

Abstract
Estrogen is neuroprotective against a variety of insults, including β-amyloid peptide (Aβ); however, the underlying mechanism(s) is not fully understood. Here, we report that 17β-estradiol (E2) selectively regulates neuronal expression of the Bcl-2 family (bcl-2, bcl-x, bcl-w, bax, bak, bad, bik, bnip3, bid, and bim). In primary cerebrocortical neuron cultures under basal conditions, we observe that E2 upregulates expression of antiapoptotic Bcl-w and downregulates expression of proapoptotic Bim in an estrogen receptor (ER)-dependent manner. In the presence of toxic levels of Aβ, we observe that E2 attenuates indices of neuronal apoptosis: c-Jun N-terminal kinase (JNK)-dependent downregulation of Bcl-w and upregulation of Bim, mitochondrial release of cytochrome c and Smac, and cell death. These neuroprotective effects of E2 against Aβ-induced apoptosis are mimicked by the JNK inhibitor SP600125 (anthra[1,9-cd]pyrazol-6(2H)-one). In addition, E2 attenuates Aβ-induced JNK phosphorylation in an ER-dependent manner, but does not affect basal levels of JNK phosphorylation. These results suggest that E2 may reduce Aβ-induced neuronal apoptosis at least in part by two complementary pathways: (1) ER-dependent, JNK-independent upregulation of Bcl-w and downregulation of Bim under basal conditions, and (2) ER-dependent inhibition of Aβ-induced JNK activation and subsequent JNK-dependent downregulation of Bcl-w and upregulation of Bim, resulting in mitochondrial release of cytochrome c and Smac and eventual cell death. These data provide new understanding into the mechanisms contributing to estrogen neuroprotection, a neural function with potential therapeutic relevance to Alzheimer's disease.
Keywords: β-amyloid, apoptosis, Bcl-w, Bim, c-Jun N-terminal kinase, estrogen
Introduction
Estrogen is an established modulator of neuron viability. Elevated estrogen levels are associated with decreased neuron death in specific sexually dimorphic nuclei during development (Forger, 2006) and in adulthood after toxic challenge (Wise et al., 2001). In cell culture models, 17β-estradiol (E2) is neuroprotective against a variety of insults, including serum deprivation (Green et al., 1997), oxidative stress (Behl et al., 1995), excitotoxicity (Goodman et al., 1996; Singer et al., 1996), and β-amyloid peptide (Aβ) (Behl et al., 1995; Goodman et al., 1996; Green et al., 1996; Mook-Jung et al., 1997; Pike, 1999). The mechanisms underlying estrogen neuroprotection are not fully understood; however, several candidates have been identified. One putative mechanism of estrogen neuroprotection is regulation of the Bcl-2 family, pivotal regulators of apoptosis that include both proteins that promote cell survival (e.g., Bcl-2, Bcl-xL, and Bcl-w) and others that antagonize it (e.g., Bax, Bak, Bad, Bik, BNIP3, Bid, and Bim) (Antonsson and Martinou, 2000). Upregulation of antiapoptotic proteins, such as Bcl-2 (Garcia-Segura et al., 1998; Singer et al., 1998; Dubal et al., 1999; Nilsen and Diaz Brinton, 2003) and Bcl-xL (Patrone et al., 1999; Pike, 1999; Koski et al., 2004), or downregulation of proapoptotic Bad (Gollapudi and Oblinger, 1999) may contribute to the protective effects of estrogen. However, the relationship between estrogen and neuronal expression of Bcl-2 family members remains incompletely defined.
If members of the Bcl-2 family are important mediators of estrogen neuroprotection, then elucidating the underlying mechanism(s) will require identification of not only estrogen-regulated Bcl-2 family members but also the upstream and downstream signaling components in this pathway. One putative upstream component of regulation by estrogen of Bcl-2 family expression is c-Jun N-terminal kinase (JNK) signaling. JNK signaling is linked to transcriptional regulation of many genes (Ip and Davis, 1998), including members of the Bcl-2 family (Harris and Johnson, 2001; Bae and Song, 2003). Interestingly, JNK activation is observed in cultured neurons after Aβ exposure, and its inhibition significantly attenuates Aβ toxicity (Bozyczko-Coyne et al., 2001; Morishima et al., 2001; Troy et al., 2001; Yao et al., 2005). The mitochondrial localization of Bcl-2 proteins suggests that a downstream component of this pathway includes regulation of the proapoptotic molecules released from mitochondria, such as cytochrome c (Liu et al., 1996) and second mitochondrion-derived activator of caspase (Smac/DIABLO) (Du et al., 2000; Verhagen et al., 2000). Previous studies indicate that Aβ causes mitochondrial release of both cytochrome c (Wang et al., 2001; Agostinho and Oliveira, 2003) and Smac (Yao et al., 2005) in cultured neurons. Whether the mechanism of estrogen neuroprotection against Aβ involves regulation of JNK activation and its downstream effectors is unclear.
In this study, we examined the regulatory effects of E2 on neuronal expression of members of the Bcl-2 family under basal conditions and after insult with Aβ. Furthermore, we investigated both the upstream (e.g., JNK signaling) and downstream (e.g., cytochrome c and Smac release) components of this antiapoptotic pathway.
Materials and Methods
Cell culture.
Primary cultures of cerebrocortical neurons were prepared from embryonic (gestational day 18) Sprague Dawley rat pups, with minor modifications of a previously described protocol (Nguyen et al., 2005). In brief, dissected cerebral cortices were incubated 5 min in 0.125% trypsin at 37°C, followed by trypsin quenching with 1 vol of DMEM containing 20% fetal bovine serum. Cell suspensions were centrifuged (5 min at 200 × g), resuspended in serum-free DMEM, mechanically dissociated by repeated passage through a fire-polished Pasteur pipette, and then filtered through a sterile 40 μm nylon mesh (Falcon, Franklin Lakes, NJ). Cells were plated on to poly-l-lysine (0.05 mg/ml)-coated multiwell plates (Nunc, Naperville, IL) at either 5 × 104 cells/cm2 (cell viability analysis) or 1.5 × 105 cells/cm2 (Western blot and RT-PCR) in serum-free, phenol red-free DMEM buffered with 26 mm bicarbonate, 20 mm HEPES, and supplemented with 100 μg/ml transferrin, 5 μg/ml insulin, 100 μm putrescine, and 30 nm selenium. Cultures were maintained in a humidified incubator at 37°C with room air supplemented to 5% CO2. These studies were conducted under a protocol approved by the Institutional Animal Care and Use Committee of the University of Southern California.
Experimental treatment of cultures.
Cortical neuron cultures were used for experimentation 3–6 d in vitro after plating. Cultures were treated with 0.001–1000 or 10 nm E2 (solubilized in 100% ethanol) (Sigma, St. Louis, MO), which in some experiments was followed 1 h later by exposure to 25 μm aggregated Aβ25–35 (Biochem, Torrance, CA) prepared as described previously (Pike et al., 1993). Estrogen receptor (ER) antagonist 7α,17β-[9-[(4,4,5,5,5-pentafluoropentyl)sulfinyl]nonyl]estra-1,3,5(10)-triene-3,17-diol (ICI 182,780) (1 μm; solubilized in 100% ethanol) (Tocris, Ellisville, MO) was added to cultures 1 h before E2. The JNK inhibitor anthra[1,9-cd]pyrazol-6(2H)-one (SP600125) (100 nm; solubilized in DMSO) (Calbiochem, La Jolla, CA) was added to cultures 1 h before Aβ or E2. Final concentrations of drug vehicles were ≤0.1%; vehicle controls were added as appropriate.
Assessment of cell viability.
Cell viability was assessed using calcein-AM and ethidium homodimer fluorescent staining (Invitrogen, Eugene, OR) as described previously (Pike, 1999). Briefly, live cells were counted in four fields per well, four to six wells per condition, in three or more independent culture preparations. The number of live cells counted per well in vehicle-treated controls ranged from 200 to 300. Cell viability is presented graphically as a percentage of live cells in the vehicle-treated, control condition.
RT-PCR.
RT-PCR was performed using a standard protocol, as described previously (Yao et al., 2005). In brief, total cellular RNA was isolated using Trizol reagent (Invitrogen) and reverse transcribed into the cDNA using the Superscript first-strand synthesis system (Invitrogen). Next, 1 μl of reverse transcription product was mixed with 2.5 U of JumpStart TaqDNA polymerase (Sigma), 20 pmol each of sense and antisense primers in a buffer containing 10 mm Tris-HCl, pH 8.3, 50 mm KCl, 2.5 mm MgCl2, and 0.2 mm of each dNTP in a volume of 50 μl. The primers used in this experiment were as follows: 5′-CCGGGAGAACAGGGTATGAT-3′, 5′-CAGGTATGCACCCAGAGTGA-3′ for bcl-2; 5′-AGGCTGGCGATGAGTTTGAA-3′, 5′-CGGCTCTCGGCTGCTGCATT-3′ for bcl-x; 5′-AGCCTCAACCCCAGACACAC-3′, 5′-AAGGCCCCTACAGTTACCAG-3′ for bcl-w; 5′-TCAGCCCATCTTCTTCCAGATGGT-3′, 5′-CCACCAGCTCTGAACAGATCATGA-3′ for bax; 5′-ACTGCGATGAGGCCCTGTCT-3′, 5′-GGCCCAACAGAACCACACCA-3′ for bak; 5′-ATGGGAACCCCAAAGCAGCC-3′, 5′-TCACTGGGAGGGAGTGGAGC-3′ for bad; 5′-ATTTCATGAGGTGCCTGGAG-3′, 5′-GGCTTCCAATCAAGCTTCTG-3′ for bik; 5′-GAATCTGGACGAAGCAGCTC-3′, 5′-AACATTTTCTGGCCGACTTG-3′ for bnip3; 5′-ACTCTGAGGTCAGCAACGGT-3′, 5′-CTAACCAAGTCCCTCACGTA-3′ for bid; 5′-GCCCCTACCTCCCTACAGAC-3′, 5′-CAGGTTCCTCCTGAGACTGC-3′ for bim; and 5′-AGCCATGTACGTAGCCATCC-3′, 5′-CTCTCAGCTGTGGTGGTGAA-3′ for β-actin (internal control). Primers were chemically synthesized (Integrated DNA Technologies, Coralville, IA). The PCR cycles consisted of initial incubation at 94°C for 1 min; denaturation at 94°C for 30 s; annealing at 52°C for 30 s; and extension at 72°C for 1 min, for 30 cycles, and final extension at 72°C for 3 min. RT-PCR products were electrophoresed on 1.7% agarose gels and visualized under UV light after ethidium bromide staining. Analysis of RT-PCR products for the bcl-x and bim primer sets was limited to the 337 bp bcl-xL band and the 319 bp bimEL band.
Design and transfection of small interfering RNAs.
Small interfering RNA (siRNA) that targets bim was designed using the target finder and design tool (Ambion, Austin, TX). The target mRNA sequence of the siRNA is 5′-AAGAUCUUCUCUGCUGUCCCG-3′, corresponding to nucleotides 240–260 of bim gene. As a negative control, a scrambled siRNA was designed consisting of the same nucleotide composition as the specific bim siRNA but lacking significant homology to the genome. As an additional negative control, a mismatched siRNA was used in which two bases in the specific bim siRNA were modified to make them noncomplementary to the target mRNA. The antisense and sense template DNA oligonucleotides for each siRNA, plus T7 promoter 5′-CCTGTCTC-3′ to the 3′ end, were chemically synthesized (Integrated DNA Technologies) and were as follows: siRNA targeting bim (sibim), 5′-AAGATCTTCTCTGCTGTCCCG-3′, 5′-AACGGGACAGCAGAGAAGATC-3′; scrambled siRNA (ncbim), 5′-AATGCCTCCGCTTGTCATCTG-3′, 5′-CAGATGACAAGCGGAGGCA-3′; mismatched siRNA (mmbim), 5′-AAGATCTTCTCGTCTGTCCCG-3′, 5′-AACGGGACAGACGAGAAGATC-3′. The synthesized template DNA was in vitro transcribed into double-strand siRNA using the Silencer siRNA construction kit (Ambion). siRNA transfection with siPORT Amine (Ambion) was performed according to the manufacturer's instructions.
Western blot.
Total cell lysates and mitochondrial and cytosolic extracts (prepared using a mitochondria/cytosol fractionation kit; Biovision, Mountain View, CA) were processed for Western blots using a standard protocol described previously (Pike, 1999). Briefly, lysate and extract samples were diluted into reducing sample buffer, electrophoresed for ∼1.5 h at 120 V in 15% polyacrylamide gels, and then transferred onto a polyvinylidene difluoride membrane (Millipore, Medford, MA) at constant voltage (100 V) for 1 h. After blocking of nonspecific binding (1 h incubation in 10 mm Tris, 100 mm NaCl, 0.1% Tween, 3% bovine serum albumin), membranes were incubated with primary antibody, which included goat anti-Bcl-w (Santa Cruz Biotechnology, Santa Cruz, CA), goat anti-Bim (Santa Cruz Biotechnology), mouse anti-cytochrome c (Santa Cruz Biotechnology), goat-anti-Smac (Santa Cruz Biotechnology), or mouse anti-phospho-JNK (Thr183/Tyr185) (Cell Signaling Technology, Beverly, MA). After rinsing (5 min; six times; in 10 mm Tris, 100 mm NaCl, 0.1% Tween 20), membranes were incubated in the appropriate horseradish peroxidase-conjugated secondary antibody, followed by enhanced chemiluminescence detection (Amersham Biosciences, Arlington Heights, IL). To detect total JNK or verify equal loading of protein across conditions, membranes were stripped (5 min in 100 mm glycine, pH 2.5; and then 5 min in 62.5 mm Tris, 2% SDS, 0.7% 2-mercaptoethanol, pH 6.7, at 60°C) and reprobed with rabbit anti-JNK (Cell Signaling Technology) or mouse anti-β-tubulin (Chemicon, Temecula, CA) antibody. Blots were quantified by band densitometry of scanned films using NIH Image 1.61 software. For JNK blots, the ratio of phospho:total JNK was determined and normalized to the vehicle control condition. For anti-Bim blots, the ∼24 kDa band corresponding to BimEL was quantified. Data are presented graphically as a percentage of control values.
Statistical analyses.
All experiments were repeated at least three times using independent culture preparations. Quantitative data were statistically analyzed by one-way ANOVA, followed by between-group comparisons using Fisher's least significant difference test. Statistical significance was concluded with a value of p < 0.01 for all analyses.
Results
Estrogen reduces Aβ-induced neuronal death
To confirm the established neuroprotective effect of E2 against Aβ-induced neuronal death in our experimental system (Pike, 1999; Cordey et al., 2003; Cordey and Pike, 2006), primary cerebrocortical neuron cultures were pretreated with increasing concentrations (0.001–1000 nm) of E2 for 60 min, followed by exposure to 25 μm aggregated Aβ25–35 for 6–48 h. Cell viability assays revealed that nanomolar levels of E2 significantly reduced neuronal death induced by Aβ25–35 in a dose-dependent (Fig. 1A) and time-dependent (Fig. 1B) manner. The 10 nm dose of E2 yielded maximal protection and thus was the concentration used in subsequent experiments.
Figure 1.

E2 reduces Aβ-induced neuronal death in a dose- and time-dependent manner. A, Neuron cultures were pretreated with increasing concentrations (0.001–1000 nm) of E2 for 60 min, followed by exposure to 25 μm Aβ25–35 for 48 h, and then assayed for cell viability. Data show mean cell viability (+SEM) from a representative experiment (n = 6). Significance is defined as follows: *p < 0.01 compared with Aβ25–35 condition. B, Neuron cultures were pretreated with 0 nm (black bars) or 10 nm (gray bars) E2 for 60 min, followed by exposure to 25 μm Aβ25–35 for the indicated times, and then assayed for cell viability. Data show mean cell viability (+SEM) from a representative experiment (n = 6). Significance is defined as follows: *p < 0.01 compared with Aβ treatment at the matched time point.
Estrogen upregulates Bcl-w and downregulates Bim expression
To investigate the contributions of the Bcl-2 family to estrogen neuroprotection, we first assessed the effect of E2 on expression of bcl-2, bcl-x, bcl-w, bax, bak, bad, bik, bnip3, bid, and bim under basal culture conditions (i.e., in the absence of Aβ challenge). Exposure of neuron cultures to 10 nm E2 for 6–72 h did not induce detectable changes in mRNA levels of several Bcl-2 family members, including bak, bad, bik, and bid. Modest alterations in mRNA levels were consistently observed for four genes 12–72 h after E2 exposure: bcl-x and to a lesser extent bcl-2 were mildly increased, and bax and bnip3 were slightly decreased. The most robust findings were that E2 increased expression of antiapoptotic bcl-w and decreased expression of proapoptotic bim at 12–72 h (Fig. 2A). Because bcl-w and bim were clearly the most strongly E2-regulated Bcl-2 family genes in our system, all subsequent studies focused on these two genes.
Figure 2.

Estrogen upregulates Bcl-w and downregulates Bim expression. A, E2 induces a time-dependent increase of bcl-w and decrease of bim. Neuron cultures were treated with 10 nm E2 for 6–72 h, and mRNA expression levels of Bcl-2 family members (bcl-2, bcl-x, bcl-w, bax, bak, bad, bik, bnip3, bid, and bim) were detected by RT-PCR, followed by agarose gel electrophoresis. β-actin served as an internal control. B, Representative Western blots show E2 regulation of Bcl-w (top panel) and Bim (middle panel) protein levels; β-tubulin (bottom panel) was used as an internal control. C, D, Relative amounts of Bcl-w (C) and Bim (D) protein levels were determined by densitometric scanning of Western blots from three to five independent experiments. Data are represented as a mean (+SEM) percentage of control values. *p < 0.01 relative to vehicle-treated control group (Ctrl).
To confirm E2 regulation of bcl-w and bim gene expression at the protein level, Bcl-w and Bim were analyzed by Western blot. Results revealed a similar time-dependent upregulation of Bcl-w (Fig. 2B, top panel) and downregulation of Bim (Fig. 2B, middle panel). Densitometry measures indicated that 48 h after E2 treatment Bcl-w was increased to ∼175% of basal level (Fig. 2C), whereas Bim was decreased to ∼35% (Fig. 2D).
Estrogen attenuates Aβ-induced Bcl-w downregulation and Bim upregulation
After determining the regulatory effects of E2 on expression of Bcl-w and Bim in neurons under basal conditions, we extended our investigation to evaluate E2 effects under Aβ challenge. First, in agreement with our recent observations (Yao et al., 2005), we found that exposure of neuron cultures to 25 μm Aβ25–35 in the absence of E2 resulted in significant, time-dependent decrease of bcl-w mRNA (Fig. 3A) and modest increase of bim mRNA (Fig. 3B). Next, to explore whether E2 affects the Aβ-induced changes in bcl-w and bim mRNAs, neuron cultures were pretreated with 10 nm E2 for 60 min, followed by exposure to 25 μm Aβ25–35 for 6–48 h. The results showed that E2 pretreatment attenuated both Aβ-induced bcl-w downregulation (Fig. 3A) and bim upregulation (Fig. 3B). These mRNA observations were confirmed at the protein level 48 h after Aβ exposure by Western blot using antibodies that detect Bcl-w (Fig. 3C, top panel) and Bim (Fig. 3C, middle panel). Densitometric analyses of Western blots showed that Aβ significantly reduced Bcl-w expression to ∼45% of basal levels (Fig. 3D) and increased Bim expression to ∼125% of basal levels (Fig. 3E) (p < 0.01 in comparison with vehicle-treated control group). Importantly, E2 pretreatment significantly attenuated the effects of Aβ on expression of Bcl-w (Fig. 3D) and Bim (Fig. 3E) (p < 0.01 relative to Aβ25–35-treated condition).
Figure 3.

Estrogen attenuates Aβ-induced Bcl-w downregulation and Bim upregulation. A, B, Neuron cultures were pretreated with 10 nm E2 for 60 min, followed by exposure to 25 μm Aβ25–35 for indicated times, and analysis of bcl-w (A) and bim (B) mRNA expression by RT-PCR. β-actin served as an internal control. C, Neuron cultures were pretreated with 10 nm E2 for 60 min followed by exposure to 25 μm Aβ25–35 for 48 h. A representative Western blot of culture lysates shows protein levels of Bcl-w (top panel) and Bim (middle panel); β-tubulin (bottom panel) was used as a control. D, E, Relative amounts of Bcl-w (D) and Bim (E) protein levels were determined by densitometric scanning of Western blots from three to four independent experiments. Data are represented as a mean (+SEM) percentage of control values. #p < 0.01 compared with vehicle-treated control group; *p < 0.01 relative to Aβ25–35-treated condition.
Reduction of Bim expression attenuates Aβ toxicity
If regulation by estrogen of Bcl-w and Bim expression is important to estrogen neuroprotection against Aβ, then levels of both Bcl-w and Bim should significantly contribute to Aβ toxicity. We recently demonstrated that Aβ toxicity is reduced by increased Bcl-w expression and potentiated by decreased Bcl-w expression (Yao et al., 2005). Previous research indicates that the proapoptotic Bim can contribute to Aβ toxicity (Yin et al., 2002). To confirm this finding in our culture paradigm, we evaluated the prediction that decreasing Bim expression using specific siRNA should reduce Aβ toxicity. Cultures were transfected for 1 or 3 d with siRNA directed against bim or with mismatched or scrambled bim siRNA. RT-PCR analyses show that bim mRNA was significantly reduced by the specific siRNA but by neither of the control siRNAs (Fig. 4A). Western blot analysis revealed similar effects of the siRNA on Bim protein expression (Fig. 4B). Quantitation of Western blots indicated that bim siRNA decreased Bim levels by 76% 1 d after transfection and by up to 81% 3 d after transfection in comparison with scrambled siRNA control (Fig. 4C). These results demonstrate that the designed bim siRNA had strong inhibitory effects on Bim expression at the mRNA and protein levels.
Figure 4.
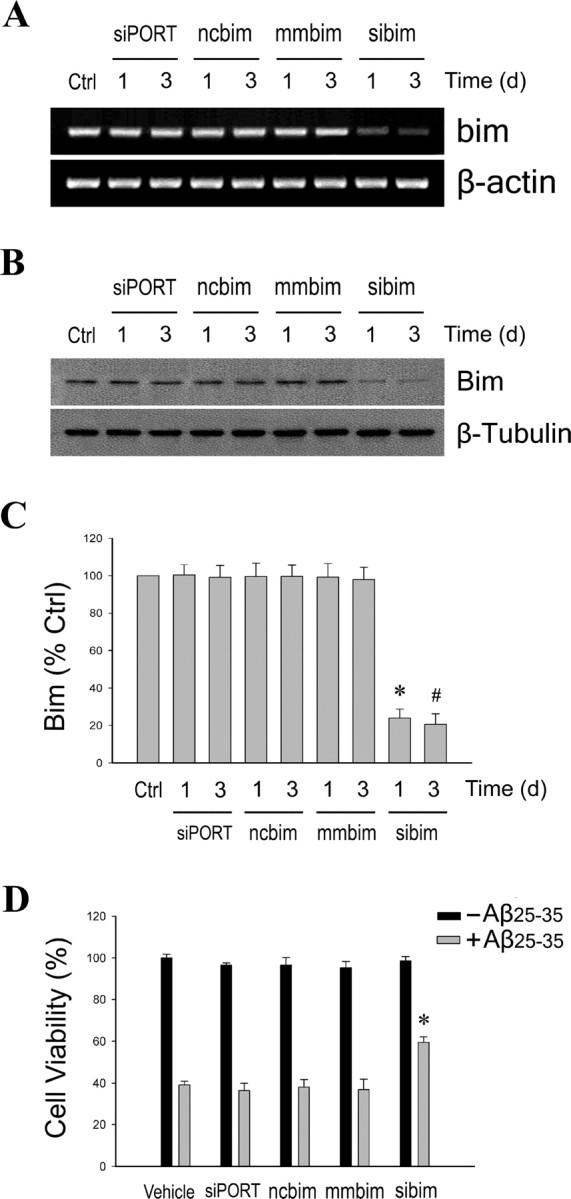
Suppression of endogenous Bim expression decreases Aβ-induced neuronal death. Primary neuron cultures were transfected for 1 or 3 d with bim-specific siRNA (sibim), scrambled siRNA (ncbim), mismatched siRNA (mmbim), or siPORT amine vehicle (siPORT), or were untreated (Ctrl). A, Representative agarose gels of RT-PCR products show endogenous bim mRNA expression decreased only by specific bim siRNA (top) with no change in levels of β-actin, an internal control (bottom). B, Protein levels of Bim were similarly affected by the siRNA treatments, as determined by Western blot (top), with no change in β-tubulin levels (bottom). C, Relative amounts of Bim were determined by densitometric scanning of Western blots from three independent experiments. Data are represented as a mean (+SEM) percentage of vehicle-treated control (Ctrl) values. *p < 0.01 relative to 1 d ncbim control group; #p < 0.01 relative to 3 d ncbim control group. D, Neuron cultures were exposed for 48 h to 25 μm Aβ25–35 24 h after transfection with siRNA. Data show mean (+SEM) cell viability from a representative experiment (n = 4). *p < 0.01 relative to respective ncbim condition.
To examine the effect of Bim suppression on Aβ-induced neuron death, cultures were treated with 25 μm Aβ25–35 1 d after siRNA transfection, and cell viability was assessed 2 d later. In comparison with both the mismatched and scrambled siRNA conditions, cultures transfected with the bim siRNA showed significantly decreased Aβ-induced cell death (p < 0.01, compared with scrambled siRNA) (Fig. 4D). This finding suggests that Bim plays a significant role in regulating apoptosis pathways involved in Aβ toxicity.
Estrogen regulation of Bcl-w and Bim expression is ER dependent
To determine whether the regulatory effects of E2 on Bcl-w and Bim expression are dependent on ERs, neuron cultures were pretreated with ICI 182,780, an ER antagonist (Wakeling et al., 1991). ICI 182,780 (1 μm; effective concentration determined in Fig. 9A) exposure had no effect on basal expression of either bcl-w (Fig. 5A) or bim (Fig. 5C), but blocked E2 regulation of both bcl-w (Fig. 5A) and bim expression (Fig. 5C). Furthermore, ICI 182,780 also blocked the inhibitory effects of E2 on Aβ-induced bcl-w downregulation (Fig. 5B) and bim upregulation (Fig. 5D). These mRNA observations were confirmed at the protein level 48 h after Aβ exposure by Western blot using antibodies directed against Bcl-w (Fig. 5E, top panel) and Bim (Fig. 5E, middle panel). Quantitative analysis of blots confirmed that ICI 182,780 not only blocked E2 regulation of basal Bcl-w (Fig. 5F) and Bim expression (Fig. 5G), but also E2 inhibition of Aβ-induced Bcl-w downregulation (Fig. 5F) and Bim upregulation (Fig. 5G).
Figure 9.
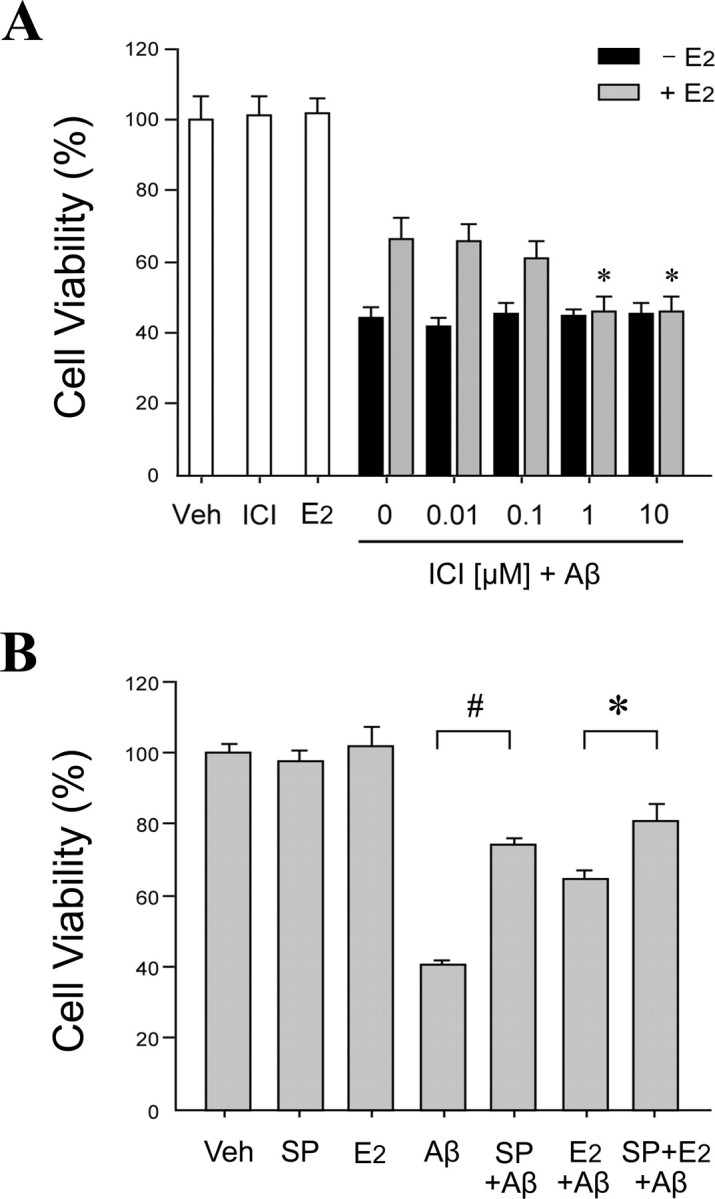
Aβ toxicity is JNK dependent and attenuated by ER-dependent estrogen actions. A, E2-induced neuroprotection against Aβ is ER dependent. Primary neuron cultures were pretreated with ICI 182,780 (ICI) at concentrations ranging from 0.01 to 10 μm for 60 min, followed by treatment with 0 nm (black bars) or 10 nm (gray bars) E2 for 60 min, and then exposed to 25 μm Aβ25–35 for 48 h. Controls groups with vehicle treatment (Veh), 10 nm E2 alone, and 1 μm ICI alone are shown in white bars. Data show mean cell viability (+SEM) from a representative experiment (n = 4). *p < 0.01 compared with E2 plus Aβ25–35 and 0 μm ICI treatments. B, Inhibition of JNK signaling reduces Aβ toxicity and increases estrogen neuroprotection. Neuron cultures were pretreated with 100 nm JNK inhibitor SP600125 (SP) for 60 min, followed by 10 nm E2 for 60 min, and then exposure to 25 μm Aβ25–35 for 48 h. Data show mean cell viability (+SEM) from a representative experiment (n = 4). Significance is defined as follows: #p < 0.01 compared with Aβ treatment; *p < 0.01 compared with E2 plus Aβ25–35 treatment.
Figure 5.

Estrogen regulation of Bcl-w and Bim expression is ER dependent. A, C, Representative agarose gels of RT-PCR products using bcl-w (A) or bim (C) primers show that the ER antagonist ICI 182,780 blocks E2 regulation of bcl-w and bim under basal conditions. Neuron cultures were pretreated with 1 μm ICI 182,780 for 60 min, followed by 10 nm E2 for 24 and 48 h, respectively. β-actin served as an internal control. B, D, Representative agarose gels of RT-PCR products using bcl-w (B) or bim (D) primers show that the ER antagonist ICI 182,780 blocks E2 inhibition of Aβ-induced changes in bcl-w and bim expression. Neuron cultures were pretreated with 1 μm ICI 182,780 for 60 min, followed by 10 nm E2 for 60 min, and then exposed to 25 μm Aβ25–35 for 24 and 48 h. β-actin served as an internal control. E, ER dependence of E2 bcl-w and bim regulation was extended to protein expression using Western blots. A representative Western blot shows Bcl-w (top panel), Bim (middle panel), and β-tubulin (bottom panel) expression in lysates from neuron cultures that were pretreated with 1 μm ICI 182,780, followed 60 min later with 10 nm E2 treatment, and then 60 min later exposed to 25 μm Aβ25–35 for 48 h. F, G, Relative protein levels of Bcl-w (F) and Bim (G) were determined by densitometric scanning of Western blots from four independent experiments. Data show mean (+SEM) percentages of control values. *p < 0.01 relative to E2-treated condition; #p < 0.01 compared with E2 plus Aβ25–35 treatment.
Effect of JNK inhibition on estrogen regulation of Bcl-w and Bim expression
JNK signaling is an established signaling pathway in the regulation of Bcl-2 family expression (Harris and Johnson, 2001; Bae and Song, 2003; Yao et al., 2005). To begin investigating the role of JNK signaling in E2 regulation of Bcl-2 family members, we assessed the effect of the specific JNK inhibitor SP600125 (Bennett et al., 2001) on E2 induced bcl-w upregulation and bim downregulation under nonchallenged conditions. Neuron cultures were pretreated for 60 min with 100 nm SP600125 followed by treatment with 10 nm E2 for 24 and 48 h. RT-PCR analyses showed that basal mRNA levels of bcl-w (Fig. 6A) and bim (Fig. 6C) were not affected by pharmacological inhibition of JNK. Similarly, E2-induced bcl-w upregulation (Fig. 6A) and bim downregulation (Fig. 6C) also were not altered by JNK inhibition, suggesting that the regulation of E2 on basal expression of bcl-w and bim is not dependent on JNK signaling.
Figure 6.

Estrogen regulation of Bcl-w and Bim expression is JNK independent under basal conditions but involves JNK signaling under Aβ challenge. A, C, Representative agarose gels of RT-PCR products using bcl-w (A) or bim (C) primers show that, under basal conditions, the JNK inhibitor SP600125 neither blocks E2 regulation of bcl-w and bim nor independently affects expression of bcl-w and bim. Neuron cultures were pretreated with 100 nm SP600125 for 60 min, followed by 10 nm E2 for 24 and 48 h. β-actin served as an internal control. B, D, Representative agarose gels of RT-PCR products using bcl-w (B) or bim (D) primers show that the JNK inhibitor SP600125 both independently and additively with E2 blocks Aβ-induced changes in bcl-w and bim expression. Neuron cultures were pretreated with 100 nm SP600125 for 60 min, followed by 10 nm E2 for 60 min, and then exposed to 25 μm Aβ25–35 for 24 and 48 h. β-actin served as an internal control. Effects of JNK inhibition on E2 regulation of bcl-w and bim were extended to protein expression using Western blots. E, F, Representative blots show Bcl-w (E) and Bim (F) and expression in lysates from neuron cultures that were pretreated with 100 nm SP600125, followed 60 min later with 10 nm E2 treatment, and then 60 min later exposed to 25 μm Aβ25–35 for 48 h. G, H, Relative protein levels of Bcl-w (G) and Bim (H) were determined by densitometric scanning of Western blots from three independent experiments. *p < 0.01 relative to Aβ25–35 condition; #p < 0.01 compared with E2 plus Aβ25–35 treatment; Δp < 0.01 relative to Aβ25–35 plus SP600125 condition.
Our recent data indicate that JNK signaling contributes to the mechanism by which Aβ regulates expression of Bcl-2 family members (Yao et al., 2005). We confirm here that inhibition of JNK signaling by pretreatment with 100 nm SP600125 mostly prevents Aβ-induced downregulation of bcl-w (Fig. 6B) and upregulation of bim (Fig. 6D). Although the regulation of E2 on basal expression of bcl-w and bim is not dependent on JNK signaling (Fig. 6A,C), we investigated whether JNK signaling may contribute to the inhibitory effect of E2 on Aβ-induced regulation of bcl-w and bim. Neuron cultures were pretreated with 100 nm SP600125 for 60 min, followed by treatment with 10 nm E2 for 60 min and exposure to 25 μm Aβ25–35 for 48 h. RT-PCR analyses showed that the inhibitory effects of E2 on Aβ-induced downregulation of bcl-w (Fig. 6B) and upregulation of bim (Fig. 6D) appeared to be enhanced by cotreatment with SP600125. These mRNA observations were evaluated at the protein level with Bcl-w (Fig. 6E) and Bim (Fig. 6F) Western blots. Densitometric analyses of blots confirmed that JNK inhibition attenuated Aβ-induced downregulation of Bcl-w (Fig. 6G) (*p < 0.01 relative to Aβ treatment) and upregulation of Bim (Fig. 6H) (*p < 0.01 relative to Aβ treatment). However, the effect of estrogen and SP600125 cotreatment was not significantly greater than the effect of SP600125 alone for either Aβ-induced downregulation of Bcl-w (Fig. 6G) or upregulation of Bim (Fig. 6H). These data suggest the possibility that the inhibitory effect of E2 on Aβ-induced changes of Bcl-w and Bim may involve not only a JNK-independent regulatory action on Bcl-w and Bim expression, but also inhibition of Aβ-induced JNK signaling.
Estrogen reduces Aβ-induced JNK activation
To explore whether E2 regulates JNK signaling, we first determined the effect of E2 on activation of JNK under basal conditions. Neuron cultures were treated with 10 nm E2 for 5 min to 48 h and whole-cell extracts were analyzed by Western blot using antibodies that recognize total JNK and phosphorylated JNK, an indicator of activation. Results showed that two isoforms of JNK (46 and 54 kDa) were detected. E2 treatment affected neither phosphorylated (Fig. 7A, top panel) nor total (Fig. 7A, bottom panel) JNK protein levels, indicating that E2 does not significantly affect JNK signaling under nonchallenge conditions (Fig. 7B).
Figure 7.

Estrogen reduces Aβ-induced JNK activation. A, Neuron cultures were treated with 10 nm E2 for the indicated times and then assessed by Western blot with phospho-JNK (p-JNK) (top panel) and pan-JNK (bottom panel) antibodies. B, Blots were quantified by band densitometry for both the 54 kDa (black bars) and 46 kDa (gray bars) JNK forms. C, D, E2 reduces Aβ-induced JNK activation, as shown by both representative Western blots (C) and densitometric quantification of blots (D), in neuron cultures that were pretreated with 10 nm E2 for 60 min, followed by exposure to 25 μm Aβ25–35 for the indicated times. E, F, Representative Western blots probed with phospho-JNK (top panel) and pan-JNK (bottom panel) antibodies (E) and densitometric quantification of blots (F) show that pretreatment of neuron cultures with 1 μm ICI 182,780 for 60 min attenuates the inhibitory effect of 10 nm E2 treatment on JNK phosphorylation induced by exposure to 25 μm Aβ25–35 for 6 h. G, H, The JNK inhibitor SP600125 reduces Aβ-induced JNK activation, as shown by representative Western blots (G) and densitometric quantification of blots (H) from lysates of neuron cultures that were pretreated with 100 nm SP600125 for 60 min, followed by exposure to 25 μm Aβ25–35 for the indicated times. All experiments were repeated in three or more independent culture preparations. Data show mean phospho-JNK:total JNK ratios, normalized to the vehicle control condition. *p < 0.01 relative to matched Aβ25–35 condition at the same time point; #p < 0.01 relative to vehicle control (Ctrl) condition.
Next, we determined the effect of E2 on Aβ-induced JNK activation. Neuron cultures were pretreated with 10 nm E2 for 60 min, followed by exposure to 25 μm Aβ25–35. Western blot results show that Aβ25–35 triggers JNK phosphorylation that was detectable as early as 3 h after Aβ treatment and persisted at least through 48 h (Fig. 7C). Both the 46 and 54 kDa JNK bands showed an Aβ-induced increase in phosphorylation, with the 54 kDa band showing the more robust increase (Fig. 7D). E2 pretreatment incompletely blocked the Aβ-induced increase in JNK activation at most time points (Fig. 7C, top panel; D) but did not alter total JNK protein levels (Fig. 7C, bottom panel). E2 similarly inhibited Aβ-induced phosphorylation of the 46 and 54 kDa JNK bands. To assess the role of ER in this effect, neuron cultures were pretreated with ER antagonist ICI 182,780 (1 μm) for 60 min, followed by treatment with 10 nm E2 for 60 min, and then exposed to 25 μm Aβ25–35 for 6 h. Western blots indicated that ICI 182,780 blocked the inhibitory effect of E2 on Aβ-induced JNK phosphorylation (Fig. 7E,F), demonstrating that the regulatory effect of E2 on Aβ-induced JNK activation occurs in an ER-dependent manner. For comparison, we confirmed our previous finding (Yao et al., 2005) that the JNK inhibitor SP600125 both reduces basal levels of JNK phosphorylation and blocks Aβ-induced JNK phosphorylation of the 46 and 54 kDa JNK bands (Fig. 7G,H).
Estrogen attenuates JNK-dependent mitochondrial cytochrome c and Smac release induced by Aβ
Bcl-2 family members exert their effects in part through regulating mitochondrial release of cytochrome c and Smac into the cytosol (Kuwana and Newmeyer, 2003), an event that is a general feature of the mitochondrial pathway of apoptosis (Liu et al., 1996; Du et al., 2000; Verhagen et al., 2000). In this paradigm, we previously reported that Bcl-w overexpression effectively inhibited Aβ-induced Smac release from mitochondria, whereas knock-down of Bcl-w expression increased Smac release (Yao et al., 2005). On the contrary, Bim can increase mitochondrial cytochrome c (Putcha et al., 2001; Whitfield et al., 2001) and Smac (Yin et al., 2002) release. Because we observed that E2 increases Bcl-w and decreases Bim expression, E2 may regulate Aβ-induced mitochondrial cytochrome c and Smac release. To investigate this possibility and the potential involvement of JNK signaling, we analyzed by Western blot mitochondrial and cytosolic extracts of neuron cultures treated with 25 μm Aβ25–35 in the presence and absence of either 100 nm SP600125 or 10 nm E2. We observed that, 48 h after Aβ25–35 treatment, cytochrome c (Fig. 8A) and Smac (Fig. 8B) levels decreased in the mitochondrial fraction and increased in the cytosolic fraction. Pretreatment with JNK inhibitor SP600125 mostly prevented Aβ-induced cytochrome c and Smac release, suggesting a JNK-dependent mechanism. Similarly, E2 also attenuated Aβ-induced mitochondrial release of both cytochrome c (Fig. 8C) and Smac (Fig. 8D). Densitometric analyses of blots showed that E2 significantly reduced Aβ-induced depletion of cytochrome c (Fig. 8E) and Smac (Fig. 8F) from mitochondria and accumulation of cytochrome c (Fig. 8G) and Smac (Fig. 8H) in cytosol (*p < 0.01 relative to 24 h Aβ treatment; #p < 0.01 relative to 48 h Aβ treatment).
Figure 8.

Estrogen reduces JNK-dependent mitochondrial cytochrome c and Smac release induced by Aβ. A, B, Neuron cultures were pretreated with 100 nm JNK inhibitor SP600125 for 60 min, followed by exposure to 25 μm Aβ25–35 for 48 h, and then analyzed for cytochrome c (Cyt C) (A) and Smac (B) content in mitochondrial (Mt) and cytosolic (Cyt) extracts by Western blot. Neuron cultures were pretreated with 10 nm E2 for 60 min, followed by exposure to 25 μm Aβ25–35 for 24 and 48 h. C, D, Levels of cytochrome c (C) and Smac (D) in mitochondrial and cytosolic extracts were analyzed by Western blot. E–H, Relative protein levels of cytochrome c, in the mitochondrial fraction (E) and cytosolic fraction (G), and Smac, in the mitochondrial fraction (F) and cytosolic fraction (H), were determined by densitometric scanning of Western blots from three independent experiments. Data are presented as mean (+SEM) percentages of vehicle-treated control (Ctrl) values. *p < 0.01 relative to Aβ25–35 condition at 24 h; #p < 0.01 relative to Aβ25–35 condition at 48 h.
Estrogen neuroprotection against JNK-dependent Aβ toxicity is mediated by estrogen receptor
The above studies suggest that E2 antagonizes Aβ-induced apoptosis signaling by an ER-dependent mechanism that operates at least in part by inhibiting JNK activation. Next, we investigated the effects of ER antagonism on the ability of E2 to attenuate Aβ-induced neuron death. To verify that the neuroprotective effect of E2 on Aβ toxicity is mediated by ER, neuron cultures were pretreated with increasing concentrations (0.01–10 μm) of the ER antagonist ICI 182,780, followed by 48 h exposure to 25 μm Aβ25–35 in the presence or absence of 10 nm E2. ICI 182,780 alone had no effect on cell viability under basal conditions and after Aβ challenge (Fig. 9A). However, at concentrations of 1 μm and above, ICI 182,780 almost completely blocked E2-mediated neuroprotection against Aβ toxicity (p < 0.01 in comparison with E2 plus Aβ treatment) (Fig. 9A).
We observed that E2 inhibits JNK signaling and that JNK signaling contributes to Aβ-induced changes in Bcl-w and Bim expression and mitochondrial release of cytochrome c and Smac,. Here, we extended these observations to neuronal survival. Neuron cultures were exposed for 48 h to 25 μm Aβ25–35 with or without pretreatment with 100 nm SP600125 and or 10 nm E2. Cell viability assays showed that independent treatment with either SP600125 or E2 significantly attenuated neuronal death induced by Aβ25–35 (Fig. 9B). Cotreatment with SP600125 and E2 resulted in a modest increase in neuroprotection that was significantly greater than E2 treatment alone but not significantly greater than SP600125 treatment alone (Fig. 9B).
Discussion
Although abundant evidence has established estrogen as a neuroprotective factor that is relevant across the life span, elucidation of its protective mechanisms has proven challenging. One compelling mechanism involves regulation of the Bcl-2 family. Findings from this study not only further define regulation by estrogen of Bcl-2 family members, they also identify two separate pathways of regulation: (1) increased expression of antiapoptotic Bcl-w and decreased expression of proapoptotic Bim under basal conditions, and (2) attenuation of JNK-dependent changes in expression of Bcl-2 family that occurs under apoptotic challenge (Fig. 10).
Figure 10.

Estrogen neuroprotection may involve two complementary pathways. Under basal conditions (solid lines), E2 increases expression of antiapoptotic Bcl-w and decreases expression of proapoptotic Bim, events that counteract the mitochondrial pathway of apoptosis. This may represent an estrogen maintenance pathway of neuron survival. Under toxic challenges such as Aβ exposure (dashed lines), E2 can inhibit activation of JNK, functionally mimicking the pharmacological JNK inhibitor SP600125. JNK activation induces the mitochondrial pathway of apoptosis involving decreased expression of Bcl-w and increased expression of Bim, followed by mitochondrial release of cytochrome c and Smac, and eventually neuron death. Because E2 does not alter basal JNK activity, such a protective mechanism may represent an estrogen response pathway that is activated after injury.
Consistent with an emerging literature, we found that, in the absence of toxic challenge, estrogen regulates Bcl-2 family members in a manner that favors antagonism of apoptosis. Our most robust findings are the novel observations that E2 significantly increases basal expression of antiapoptotic Bcl-w and decreases basal expression of proapoptotic Bim. Bcl-w is widely expressed in mammalian tissues, including CNS (Print et al., 1998; Hamnér et al., 1999; O'Reilly et al., 2001), and functions as a negative regulator of neuronal apoptosis (Gibson et al., 1996; Hamnér et al., 2001; Middleton et al., 2001). Neural expression of Bcl-w is highest in the mature brain (Hamnér et al., 1999), suggesting that Bcl-w function may be particularly important in adulthood. In a previous study, we reported that Aβ-induced neuronal death was reduced by Bcl-w overexpression and potentiated by Bcl-w suppression (Yao et al., 2005), findings that suggest a critical role for Bcl-w in Aβ-induced apoptosis.
In contrast to Bcl-w, Bim is a proapoptotic protein that exists in several isoforms: BimEL, BimL, BimS, and BimAD (O'Connor et al., 1998; U et al., 2001; Marani et al., 2002). In the CNS, Bim expression is localized primarily in neurons (O'Reilly et al., 2000) and is upregulated in a variety of neuron death paradigms (Putcha et al., 2001; Whitfield et al., 2001; Becker et al., 2004; Biswas et al., 2005). Inhibition of Bim by antisense and genetic knock-out approaches can significantly reduce neuronal apoptosis (Whitfield et al., 2001; Becker et al., 2004). In cultured cerebral endothelial cells, Aβ toxicity is associated with increased Bim expression and is suppressed by Bim knock-down (Yin et al., 2002). Consistent with our observations, these results implicate Bim in the mechanism of Aβ-induced apoptosis. Bim produces its proapoptotic effects by interacting with and neutralizing antiapoptotic Bcl-2 family proteins such as Bcl-w (O'Connor et al., 1998; Puthalakath et al., 1999; Wilson-Annan et al., 2003; Shinoda et al., 2004). Thus, E2-induced upregulation of Bcl-w and downregulation of Bim should result in complementary inhibition of neuronal apoptosis induced by toxins such as Aβ.
In addition to E2 regulation of Bcl-w and Bim, we observed relatively modest elevations in mRNA levels of antiapoptotic Bcl-2 family members bcl-2 and bcl-x, and small but consistent reductions in mRNA levels of proapoptotic bax and bnip3. Consistent with our findings are previous results in neuronal cultures showing that estrogen increases basal expression of Bcl-2 (Singer et al., 1998; Honda et al., 2001; Nilsen and Diaz Brinton, 2003; Zhao et al., 2004) and Bcl-xL (Patrone et al., 1999; Pike, 1999; Koski et al., 2004) and decreases expression of proapoptotic bnip2 (Belcredito et al., 2001). Similar results have been observed in rodent brain under nonchallenge conditions. For example, accumulating evidence links developmental sex differences in E2 exposure to differences in Bcl-2 family expression and neuron survival in sexually dimorphic brain regions (for review, see Forger, 2006). In adult female rats, E2 positively regulates expression of Bcl-2 in hypothalamus (Garcia-Segura et al., 1998) and bcl-x in hippocampus (Stoltzner et al., 2001).
One interpretation of the current data is that estrogen functions as an endogenous, homeostatic regulator of apoptosis signaling. Under normal conditions in estrogen-responsive brain regions, estrogen may help maintain long-term neuronal viability by regulating the expression of several Bcl-2 family members. Because apoptosis is affected by the net interactions of proapoptotic and antiapoptotic Bcl-2 members, we speculate that neuroprotective actions of estrogen reflect additive regulatory effects on Bcl-w, Bim, and other Bcl-2 proteins. This “E2 maintenance pathway” (Fig. 10) appears to be ER dependent, but it is unclear whether the mechanism involves predominantly classic genomic pathways (i.e., interaction of activated ER with estrogen response elements on target genes) (Pike, 1999; Perillo et al., 2000) and/or indirect genomic pathways activated by cell signaling pathways [e.g., CREB (cAMP response element-binding protein) signaling] (Honda et al., 2001; Wu et al., 2005). Although activation of JNK signaling is linked to regulation of Bcl-2 family members (Harris and Johnson, 2001; Bae and Song, 2003), our data do not implicate JNK signaling in the E2 maintenance pathway, because under basal conditions E2 did not affect JNK phosphorylation and JNK inhibition did not affect E2 regulation of Bcl-w and Bim.
In addition to regulating expression of Bcl-2 family members under basal conditions, our data show that E2 antagonizes proapoptotic changes in Bcl-2 family expression induced by toxic challenge. Consistent with our previous report (Yao et al., 2005), we found that Aβ-induced neuronal apoptosis involves JNK-dependent alterations in Bcl-2 family expression: downregulation of Bcl-w and upregulation of Bim. Other members of the Bcl-2 family may also play a significant role in Aβ toxicity. We observed that E2 significantly attenuated Aβ-induced changes in Bcl-w and Bim expression. Notably, E2 also significantly reduced Aβ-induced JNK phosphorylation, suggesting that E2 attenuation of Aβ-induced changes in Bcl-w and Bim expression involves inhibition of JNK activation.
Activation of JNK signaling has been closely linked to a variety of apoptotic stimuli, whereas inhibition of JNK signaling provides protection against neuronal apoptosis in multiple paradigms, including Aβ neurotoxicity (Bozyczko-Coyne et al., 2001; Morishima et al., 2001; Troy et al., 2001). It appears that JNK signaling promotes apoptosis at least in part via both transcriptional and posttranslational regulation of Bcl-2 family members (Sanchez and Yuan, 2001). Consequently, JNK inhibition can block alterations in Bcl-2 family expression induced during apoptosis (Harris and Johnson, 2001; Linseman et al., 2002; Schuster et al., 2002; Lei and Davis, 2003; Okuno et al., 2004; Yao et al., 2005; Papadakis et al., 2006). Our results show that inhibition of JNK phosphorylation by both the pharmacological inhibitor SP600125 and E2 attenuated Aβ-induced changes in Bcl-w and Bim expression, mitochondrial cytochrome c and Smac release, and neuron death. Consistent with our data are findings in non-neural cell types that E2 can reduce JNK activation, which results in attenuation of JNK-dependent gene expression (Srivastava et al., 1999, 2001) and/or increased cell survival (Razandi et al., 2000; Eckhoff et al., 2003). Interestingly, despite significantly attenuating indices of apoptosis, E2 provided only partial protection against Aβ-induced cell death. Incomplete neuroprotection in primary neuron cultures is commonly observed with not only estrogen (Singer et al., 1996; Pike, 1999; Harms et al., 2001; Honda et al., 2001) but also androgens (Ahlbom et al., 2001; Hammond et al., 2001; Pike, 2001) and progesterone (Nilsen and Brinton, 2002), suggesting that sex steroid hormones act as partial modulators of neuronal apoptosis pathways.
Thus, E2 may regulate Bcl-2 family members not only by an E2 maintenance pathway but also by an “E2 response pathway” (Fig. 10). In the latter case, E2 can respond to at least some toxic challenges by an ER-dependent pathway to inhibit activation of cell signaling pathways that induce proapoptotic changes in Bcl-2 family expression (e.g., JNK signaling). Importantly, the literature indicates several mechanisms that contribute to estrogen neuroprotection (Green and Simpkins, 2000). We speculate that the pathways identified in this study likely function in conjunction with other mechanisms of estrogen neuroprotection.
Recent observations in animal models are consistent with an E2 response pathway in which E2 responds to neural injury by regulating expression of Bcl-2 family members in a way that reduces apoptosis. For example, in ischemic brain injury models, E2 is neuroprotective and attenuates injury-induced downregulation of Bcl-2 (Dubal et al., 1999; Alkayed et al., 2001; Zhang et al., 2004) and, in some reports, upregulation of Bax (Won et al., 2006). Similarly, E2 neuroprotection is associated with increased Bcl-2 expression after traumatic brain injury (Soustiel et al., 2005) and bcl-2 and bcl-x levels after spinal cord injury (Yune et al., 2004). Such an E2 response pathway may be particularly important for resistance to neuronal death associated with stroke, Alzheimer's disease, and other age-related disorders.
Together, our results are consistent with the hypothesis that E2 attenuates Aβ-induced neuronal death, at least in part, by ER-dependent upregulation of Bcl-w and downregulation of Bim, as well as by inhibition of Aβ-induced JNK activation, subsequent JNK-dependent downregulation of Bcl-w and upregulation of Bim, and mitochondrial release of cytochrome c and Smac. Furthermore, our findings suggest that these neuroprotective actions of E2 reflect concurrent effects of at least two different, ER-dependent signaling pathways: (1) an E2 maintenance pathway of JNK-independent regulation of Bcl-2 family members under basal, nonchallenge conditions, and (2) an E2 response pathway of inhibition of JNK-dependent regulation of Bcl-2 family members associated with neural injury. These data provide new understanding into the mechanisms contributing to estrogen neuroprotection, a function with potential therapeutic relevance to Alzheimer's disease and other age-related neurodegenerative disorders.
Footnotes
This work was supported by National Institutes of Health Grants AG26752 and AG23739.
References
- Agostinho P, Oliveira CR. Involvement of calcineurin in the neurotoxic effects induced by amyloid-beta and prion peptides. Eur J Neurosci. 2003;17:1189–1196. doi: 10.1046/j.1460-9568.2003.02546.x. [DOI] [PubMed] [Google Scholar]
- Ahlbom E, Prins GS, Ceccatelli S. Testosterone protects cerebellar granule cells from oxidative stress-induced cell death through a receptor mediated mechanism. Brain Res. 2001;892:255–262. doi: 10.1016/s0006-8993(00)03155-3. [DOI] [PubMed] [Google Scholar]
- Alkayed NJ, Goto S, Sugo N, Joh HD, Klaus J, Crain BJ, Bernard O, Traystman RJ, Hurn PD. Estrogen and Bcl-2: gene induction and effect of transgene in experimental stroke. J Neurosci. 2001;21:7543–7550. doi: 10.1523/JNEUROSCI.21-19-07543.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonsson B, Martinou JC. The Bcl-2 protein family. Exp Cell Res. 2000;256:50–57. doi: 10.1006/excr.2000.4839. [DOI] [PubMed] [Google Scholar]
- Bae MA, Song BJ. Critical role of c-Jun N-terminal protein kinase activation in troglitazone-induced apoptosis of human HepG2 hepatoma cells. Mol Pharmacol. 2003;63:401–408. doi: 10.1124/mol.63.2.401. [DOI] [PubMed] [Google Scholar]
- Becker EB, Howell J, Kodama Y, Barker PA, Bonni A. Characterization of the c-Jun N-terminal kinase-BimEL signaling pathway in neuronal apoptosis. J Neurosci. 2004;24:8762–8770. doi: 10.1523/JNEUROSCI.2953-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behl C, Widmann M, Trapp T, Holsboer F. 17-β Estradiol protects neurons from oxidative stress-induced cell death in vitro. Biochem Biophys Res Commun. 1995;216:473–482. doi: 10.1006/bbrc.1995.2647. [DOI] [PubMed] [Google Scholar]
- Belcredito S, Vegeto E, Brusadelli A, Ghisletti S, Mussi P, Ciana P, Maggi A. Estrogen neuroprotection: the involvement of the Bcl-2 binding protein BNIP2. Brain Res Brain Res Rev. 2001;37:335–342. doi: 10.1016/s0165-0173(01)00138-2. [DOI] [PubMed] [Google Scholar]
- Bennett BL, Sasaki DT, Murray BW, O'Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, Bhagwat SS, Manning AM, Anderson DW. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci USA. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas SC, Liu DX, Greene LA. Bim is a direct target of a neuronal E2F-dependent apoptotic pathway. J Neurosci. 2005;25:8349–8358. doi: 10.1523/JNEUROSCI.1570-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozyczko-Coyne D, O'Kane TM, Wu ZL, Dobrzanski P, Murthy S, Vaught JL, Scott RW. CEP-1347/KT-7515, an inhibitor of SAPK/JNK pathway activation, promotes survival and blocks multiple events associated with Abeta-induced cortical neuron apoptosis. J Neurochem. 2001;77:849–863. doi: 10.1046/j.1471-4159.2001.00294.x. [DOI] [PubMed] [Google Scholar]
- Cordey M, Pike CJ. Conventional protein kinase C isoforms mediate neuroprotection induced by phorbol ester and estrogen. J Neurochem. 2006;96:204–217. doi: 10.1111/j.1471-4159.2005.03545.x. [DOI] [PubMed] [Google Scholar]
- Cordey M, Gundimeda U, Gopalakrishna R, Pike CJ. Estrogen activates protein kinase C in neurons: role in neuroprotection. J Neurochem. 2003;84:1340–1348. doi: 10.1046/j.1471-4159.2003.01631.x. [DOI] [PubMed] [Google Scholar]
- Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- Dubal DB, Shughrue PJ, Wilson ME, Merchenthaler I, Wise PM. Estradiol modulates bcl-2 in cerebral ischemia: A potential role for estrogen receptors. J Neurosci. 1999;19:6385–6393. doi: 10.1523/JNEUROSCI.19-15-06385.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckhoff DE, Smyth CA, Eckstein C, Bilbao G, Young CJ, Thompson JA, Contreras JL. Suppression of the c-Jun N-terminal kinase pathway by 17beta-estradiol can preserve human islet functional mass from proinflammatory cytokine-induced destruction. Surgery. 2003;134:169–179. doi: 10.1067/msy.2003.219. [DOI] [PubMed] [Google Scholar]
- Forger NG. Cell death and sexual differentiation of the nervous system. Neuroscience. 2006;138:929–938. doi: 10.1016/j.neuroscience.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Garcia-Segura LM, Cardona-Gomez P, Naftolin F, Chowen JA. Estradiol upregulates Bcl-2 expression in adult brain neurons. NeuroReport. 1998;9:593–597. doi: 10.1097/00001756-199803090-00006. [DOI] [PubMed] [Google Scholar]
- Gibson L, Holmgreen SP, Huang DC, Bernard O, Copeland NG, Jenkins NA, Sutherland GR, Baker E, Adams JM, Cory S. Bcl-w, a novel member of the bcl-2 family, promotes cell survival. Oncogene. 1996;13:665–675. [PubMed] [Google Scholar]
- Gollapudi L, Oblinger MM. Estrogen and NGF synergistically protect terminally differentiated, ERalpha-transfected PC12 cells from apoptosis. J Neurosci Res. 1999;56:471–481. doi: 10.1002/(SICI)1097-4547(19990601)56:5<471::AID-JNR3>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Goodman Y, Bruce AJ, Cheng B, Mattson MP. Estrogens attenuate and corticosterone exacerbates excitotoxicity, oxidative injury, and amyloid beta-peptide toxicity in hippocampal neurons. J Neurochem. 1996;66:1836–1844. doi: 10.1046/j.1471-4159.1996.66051836.x. [DOI] [PubMed] [Google Scholar]
- Green PS, Simpkins JW. Neuroprotective effects of estrogens: potential mechanisms of action. Int J Dev Neurosci. 2000;18:347–358. doi: 10.1016/s0736-5748(00)00017-4. [DOI] [PubMed] [Google Scholar]
- Green PS, Gridley KE, Simpkins JW. Estradiol protects against β-amyloid (25–35)-induced toxicity in SK-N-SH human neuroblastoma cells. Neurosci Lett. 1996;218:165–168. doi: 10.1016/s0304-3940(96)13148-7. [DOI] [PubMed] [Google Scholar]
- Green PS, Bishop J, Simpkins JW. 17α-estradiol exerts neuroprotective effects on SK-N-SH cells. J Neurosci. 1997;17:511–515. doi: 10.1523/JNEUROSCI.17-02-00511.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond J, Le Q, Goodyer C, Gelfand M, Trifiro M, LeBlanc A. Testosterone-mediated neuroprotection through the androgen receptor in human primary neurons. J Neurochem. 2001;77:1319–1326. doi: 10.1046/j.1471-4159.2001.00345.x. [DOI] [PubMed] [Google Scholar]
- Hamnér S, Skoglösa Y, Lindholm D. Differential expression of Bcl-w and Bcl-x messenger RNA in the developing and adult rat nervous system. Neuroscience. 1999;91:673–684. doi: 10.1016/s0306-4522(98)00642-3. [DOI] [PubMed] [Google Scholar]
- Hamnér S, Arumae U, Li-Ying Y, Sun YF, Saarma M, Lindholm D. Functional characterization of two splice variants of rat bad and their interaction with Bcl-w in sympathetic neurons. Mol Cell Neurosci. 2001;17:97–106. doi: 10.1006/mcne.2000.0905. [DOI] [PubMed] [Google Scholar]
- Harms C, Lautenschlager M, Bergk A, Katchanov J, Freyer D, Kapinya K, Herwig U, Megow D, Dirnagl U, Weber JR, Hortnagl H. Differential mechanisms of neuroprotection by 17β-estradiol in apoptotic versus necrotic neurodegeneration. J Neurosci. 2001;21:2600–2609. doi: 10.1523/JNEUROSCI.21-08-02600.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris CA, Johnson EM., Jr BH3-only Bcl-2 family members are coordinately regulated by the JNK pathway and require Bax to induce apoptosis in neurons. J Biol Chem. 2001;276:37754–37760. doi: 10.1074/jbc.M104073200. [DOI] [PubMed] [Google Scholar]
- Honda K, Shimohama S, Sawada H, Kihara T, Nakamizo T, Shibasaki H, Akaike A. Nongenomic antiapoptotic signal transduction by estrogen in cultured cortical neurons. J Neurosci Res. 2001;64:466–475. doi: 10.1002/jnr.1098. [DOI] [PubMed] [Google Scholar]
- Ip YT, Davis RJ. Signal transduction by the c-Jun N-terminal kinase (JNK)—from inflammation to development. Curr Opin Cell Biol. 1998;10:205–219. doi: 10.1016/s0955-0674(98)80143-9. [DOI] [PubMed] [Google Scholar]
- Koski CL, Hila S, Hoffman GE. Regulation of cytokine-induced neuron death by ovarian hormones: involvement of antiapoptotic protein expression and c-JUN N-terminal kinase-mediated proapoptotic signaling. Endocrinology. 2004;145:95–103. doi: 10.1210/en.2003-0803. [DOI] [PubMed] [Google Scholar]
- Kuwana T, Newmeyer DD. Bcl-2-family proteins and the role of mitochondria in apoptosis. Curr Opin Cell Biol. 2003;15:691–699. doi: 10.1016/j.ceb.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Lei K, Davis RJ. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc Natl Acad Sci USA. 2003;100:2432–2437. doi: 10.1073/pnas.0438011100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linseman DA, Phelps RA, Bouchard RJ, Le SS, Laessig TA, McClure ML, Heidenreich KA. Insulin-like growth factor-I blocks Bcl-2 interacting mediator of cell death (Bim) induction and intrinsic death signaling in cerebellar granule neurons. J Neurosci. 2002;22:9287–9297. doi: 10.1523/JNEUROSCI.22-21-09287.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- Marani M, Tenev T, Hancock D, Downward J, Lemoine NR. Identification of novel isoforms of the BH3 domain protein Bim which directly activate Bax to trigger apoptosis. Mol Cell Biol. 2002;22:3577–3589. doi: 10.1128/MCB.22.11.3577-3589.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton G, Wyatt S, Ninkina N, Davies AM. Reciprocal developmental changes in the roles of Bcl-w and Bcl-xL in regulating sensory neuron survival. Development. 2001;128:447–457. doi: 10.1242/dev.128.3.447. [DOI] [PubMed] [Google Scholar]
- Mook-Jung I, Joo I, Sohn S, Kwon HJ, Huh K, Jung MW. Estrogen blocks neurotoxic effects of β-amyloid (1–42) and induces neurite extension on B103 cells. Neurosci Lett. 1997;235:101–104. doi: 10.1016/s0304-3940(97)00632-0. [DOI] [PubMed] [Google Scholar]
- Morishima Y, Gotoh Y, Zieg J, Barrett T, Takano H, Flavell R, Davis RJ, Shirasaki Y, Greenberg ME. β-Amyloid induces neuronal apoptosis via a mechanism that involves the c-Jun N-terminal kinase pathway and the induction of Fas ligand. J Neurosci. 2001;21:7551–7560. doi: 10.1523/JNEUROSCI.21-19-07551.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TV, Yao M, Pike CJ. Androgens activate mitogen-activated protein kinase signaling: role in neuroprotection. J Neurochem. 2005;94:1639–1651. doi: 10.1111/j.1471-4159.2005.03318.x. [DOI] [PubMed] [Google Scholar]
- Nilsen J, Brinton RD. Impact of progestins on estrogen-induced neuroprotection: synergy by progesterone and 19-norprogesterone and antagonism by medroxyprogesterone acetate. Endocrinology. 2002;143:205–212. doi: 10.1210/endo.143.1.8582. [DOI] [PubMed] [Google Scholar]
- Nilsen J, Diaz Brinton R. Mechanism of estrogen-mediated neuroprotection: regulation of mitochondrial calcium and Bcl-2 expression. Proc Natl Acad Sci USA. 2003;100:2842–2847. doi: 10.1073/pnas.0438041100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor L, Strasser A, O'Reilly LA, Hausmann G, Adams JM, Cory S, Huang DC. Bim: a novel member of the Bcl-2 family that promotes apoptosis. EMBO J. 1998;17:384–395. doi: 10.1093/emboj/17.2.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuno S, Saito A, Hayashi T, Chan PH. The c-Jun N-terminal protein kinase signaling pathway mediates Bax activation and subsequent neuronal apoptosis through interaction with Bim after transient focal cerebral ischemia. J Neurosci. 2004;24:7879–7887. doi: 10.1523/JNEUROSCI.1745-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Reilly LA, Cullen L, Visvader J, Lindeman GJ, Print C, Bath ML, Huang DC, Strasser A. The proapoptotic BH3-only protein bim is expressed in hematopoietic, epithelial, neuronal, and germ cells. Am J Pathol. 2000;157:449–461. doi: 10.1016/S0002-9440(10)64557-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Reilly LA, Print C, Hausmann G, Moriishi K, Cory S, Huang DC, Strasser A. Tissue expression and subcellular localization of the pro-survival molecule Bcl-w. Cell Death Differ. 2001;8:486–494. doi: 10.1038/sj.cdd.4400835. [DOI] [PubMed] [Google Scholar]
- Papadakis ES, Finegan KG, Wang X, Robinson AC, Guo C, Kayahara M, Tournier C. The regulation of Bax by c-Jun N-terminal protein kinase (JNK) is a prerequisite to the mitochondrial-induced apoptotic pathway. FEBS Lett. 2006;580:1320–1326. doi: 10.1016/j.febslet.2006.01.053. [DOI] [PubMed] [Google Scholar]
- Patrone C, Andersson S, Korhonen L, Lindholm D. Estrogen receptor-dependent regulation of sensory neuron survival in developing dorsal root ganglion. Proc Natl Acad Sci USA. 1999;96:10905–10910. doi: 10.1073/pnas.96.19.10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perillo B, Sasso A, Abbondanza C, Palumbo G. 17beta-estradiol inhibits apoptosis in MCF-7 cells, inducing bcl-2 expression via two estrogen-responsive elements present in the coding sequence. Mol Cell Biol. 2000;20:2890–2901. doi: 10.1128/mcb.20.8.2890-2901.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike CJ. Estrogen modulates neuronal Bcl-xL expression and β-amyloid-induced apoptosis: relevance to Alzheimer's disease. J Neurochem. 1999;72:1552–1563. doi: 10.1046/j.1471-4159.1999.721552.x. [DOI] [PubMed] [Google Scholar]
- Pike CJ. Testosterone attenuates β-amyloid toxicity in cultured hippocampal neurons. Brain Res. 2001;919:160–165. doi: 10.1016/s0006-8993(01)03024-4. [DOI] [PubMed] [Google Scholar]
- Pike CJ, Burdick D, Walencewicz AJ, Glabe CG, Cotman CW. Neurodegeneration induced by β-amyloid peptides in vitro: the role of peptide assembly state. J Neurosci. 1993;13:1676–1687. doi: 10.1523/JNEUROSCI.13-04-01676.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Print CG, Loveland KL, Gibson L, Meehan T, Stylianou A, Wreford N, de Kretser D, Metcalf D, Kontgen F, Adams JM, Cory S. Apoptosis regulator bcl-w is essential for spermatogenesis but appears otherwise redundant. Proc Natl Acad Sci USA. 1998;95:12424–12431. doi: 10.1073/pnas.95.21.12424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putcha GV, Moulder KL, Golden JP, Bouillet P, Adams JA, Strasser A, Johnson EM. Induction of BIM, a proapoptotic BH3-only BCL-2 family member, is critical for neuronal apoptosis. Neuron. 2001;29:615–628. doi: 10.1016/s0896-6273(01)00238-0. [DOI] [PubMed] [Google Scholar]
- Puthalakath H, Huang DC, O'Reilly LA, King SM, Strasser A. The proapoptotic activity of the Bcl-2 family member Bim is regulated by interaction with the dynein motor complex. Mol Cell. 1999;3:287–296. doi: 10.1016/s1097-2765(00)80456-6. [DOI] [PubMed] [Google Scholar]
- Razandi M, Pedram A, Levin ER. Plasma membrane estrogen receptors signal to antiapoptosis in breast cancer. Mol Endocrinol. 2000;14:1434–1447. doi: 10.1210/mend.14.9.0526. [DOI] [PubMed] [Google Scholar]
- Sanchez I, Yuan J. A convoluted way to die. Neuron. 2001;29:563–566. doi: 10.1016/s0896-6273(01)00232-x. [DOI] [PubMed] [Google Scholar]
- Schuster N, Dunker N, Krieglstein K. Transforming growth factor-beta induced cell death in the developing chick retina is mediated via activation of c-jun N-terminal kinase and downregulation of the anti-apoptotic protein Bcl-XL. Neurosci Lett. 2002;330:239–242. doi: 10.1016/s0304-3940(02)00801-7. [DOI] [PubMed] [Google Scholar]
- Shinoda S, Schindler CK, Meller R, So NK, Araki T, Yamamoto A, Lan JQ, Taki W, Simon RP, Henshall DC. Bim regulation may determine hippocampal vulnerability after injurious seizures and in temporal lobe epilepsy. J Clin Invest. 2004;113:1059–1068. doi: 10.1172/JCI19971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer CA, Rogers KL, Strickland TM, Dorsa DM. Estrogen protects primary cortical neurons from glutamate toxicity. Neurosci Lett. 1996;212:13–16. doi: 10.1016/0304-3940(96)12760-9. [DOI] [PubMed] [Google Scholar]
- Singer CA, Rogers KL, Dorsa DM. Modulation of Bcl-2 expression: a potential component of estrogen neuroprotection. NeuroReport. 1998;9:2565–2568. doi: 10.1097/00001756-199808030-00025. [DOI] [PubMed] [Google Scholar]
- Soustiel JF, Palzur E, Nevo O, Thaler I, Vlodavsky E. Neuroprotective anti-apoptosis effect of estrogens in traumatic brain injury. J Neurotrauma. 2005;22:345–352. doi: 10.1089/neu.2005.22.345. [DOI] [PubMed] [Google Scholar]
- Srivastava S, Weitzmann MN, Cenci S, Ross FP, Adler S, Pacifici R. Estrogen decreases TNF gene expression by blocking JNK activity and the resulting production of c-Jun and JunD. J Clin Invest. 1999;104:503–513. doi: 10.1172/JCI7094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Toraldo G, Weitzmann MN, Cenci S, Ross FP, Pacifici R. Estrogen decreases osteoclast formation by down-regulating receptor activator of NF-kappa B ligand (RANKL)-induced JNK activation. J Biol Chem. 2001;276:8836–8840. doi: 10.1074/jbc.M010764200. [DOI] [PubMed] [Google Scholar]
- Stoltzner SE, Berchtold NC, Cotman CW, Pike CJ. Estrogen regulates bcl-x expression in rat hippocampus. NeuroReport. 2001;12:2797–2800. doi: 10.1097/00001756-200109170-00009. [DOI] [PubMed] [Google Scholar]
- Troy CM, Rabacchi SA, Xu Z, Maroney AC, Connors TJ, Shelanski ML, Greene LA. β-Amyloid-induced neuronal apoptosis requires c-Jun N-terminal kinase activation. J Neurochem. 2001;77:157–164. doi: 10.1046/j.1471-4159.2001.t01-1-00218.x. [DOI] [PubMed] [Google Scholar]
- U M, Miyashita T, Shikama Y, Tadokoro K, Yamada M. Molecular cloning and characterization of six novel isoforms of human Bim, a member of the proapoptotic Bcl-2 family. FEBS Lett. 2001;509:135–141. doi: 10.1016/s0014-5793(01)03145-3. [DOI] [PubMed] [Google Scholar]
- Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, Moritz RL, Simpson RJ, Vaux DL. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000;102:43–53. doi: 10.1016/s0092-8674(00)00009-x. [DOI] [PubMed] [Google Scholar]
- Wakeling AE, Dukes M, Bowler J. A potent specific pure antiestrogen with clinical potential. Cancer Res. 1991;51:3867–3873. [PubMed] [Google Scholar]
- Wang CN, Chi CW, Lin YL, Chen CF, Shiao YJ. The neuroprotective effects of phytoestrogens on amyloid beta protein-induced toxicity are mediated by abrogating the activation of caspase cascade in rat cortical neurons. J Biol Chem. 2001;276:5287–5295. doi: 10.1074/jbc.M006406200. [DOI] [PubMed] [Google Scholar]
- Whitfield J, Neame SJ, Paquet L, Bernard O, Ham J. Dominant-negative c-Jun promotes neuronal survival by reducing BIM expression and inhibiting mitochondrial cytochrome c release. Neuron. 2001;29:629–643. doi: 10.1016/s0896-6273(01)00239-2. [DOI] [PubMed] [Google Scholar]
- Wilson-Annan J, O'Reilly LA, Crawford SA, Hausmann G, Beaumont JG, Parma LP, Chen L, Lackmann M, Lithgow T, Hinds MG, Day CL, Adams JM, Huang DC. Proapoptotic BH3-only proteins trigger membrane integration of prosurvival Bcl-w and neutralize its activity. J Cell Biol. 2003;162:877–887. doi: 10.1083/jcb.200302144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise PM, Dubal DB, Wilson ME, Rau SW, Bottner M, Rosewell KL. Estradiol is a protective factor in the adult and aging brain: understanding of mechanisms derived from in vivo and in vitro studies. Brain Res Brain Res Rev. 2001;37:313–319. doi: 10.1016/s0165-0173(01)00136-9. [DOI] [PubMed] [Google Scholar]
- Won CK, Kim MO, Koh PO. Estrogen modulates Bcl-2 family proteins in ischemic brain injury. J Vet Med Sci. 2006;68:277–280. doi: 10.1292/jvms.68.277. [DOI] [PubMed] [Google Scholar]
- Wu TW, Wang JM, Chen S, Brinton RD. 17Beta-estradiol induced Ca2+ influx via L-type calcium channels activates the Src/ERK/cyclic-AMP response element binding protein signal pathway and BCL-2 expression in rat hippocampal neurons: a potential initiation mechanism for estrogen-induced neuroprotection. Neuroscience. 2005;135:59–72. doi: 10.1016/j.neuroscience.2004.12.027. [DOI] [PubMed] [Google Scholar]
- Yao M, Nguyen TV, Pike CJ. β-Amyloid-induced neuronal apoptosis involves c-Jun N-terminal kinase-dependent downregulation of Bcl-w. J Neurosci. 2005;25:1149–1158. doi: 10.1523/JNEUROSCI.4736-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin KJ, Lee JM, Chen SD, Xu J, Hsu CY. Amyloid-β induces Smac release via AP-1/Bim activation in cerebral endothelial cells. J Neurosci. 2002;22:9764–9770. doi: 10.1523/JNEUROSCI.22-22-09764.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yune TY, Kim SJ, Lee SM, Lee YK, Oh YJ, Kim YC, Markelonis GJ, Oh TH. Systemic administration of 17beta-estradiol reduces apoptotic cell death and improves functional recovery following traumatic spinal cord injury in rats. J Neurotrauma. 2004;21:293–306. doi: 10.1089/089771504322972086. [DOI] [PubMed] [Google Scholar]
- Zhang L, Nair A, Krady K, Corpe C, Bonneau RH, Simpson IA, Vannucci SJ. Estrogen stimulates microglia and brain recovery from hypoxia-ischemia in normoglycemic but not diabetic female mice. J Clin Invest. 2004;113:85–95. doi: 10.1172/JCI200418336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Wu TW, Brinton RD. Estrogen receptor subtypes alpha and beta contribute to neuroprotection and increased Bcl-2 expression in primary hippocampal neurons. Brain Res. 2004;1010:22–34. doi: 10.1016/j.brainres.2004.02.066. [DOI] [PubMed] [Google Scholar]