

Abstract
Lewy bodies, the pathological hallmark of dementia with Lewy bodies (DLB), are large juxtanuclear inclusions of aggregated α-synuclein. However, the small number of cortical Lewy bodies relative to the total neuron count does not correlate with the extent of cognitive impairment. In contrast to dopaminergic neurons in Parkinson's disease, nerve cell loss is usually less prevalent in the cortex of DLB, suggesting a different mechanism of neurodegeneration.
Because antibodies used for immunodetection per se do not generally differentiate the aggregated from the physiological and monomeric isoform of α-synuclein, we developed the paraffin-embedded tissue (PET) blot and the protein aggregate filtration (PAF) assay for the sensitive and selective detection of α-synuclein aggregates in tissue slides and brain homogenates, respectively.
In contrast to common immunohistochemistry, the PET blot detected an enormous number of small α-synuclein aggregates, which, in contrast to the few Lewy bodies, may explain the cognitive impairment in DLB. Using the PAF assay, we demonstrate that the absolute majority of α-synuclein aggregates are located at presynaptic terminals, suggesting a severe pathological impact on synaptic function. Indeed, parallel to the massive presynaptic accumulation of α-synuclein aggregates, we observed significant synaptic pathology with almost complete loss of dendritic spines at the postsynaptic area.
Our results provide strong evidence for a novel concept of neurodegeneration for DLB in which synaptic dysfunction is caused by presynaptic accumulation of α-synuclein aggregates. This concept may also be valid for Parkinson's disease.
Keywords: α-synuclein, dementia with Lewy bodies, Parkinson's disease, dendritic spines, neurodegeneration, Lewy bodies, presynaptic
Introduction
Dementia with Lewy bodies (DLB) is the second most common cause of dementia (∼25%) after Alzheimer's disease (Perry et al., 1990). DLB is characterized by cognitive impairment and motor dysfunction (Burn, 2004). Parkinson patients primarily affected by progressive motor dysfunction may develop dementia indistinguishable from DLB with an incidence of 20–40% (Burn, 2004). However, in contrast to Parkinson's disease (PD), which is thought to be caused by the loss of dopaminergic neurons in the substantia nigra (Olanow and Tatton, 1999), nerve cell loss is usually less prevalent in the cortex of DLB, suggesting a different mechanism of neurodegeneration.
Both DLB and PD are diagnosed postmortem by the histological detection of Lewy bodies. These are large intraneuronal protein aggregates localized near the nucleus and mainly consisting of α-synuclein (Spillantini et al., 1997). However, although Lewy bodies are pathological hallmarks of DLB, they are often found in cases without any clinical signs of dementia (Hughes et al., 1992; Parkkinen et al., 2005). Moreover, the number of cortical Lewy bodies relative to the total neuron count does not correlate with the severity of clinical symptoms and disease duration (Gomez-Tortosa et al., 1999). We therefore hypothesized that DLB might be caused by α-synuclein aggregates other than Lewy bodies, which are only detectable by more sensitive and specific methods.
The few small α-synuclein aggregates observed in axonal terminals preceding the formation of Lewy bodies in DLB (Marui et al., 2002) gave rise to the idea that Lewy bodies are formed by an aggresome-related process as a general cytoprotective measure in which smaller α-synuclein aggregates are sequestered from the neuronal periphery by active retrograde transport on microtubules (Kopito, 2000; McNaught et al., 2002). In contrast to juxtanuclear Lewy bodies, native α-synuclein is localized at presynaptic terminals (Hsu et al., 1998; Murphy et al., 2000) where it exerts its physiological function (Chandra et al., 2005). Together with the fact that presynaptic terminals are the physiologically most active compartments of neurons, it would seem to suggest that presynaptic terminals are the sites of α-synuclein aggregation.
Materials and Methods
Paraffin-embedded tissue blotting.
Deparaffinized and dried tissue sections layered on nitrocellulose membranes were prewetted and digested with 250 μg/ml proteinase-K in digestion buffer (10 mm Tris-HCl, pH 7.8, 100 mm NaCl, and 0.1% Brij 35) for 8 h at 55°C. After washing, the membranes were treated for 10 min with 3 m guanidine isothiocyanate in 10 mm Tris-HCl, pH 7.8, for optimal epitope retrieval. Immunodetection of α-synuclein aggregates (LB509, epitope 115-122, 1:10,000 for 1 h; Zymed, Berlin, Germany) was performed with alkaline phosphatase-coupled rabbit anti-mouse antibody as a secondary antibody. Aggregates were visualized by the formazan reaction using 0.0675% (w/v) nitro-blue tetrazolium chloride (Roche, Mannheim, Germany) and 0.033% (w/v) 5-bromo-4-chloro-3′-indoyl-phosphate p-toluidine salt (Roche) in NTM buffer (10 mm Tris-HCl, pH 9.5, 100 mm NaCl, and 50 mm MgCl2).
Protein aggregate filtration assay.
Brain samples from the gyrus frontales medialis were homogenized in 9 vol (w/v) of MSE buffer (10 mm MOPS/KOH, pH 7.4, 0.32 m sucrose, 1 mm EGTA, and 1 mm EDTA) including protease inhibitors (1 mm PMSF, 0.2 mmN-tosyl-l-phenylalanine chloromethyl ketone, and 0.2 mmN-tosyl-l-lysine chloromethyl ketone). After centrifugation at 16,000 × g for 5 min, the pellet fractions were subjected to digestion with DNase I in 4% SB14 (N-tetradecyl-N,N-dimethyl-3-ammonio-1-propane sulfonate) for 15 min at 37°C. All samples were adjusted to a final concentration of 5% sodium lauryl sulfate (SLS) and sucked through a blocked nitrocellulose membrane (0.2 μm pore size). After filtration, each slot was washed with 0.1% SDS. Immunoreactivity of α-synuclein aggregates (LB509, 1:10,000 for 1 h) was visualized by chemiluminescence using a secondary HRP-coupled goat anti-mouse antibody.
Sucrose gradient analysis of α-synuclein aggregates.
For the separation of α-synuclein aggregates of cortical brain homogenates from the gyrus frontales medialis, a sucrose step-gradient was prepared overlaying 2.2 m with 1.4 m and finally with 1.2 m sucrose in volume ratios of 3.5:8:8 (v/v) (Iwatsubo et al., 1996). The tissue was rapidly thawed and homogenized in ice-cold MSE buffer including protease inhibitors by 12 strokes at 900 rpm with a Potter-Elvehjem tissue grinder (Wheaton Science Products, Millville, NJ). The homogenate was adjusted to 1.0 m sucrose and carefully layered on the gradient. The gradients were centrifuged at 160,000 × g for 3 h using a SW32.1 rotor (Beckman, Fullerton, CA). Fractions of the gradient were collected from the bottom.
For more detailed analysis of the synaptosomal fraction, the corresponding fractions were diluted threefold and centrifuged at 16,000 × g for 15 min. The pellet was resuspended in cold 320 mm sucrose containing 5 mm HEPES/NaOH, pH 7.4. For hypotonic lysis, the suspension was diluted 10-fold with cold water and incubated for 45 min on ice. The suspension was homogenized and applied to a sucrose step-gradient containing 2.2, 1.4, 1.2, 1.1, and 0.8 m sucrose at a ratio of 2.5:6:6:6:6 (v/v). The gradient was centrifuged in a SW32.1 rotor at 160,000 × g for 5 h.
The gradients were fractionated from the bottom and analyzed for α-synuclein aggregates with the protein aggregate filtration (PAF) method. Fractions were adjusted to 5% SLS, sucked through a blocked nitrocellulose membrane, and developed as described above.
For additional methods, see the supplemental material (available at www.jneurosci.org).
Results
Detection of α-synuclein aggregates other than Lewy bodies
Because immunohistochemistry is used for the morphological analysis of native α-synuclein (Hsu et al., 1998), as well as for aggregates, existing α-synuclein aggregates could not be differentiated from physiological α-synuclein at synaptic terminals. We therefore developed a method originally applied to prion diseases for the selective and sensitive detection of protein aggregates in tissue slides, the so-called paraffin-embedded tissue (PET) blot (Schulz-Schaeffer et al., 2000). The PET blotting method allows the selective detection of protein aggregates, because non-aggregated proteins are degraded by a proteinase-K treatment.
At present, the neuropathological diagnosis of DLB depends on histological detection of Lewy bodies in the cortex and brain stem. These are best visualized by immunohistochemistry using α-synuclein antibodies (Fig. 1A). In contrast to immunohistochemical staining, the PET blot method adapted to the detection of α-synuclein aggregates additionally shows massive deposits of diffuse aggregates in the cortical and subcortical gray matter (Fig. 1C). Here, the cingulate gyrus and gyrus frontales medialis were used because they are scoring areas for the neuropathological diagnosis (McKeith et al., 1996). Magnified, the diffuse aggregates become visible as fine granular deposits (Fig. 1D), which are smaller than the also-visible Lewy bodies (Fig. 1E). This pattern is significantly different from immunohistochemistry even using an antibody against phosphorylated α-synuclein (Fujiwara et al., 2002). As opposed to Lewy bodies, the enormous number of small α-synuclein aggregates may explain the cognitive impairment in DLB. However, the origin of these small granular aggregates and how they induce neuronal degeneration remains enigmatic. The distribution of the fine granular α-synuclein deposits of the PET blot strongly resembled the distribution of prion protein aggregates in the majority of Creutzfeldt–Jakob diseases, which are known to be located at presynaptic terminals (Kitamoto et al., 1992), suggesting a presynaptic localization of the small α-synuclein aggregates.
Figure 1.
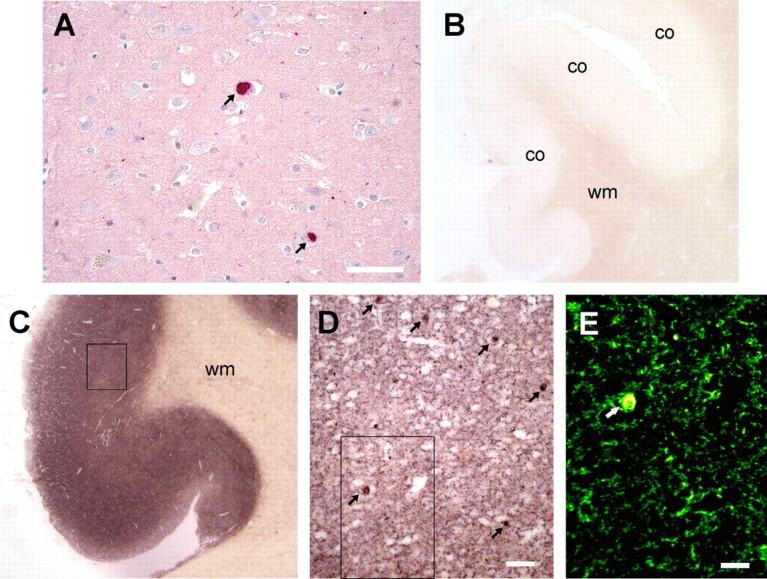
Revealing previously undetectable, small granular α-synuclein aggregates with the PET blot. A, Common immunohistochemistry (LB509) identifies only α-synuclein deposits (dark red) as Lewy bodies (arrows) and few Lewy neurites (dots or threads). B, C, Compared with a control (B), the PET blot method detects diffuse α-synuclein aggregates in the frontal cortex (co) of the gyrus cinguli but not in the white matter (wm). The enormous number of α-synuclein aggregates is visible as a dark brown color reaction throughout the cortex in DLB. D, The magnification (see box in C) shows that the majority of α-synuclein aggregates other than Lewy bodies (arrows) form fine granular deposits. E, The virtual fluorescence image of the PET blot section (box in D) created by digital processing clearly demonstrates that the fine granular α-synuclein aggregates are much smaller compared with Lewy bodies (arrow). Scale bars: A, 50 μm; D, 20 μm; E, 10 μm.
Presynaptic accumulation of small α-synuclein aggregates
To demonstrate a possible localization of α-synuclein aggregates at presynaptic terminals, we used brain homogenates of confirmed human DLB cases because, unlike for PD, there is no mouse model for DLB. It has been shown previously that active synaptosomes representing presynaptic terminals can be prepared from frozen human brains (Hardy et al., 1983) to measure their neurotransmitter uptake (Dodd et al., 1989). Brain homogenates were separated by subcellular fractionation using sucrose density gradient centrifugation. The biochemical detection of α-synuclein aggregates in suspensions by means of their selective insolubility in detergents, followed by Western blot analysis, turned out to be difficult and laborious. We therefore developed the PAF assay as a new and simple biochemical method to screen liquid samples for the presence of α-synuclein aggregates, thus avoiding any ultracentrifugation step. This method is based on the separation of aggregates from their native isoform by microfiltration using a membrane with 0.2 μm pore size and subsequent immunodetection. The PAF assay specifically detects α-synuclein aggregates in brain homogenates of DLB cases, because no immunoreactivity was observed in cases with isolated tau pathology or in non-dementia controls (Fig. 2A).
Figure 2.

Small granular α-synuclein deposits represent the absolute majority of α-synuclein aggregates in DLB. A, The new developed PAF assay specifically detects α-synuclein aggregates in brain homogenates of DLB compared with a case with tau pathology (TP) and non-dementia controls (C1 and C2). B, Sucrose gradient with the indicated molar concentrations for the separation of α-synuclein aggregates in brain homogenates by ultracentrifugation, which was originally used for purification of Lewy bodies (Iwatsubo et al., 1996). C, PAF assay of the sucrose gradient fraction with the plot of its densitometric analysis. D, Identification of small granular α-synuclein (α-Syn) aggregates in the three sucrose gradient peaks by immunofluorescence (LB509; red) and thioflavin S (ThS) fluorescence (green). Scale bars, 20 μm.
For sucrose gradient analysis of brain homogenates, we followed a protocol originally used for the isolation of Lewy bodies (Fig. 2B) (Iwatsubo et al., 1996). Surprisingly, the densitometric analysis of the gradient fractions with the PAF assay resulted in three different α-synuclein aggregate peaks (Fig. 2C, peaks I–III). This was one peak more than expected from the PET blot showing Lewy bodies and the fine granular α-synuclein deposits (Fig. 1). The first peak corresponded to the Lewy body fraction from which the Lewy bodies were previously isolated for further purification (Iwatsubo et al., 1996). It is evident that the Lewy body peak is only a minor fraction representing 0.02–11% of the α-synuclein aggregates in DLB.
To locate the fine granular aggregates within the sucrose gradient by their size, the α-synuclein aggregate peaks were analyzed by immunofluorescence using thioflavin S as a fluorescent marker for amyloidogenic aggregates. The α-synuclein immunoreactivities of all peaks showed a costaining with thioflavin S, suggesting a fibrillar fine structure of these aggregates (Fig. 2D). Very large structures, typical for Lewy bodies, were only observed within peak I, with diameters from 12 to 19 μm. Because of the fluorescence pattern (Fig. 2D), it appeared that the Lewy bodies were made up of smaller aggregate structures. Indeed, smaller but dense α-synuclein aggregates probably resulting from fragmentation of Lewy bodies were observed in peak I. However, cortical Lewy bodies in DLB are known to be less compact than midbrain Lewy bodies in PD. Much smaller α-synuclein aggregates are visible in peaks II and III (Fig. 2D), ranging from 1.0 to 4.3 μm (peak II) and from 0.7 to 3.9 μm (peak III), respectively. The average diameters of 2.4 μm (peak II) and 1.8 μm (peak III) indicate that these fractions represent the fine granular α-synuclein aggregates detected with the PET blot (Fig. 1C–E).
To demonstrate a possible association between α-synuclein aggregates and synaptosomes representing presynaptic terminals, sucrose gradient fractions were analyzed for syntaxin, a marker for synaptic plasma membrane, and synaptophysin, a synaptic vesicle marker (Fig. 3A). The signals of both markers were almost exclusively associated with the α-synuclein aggregate peak III (Fig. 3B). Moreover, the densitometric signals of syntaxin and synaptophysin within the fractions 17 and 19 also correlated quantitatively (Fig. 3B), suggesting that this peak contained synaptosomes. Indeed, synaptosomes corresponding to peak III have previously been isolated from the 1.0 m/1.2 m sucrose interface (IF) (Ueda et al., 1979). Because the synaptosomal fractions (peak III) represent the majority of α-synuclein aggregates ranging from 50 to 92% (fractions 15–19), they confirm the results of PET blot analysis.
Figure 3.

Small granular α-synuclein aggregates are trapped in synaptosomes, indicating a presynaptic localization. A, Location of synaptosomes by Western blot analysis of sucrose gradient fractions for syntaxin (STX; HPC-1) as the synaptic plasma membrane marker and synaptophysin (SPH; SY38) as the synaptic vesicle marker. B, Plot of the relative densitometric intensities for α-synuclein aggregates (filled circles), syntaxin (gray bars), and synaptophysin (black bars). C, Synaptosomes of peak III were subjected to hypotonic shock and applied to a more differentiated sucrose gradient. D, Western blot analysis of the gradient in C for synaptophysin and syntaxin. E, PAF blot analysis of the gradient in C for α-synuclein aggregates and their densitometric quantification (filled circles) together with synaptophysin (black bars) and syntaxin (gray bars). The fractions corresponding to the IFs of the gradient in C are marked by inverted triangles.
To determine whether the α-synuclein aggregates were localized inside the synaptosomes, the corresponding fractions were subjected to hypotonic lysis according to standard protocols (Huttner et al., 1983) and analyzed by a more differentiated sucrose gradient (Fig. 3C). Releasing α-synuclein aggregates from synaptosomes might result in a different localization compared with the results of Lewy body sucrose gradient analysis (Fig. 2 B). Indeed, analysis of the fractions with the PAF blot showed a clear shift of the α-synuclein aggregates from the synaptosomal IF (peak III or IF III) to the IF above 1.4 m sucrose (peak II or IF II), but no aggregates were found in the Lewy body fraction above 2.2 m sucrose (Fig. 3E). The different gradient localizations of syntaxin (Fig. 3E, IF III–IF V) and synaptophysin (Fig. 3E, IF II) confirmed lysis of synaptosomes. This clearly indicates that the aggregates were trapped in synaptosomes. Moreover, it became evident that the aggregates of peak II of the first sucrose gradient (Fig. 2B) were released either from synaptosomes during homogenization or are the result of a destroyed presynaptic membrane structure. This would mean that 89% to almost 100% of α-synuclein aggregates are located at presynaptic terminals.
Presynaptic aggregate accumulation is linked to loss of dendritic spines
The massive accumulation of α-synuclein aggregates at presynaptic terminals suggested a severe pathological impact on synaptic function. Therefore, we compared the levels of presynaptic and postsynaptic marker proteins of confirmed DLB cases with an age-matched, non-dementia control group using Western blot analysis for synaptophysin, syntaxin, postsynaptic density protein (PSD95), and drebrin. There was a clear reduction in the synaptic vesicle marker synaptophysin, which seems to correlate with the reduction in the presynaptic plasma membrane marker syntaxin compared with controls (Fig. 4A). Levels of synaptophysin and syntaxin were decreased by 52 and 57%, respectively (Fig. 4B). The reduction in presynaptic markers also appeared to correlate with the decrease in PSD95 as a postsynaptic marker protein by 48% (Fig. 4C,D). However, the most significant changes were observed for the actin-binding protein drebrin, which is involved in the formation and maintenance of dendritic spines (Takahashi et al., 2003). Actin polymerizes to a network responsible for the shape of dendritic spines. In DLB, drebrin was either totally absent or beyond the detection limit (Fig. 4C,D). This dramatic reduction in drebrin prompted us to look for changes in dendritic spine morphology. Indeed, Golgi–Cox–Davenport staining of cortical neurons revealed a dramatic reduction in dendritic spines in DLB (Fig. 4F,H) compared with controls (Fig. 4E,G). Thus, spine retraction rather than complete synaptic degradation appears to be the result of the massive presynaptic accumulation of α-synuclein aggregates, because the levels of synaptic markers (synaptophysin, syntaxin, and PSD95) are decreased only by ∼50%.
Figure 4.

Pathophysiological changes at synapses in DLB. A–D, Brain homogenates of six confirmed DLB cases (D1–D6) are compared with age-matched non-dementia controls (C1–C6). A, C, Western blot analysis for presynaptic markers (A) synaptophysin (SPH; SY38) and syntaxin (STX; HPC-1) and postsynaptic markers (C) postsynaptic density protein (PSD95, rabbit anti-PSD95) and drebrin (Dreb.; M2F6), including the loading control β-actin (β-Act.). B, D, Plot of the β-actin normalized average intensities for presynaptic marker (B) and postynaptic marker (D) proteins of non-dementia controls (black bars) and DLB cases (gray bars) ± SE. E–H, Golgi–Cox–Davenport staining of brain slices for visualization of dendrites and their spines in non-dementia control (E, G) and DLB (F, H). G and H show the magnification of the black boxes in E and F. Scale bars, 50 μm.
Discussion
Lewy body formation as cellular protective mechanism
Our results strongly suggest that presynaptic terminals in neurons are the loci of α-synuclein aggregate formation and that the subcellular localization is important for their neurodegenerative effect. It appears that aggresome formation resulting in Lewy bodies is a protective mechanism because of axonal retrograde transport of small, probably presynaptic α-synuclein aggregates (Kopito, 2000; McNaught et al., 2002). This reduces not only the accessible toxic surface compared with small aggregates but also removes them from their most hazardous location at presynaptic terminals.
Dementia in DLB by loss of dendritic spines
The accumulation of presynaptic α-synuclein aggregates is associated with the observed almost complete loss of dendritic spines. The depletion of the dendritic spine protein drebrin in DLB is in line with the changes in spines because it is involved in spine morphogenesis by binding to the actin network (Takahashi et al., 2003).
Memory is associated with the formation of new dendritic spines caused by long-term potentiation (Engert and Bonhoeffer, 1999). However, the opposite effect in which long-term depression resulted in complete retraction of spines was also observed, suggesting that spine morphology is an activity-dependent bidirectional process (Nagerl et al., 2004). Indeed, selective elimination of dendritic spines without apparent changes in the dendritic tree because of lack of sensory experience was observed (Trachtenberg et al., 2002). Thus, gradual increasing impairment of neurotransmitter release caused by presynaptic accumulation of α-synuclein aggregates would explain the observed pathological changes in dendritic spine morphology. The observed loss of dendritic spines is confirmed by the selective spinodendritic degeneration recently reported for DLB (Zaja-Milatovic et al., 2006). Interestingly, degeneration of dendritic spines in striatal medium spiny neurons was observed also in PD (Zaja-Milatovic et al., 2005), suggesting that our findings might also be relevant for PD.
In conclusion, our findings may serve as a basis for a novel concept of neurodegeneration in which synaptic dysfunction caused by synaptosomal α-synuclein aggregates sufficiently explains the neurodegenerative process in DLB. This concept may also be valid for PD, given that synaptic dysfunction precedes the cell death of dopaminergic neurons. Our data may pave the way toward completely new therapeutic strategies for DLB, and possibly also for PD, that focus on the maintenance of synaptic function.
Footnotes
This work was supported by the VolkswagenStiftung (ZN1294). We thank Manuela Becker and Tatjana Pfander for excellent technical assistance. We are grateful to U. Hahmann for critical discussions.
References
- Burn DJ. Cortical Lewy body disease. J Neurol Neurosurg Psychiatry. 2004;75:175–178. doi: 10.1136/jnnp.2003.025569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra S, Gallardo G, Fernandez-Chacon R, Schluter OM, Sudhof TC. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell. 2005;123:383–396. doi: 10.1016/j.cell.2005.09.028. [DOI] [PubMed] [Google Scholar]
- Dodd PR, Watson WE, Morrison MM, Johnston GA, Bird ED, Cowburn RF, Hardy JA. Uptake of gamma-aminobutyric acid and l-glutamic acid by synaptosomes from postmortem human cerebral cortex: multiple sites, sodium dependence and effect of tissue preparation. Brain Res. 1989;490:320–331. doi: 10.1016/0006-8993(89)90249-7. [DOI] [PubMed] [Google Scholar]
- Engert F, Bonhoeffer T. Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature. 1999;399:66–70. doi: 10.1038/19978. [DOI] [PubMed] [Google Scholar]
- Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T. Alpha-synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002;4:160–164. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- Gomez-Tortosa E, Newell K, Irizarry MC, Albert M, Growdon JH, Hyman BT. Clinical and quantitative pathologic correlates of dementia with Lewy bodies. Neurology. 1999;53:1284–1291. doi: 10.1212/wnl.53.6.1284. [DOI] [PubMed] [Google Scholar]
- Hardy JA, Dodd PR, Oakley AE, Perry RH, Edwardson JA, Kidd AM. Metabolically active synaptosomes can be prepared from frozen rat and human brain. J Neurochem. 1983;40:608–614. doi: 10.1111/j.1471-4159.1983.tb08024.x. [DOI] [PubMed] [Google Scholar]
- Hsu LJ, Mallory M, Xia Y, Veinbergs I, Hashimoto M, Yoshimoto M, Thal LJ, Saitoh T, Masliah E. Expression pattern of synucleins (non-Abeta component of Alzheimer's disease amyloid precursor protein/alpha-synuclein) during murine brain development. J Neurochem. 1998;71:338–344. doi: 10.1046/j.1471-4159.1998.71010338.x. [DOI] [PubMed] [Google Scholar]
- Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson's disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry. 1992;55:181–184. doi: 10.1136/jnnp.55.3.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttner WB, Schiebler W, Greengard P, De Camilli P. Synapsin I (protein I), a nerve terminal-specific phosphoprotein. III. Its association with synaptic vesicles studied in a highly purified synaptic vesicle preparation. J Cell Biol. 1983;96:1374–1388. doi: 10.1083/jcb.96.5.1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwatsubo T, Yamaguchi H, Fujimuro M, Yokosawa H, Ihara Y, Trojanowski JQ, Lee VM. Purification and characterization of Lewy bodies from the brains of patients with diffuse Lewy body disease. Am J Pathol. 1996;148:1517–1529. [PMC free article] [PubMed] [Google Scholar]
- Kitamoto T, Shin RW, Doh-ura K, Tomokane N, Miyazono M, Muramoto T, Tateishi J. Abnormal isoform of prion proteins accumulates in the synaptic structures of the central nervous system in patients with Creutzfeldt-Jakob disease. Am J Pathol. 1992;140:1285–1294. [PMC free article] [PubMed] [Google Scholar]
- Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000;10:524–530. doi: 10.1016/s0962-8924(00)01852-3. [DOI] [PubMed] [Google Scholar]
- Marui W, Iseki E, Nakai T, Miura S, Kato M, Ueda K, Kosaka K. Progression and staging of Lewy pathology in brains from patients with dementia with Lewy bodies. J Neurol Sci. 2002;195:153–159. doi: 10.1016/s0022-510x(02)00006-0. [DOI] [PubMed] [Google Scholar]
- McKeith IG, Galasko D, Kosaka K, Perry EK, Dickson DW, Hansen LA, Salmon DP, Lowe J, Mirra SS, Byrne EJ, Lennox G, Quinn NP, Edwardson JA, Ince PG, Bergeron C, Burns A, Miller BL, Lovestone S, Collerton D, Jansen EN, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology. 1996;47:1113–1124. doi: 10.1212/wnl.47.5.1113. [DOI] [PubMed] [Google Scholar]
- McNaught KS, Shashidharan P, Perl DP, Jenner P, Olanow CW. Aggresome-related biogenesis of Lewy bodies. Eur J Neurosci. 2002;16:2136–2148. doi: 10.1046/j.1460-9568.2002.02301.x. [DOI] [PubMed] [Google Scholar]
- Murphy DD, Rueter SM, Trojanowski JQ, Lee VM. Synucleins are developmentally expressed, and α-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J Neurosci. 2000;20:3214–3220. doi: 10.1523/JNEUROSCI.20-09-03214.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagerl UV, Eberhorn N, Cambridge SB, Bonhoeffer T. Bidirectional activity-dependent morphological plasticity in hippocampal neurons. Neuron. 2004;44:759–767. doi: 10.1016/j.neuron.2004.11.016. [DOI] [PubMed] [Google Scholar]
- Olanow CW, Tatton WG. Etiology and pathogenesis of Parkinson's disease. Annu Rev Neurosci. 1999;22:123–144. doi: 10.1146/annurev.neuro.22.1.123. [DOI] [PubMed] [Google Scholar]
- Parkkinen L, Kauppinen T, Pirttila T, Autere JM, Alafuzoff I. Alpha-synuclein pathology does not predict extrapyramidal symptoms or dementia. Ann Neurol. 2005;57:82–91. doi: 10.1002/ana.20321. [DOI] [PubMed] [Google Scholar]
- Perry RH, Irving D, Tomlinson BE. Lewy body prevalence in the aging brain: relationship to neuropsychiatric disorders, Alzheimer-type pathology and catecholaminergic nuclei. J Neurol Sci. 1990;100:223–233. doi: 10.1016/0022-510x(90)90037-n. [DOI] [PubMed] [Google Scholar]
- Schulz-Schaeffer WJ, Tschoke S, Kranefuss N, Drose W, Hause-Reitner D, Giese A, Groschup MH, Kretzschmar HA. The paraffin-embedded tissue blot detects PrP(Sc) early in the incubation time in prion diseases. Am J Pathol. 2000;156:51–56. doi: 10.1016/S0002-9440(10)64705-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Sekino Y, Tanaka S, Mizui T, Kishi S, Shirao T. Drebrin-dependent actin clustering in dendritic filopodia governs synaptic targeting of postsynaptic density-95 and dendritic spine morphogenesis. J Neurosci. 2003;23:6586–6595. doi: 10.1523/JNEUROSCI.23-16-06586.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trachtenberg JT, Chen BE, Knott GW, Feng G, Sanes JR, Welker E, Svoboda K. Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature. 2002;420:788–794. doi: 10.1038/nature01273. [DOI] [PubMed] [Google Scholar]
- Ueda T, Greengard P, Berzins K, Cohen RS, Blomberg F, Grab DJ, Siekevitz P. Subcellular distribution in cerebral cortex of two proteins phosphorylated by a cAMP-dependent protein kinase. J Cell Biol. 1979;83:308–319. doi: 10.1083/jcb.83.2.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaja-Milatovic S, Milatovic D, Schantz AM, Zhang J, Montine KS, Samii A, Deutch AY, Montine TJ. Dendritic degeneration in neostriatal medium spiny neurons in Parkinson disease. Neurology. 2005;64:545–547. doi: 10.1212/01.WNL.0000150591.33787.A4. [DOI] [PubMed] [Google Scholar]
- Zaja-Milatovic S, Keene CD, Montine KS, Leverenz JB, Tsuang D, Montine TJ. Selective dendritic degeneration of medium spiny neurons in dementia with Lewy bodies. Neurology. 2006;66:1591–1593. doi: 10.1212/01.wnl.0000216137.09685.c1. [DOI] [PubMed] [Google Scholar]