to pups. Animals were injected once daily from P0 to P5. (−)-2,5-Dimethoxy-4-iodoamphetamine hydrochloride (DOI) (20 mg/kg; Sigma/RBI) or physiological saline was administered twice at 18 h intervals at P0 and P1, and mice were killed 2 h after the second injection.
Light microscopy on plastic sections and electron microscopy.
Pups were anesthetized and perfused transcardially with 1% glutaraldehyde and 4% paraformaldehyde (PFA). Brains were sliced in 400-μm-thick sections, postfixed 2 h in 1% osmium tetroxide, dehydrated in alcohol, cleared in acetone, and embedded in Araldite. For light microscopy, transverse serial sections (1 μm) were cut, heat dried, and stained with toluidine blue. Ultrathin sections were cut and stained with uranyl acetate/lead citrate and examined with a Philips (Aachen, Germany) CM100 electron microscope.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling staining.
Pups were anesthetized, and their brains were immediately removed and frozen in isopentane. To visualize nuclei with DNA cleavage, serial coronal brain sections (20 μm) of the brain were cut on a cryostat, and residues of fluorescein-labeled nucleotides were catalytically added to DNA fragments by terminal deoxy-nucleotidyl-transferase (TdT). Briefly, sections were fixed in fresh 4% PFA/PBS at room temperature for 15 min, washed in PBS three times for 5 min, equilibrated at room temperature for 10 min, and incubated with nucleotide mix and TdT (ApoAlert DNA fragmentation kit; Clontech, Mountain View, CA) at 37°C for 1 h. Tailing reaction was stopped by incubating sections in 2× SSC at room temperature for 15 min. Apoptotic cells exhibit strong, nuclear green fluorescence.
Immunocytochemistry.
Pups were anesthetized and perfused transcardially with 4% PFA in 0.12 m phosphate buffer, pH 7.4. After perfusion, brains were removed from the skull and postfixed overnight in fresh fixative. Brains were cryoprotected in 30% sucrose phosphate buffer. Serial, 48-μm-thick, frozen sections were obtained in the coronal plane. Alternate series of sections were used for immunostaining and Nissl (0.05% thionin in acetate buffer, pH 5.5). For immunostaining, rat monoclonal antibodies against serotonin (1:50; Chemicon, Temecula, CA), GABA (Sigma, St. Louis, MO), and Bcl-2 (Boehringer Mannheim, Mannheim, Germany), as well as rabbit polyclonal antibodies recognizing apoptosis-inducing factor (AIF) (1:200; a kind gift from G. Kroemer, IGR, Villejuif, France), Bcl-XL (1:500; Santa Cruz Biotechnology, Santa Cruz, CA), Bax (1:500; Santa Cruz Biotechnology), c-jun sc-45 (1:500; Santa Cruz Biotechnology), the activated caspase-3 (1:200; BD Biosciences PharMingen, San Diego, CA) and caspase-9 (1:1000; Cell Signaling Technology, Beverly, MA), the serotonin transporter (5-HTT) (1:5000; Calbiochem, La Jolla, CA), and the calcium binding proteins calbindin (1:8000; Swant, Bellinzona, Switzerland) and calretinin (1:10,000; Swant), and guinea pig polyclonal antibodies against the vesicular glutamate transporter type 1 (VgluT1) (1:16,000; Chemicon) and type 2 (VgluT2) (1:3000; Chemicon) were diluted in 0.02 m PBS with 0.2% gelatin and 0.25% Triton X-100 (PBS+).
Sections were rinsed in PBS+, incubated overnight at room temperature with the primary antibody, rinsed in PBS+, incubated 1.5 h in the appropriate 1:200 biotinylated secondary antibody [biotinylated goat anti-rabbit, goat anti-guinea pig (Sigma), and rabbit anti-rat (DakoCytomation, Carpinteria, CA), rinsed in PBS+, incubated 1.5 h in 1:400 streptavidin peroxidase complex (Amersham Biosciences, Arlington Heights, IL), rinsed in Tris buffer (0.05 m), pH 7.8, and revealed in 0.002% diaminobenzidine, 0.003% H2O2, and 0.6% nickel ammonium sulfate.
For immunofluorescence, sections were rinsed in PBS+, incubated overnight at room temperature with the primary antibody, rinsed in PBS+, incubated 2 h with secondary antibody [FITC anti-rat, 1:200 (Sigma); cyanine 3 anti-rabbit, 1:200 (Jackson ImmunoResearch, West Grove, PA)]. Sections were washed and analyzed with a Leica (Nussloch, Germany) microscope.
Western blotting.
Tissues were homogenized in 50 mm HEPES buffer, pH 7, 0.1% Triton X-100 supplemented with a protease inhibitor cocktail (Sigma), incubated on ice for 20 min, and centrifuged for 30 min at 12,000 × g at 4°C. Supernatants were collected and quantified using the Bradford technique, and aliquots were kept at −80°C until use. For immunoblotting experiments, samples (60 μg) were diluted in loading buffer (0.1 m Tris-HCl, pH 6.8, 15% glycerol, 3% SDS, 3% β-mercaptoethanol, and 0.02% bromophenol blue), subjected to SDS/PAGE on 10% polyacrylamide gels (Bio-Rad, Hercules, CA), and transferred to polyvinylidene difluoride membranes (Amersham Biosciences). After 1 h in Tris-buffered saline (0.1 m Tris-HCl and 0.9% NaCl, pH 7.4) containing 0.2% Tween 20 and 5% bovine serum albumin, blots were incubated with either Bax (1:200), Bcl-XL (1:200), or caspase-8 (1:100; Santa Cruz Biotechnology) specific polyclonal antibody for 16 h at 4°C. Immunoblots were washed in TBS–0.02% Tween 20 (TBST), incubated for 1 h with the appropriate horseradish peroxidase-coupled IgG (diluted 1:5000), washed in TBS–0.02% Tween 20, revealed using Super Signal (Pierce, Rockford, IL), and exposed to Hyperfilm ECL (Amersham Biosciences). For quantification with actin, immunoblots were washed six times for 30 min in TBST and incubated with β-actin (1:10,000; Santa Cruz Biotechnology) monoclonal antibody for 1 h at room temperature. Sample densities were analyzed using a Bio-Rad densitometer and the Bio-Rad Analyst software. Densities of immunoreactive (IR) protein bands were quantified, and background was subtracted. Data were graphed as the ratio of the protein of interest/β-actin. The identity of the measured bands was unknown to the person who performed the densitometry.
In situ hybridization.
Pups were anesthetized, and brains were immediately removed and frozen in isopentane. Coronal sections (16 μm) were cut on a cryostat, collected onto SuperFrost slides, and stored at −80°C. cDNAs encoding murine 5-HT2A, 5-HT2C, FasL (gift from S. Nagata, Osaka, Japan), FasR (gift from P. Schneider, University of Lausanne, Lausanne, Switzerland), BDNF, trkB, NT3, Bax, Bid, Bak, Bcl-2, or human Bcl-XL (gifts from Stan Krajewski, Burnham Institute, La Jolla, CA) were used as templates, and 35S-labeled ([35S]UTP; >1000 Ci/mmol; Amersham Biosciences) antisense RNA probes were made by in vitro transcription.
Tissue sections were fixed for 15 min in 4% paraformaldehyde, washed in PBS, acetylated, dehydrated, and air dried. Sections were covered with hybridization buffer (50% formamide, 0.3 m NaCl, 20 mm Tris-Hcl, pH 7.4, 5 mm EDTA, 1× Denhardt's, 10% dextran sulfate, 10 mm DTT, 10 mm NaH2PO4, and 250 μg/ml yeast tRNA) containing 106 cpm of riboprobes. Slides were hybridized overnight in a humid chamber at 48°C. Sections were washed in 5× SSC and 0.15% DTT at 42°C and for 20 min in 50% formamide, 2× SSC, and 1.25% DTT at 60°C. Sections were then treated for 30 min at 37°C in 0.02% RNase A (Roche Diagnostics, Indianapolis, IN) in 10 mm Tris-HCl, pH 7.6, 30 mm NaCl, and 10 mm EDTA. Sections were sequentially washed for 15 min at 37°C in 10 mm Tris-HCl, pH 7.6, 30 mm NaCl, and 10 mm EDTA, in 2× SSC and in 0.1× SSC. The slides were dehydrated and air dried. Autoradiograms were obtained by apposing the sections to Hyperfilms (β-max; Amersham Biosciences) for 4 d. Autoradiographic films were developed in D19 (Eastman Kodak, Rochester, NY) for 4 min and fixed in Al-4 (Ilford, Cheshire, UK). Some slides were dipped in photographic emulsion (NTB2; Eastman Kodak) and exposed for ∼10 d. After development of the emulsion, the sections were counterstained in cresyl violet.
Quantification of gene expression was performed by densitometry. Care was taken to group the sections from the different ages and the different conditions (normal and knock-out mice) on the same Hyperfilm. Images of the individual autoradiograms were captured digitally using a CCD camera coupled to a digitization board, both driven by Samba software (Unilog, Paris, France). The mean optical density per pixel (pixel size, 23 × 23 μm) was obtained from areas of the cortex in three consecutive sections per individual (two to three mice per age).
Morphometric analysis: counts of pyknotic profiles.
For all quantification, slides were coded and counts were performed with the examiner blind to the time point, genotype, and treatment. Coronal sections (48 μm thick) of wild-type, VMAT2 KO, and VMAT2-MAOA double knock-out (DKO) pups were Nissl stained and analyzed using a 40× objective and a millimetric eyepiece. Pyknotic cells were identified by their densely condensed and fragmented nuclei. For each pup (P1, P2, P3, and P4), the number of pyknotic cells was counted on the two hemispheres in three sections spaced by 240 μm. Pyknotic profiles were counted in an area of 40,000 μm2 in the motor (M) and somatosensory (S) cortices (Fig. 1). In the cingulate (Cg) and retrosplenial granular (RSG) cortices, the number of pyknotic profiles was counted within the entire cortical area (90,000–140,000 μm2 for layers II/III and 220,000–440,000 μm2 in layers V/VI, according to the ages examined). Statistical comparisons between wild-type, VMAT2 KO, and VMAT2-MAOA DKO were done using the unpaired Student's t test.
Figure 1.
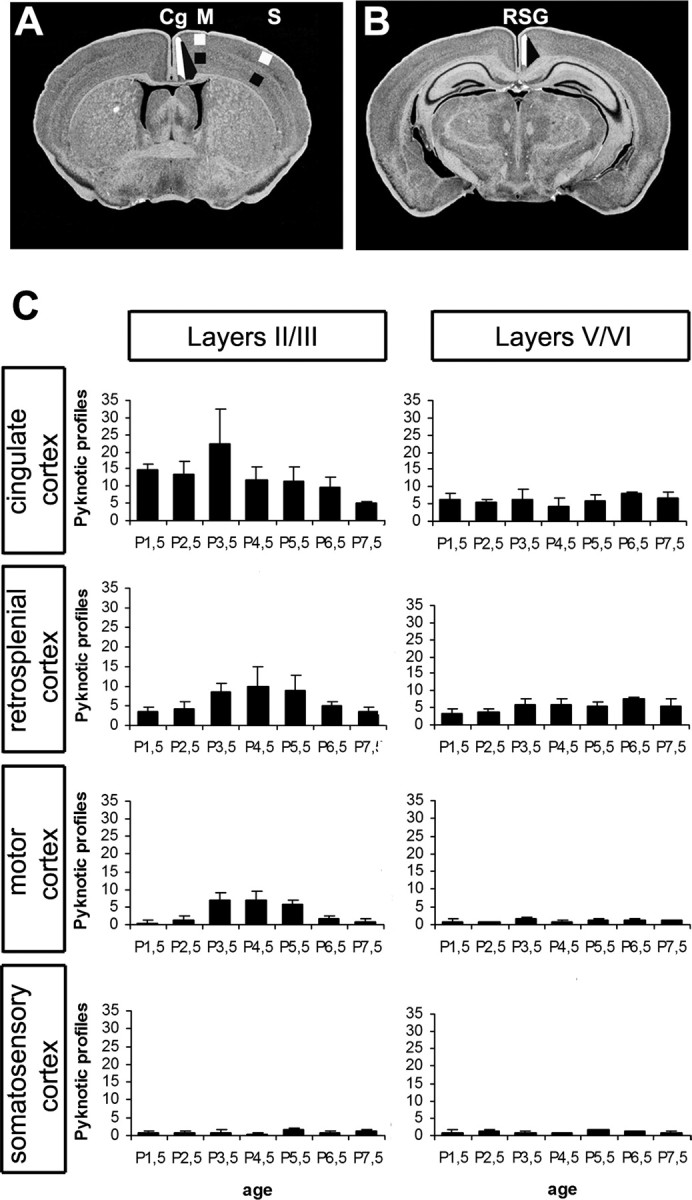
Spatiotemporal pattern of cell death during the first postnatal week of development. A, B, Representative coronal levels on which pyknotic profiles were counted [according to the mouse brain atlas by Paxinos and Franklin (2001)]. The white and black areas delimit the counting zones in layers II/III and V/VI. C, Number of pyknotic profiles in layers II/III and V/VI of cingulate and retrosplenial, primary motor, and somatosensory cortices at P1.5 (n = 6), P2.5 (n = 6), P3.5 (n = 5), P4.5 (n = 5), P5.5 (n = 6), P6.5 (n = 3) and P7.5 (n = 3).
Results
We examined four areas of the developing cerebral cortex: Cg, RSG, primary motor (M), and primary somatosensory (S1). Pyknotic profiles identified with Nissl staining were counted in the superficial layers (II–III) and infragranular layers (V–VI) during the first postnatal week (Fig. 1). In control mice, cell death predominated in the superficial layers of the limbic cortical areas Cg and RSG and in the medial cortical area M1, whereas scarce pyknotic profiles were observed laterally, in S1 or in the infragranular layers (Fig. 1C). Cell death peaked at P3.5 in the cingulate cortex and at P4.5 in the retrosplenial cortex.
Genetic depletion of monoamine vesicular storage causes increased apoptosis in the developing cerebral cortex
Mice lacking the vesicular monoamine transporter have undetectable levels of monoamines in their brains, although serotoninergic and catecholaminergic cell groups and projections are present and develop normally (Fon et al., 1997; Alvarez et al., 2002). A significant increase in the number of pyknotic cells was noted in the Cg and the RSG. This increase was of 80% (p < 0.01) at P1, 57% (p < 0.05) at P2, and returned to control levels at P4 (Fig. 2A–C).
Figure 2.
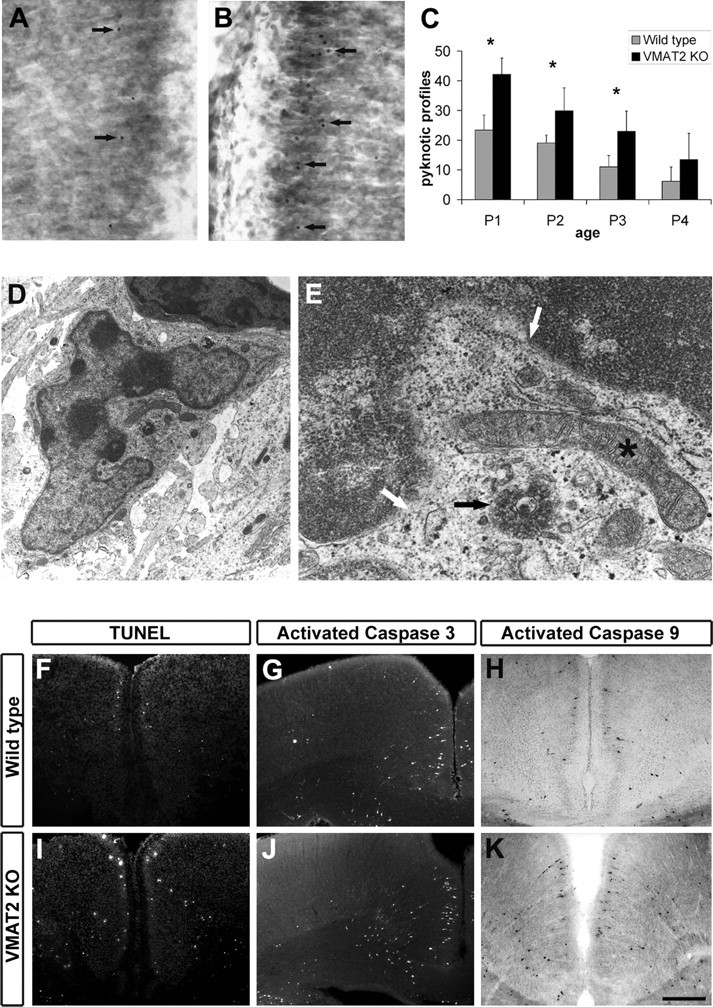
A, B, Nissl-stained sections of the Cg of wild-type (A) and VMAT2 KO (B) mice at P1. Arrows point to pyknotic profiles. C, Number of pyknotic profiles in layers II/III of the cingulate cortex (P1: WT, n = 7; VMAT2 KO, n = 5; P2: WT, n = 4; VMAT2 KO, n = 4; P3: WT, n = 3; VMAT2 KO, n = 3; P4: WT, n = 3; VMAT2 KO, n = 3). *p < 0.05 indicates that the results are statistically different. D, Electron micrograph showing an apoptotic profile in the superficial layers of Cg of a VMAT2 KO at P1. E, Magnification from D. White arrows point to the disruption of the nuclear envelope. Some chromatin is visible in the cytoplasm (black arrow), whereas organelles such as mitochondria are still preserved (*). TUNEL staining (F, I) and activation of caspase-3 (G, J) and capase-9 (H, K) immunoreactivity were performed on coronal sections of wild-type and VMAT2 KO mice at P1. Scale bar: A, B, 50 μm; D, 800 nm; E, 200 nm; F, H, I, K, 200 μm; G, J, 240 μm.
Apoptosis is the main mechanism of cell death in the developing cerebral cortex
At the electron microscopic level, each type of cell death (apoptosis, necrosis, and autophagy) can be distinguished from a series of stereotyped, morphologically well defined phases. In control and VMAT2 KO mice, neurons at different stages of apoptosis could be detected in layer II of the Cg or the RSG. We observed cells with clumping of the nuclear chromatin featuring the first unequivocal evidence of apoptosis, as well as cells in an advanced apoptotic stage when the cell and its nucleus assume an irregular shape and nuclear budding occurs to produce discrete fragment (Fig. 2D,E). Cells fragmented into membrane-bound apoptotic bodies still displaying a sharp segregation of condensed chromatin in nuclear fragments and intact organelles were occasionally observed. We did not note the presence of large autophagic vacuoles characteristic of autophagy in either control or VMAT2 KO mice.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) confirmed these observations and indicated that the main mode of cell death in the postnatal critical period of the first postnatal week is apoptosis. TUNEL-positive profiles were increased in the Cg (P1, wild type, n = 4.67 ± 0.33; VMAT2 KO, n = 9.67 ± 0.33; P3: wild type, n = 10.33 ± 0.66; VMAT2 KO, n = 20 ± 0.33; values are mean ± SEM of three animals; *p < 0.05) and RSG (P1: wild type, n = 2 ± 0.33; VMAT2 KO, n = 5 ± 0.33; P2, wild type, n = 16.3 ± 0.66; VMAT2 KO, n = 27 ± 1; values are mean ± SEM of three animals; *p < 0.05) of VMAT2 KO mice (Fig. 2F,I).
Apoptosis is induced in a mitochondria caspase-dependent cascade
The cleavage of the effector procaspase-3 is a hallmark of apoptosis. Numerous cleaved caspase-3 IR profiles were found in the cerebral cortex of control mice. A significant increase of cleaved caspase-3 IR profiles was found in the cingulate cortex of mice lacking VMAT2 between P1 and P3 (P1: wild type, n = 7.5 ± 1; VMAT2 KO, n = 19.5 ± 1; P2: wild type, n = 17.2 ± 0.75; VMAT2 KO, n = 30.5 ± 1.5; values are mean ± SEM of four animals; **p < 0.001) (Fig. 2G,J). By P5, the rate of cleaved caspase-3 IR profiles returned to normal in mice lacking VMAT2. Effector procaspase-3 can be cleaved by either the activation of the FasR–caspase-8 pathway or the apoptosome formed by the association of the apoptosis proteases-activating factor Apaf1, cytochrome c, and cleaved caspase-9. We did not detect FasR or FasL mRNA expression or activation of caspase-8 in control and VMAT2 KO mice (see Fig. 5G). In contrast, numerous cells containing cleaved caspase-9 IR were present in the Cg and the RSG during the P0–P4 period. In VMAT2 KO mice, an increase of cleaved caspase-9-immunoreactive cells was noted in these cortical areas (P1: wild type, n = 3.33 ± 0.33; VMAT2 KO, n = 7.67 ± 0.33; P2: wild type, n = 9 ± 0.66; VMAT2 KO, n = 20.33 ± 0.66; values are mean ± SEM of three animals; *p < 0.001) (Fig. 3H,K). Apoptosis-inducing factor AIF, an intramitochondrial protein that promotes cell death in a caspase-independent manner on release into nonmitochondrial compartments, was located in the mitochondrial compartment in both control and VMAT2 KO mice (data not shown). Together, these results suggest that, in both wild-type and VMAT2 KO mice, apoptosis is induced via a mitochondria-dependent cascade involving the activation of the caspase-9 apoptosome.
Figure 5.

Number of pyknotic profiles in layers II/III in the cingulate cortex in pups treated with physiological saline (A), PCPA (B), a selective inhibitor of serotonin synthesis, αMT (C), and a selective inhibitor of catecholamine synthesis or the combined PCPA and αMT (D) during the first postnatal days. E, Number of pyknotic profiles in layers II/III of the cingulate cortex between normal and Pet-1 KO mice. Serotonin immunostaining at the level of the cingulate cortex of a normal (F) and PCPA-treated (G) pup at P2. Note the disappearance of the serotonin-containing fibers (G). H, I, Serotonin immunostaining at the level of the cingulate cortex of wild-type (H) and Pet-1 KO (I) mice at P2. Note the disappearance of 5-HT-containing fibers in the superficial layers. Scale bars, 60 μm.
Figure 3.
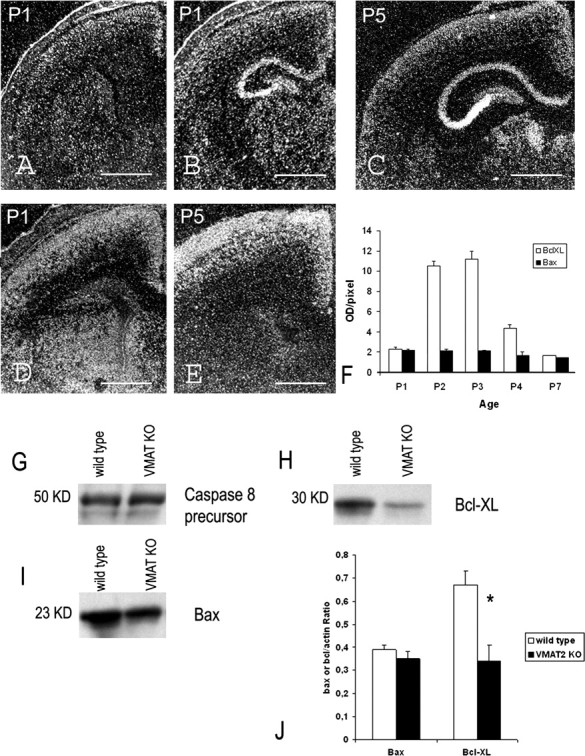
Spatiotemporal pattern of Bax and Bcl-XL mRNAs during the first postnatal week. A–C, Detection of Bax mRNAs at the level of the cingulate (A) and retrosplenial (B, C) cortex at P1 (A, B) and P5 (C). D, E, Detection of Bcl-XL mRNAs at the level of the cingulate cortex at P1 (D) and P5 (E). F, Quantification of Bax and Bcl-XL mRNA level in the superficial layers of the cingulate cortex of normal mice during the first postnatal week. G–I, Western blot analysis of caspase-8 (G), Bcl-XL (H), and Bax (I) in the cerebral cortex of normal and VMAT2 KO mice at P2. G, Caspase-8 antibody recognized a 50 kDa band corresponding to the caspase-8 precursor. The activated caspase-8 p20 was not present both in normal and VMAT2 KO. H, Bcl-XL antibody recognized a 30 kDa band. I, Bax antibody recognized a 23 kDa band. J, Quantification of Bcl-XL and Bax protein. Data were expressed as ratio of Bcl-XL/actin and Bax/actin (n = 2, wild type; n = 3, VMAT2 KO). Values are mean ± SEM of sample animals. *p < 0.05 indicates that the results are statistically different. Scale bars: A, B, D, 300 μm; C, 250 μm; E, 200 μm.
In several studies, c-jun immunoreactivity as revealed with the sc45 polyclonal antibody was found to be a visible hallmark of apoptotic cell death in the developing cerebral cortex (Ferrer et al., 1996, 2000). This was now found to be a poor detector of the c-jun protein “per se” but a very useful marker of caspase-3 proteolysis (Casas et al., 2001; Ribera et al., 2002). In normal mice, few c-jun IR profiles were found in supragranular layers of the cortex. In mice lacking VMAT2, a significant increase of c-jun IR profiles was found in layer II of the cingulate cortex (P1: wild type, n = 5.75 ± 0.75; VMAT2 KO, n = 15.5 ± 0.5; values are mean ± SEM of four animals; **p < 0.001), further demonstrating the caspase-3-mediated apoptosis in the cerebral cortex of normal and VMAT2 KO mice.
Proapoptotic/anti-apoptotic factors
Cell death induced by apoptosome activation depends on the ratio of proapoptotic/anti-apoptotic factors expressed by mitochondria. During the first postnatal week, we found that cortical neurons express a combination of the proapoptotic factor, Bax (Fig. 3A–C) and the anti-apoptotic factor Bcl-XL. We did not detect by in situ hybridization or immunocytochemistry the proapoptotic factors Bid and Bak or the anti-apoptotic Bcl-2 (data not shown). The levels of Bax mRNA were higher in the supragranular layers than the infragranular layers, and they did not change during the 5 postnatal days. Bcl-XL mRNA levels were higher in the supragranular layers and the upper part of layer V than in layers IV and VI. Interestingly, Bcl-XL mRNA expression increased during early postnatal life, reaching a peak at P3, then declined rapidly (Fig. 3D–F). We found a decrease in Bcl-XL mRNA and protein levels at P3 in the supragranular layers of VMAT2 KO cerebral cortex (Fig. 3H–J) (see Fig. 7F), and Bax mRNA levels were unchanged (Fig. 3I,J). Thus, the increase of apoptotic mitochondrial-dependent cell death observed in VMAT2 KO is correlated to a decrease in the Bcl-XL/Bax mRNA expression ratio.
Figure 7.

Pattern and level of NT3 (A–C) and Bcl-XL (D–F) mRNA in the cingulate cortex of wild-type (A–F), VMAT2 KO (C, F), and VMAT2-MAOA DKO (C, F) mice. A, At P1, moderate levels of NT3 mRNA were detected in layer II of the Cg. A strong expression of NT3 mRNA was also detected in the indusium griseum (arrow, IG). C, From P3, high levels of NT3 mRNAs were detected in Cg and indusium griseum. C, Quantification of NT3 mRNA levels in the cingulate cortex of wild type (P2, n = 3; P3, n = 3), VMAT2 KO (P2, n = 4; P3, n = 4), and VMAT2-MAOA DKO (P2, n = 3; P3, n = 3). Values are mean ± SEM of sample animals. D, At P1, moderate levels of Bcl-XL mRNA were detected throughout the entire cerebral cortex. E, At P3, moderate levels of Bcl-XL mRNAs were detected in the supragranular layers of the cerebral cortex. Weak Bcl-XL mRNA levels were found in the upper part of layer V. F, Quantification of Bcl-XL mRNA levels in the cingulate cortex of wild type (P2, n = 3; P3, n = 3), VMAT2 KO (P2, n = 4; P3, n = 4), and VMAT2-MAOA DKO (P2, n = 3; P3, n = 3). Values are mean ± SEM of sample animals. *p < 0.05 indicates that the results are statistically different between the groups analyzed (Student's t test). G, Reversal of cell death observed in mice lacking trkB. Quantification of cell death in the cingulate cortex of wild type, trkB KO, and trkB-MAOA DKO (P1, n = 3; P2, n = 3; P3, n = 4; P4, n = 4). Values are mean ± SEM of sample animals. **p < 0.005 indicates that the results are statistically different. Scale bars, 330 μm.
Neurotransmitter phenotype
At early stages of apoptosis, caspase-3 and caspase-9 IR profiles display typical morphologies of cortical pyramidal neurons, suggesting that apoptosis could affect preferentially the glutamatergic cortical neurons (Fig. 4A–C′). Glutamatergic neurons were herein revealed using antibodies against the vesicular glutamate transporters VgluT1 and VgluT2. VgluT1 and VgluT2 expression is namely restricted to the axonal compartment of selected glutamatergic populations (Boulland et al., 2004). In our experiments by P3, VgluT1 is associated with puncta that are specifically enriched in layer V, and VgluT2 is enriched in layers I and IV. In brain sections of VMAT2 KO, numerous cell bodies presenting a degenerative profile were VgluT1 or VgluT2 immunoreactive. We did not find any apoptotic profiles with GABA-, calretinin-, or NPY-specific antibodies (data not shown).
Figure 4.

Morphological and neurotransmitter phenotypes of dying cells in mice lacking VMAT2 at P3. At early stages of apoptosis, morphological characteristics can be recognized using activated caspase-3 (A–C) or caspase-9 immunoreactivity (A′–C′). A, A′, In layer V of the cerebral cortex, activation of caspase-3 (A) or caspase-9 was only observed in large pyramidal neurons. B, C, B′, C′, In the supragranular layers of the cingulate cortex, activation of caspase-3 (B, C) or caspase-9 (B′, C′) was observed only in pyramidal neurons. Arrows indicate the apical dendrite. In normal mice, immunoreactivity to the vesicular glutamate transporter type 1 (vglut1) or type 2 (vglut2) labels axon terminals. In mice lacking VMAT2, the normal VgluT1- or VgluT2-positive punctas were observed in axons terminals (D–G). However, abnormal cytoplasmic accumulations of VgluT1 (D, E) or VgluT2 (F, G) were observed in many neurons of the cingulate cortex of VMAT2 KO. In some cases, VgluT1 (E) or VgluT2 (F) immunoreactivity was densely packed around a small round-shaped nucleus, a hallmark of pyknosis. Arrows indicate these abnormal locations. Scale bars: A, A′, C, C′, 15 μm; B, B′, 25 μm; D, F, 80 μm; E, G, 25 μm.
Pharmacological inhibition of monoamine synthesis and genetic inactivation of serotonin synthesis
PCPA, a selective inhibitor of serotonin synthesis, or αMT, a selective inhibitor of catecholamine synthesis, were administered daily to pups from P0.5 to P4.5. We also combined PCPA and αMT injections. PCPA treatments in mouse pups have been found previously to cause a 80–50% decrease of brain 5-HT levels (Rebsam et al., 2005; Dailly et al., 2006). Anti-serotonin immunoreactivity showed a reduction of serotonin-immunolabeled axons after PCPA injections (Fig. 5F,G). Cell counts in Cg, RSG, M1, and S1 were performed 24 h after the last injection. These treatments did not modify the amount of pyknotic cells in the cortical areas examined (Fig. 5A–D).
Mice lacking the ETS domain transcription factor Pet-1 display a 80% reduction in serotonin levels (Hendricks et al., 2003). Serotonin-containing fibers and serotonin transporter-immunoreactive fibers were absent in the cingulate cortex of mice lacking Pet-1 (Fig. 5H,I). However, no change in cell death was noted at P1, P3, or P5 (Fig. 5E).
Reversal of cell death in VMAT2-MAOA DKO mice
MAOA is a membrane-bound mitochondrial flavoprotein that oxidatively deaminates monoamines in neurons and glial cells (Shih et al., 1999). MAOA is the major MAO isoform expressed in neurons during development, including the raphe nucleus (Vitalis et al., 2002a). As a consequence, VMAT2-MAOA DKO display high levels of serotonin, whereas brain levels of dopamine and noradrenaline remain low or undetectable (Alvarez et al., 2002). In the VMAT2-MAOA DKO, cell death counts were similar to wild-type levels (Table 1) in the cerebral cortex during the P0–P4 period. Correspondingly, we found a normalization in TUNEL-positive (wild type, n = 8.33 ± 0.66; VMAT2 KO, n = 16.3 ± 0.66; VMAT2-MAOA DKO, n = 9.67 ± 1; values are mean ± SEM of three animals; *p < 0.05 between VMAT2 and VMAT2-MAOA samples) and activated caspase-3 (wild type, n = 12.3 ± 0.66; VMAT2 KO, n = 26.7 ± 1; VMAT2-MAOA DKO, n = 12.3 ± 1; values are mean ± SEM of three animals; **p < 0.001 between VMAT2 and VMAT2-MAOA DKO) profiles at P2 in Cg. Furthermore, the levels of the anti-apoptotic protein Bcl-XL and its mRNA were normal in VMAT2-MAOA DKO (see Fig. 7F).
Table 1.
Number of pyknotic profiles in the superficial layers (II/III) of the cingulate cortex of wild type, VMAT2 KO, and VMAT2-MAOA DKO from P1.5 to P4.5
| P1.5 | P2.5 | P3.5 | P4.5 | |
|---|---|---|---|---|
| Wild type | 23.4 ± 5.1 (n = 7) | 19.1 ± 2.6 (n = 4) | 11 ± 3.8 (n = 3) | 6.2 ± 4.8 (n = 3) |
| VMAT2 KO | 42.2 ± 5.5* (n = 7) | 29.9 ± 7.7 (n = 4) | 23 ± 6.8 (n = 3) | 13.5 ± 8.8 (n = 3) |
| VMAT2-MAOA DKO | 22 ± 4.2 (n = 3) | 22.7 ± 6 (n = 4) | 14.4 ± 4.9 (n = 3) | 8.1 ± 0.3 (n = 3) |
Data are expressed as the mean ± SD. *p < 0.05 indicates that the results are statistically different between VMAT2 KO group and VMAT2-MAOA DKO and wild-type groups.
5-HT2 receptor activation partially reverses the increased cell death in VMAT2 KO
In cortical cultures, serotonin acting through 5-HT2 receptors enhances the survival of glutamatergic neurons (Lavdas et al., 1997). We analyzed the spatiotemporal pattern of 5-HT2A and 5-HT2C mRNA expression in the cerebral cortex during the first postnatal week. The 5-HT2A gene is expressed in layer V neurons of Cg and RSG (Fig. 6C). Interestingly, the 5-HT2C gene is dynamically expressed in layer II of Cg and RSG during the early postnatal period (Fig. 6B). VMAT2 KO mice were treated with the 5-HT2A/2C agonist DOI. Counts of pyknotic profiles indicated a reduction of the massive cell death observed in VMAT2 KO treated mice (Fig. 6D).
Figure 6.

Pattern of 5-HT2A and 5-HT2C receptor mRNAs in the cingulate cortex. A–C, Alternate sections processed with Nissl (A), 5-HT2A (B), and 5-HT2C (C) mRNA detection. B, 5-HT2A mRNAs were detected in layer III, layer VI, and the subplate. Arrow indicates layer II. C, 5-HT2C mRNA expression was moderate and homogeneous in layer II. Scattered neurons expressing high levels of 5-HT2C mRNA were detected in the upper part of layer V. D, In wild type, DOI injection (n = 5) does not induce changes in the cell death ratio compared with saline (n = 5). In VMAT2 KO, DOI injection (n = 5) decreases cell death in the supragranular layers of Cg compared with VMAT2 KO injected with saline (n = 4). *p < 0.05 indicates that the results are statistically different. Scale bars, 370 μm.
Levels of neurotrophins are unchanged in the cortex of VMAT2 KO
5-HT2A receptor activation increases the expression of BDNF mRNA in the neocortex and hippocampus (Vaidya et al., 1997). However, in VMAT2 KO (Fig. 7A–C) and VMAT2-MAOA DKO (Fig. 7C), in situ hybridization showed that trkB, trkC, BDNF, and NT3 mRNAs are expressed according to a normal pattern and at unchanged levels. To determine whether changes in 5-HT levels could affect other anti-apoptotic pathways, we quantified the levels of Bcl-XL mRNA and found a significant decrease in VMAT2 KO. Bcl-XL mRNA levels were normalized in VMAT2-MAOA DKO (Fig. 7D–F).
The neuroprotectant effect of 5-HT is independent of trkB signaling
To further exclude the participation of trkB signaling pathways in the anti-apoptotic effects of increases in 5-HT levels, we examined the brains of trkB KO mice. These mice display an increase of cell death in the supragranular layers of the cingulate and retrosplenial cortices (Fig. 7G) and have normal levels of 5-HT. Mice lacking both trkB and MAOA display high levels of brain 5-HT, similar to those found in the MAOA KO mice (Vitalis et al., 2002b). Interestingly a rescue of cell death in the cortex of the trkB-MAOA DKO mice was noted during the P0–P4 period (Fig. 7G).
Discussion
Despite the essential role of the core apoptotic pathway, little is known about the principal actors and signals regulating PCD of postmitotic neurons in the cerebral cortex (Kuan et al., 2000). Here we show that cell death in the cerebral cortex is apoptotic in nature, dependent of the Bcl-XL/Bax ratio and of the subsequent activation of the apoptosome–caspase-3 cascade. The role played by neurotransmission in neuronal apoptosis appears to be essential because invalidation of Munc-18.1, a key player in membrane fusion and synaptic vesicle exocytosis, triggers massive and widespread cell death in the brain (Verhage et al., 2000). Similarly, blockade of the NMDA receptors causes an increased cell death (Ikonomidou et al., 1999). In this study, we evaluated the impact of serotoninergic and catecholaminergic neurotransmission blockade by either inhibiting their synthesis or invalidating monoamine storage. The results show that serotonin, through 5-HT2 receptor activation, can act as a neuroprotectant, controlling the expression of the anti-apoptotic protein Bcl-XL, thereby diminishing the risk of mitochondrial-dependent activation of caspases.
Apoptosis is the main PCD in the postnatal cerebral cortex
We confirm previous studies showing that PCD in the developing cortex is both area and layer specific during the first 5 postnatal days. Areas corresponding to cingulate and retrosplenial cortices display cell death ratios superior to those displayed in primary sensory areas. Cell death targets preferentially supragranular neurons, particularly in layer II (Finlay et al., 1983; Verney et al., 2000). Furthermore, our results indicate that the vast majority of the observed cell death is apoptotic in nature. Electron microscopy does not detect autophagic characteristics such as large autophagosomes. Apoptosis is caspase dependent because we do not detect AIF mitochondrial extrusion. Finally, the lack of expression of the FasL/FasR mRNAs and the lack of activation of the procaspase-8 suggest that the mechanism of apoptosis is not a type I apoptosis, independent of the mitochondria. We show the involvement of the mitochondrial-dependent pathway because caspase-9 and caspase-3 are activated. The peptide antibody directed against the amino acids 91–105 of the c-jun protein sequence reveals neoepitopes, which are products of caspase-3 proteolytic activity (Ribera et al., 2002). Using this antibody, we confirm the caspase-3 dependence of cortical apoptosis during early postnatal development. We also show the unique combination of Bcl-2 family members expressed by cortical neurons, the proapoptotic Bax and the anti-apoptotic Bcl-XL. Judging from Bcl-XL/Bax mRNA ratio, cortical neurons in the supragranular layers exhibit higher susceptibility to apoptosis than infragranular layers from P3. Finally, our observations indicate that, during this period, apoptosis concerns glutamatergic neurons and not GABAergic neurons.
Inhibition of monoamine storage increases cell death
Because monoaminergic neurotransmitter systems appear early in cerebral cortex development (Lauder, 1993; Vitalis and Parnavelas, 2003), we evaluated the potential role of monoamines, serotonin, and catecholamines in regulating PCD in early postnatal development. The brain VMAT2 stores monoamines in synaptic vesicles, protecting them from degradation. As a consequence, invalidation of the VMAT2 gene in vivo leads to a drastic depletion of monoamine levels (Fon et al., 1997), providing a model of nearly complete abrogation of monoaminergic neurotransmission. Using this model, we find a significant increase of cell death in the supragranular layers of the cingulate and retrosplenial areas. These results can further be correlated with an increase in apoptosis, as detected by electron microscopy and TUNEL. Additional characterization also indicates an increase in caspase-9-, caspase-3-, and c-jun-immunoreactive profiles, suggesting that the increase in cell death observed in the cortex of VMAT2 KO mice is attributable to an increase of apoptotic caspase-dependent mechanism. We also observe a specific decrease in the mRNA levels of the anti-apoptotic factor Bcl-XL, whereas mRNA levels of the proapoptotic factor Bax remain unchanged. This suggests that the decrease of the Bcl-XL/Bax mRNA ratio is probably the main phenomenon at play in the increased cell death in VMAT2 KO cerebral cortex. However, the precise mechanisms by which the depletion of VMAT2 leads to this change in gene expression and to exaggerated cell death remain elusive at the moment.
Inhibition of monoamine synthesis
Because VMAT2 affects the levels of all amines, to gain additional selectivity, we blocked the synthesis of a given monoamine. We depleted pharmacologically serotonin and/or catecholamine levels by inhibiting their synthesis during the first 5 postnatal days. Our data failed to show an increase in cell death in the cerebral cortex. However, these pharmacological treatments are not specific and may cause only partial monoamine depletion [80% reduction of 5-HT in the cortex (Dailly et al., 2006); 50% in the whole brain (Rebsam et al., 2005)]. To avoid any confounding effect of pharmacological treatment, we used mice lacking the transcription factor Pet-1. This transcription factor specifies the vast majority of central serotoninergic neurons, and its invalidation causes the loss of 80% of dorsal and median raphe serotoninergic neurons (Hendricks et al., 2003). As a consequence, Pet-1 KO display a 80% reduction of serotonin content in the brain (Hendricks et al., 2003) and, as shown here, a severe reduction of serotonin-positive fibers in the cingulate and retrosplenial cortices. However, there was no increase in cell death in these cortical areas. It is possible that the lowered levels of serotonin that are achieved by the pharmacological inhibition of tryptophan hydroxylase or the genetic invalidation of Pet-1 do not suffice to increase cell death. Indeed G-protein-coupled serotonin receptors have a high affinity for serotonin, and the remaining serotonin could suffice to activate them and maintain a normal level of apoptotic cell death. Alternatively, it is possible that, in the VMAT2 KO, it is the combination of a severe depletion of all monoamines that is responsible for the increase in cell death. Thus, other genetic models with more severe serotoninergic or catecholaminergic (dopaminergic and noradrenergic) depletion are warranted to definitively settle this question.
High serotonin levels have a neuroprotectant action in early development
In synaptic vesicles, monoamines are protected from degradation by MAO activity. MAOA is the major form expressed in specific neuronal populations such as serotoninergic neurons during early postnatal life (Vitalis et al., 2002a). Thus, invalidating MAOA in a VMAT2 KO genetic background should maintain a pool of cytoplasmic 5-HT by preventing its degradation. Indeed, high serotonin levels are detected in the brain of VMAT2-MAOA DKO mice, whereas dopamine and norepinephrine levels remain low or undetectable (Alvarez et al., 2002). Interestingly, the increase in cell death observed in the VMAT2 KO is reversed in the cerebral cortex of VMAT2-MAOA DKO, indicating that the high serotonin levels observed in VMAT2-MAOA KO could act as a neuroprotectant on cortical neurons. This effect confirms a previous report by Persico et al. (2003) in the brain of mice lacking 5-HTT. 5-HTT KO mice display high serotonin levels and reduced PCD, as judged by the decrease of TUNEL-positive cells in their brain. Interestingly, we find that Bcl-XL mRNA levels return to normal in the cerebral cortex of VMAT2-MAOA DKO, and we do not detect a decrease in expression of the proapoptotic factor Bax. This suggests that high serotonin levels maintain a normal Bcl-XL/Bax mRNA ratio. As a consequence, a normal level of cell death is observed in the cerebral cortex of VMAT2-MAOA DKO. The mechanisms that allow this normalization at both the cellular and molecular levels are not fully understood but likely involve one or several 5-HT receptors subtypes. We find that 5-HT2 receptor activation leads to a partial restoration of the increased cell death phenotype in VMAT2 KO. These results are consistent with the prosurvival activity of 5-HT2 receptor activation that was noted previously in vitro (Dooley et al., 1997). Because activation of 5-HT2A/2C receptors leads to an augmentation of the trophic factor BDNF (Vaidya et al., 1997) and mice lacking the catalytic domain of trkB, the high-affinity receptor of BDNF, display an increase in cell death in the cingulate cortex (Alcantara et al., 1997; our study), we hypothesized that trkB-dependent mechanisms could be involved in the anti-apoptotic cascade triggered by high serotonin levels. However, quantification of BDNF mRNA levels showed no differences between normal, VMAT2 KO, or VMAT2-MAOA DKO in the cingulate cortex. Furthermore, trkB KO show a rescue of cell death in conditions of increased 5-HT levels. These results suggest that cell death in the VMAT2 KO is not attributable to a diminution of trkB signaling. However, we cannot rule out signaling via p75 neurotrophin receptor (Kalb, 2005). Finally we analyzed the neurotrophin NT3 that is selectively expressed in the cingulate cortex and observed no change of mRNA expression in VMAT2 KO or VMAT2-MAOA DKO.
In conclusion, severe depletion of serotonin and catecholamines increases PCD in specific areas of the cerebral cortex, whereas partial depletion of these amines has no visible consequences on PCD. Conversely, high serotonin levels appear to have a potent neuroprotectant effect in vivo. Part of this neuroprotectant effect is mediated by 5-HT2A/2C receptor signaling and involves the control of Bcl-XL/Bax mRNA ratios.
Footnotes
This work was supported by Institut National de la Santé et de la Recherche Médicale, Centre National de la Recherche Scientifique (O.C.), the Ecole Pratique des Hautes Etudes (K.H.E.-H.), University Paris 6, and Fondation Lejeune. We thank Edward Fon and Robert Edwards of the gift of the VMAT2 KO mice, Denis Lecren for photographic assistance, and Isabelle Seif and Constantino Sotelo for their help and support.
References
- Alcantara S, Frisen J, Del Rio JA, Soriano E, Barbacid M, Silos-Santiago I. TrkB signalling is required for postnatal survival of CNS neurons and protects hippocampal and motor neurons from axotomy-induced cell. J Neurosci. 1997;17:3623–3633. doi: 10.1523/JNEUROSCI.17-10-03623.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez C, Vitalis T, Fon EA, Hanoun N, Hamon M, Seif I, Edwards R, Gaspar P, Cases O. Effects of genetic depletion of monoamines on somatosensory cortical development. Neuroscience. 2002;115:753–764. doi: 10.1016/s0306-4522(02)00484-0. [DOI] [PubMed] [Google Scholar]
- Blaschke AJ, Staley K, Chun J. Widespread programmed cell death in proliferative and postmitotic regions of the fetal cerebral cortex. Development. 1996;122:1165–1174. doi: 10.1242/dev.122.4.1165. [DOI] [PubMed] [Google Scholar]
- Boulland JL, Qureshi T, Seal RP, Rafiki A, Gundersen V, Bergersen LH, Fremeau RT, Jr, Edwards RH, Storm-Mathisen J, Chaudhry FA. Expression of the vesicular glutamate transporters during development indicates the widespread corelease of multiple neurotransmitters. J Comp Neurol. 2004;480:264–280. doi: 10.1002/cne.20354. [DOI] [PubMed] [Google Scholar]
- Casas C, Ribera J, Esquerda JE. Antibodies against c-Jun N-terminal peptide cross-react with neo-epitopes emerging after caspase-mediated proteolysis during apoptosis. J Neurochem. 2001;77:904–915. doi: 10.1046/j.1471-4159.2001.00314.x. [DOI] [PubMed] [Google Scholar]
- Cases O, Seif I, Grimsby J, Gaspar P, Chen K, Pournin S, Muller U, Aguet M, Babinet C, Shih JC, De Maeyer E. Aggressive behavior and altered amounts of brain serotonin and norepinephrine in mice lacking MAOA. Science. 1995;268:1763–1766. doi: 10.1126/science.7792602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catapano LA, Arnold MW, Perez FA, Macklis JD. Specific neurotrophic factors support the survival of cortical projection neurons at distinct stages of development. J Neurosci. 2001;21:8863–8872. doi: 10.1523/JNEUROSCI.21-22-08863.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dailly E, Chenu F, Petit-Demouliere B, Bourin M. Specificity and efficacy of noradrenaline, serotonin depletion in discrete brain areas of Swiss mice by neurotoxins. J Neurosci Methods. 2006;150:111–115. doi: 10.1016/j.jneumeth.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Dikranian K, Ishimaru MJ, Tenkova T, Labruyere J, Qin YQ, Ikonomidou C, Olney JW. Apoptosis in the in vivo mammalian forebrain. Neurobiol Dis. 2001;8:359–379. doi: 10.1006/nbdi.2001.0411. [DOI] [PubMed] [Google Scholar]
- Dooley AE, Pappas IS, Parnavelas JG. Serotonin promotes the survival of cortical glutamatergic neurons in vitro. Exp Neurol. 1997;148:205–214. doi: 10.1006/exnr.1997.6633. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Bernet E, Soriano E, del Rio T, Fonseca M. Naturally occuring cell death in the cerebral cortex of the rat and removal of dead cells by transitory phagocytes. Neuroscience. 1990;39:451–458. doi: 10.1016/0306-4522(90)90281-8. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Soriano E, del Rio JA, Alcantara S, Audadell C. Cell death and removal in the cerebral cortex during development. Prog Neurobiol. 1992;39:1–43. doi: 10.1016/0301-0082(92)90029-e. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Segui J, Olive M. Strong c-Jun immunoreactivity is associated with apoptotic cell death in human tumors of the central nervous system. Neurosci Lett. 1996;214:49–52. [PubMed] [Google Scholar]
- Ferrer I, Pozas E, Planas AM. c-Jun/AP-1 (N) expression and apoptosis. Neuroscience. 2000;96:447–448. doi: 10.1016/s0306-4522(99)00551-5. [DOI] [PubMed] [Google Scholar]
- Finlay BL, Slattery M. Local differences in the amount of early cell death in neocortex predict adult local specializations. Science. 1983;219:1349–1351. doi: 10.1126/science.6828866. [DOI] [PubMed] [Google Scholar]
- Fon EA, Pothos EN, Sun BC, Killeen N, Sulzer D, Edwards RH. Vesicular transport regulates monoamine storage and release but is not essential for amphetamine action. Neuron. 1997;19:1271–1283. doi: 10.1016/s0896-6273(00)80418-3. [DOI] [PubMed] [Google Scholar]
- Gaspar P, Cases O, Maroteaux L. The developmental role of serotonin: news from mouse molecular genetics. Nat Rev Neurosci. 2003;4:1002–1012. doi: 10.1038/nrn1256. [DOI] [PubMed] [Google Scholar]
- Hendricks TJ, Fyodorov DV, Wegman LJ, Letutiu NB, Pehek EA, Yamamoto B, Silver J, Weeber EJ, Sweatt JD, Deneris ES. Pet-1 ETS gene plays a critical role in 5-HT neuron development and is required for normal anxiety-like and aggressive behavior. Neuron. 2003;37:233–247. doi: 10.1016/s0896-6273(02)01167-4. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Bosch F, Miksa M, Bittigau P, Vockler J, Dikranian K, Tenkova TI, Stefovska V, Turski L, Olney JW. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. 1999;283:70–74. doi: 10.1126/science.283.5398.70. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Bittigau P, Koch C, Genz K, Hoerster F, Felderhoff-Mueser U, Tenkova T, Dikranian K, Olney JW. Neurotransmitters and apoptosis in the developing brain. Biochem Pharmacol. 2001;62:401–405. doi: 10.1016/s0006-2952(01)00696-7. [DOI] [PubMed] [Google Scholar]
- Kalb R. The protean actions of neurotrophins and their receptors on the life and death of neurons. Trends Neurosci. 2005;28:5–11. doi: 10.1016/j.tins.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Klein R, Smeyne RJ, Wurst W, Long LK, Auerbach BA, Joyner AL, Barbacid M. Targeted disruption of the trkB neurotrophin receptor gene results in nervous system lesions and neonatal death. Cell. 1993;75:113–122. [PubMed] [Google Scholar]
- Kuan CY, Roth KA, Flavell RA, Rakic P. Mechanisms of programmed cell death in the developing brain. Trends Neurosci. 2000;23:291–297. doi: 10.1016/s0166-2236(00)01581-2. [DOI] [PubMed] [Google Scholar]
- Lauder JM. Neurotransmitters as growth regulatory signals: role of receptors and second messengers. Trends Neurosci. 1993;16:233–240. doi: 10.1016/0166-2236(93)90162-f. [DOI] [PubMed] [Google Scholar]
- Lavdas AA, Blue ME, Lincoln J, Parnavelas JG. Serotonin promotes the differentiation of glutamate neurons in organotypic slice cultures of the developing cerebral cortex. J Neurosci. 1997;17:7872–7880. doi: 10.1523/JNEUROSCI.17-20-07872.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin K. New York: Academic; 2001. The mouse brain in stereotaxic coordinates. [Google Scholar]
- Persico AM, Baldi A, Dell'Acqua ML, Moessner R, Murphy DL, Lesch KP, Keller F. Reduced programmed cell death in brains of serotonin transporter knockout mice. NeuroReport. 2003;14:341–344. doi: 10.1097/00001756-200303030-00009. [DOI] [PubMed] [Google Scholar]
- Pohl D, Bittigau P, Ishimaru MJ, Stadthaus D, Hubner C, Olney JW, Turski L, Ikonomidou C. N-Methyl-d-aspartate antagonists and apoptotic cell death triggered by head trauma in developing rat brain. Proc Natl Acad Sci USA. 1999;96:2508–2513. doi: 10.1073/pnas.96.5.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebsam A, Seif I, Gaspar P. Dissociating barrel development and lesion-induced plasticity in the mouse somatosensory cortex. J Neurosci. 2005;25:706–710. doi: 10.1523/JNEUROSCI.4191-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribera J, Ayala V, Esquerda JE. c-Jun-like immunoreactivity in apoptosis is the result of a crossreaction with neoantigenic sites exposed by caspase-3-mediated proteolysis. J Histochem Cytochem. 2002;50:961–972. doi: 10.1177/002215540205000710. [DOI] [PubMed] [Google Scholar]
- Shih JC, Chen K, Ridd MJ. Monoamine oxidase: from genes to behavior. Annu Rev Neurosci. 1999;22:197–217. doi: 10.1146/annurev.neuro.22.1.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomaidou D, Mione MC, Cavanagh JF, Parnavelas JG. Apoptosis and its relation to the cell cycle in the developing cerebral cortex. J Neurosci. 1997;17:1075–1085. doi: 10.1523/JNEUROSCI.17-03-01075.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidya VA, Marek GJ, Aghajanian GK, Duman RS. 5-HT2A receptor-mediated regulation of brain-derived neurotrophic factor mRNA in the hippocampus and the neocortex. J Neurosci. 1997;17:2785–2795. doi: 10.1523/JNEUROSCI.17-08-02785.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, Toonen RF, Hammer RE, van den Berg TK, Missler M, Geuze HJ, Sudhof TC. Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science. 2000;287:864–869. doi: 10.1126/science.287.5454.864. [DOI] [PubMed] [Google Scholar]
- Verney C, Takahashi T, Bhide PG, Nowakowski RS, Caviness VS., Jr Independent controls for neocortical neuron production and histogenetic cell death. Dev Neurosci. 2000;22:125–138. doi: 10.1159/000017434. [DOI] [PubMed] [Google Scholar]
- Vitalis T, Parnavelas JG. The role of serotonin in early cortical development. Dev Neurosci. 2003;25:245–256. doi: 10.1159/000072272. [DOI] [PubMed] [Google Scholar]
- Vitalis T, Fouquet C, Alvarez C, Seif I, Price D, Gaspar P, Cases O. Developmental expression of monoamine oxidases A and B in the central and peripheral nervous systems of the mouse. J Comp Neurol. 2002a;442:331–347. doi: 10.1002/cne.10093. [DOI] [PubMed] [Google Scholar]
- Vitalis T, Cases O, Gillies K, Hanoun N, Hamon M, Seif I, Gaspar P, Kind P, Price DJ. Interactions between TrkB signaling and serotonin excess in the developing murine somatosensory cortex: a role in tangential and radial organization of thalamocortical axons. J Neurosci. 2002b;22:4987–5000. doi: 10.1523/JNEUROSCI.22-12-04987.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]