

Abstract
The inositol 1,4,5-trisphosphate (InsP3) receptor type I (InsP3R-I) is the principle channel for intracellular calcium (Ca2+) release in many cell types, including central neurons. It is regulated by endogenous compounds like Ca2+ and ATP, by protein partners, and by posttranslational modification. We report that the InsP3R-I is modified by O-linked glycosylation of serine or threonine residues with β-N-acetylglucosamine (O-GlcNAc). The level of O-GlcNAcylation can be altered in vitro by the addition of the enzymes which add [OGT (O-GlcNActransferase)] or remove (O-GlcNAcase) this sugar or by loading cells with UDP-GlcNAc. We monitored the effects of this modification on InsP3R function at the single-channel level and on intracellular Ca2+ transients. Single-channel activity was monitored with InsP3R incorporated into bilayers; Ca2+ signaling was monitored using cells loaded with a Ca2+-sensitive fluorophore. We found that channel activity was decreased by the addition of O-GlcNAc and that this decrease was reversed by removal of the sugar. Similarly, cells loaded with UDP-GlcNAc had an attenuated response to uncaging of InsP3. These results show that O-GlcNAcylation is an important regulator of the InsP3R-I and suggest a mechanism for neuronal dysfunction under conditions in which O-GlcNAc is high, such as diabetes or physiological stress.
Keywords: calcium imaging; calcium-sensitive dye; β-N-acetylglucosamine; O-GlcNActransferase; O-GlcNAcase; cerebellum; interneurons; inositol 1,4,5-trisphosphate receptor
Introduction
Inositol 1,4,5-trisphosphate (InsP3)-mediated Ca2+ release from intracellular stores controls a variety of cellular processes including cell growth, fertilization, secretion, smooth muscle contraction, and neuronal signaling (Berridge, 1993). The InsP3 receptor type I (InsP3R-I), the most well studied of the three InsP3R isoforms, is involved in the regulation of neuronal functions such as long-term potentiation and depression (Nagase et al., 2003).
The InsP3R-I is regulated by endogenous compounds such as InsP3, Ca2+, and ATP, and modulated by associated proteins such as calmodulin and chromogranin (Ehrlich et al., 1994; Thrower et al., 2003; Bezprozvanny, 2005; Choe and Ehrlich, 2006). The InsP3R-I is also regulated by posttranslational modifications. For example, phosphorylation by PKA (protein kinase A) or tyrosine kinases increases channel activity, and the functional sites of phosphorylation have been identified (Jayaraman et al., 1996; Wojcikiewicz and Luo, 1998; Tang et al., 2003). The InsP3R-I is also posttranslationally modified by N-glycosylation, and the sites of modification are known (Michikawa et al., 1994). However, functional changes have not been identified with this modification.
Another class of posttranslational modification is O-GlcNAcylation, a type of O-linked glycosylation in which β-N-acetylglucosamine (O-GlcNAc) is added to serine or threonine residues (Wells and Hart, 2003). Classical O-linked and N-linked glycosylation occurs in the secretory pathway, with the enzymes involved residing in the endoplasmic reticulum and the Golgi apparatus, and glycosylation occurring on lumenal residues. This irreversible type of glycosylation regulates protein folding, targeting, and turnover (Helenius and Aebi, 2004). In the case of O-GlcNAcylation, O-GlcNActransferase (OGT) and O-GlcNAcase, the enzymes that add and remove O-GlcNAc to proteins, reside in the cytoplasm and the nucleus, and O-GlcNAcylation occurs on the nucleoplasmic and cytoplasmic domains of proteins. OGT and O-GlcNAcase have been cloned, and O-GlcNAcase has been characterized for in vitro use (Kreppel et al., 1997; Gao et al., 2001).
O-GlcNAcylation is an abundant, dynamic, and inducible modification that shares common characteristics with protein phosphorylation. Proteins modified by O-GlcNAc include transcription factors, signaling components, and metabolic enzymes. Changes in the level of O-GlcNAcylation of several of these proteins are known to regulate their activity by modulating either protein phosphorylation, protein–protein interactions, or subcellular localization (Wells and Hart, 2003). O-GlcNAcylation has also been shown to play a role in the regulation of InsP3-mediated Ca2+ signaling. Cardiomyocytes of diabetic rats show altered Ca2+ transients that can be restored to normal by decreasing the cellular O-GlcNAcylation level (Clark et al., 2003). Cardiomyocytes show an elevation of cytoplasmic Ca2+ in response to the InsP3-generating agonist angiotensin II. This response can be blocked by inducing an increase in protein O-GlcNAcylation (Nagy et al., 2006).
Here, we demonstrate for the first time that the InsP3R-I is modified by O-GlcNAc. Moreover, we show that the O-GlcNAc modification regulates the InsP3R-I at the single-channel level: addition of the O-GlcNAc group has a functional effect on the channel. InsP3-induced Ca2+ release decreases with O-GlcNAcylation and increases with de-O-GlcNAcylation. This effect is specific for InsP3R-I, because there is no functional effect of O-GlcNAc on the ryanodine receptor (RyR). We also show for the first time that cerebellar interneurons have an InsP3-dependent increase in intracellular Ca2+ and that this increase can be modulated by O-GlcNAc. We speculate that this may be partially responsible for improper neuronal signaling under conditions such as diabetes, when cellular UDP-GlcNAc levels are elevated (Robinson et al., 1995).
Materials and Methods
All procedures for animal use were in accordance with guidelines approved by the host institutions where experimental work was performed, namely the University of Paris 5 and Yale University.
InsP3R-I immunoprecipitation and purification.
Mice cerebella (Pel-Freez Biologicals, Rogers, AR) were homogenized in 10 vol of buffer A (50 mm Tris-HCl, 1 mm EDTA, 1 mm 2-β-mercaptoethanol, pH 8.3, and protease inhibitors). After centrifugation (1000 × g for 10 min at 4°C), the supernatant was further centrifuged at 100,000 × g for 30 min. The pellet was then resuspended in lysis buffer (50 mm Tris-HCl, 100 mm NaCl, 1 mm EDTA, 1 mm DTT, 1% Triton X-100, pH 8.3, and protease inhibitors) and incubated for 30 min at 4°C. After centrifugation (16,000 × g for 10 min at 4°C) the supernatant containing the solubilized receptors was incubated at 4°C with anti-InsP3R-I (affinity purified from rabbit polyclonal antiserum directed against the 19 C-terminal residues of the mouse InsP3R-I) for 1 h, and then for an additional 12–24 h with protein A-Sepharose beads. The immune complexes were then isolated by centrifugation (500 × g for 2 min) and washed three times with ice-cold PBS. The pellet of beads was then resuspended in sample buffer, and the protein was analyzed by SDS-PAGE.
When immunoprecipitating the receptor from cultured cells, the cells were harvested with HBSS (Invitrogen, Carlsbad, CA) and centrifuged at 750 × g for 3 min at 4°C. The cells were then incubated in lysis buffer, and the procedure described above was followed from that point on.
To partially purify the InsP3R-I from mouse cerebella, tissue was solubilized in 1% CHAPS (3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate), and then this mixture was passed over a heparin affinity column as described previously (Thrower et al., 2003).
Culture and treatments of SH-SY5Y and DT40 B cells.
SH-SY5Y cells were cultured in a 1:1 mixture of F-12 Ham's and Minimum Essential Medium, supplemented with 10% fetal bovine serum, 1% Minimum Essential Medium nonessential amino acids, and penicillin/streptomycin (Invitrogen). DT40 B cells were grown in RPMI 1640 supplemented with 10% fetal calf serum, 0.05 mm 2-mercaptoethanol, 1% chicken serum, glutamine, and penicillin/streptomycin. Cells were cultured in a water-saturated atmosphere at 37°C and 5% CO2. To induce an increase in O-GlcNAc, n-acetylglucosamine (GlcNAc) or O-(2-acetamido-2-deoxy-d-glucopyranosylidene)amino-N-phenylcarbamate (PUGNAc) (kindly provided by G. W. Hart, Johns Hopkins University, Baltimore, MD) was added to the growth medium. PUGNAc was added at a final concentration of 200 μm for 24 h. GlcNAc was added at a final concentration of 8 mm for 72 h or with 8 mm mannitol as an osmotic protector.
Cerebellar and cellular microsomal preparation.
Mouse or rat cerebella (Pel-Freez Biologicals) were homogenized using a Teflon-glass homogenizer in a homogenization buffer containing 250 mm sucrose, 5 mm HEPES/KOH, pH 7.4, 1 mm EGTA, 1 mm DTT, and protease inhibitors. The homogenate was then centrifuged at 1000 × g for 5 min at 4°C. The supernatant was saved and the pellet was resuspended in homogenization buffer and centrifuged again at 1000 × g for 5 min. The pellet was then discarded, and the pooled supernatants were centrifuged at 8000 × g for 10 min at 4°C. The pellet was discarded, and the supernatant was centrifuged at 100,000 × g for 75 min. The final pellet was resuspended in homogenization buffer without EGTA and snap frozen in liquid nitrogen.
When preparing microsomes from cells, the cells were homogenized by passage through a 27 gauge needle. Then, the procedure described above was followed starting with the centrifugation at 8000 × g for 10 min.
Western blot analysis.
Protein samples were resolved by SDS-PAGE and transferred to a polyvinylidene difluoride membrane. The blots were then incubated overnight at 4°C with monoclonal antibodies that recognized the O-linked sugar bond; RL2 (isotype IgG1 from Affinity BioReagents, Golden, CO) or CTD 110.6 (isotype IgM from Covance, Richmond, CA). After washing, blots were incubated for 1 h at room temperature with HRP-linked goat anti-mouse antibody (Bio-Rad, Hercules, CA). Blots were then visualized using ECL (Pierce Biotechnology, Rockford, IL). Blots to be reprobed were previously stripped of antibodies using Re-Blot Plus kit (Chemicon International, Temecula, CA). One hour incubation at room temperature with polyclonal antibodies that recognized InsP3R-I (Johenning et al., 2002) was followed by washing and incubation for 1 h at room temperature with HRP-linked goat anti-rabbit antibody (Bio-Rad). ECL was again used to visualize the blot. In some cases, the receptor was identified before the O-linked sugar bond; the order of visualization did not alter the results.
Removal of N-linked sugars or O-GlcNAc.
Heparin-purified InsP3R-I was treated with N-glycosidase F, endoglycosidase H (New England BioLabs, Beverly, MA), or recombinant O-GlcNAcase (β-N-acetylglucosaminidase; product no. A6805; Sigma, St. Louis, MO) following the manufacturer's protocol. The ability to remove O-linked sugars was assessed by examining the treated samples with antibodies that recognized the O-linked sugar bond as described above (see Western blot analysis).
Preparation of mouse cerebellar cytosol.
Cerebellar cytosol was isolated by homogenization and centrifugation as described previously (Marshall et al., 2003). Briefly, mouse cerebella were homogenized in ice-cold buffer consisting of 250 mm sucrose, 10 mm Tris, 1 mm MgCl2, pH 7.6, and protease inhibitors. The homogenate was then centrifuged at 600 × g for 10 min. The pellet was discarded, and the supernatant was centrifuged at 20,000 × g for 10 min to obtain the crude cytosol. The cytosol was then desalted by polyethylene glycol (PEG) precipitation to increase the OGT activity of the sample. The cytosol was mixed with 2 vol of 30% ice-cold PEG solution (25 mm HEPES, 10 mm MgCl2, pH 7.2), and after being vortexed, the sample was centrifuged at 20,000 × g for 20 min to precipitate proteins. The supernatant was discarded, and the pellet was resuspended in transferase assay buffer containing 25 mm HEPES, 10 mm MgCl2, and 1 mm EDTA, pH 7.
Single-channel recordings.
InsP3R-I from cerebella microsomes were incorporated into planar lipid bilayers and recorded as described previously (Bezprozvanny et al., 1991; Thrower et al., 2003). The experiments were performed with a 250 mm HEPES-Tris solution, pH 7.35, on the cis- and a 250 mm HEPES, 53 mm Ba(OH)2 solution, pH 7.35, on the trans-side of the bilayer (1 ml chamber). For the InsP3R-I, single-channel activity was recorded in the presence of 0.5 mm ATP, 2 μm InsP3, 8 μm ruthenium red, and 0.3 μm free Ca2+ on the cytoplasmic side of the InsP3R (cis-side of the bilayer; 1 ml chamber), unless otherwise specified. For the RyR, single-channel activity was recorded in the presence of 0.5 mm ATP and 0.3 μm free Ca2+, but in the absence of InsP3 (which activates the InsP3R) and ruthenium red (which blocks RyR). To elicit channel de-O-GlcNAcylation, a mixture of 4 μl of O-GlcNAcase (Sigma) plus 1 μl of 5× manufacturer reaction buffer was added directly to the cis-side of the bilayer without stirring. To elicit O-GlcNAcylation of the channel, a mixture of 1 μl of UDP-GlcNAc plus 4 μl of desalted cerebella cytosol was added directly over the membrane at the cis-side. After 1 min, the reactions were stopped by stirring for 30 s. Experiments were recorded under voltage-clamp conditions, the data were amplified (BC-525C; Warner Instrument, Hamden, CT), filtered at 1 kHz, digitized, and directly transferred to a computer. The data were acquired and analyzed with pClamp (Molecular Devices, Union City, CA). At least 3 min of recordings at each condition in each experiment were used for calculation of open probability, open and closed times.
Calcium imaging of SH-SY5Y cells.
SH-SY5Y cells were plated on 22 mm2 glass coverslips, at 3–8 × 105 cells per well of a six-well plate, and grown overnight. Cultures were supplemented for 72 h with 20 mm mannitol, 8 mm GlcNAc, or 20 mm glucose, as indicated.
Coverslips were incubated for 30 min in HEPES-buffered saline (HBS) containing 130 mm NaCl, 5 mm KCl, 1.25 mm CaCl2, 1.2 mm KH2PO4, 1 mm MgSO4, 20 mm HEPES, 6.9 mm glucose, pH 7.4, with NaOH, supplemented with 6 μm fluo-4 AM (Invitrogen). Cells were washed with HBS, and placed in an imaging chamber (Warner Instrument) perfused with HBS on the stage of a Nikon (Tokyo, Japan) Diaphot fluorescence microscope. Fluo-4 was illuminated through a 40× air objective from an Hg arc lamp and emission detected through a GFP (green fluorescent protein) filter cube to a cooled CCD camera (NeuroCCD-SM256; Red Shirt Imaging, Decatur, GA). After a baseline was established in HBS, InsP3-dependent Ca2+ release was stimulated by perfusion of MgATP-containing HBS, as indicated, to the chamber. Throughout fluo-4 loading and perfusion, HBS was supplemented with 20 mm mannitol (control), 8 mm GlcNAc, or 20 mm glucose (26.9 mm total), as indicated.
Data were collected using Neuroplex 7.0 (Red Shirt Imaging) at 1 Hz and analyzed using IGOR Pro 5.0. Cells were considered to have responded to ATP if ΔF/Fo was >0.1 after stimulation. Data are displayed as mean ± SE.
Cerebellar slice preparation and electrophysiological recording of slices.
Slices (180 μm thick) were prepared from the vermis of cerebella taken from rats aged 11–15 d. Interneurons of the cerebellar molecular layer (MLIs) were identified as described previously (Llano and Gerschenfeld, 1993). During recordings, the slices were perfused (1.5 ml/min) with a saline solution containing 125 mm NaCl, 2.5 mm KCl, 1.25 mm NaH2PO4, 26 mm NaHCO3, 2 mm CaCl2, 1 mm MgCl2, and 10 mm glucose, equilibrated with a 95% O2/5% CO2 mixture, pH 7.3. All experiments were performed at room temperature. Whole-cell recordings were performed with an intracellular recording solution containing the following (in mm): 140 K-gluconate, 5.4 KCl, 4.1 MgCl2, 9.9 HEPES-K, 0.36 Na-GTP, and 3.6 Na-ATP, pH 7.3. MLIs were held in whole-cell recording for <1 min using the above intracellular recording solution, supplemented with 200 μm K+ salt of Oregon Green 488 BAPTA-1 (OG1) (Invitrogen) and 400 μm caged Ins(1,4,5)P3 (Calbiochem, La Jolla, CA). The patch pipette was then withdrawn, and fluorescence recording could begin after a few minutes of equilibration.
InsP3 uncaging and calcium imaging in cerebellar slices.
Digital fluorescence images were obtained using an excitation-acquisition system from TILL Photonics (Planegg, Germany). Briefly, to excite fluorescence of the Ca2+ dye OG1, light from a 75 W Xenon lamp was focused on a scanning monochromator set at 488 nm and coupled, by a quartz fiber and a lens, to the microscope, equipped with a dichroic mirror and a high-pass emission filter centered at 505 and 507 nm, respectively. Images were acquired by a Peltier-cooled CCD camera (IMAGO QE; 1376 × 1040 pixels; pixel size: 244 nm after 53× magnification and 2 × 2 binning) connected to a 12 bit A/D converter. Photolysis of d-myo-InsP3 P4(5)-1-(2-nitrophenyl)ethyl ester (caged InsP3) was achieved using a pulsed xenon arc lamp (Till Photonics). A high intensity (0.5–5 ms duration; 80 J) discharge of UV light (360 ± 7.5 nm) was reflected onto the plane of focus using a dichroic mirror and Olympus (Tokyo, Japan) 60× water immersion objective (numerical aperture, 0.9). Analysis of images was performed as described previously (Llano et al., 1997) using homemade routines within the IGOR programming environment (Wavemetrics, Lake Oswego, OR).
Results
The InsP3R-I is modified by O-GlcNAc
To determine whether O-GlcNAcylation is involved in regulation of the InsP3R-I channel, we first established that the InsP3R-I is O-GlcNAcylated using biochemical techniques. We immunoprecipitated the InsP3R-I from mouse cerebella and from SH-SY5Y cells using an antibody produced to recognize the InsP3R-I (Johenning et al., 2002). Western blot analysis was performed using an antibody that recognizes O-GlcNAcylated proteins [RL2 (Snow et al., 1987)]. The InsP3R-I band was identified from both samples by RL2 indicating that the receptor is O-GlcNAcylated (Fig. 1A). This immunoreactivity was specifically inhibited in the presence of 0.3 m GlcNAc (data not shown). Another antibody that recognizes GlcNAc in a β-O-glycosidic linkage to both serine and threonine, CTD110.6 (Comer et al., 2001), also identified InsP3R-I bands (data not shown).
Figure 1.

The InsP3R-I is O-GlcNAc modified. A, Western blot analysis of InsP3R-I immunoprecipitated from mouse cerebellum (lane 1) or from SH-SY5Y cells (lane 2) and then probed with anti O-GlcNAc. B, Heparin-purified InsP3R-I from mouse cerebellum was treated at 37°C with enzymes that remove N-linked glycosylation or O-linked GlcNAc as follows: lane 1, 1 h incubation with no treatment; lane 2, 1 h incubation with N-glycosidase F (PNGase F); lane 3, overnight incubation with PNGase F; lane 4, 1 h incubation with endoglycosidase H (Endo H); lane 5, overnight incubation with Endo H; lane 6, 1 h incubation with O-GlcNAcase. The membrane was first probed with anti-O-GlcNAc. This antibody was then removed, and the membrane was reprobed with anti-InsP3R-I. WB, Western blot.
Treatment of the InsP3R-I with O-GlcNAcase removes anti-O-GlcNAc immunoreactivity
To confirm that the immunoreactivity detected was attributable to O-GlcNAcylation and not to N-glycosylation, which is known to be present on the InsP3R-I (Michikawa et al., 1994), we partially purified the InsP3R-I by heparin affinity chromatography and treated it with N-glycosidase F, endoglycosidase H, or O-GlcNAcase. N-Glycosidase F and endoglycosidase H remove oligosaccharides from N-linked glycoproteins. After treating the purified proteins with N-glycosidase F or endoglycosidase H, Western blot analysis showed that the protein sample was still stained by RL2 (Fig. 1B, lanes 2–5). In contrast, treatment with O-GlcNAcase removed the anti-O-GlcNAc immunoreactivity (Fig. 1B, lane 6). The anti-O-GlcNAc antibody was removed and the membrane was reprobed with anti-InsP3R-I showing homogeneous InsP3R-I staining in all lanes (Fig. 1B).
O-GlcNAc modification of the InsP3R-I can be modulated in vivo
When cells were incubated with the O-GlcNAcase inhibitor PUGNAc or with the hexosamine pathway intermediate GlcNAc, there was an increase in O-GlcNAcylation of numerous proteins (Haltiwanger et al., 1998). We treated SH-SY5Y and DT40 B cells with PUGNAc, prepared microsomes, and analyzed the O-GlcNAc level with Western blot analysis. Initial staining with anti-InsP3R-I showed equivalent levels of protein on the sample from SH-SY5Y cells in control conditions and from cells incubated with PUGNAc (Fig. 2A). The membrane was then stripped of antibodies and reprobed with anti-O-GlcNAc. PUGNAc increased O-GlcNAcylation of many microsomal proteins including the InsP3R-I. This effect was also seen when examining the effects in DT40 B cells (Fig. 2B). Additionally, microsomes from DT40 B cells grown under control conditions were prepared with homogenization buffer supplemented with GlcNAc. It was found that this treatment also increased the level of O-GlcNAcylation (Fig. 2B), suggesting that endogenous O-GlcNAcase can remove GlcNAc from proteins during microsomal preparation. The addition of GlcNAc to the homogenization buffer provided excess substrate that was able to compete with the GlcNAc attached to proteins for binding to O-GlcNAcase, resulting in less removal of GlcNAc from proteins.
Figure 2.

InsP3R-I O-GlcNAcylation can be modulated in vivo. A, Microsomal preparation from SH-SY5Y cells grown at 37°C as follows: lane 1, no treatment; lane 2, incubation with 200 μm PUGNAc for 24 h. B, Microsomes were prepared from DT40 B cells grown at 37°C as follows: lane 1, no treatment; lane 2, incubation with 200 μm PUGNAc for 24 h; lane 3, cells were grown in control conditions but the microsomal preparation was performed with 50 mm GlcNAc present in the homogenization buffer. C, InsP3R-I was immunoprecipitated from DT40 B cells grown at 37°C as follows: lane 1, no treatment; lane 2, incubation with 8 mm GlcNAc for 72 h. For A–C, the membrane was first probed against InsP3R-I and then reprobed against O-GlcNAc. WB, Western blot.
To confirm that the increase in O-GlcNAc immunoreactivity detected at the molecular weight of the InsP3R-I was attributable to increased O-GlcNAcylation of the InsP3R-I, we immunoprecipitated the InsP3R-I from DT40 B cells grown in media supplemented with GlcNAc. Initial staining with anti-InsP3R-I showed equivalent levels of receptor on the sample from DT40 B cells in control conditions and from cells incubated with GlcNAc (Fig. 2C, left panel). After blotting for InsP3R-I, the membrane was stripped of antibodies and reprobed for O-GlcNAc. Under control conditions, the InsP3R-I is O-GlcNAcylated. The presence of multiple high molecular weight bands on the blot reflects the observation that the receptor is easily degraded and that it forms multimers under many conditions (Fig. 2C, right panel, lane 1). Incubation with GlcNAc elevated the level of O-GlcNAcylation of the receptor (Fig. 2C, right panel, lane 2). The immunoreactivities detected for the InsP3R-I were measured by densitometry, and the O-GlcNAc signal was normalized with the InsP3R-I blot. Incubation with GlcNAc produced an almost threefold increase of the relative InsP3R-I O-GlcNAcylation from 0.046 ± 0.009 to 0.13 ± 0.016 (n = 3; p < 0.009 compared with control conditions).
Addition and removal of O-GlcNAc have reciprocal functional effects on the InsP3R-I channel
To study the modulation of the InsP3R-I by O-GlcNAcylation, we incorporated mouse cerebellar InsP3R-I into planar lipid bilayers. Under control conditions, the presence of 0.5 mm ATP, 0.3 μm free Ca2+, and 8 μm ruthenium red (which blocks RyR) on the cytosolic (cis-) side of the bilayer elicited no channel activity (Fig. 3A, first trace). Addition of 2 μm InsP3 elicited channel activity (Fig. 3A, second trace) with an open probability of 1.4 ± 0.3% (n = 4). To study the functional effect of de-O-GlcNAcylation of the InsP3R-I, we applied O-GlcNAcase directly to the bilayer on the cis-side. Removal of the O-GlcNAc modification induced an increase of more than threefold in the open probability (Fig. 3A, third trace), to 5 ± 1% (n = 4; p < 0.03 compared with control conditions) (Fig. 3C). As a control, the solution containing O-GlcNAcase was boiled to denature it before addition to the bilayer chamber; no change in channel activity was detected under these conditions (data not shown).
Figure 3.

Addition and removal of O-GlcNAc have reciprocal functional effects on the InsP3R-I channel inserted into lipid bilayers. Each trace corresponds to 10 s of current recordings from the same experiment. Openings are defined as downward deflections from the baseline. A, O-GlcNAcase activates the InsP3R-I. Trace 1, Addition of 0.5 mm ATP and 0.3 μm free Ca2+ elicits no channel activity. Trace 2, Addition of 2 μm InsP3R-I elicits channel activity with an open probability of 1.4%. Trace 3, Removal of the O-GlcNAc modification with addition of O-GlcNAcase increases the open probability to 5%. B, Increased O-GlcNAcylation inhibits the InsP3R-I. Trace 1, Addition of 0.5 mm ATP and 0.3 μm free Ca2+ elicits no channel activity. Trace 2, Addition of 2 μm InsP3R-I elicits channel activity with an open probability of 1.5%. Trace 3, Application of cerebellar cytosolic extract and UDP-GlcNAc directly to the bilayer adds the O-GlcNAc modification to the channel, reducing the open probability to 0.06%. C, Mean open probability and SEM for each bilayer condition. *p < 0.05.
To study the functional effect of O-GlcNAcylation on the InsP3R-I, we isolated mouse cerebellar cytosol (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) (Marshall et al., 2003) and used it as a source of OGT to modify the InsP3R-I inserted into planar lipid bilayers. Although OGT has been cloned and partially characterized (Kreppel et al., 1997; Lubas and Hanover, 2000), the recombinant enzyme does not retain a high level of activity for in vitro use. Therefore, brain cytosol has been used as a reasonable source of OGT activity. Addition of 0.5 mm ATP and 0.3 μm free Ca2+ to the cis-side of the bilayer elicited no channel activity (Fig. 3B, first trace). Addition of 2 μm InsP3 elicited channel openings with an open probability of 1.5 ± 0.4% (n = 4) (Fig. 3B, second trace). Addition of cerebellar cytosolic extract and the GlcNAc donor UDP-GlcNAc directly to the lipid bilayer on the cis-side induced a dramatic reduction in channel activity (Fig. 3B, third trace), to 0.08 ± 0.01% (n = 4; p < 0.02 compared with control conditions) (Fig. 3C). As a control, UDP-GlcNAc without cerebella cytosol was added to the bilayer chamber; no change in channel activity was observed under these conditions (data not shown). To show the specificity of the modulation by O-GlcNAcylation, the addition of O-GlcNAcase directly to the cis-side of bilayers containing the RyR did not induce a detectable change in RyR channel activity (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
These reciprocal functional effects were also demonstrated by consecutive addition of cytosolic extract and UDP-GlcNAc followed by O-GlcNAcase to the same channel. Addition of 2 μm InsP3 in the presence of 0.5 mm ATP and 0.3 μm free Ca2+ elicits the opening of a channel with an open probability of 1.9 ± 0.6% (n = 2) (Fig. 4, first and second traces). OGT modification of the channel was produced by addition of cytosolic extract and UDP-GlcNAc directly to the lipid bilayer reducing the channel open probability to 0.1 ± 0.02% (n = 2) (Fig. 4, third trace). The channel was then modified by O-GlcNAcase, which removed O-GlcNAc from the receptor and increased the open probability to 0.28 ± 0.05% (n = 2) (Fig. 4, fourth trace). We believe the open probability did not return all the way to control values because the single addition of O-GlcNAcase to the bilayer was not able to remove all of the O-GlcNAc. It was not possible to make a second addition of O-GlcNAcase because of bilayer breakage.
Figure 4.

Consecutive addition and removal of O-GlcNAc have reciprocal functional effects on the same InsP3R-I channel. Each trace corresponds to 2.5 s of current recording from the same experiment, and openings are defined as downward deflections from the baseline. Trace 1, Addition of 0.5 mm ATP and 0.3 μm free Ca2+ elicits no channel activity. Trace 2, Addition of 2 μm InsP3R-I elicits channel activity with an open probability of 2.5%. Trace 3, Increasing the O-GlcNAcylation of the channel by addition of cerebellar cytosolic extract and UDP-GlcNAc directly to the bilayer caused the open probability to drop to 0.08%. Trace 4, Removal of O-GlcNAc by O-GlcNAcase addition increases the open probability to 0.23%.
Incubation with GlcNAc decreases whole-cell Ca2+ transients
To investigate whether O-GlcNAcylation of the InsP3R-I also regulated the release of Ca2+ in intact cells, we treated SH-SY5Y cells for 72 h with 8 mm GlcNAc to increase O-GlcNAcylation of the InsP3R-I (Fig. 5) and exposed them to extracellular ATP, known to stimulate InsP3 production via P2Y purinergic receptors. At 50 nm ATP stimulation, 86% of control cells responded, compared with 53% of GlcNAc-treated cells. At 100 nm ATP stimulation, all control cells responded, compared with only 75% of GlcNAc treated, whereas all cells of both groups responded with ΔF/Fo > 0.1 after 1 μm ATP stimulation (Fig. 5B). This suggests that, at submaximal levels of InsP3 generation, pretreatment with GlcNAc increases the threshold for InsP3-induced Ca2+ release, but that all treated cells are capable of responding, given sufficient stimulation.
Figure 5.
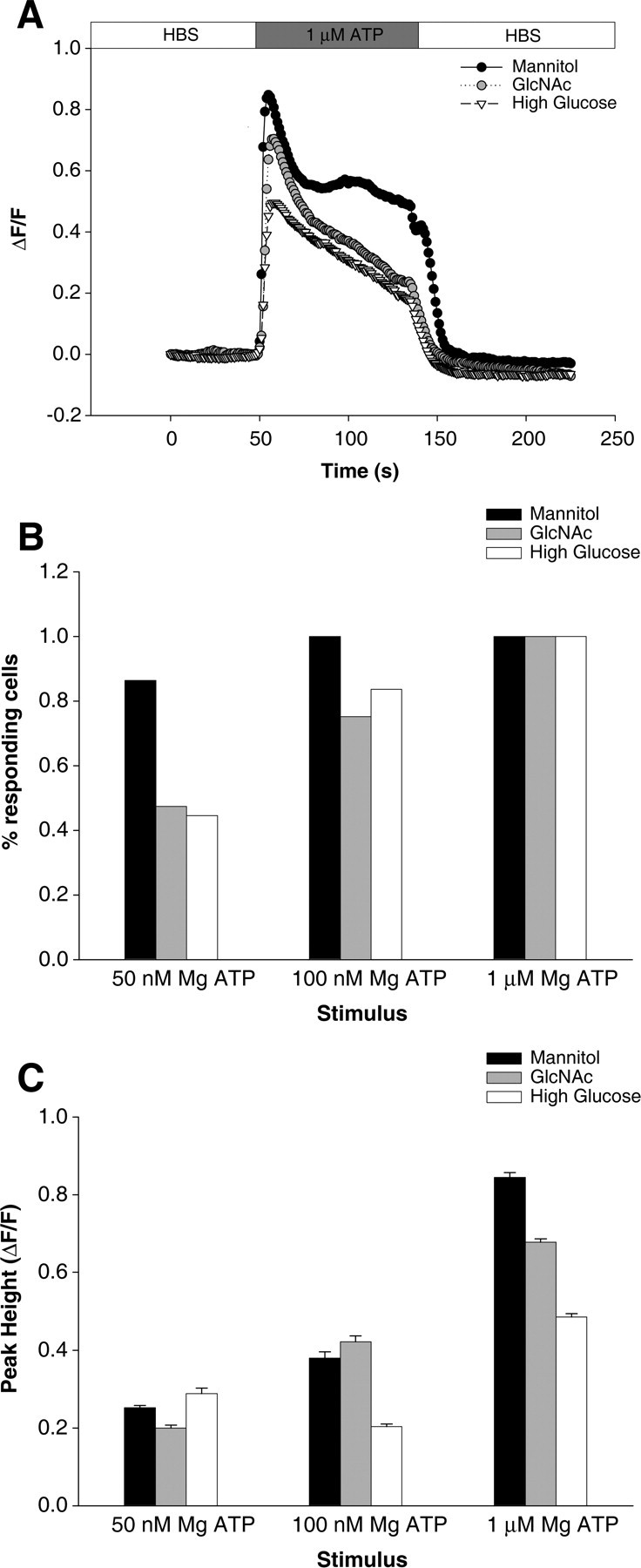
GlcNAc reduces whole-cell Ca2+ transients. A, Single traces show the time course of the Ca2+ response for mannitol-, GlcNAc-, or high-glucose-treated SH-SY5Y cells stimulated with 1 μm ATP where indicated. B, Percentage of cells responding to 50 nm, 100 nm, or 1 μm ATP application from each treatment group, as indicated. C, Peak height of response to ATP stimulation for each treatment group, as indicated. Data are displayed as mean ± SEM.
Looking at the cells that did respond under each condition, we see that the responses are different. With 50 or 100 nm ATP stimulation, control and GlcNAc-treated cells respond similarly, whereas at maximal stimulation control cells exhibit a greater peak height than GlcNAc treated (0.84 ± 0.01 vs 0.68 ± 0.01; p < 10−20) (Fig. 5C). Combined with the data above, we conclude that GlcNAcylation leads to both a decrease in the sensitivity of cells to InsP3 and to a reduced maximal Ca2+ response because of InsP3 stimulation. This suggests an effect at the cell level consistent with a decrease in InsP3R open probability as determined for single channels.
UDP-GlcNAc is generated physiologically through the hexosamine pathway, which branches off from the glycolytic intermediate fructose-6-phosphate. As such, the physiological concentration of UDP-GlcNAc is controlled by the glucose uptake of a cell, and it stands to reason that a high glucose environment can lead to increased O-GlcNAcylation of proteins (Robinson et al., 1995). To investigate the role of this more physiological agonist, cells were incubated for 72 h in 26.9 mm glucose (6.9 mm in medium, supplemented with 20 mm) to induce increased O-GlcNAcylation. Similar to GlcNAc-treated cells, fewer high-glucose-treated cells responded to low levels of ATP stimulation compared with control, whereas all cells responded at high agonist concentration (Fig. 5B). In addition, the effect on peak height at different concentrations of ATP was even more pronounced for high-glucose-treated cells, with the mean peak height of responding cells only one-half that of control cells at 100 nm ATP stimulation (0.20 ± 0.01 vs 0.49 ± 0.03) (Fig. 5C).
Addition of UDP-GlcNAc decreases calcium transients after uncaging InsP3
To verify that the effect of GlcNAc on intact cell Ca2+ signaling was attributable to InsP3R Ca2+ release, we tested the effect of GlcNAc addition on the responses to release of InsP3 in cerebellar interneurons. First, it was necessary to show that uncaging of InsP3 in these cells was capable of inducing Ca2+ release. Cells were preloaded with caged InsP3 and the Ca2+ indicator OG-1 (Fig. 6). After a 4 min equilibration period, a UV flash was produced every 3 min to release InsP3, and Ca2+ transients were monitored. The magnitude and shape of the responses were stable, diminishing by <20% over 19 min (Fig. 6B,C). The ability of uncaged InsP3 to induce Ca2+ release was inhibited by the addition of the InsP3R inhibitor 2-APB at 20 μm (data not shown).
Figure 6.
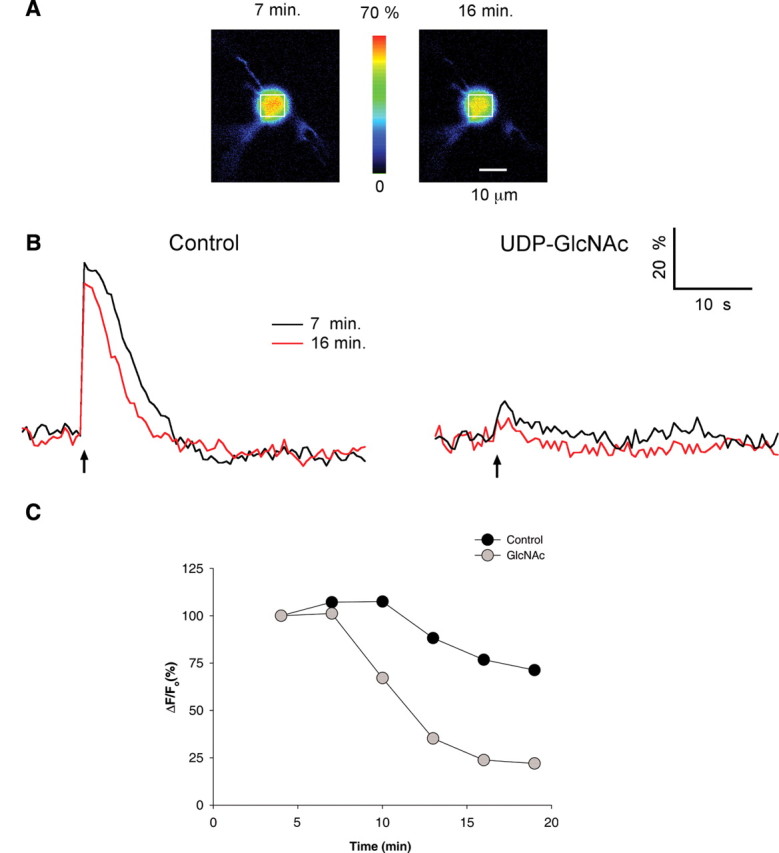
Uncaging of InsP3 elicits calcium release in cerebellar interneurons. A, Pseudocolor images of OG1 Ca2+-dependent fluorescence from a postnatal day 12 interneuron at the peak of the response to photorelease of caged InsP3. The images on the left and right were obtained at 7 and 16 min, respectively, after loading the cell with dye and caged InsP3. B, Left, Traces show the time course of the relative percentage change in fluorescence (ΔF/Fo) attributable to InsP3 uncaging at the time indicated in a region of interest over the soma (identified by the white boxes in A) of a cell preloaded with OG1 and caged InsP3. Right, Traces from an ROI at times indicated as in control, but preloaded with OG1, caged InsP3, and UDP-GlcNAc. C, Time course of the inhibition of Ca2+ release by UDP-GlcNAc compared with control cells. When the magnitudes of the Ca2+ transients were normalized to those obtained initially, responses decreased more rapidly from cells loaded with UDP-GlcNAc than control cells.
To test the effect of O-GlcNAcylation on the response to uncaging of InsP3, UDP-GlcNAc (10 mm) was added to the pipette during the preloading stage of the experiments. The first response after equilibration (at 4 min after introduction of the compound) was already reduced; the GlcNAc loaded cells showed a 72% reduction in the response to uncaging of InsP3 (n = 6; p < 0.001). The response to uncaging of InsP3 at 4 and 7 min was stable in both the control cells and in cells loaded with GlcNAc (Fig. 6C). Subsequent responses to uncaging of InsP3 in control cells declined by ∼20% in part because the amount of caged InsP3 was decreasing in the cell. In contrast, the responses to uncaging of InsP3 in GlcNAc-loaded cells declined nearly to baseline (Fig. 6B,C), confirming a reduced sensitivity to InsP3 in GlcNAc-treated cells.
Discussion
In this study, we show for the first time that the InsP3R-I is O-GlcNAcylated and that modification by O-GlcNAcylation has functional effects on the InsP3R-I channel. Addition of O-GlcNAc decreases the probability of channel opening and removal of O-GlcNAc increases channel activity, most likely by influencing the InsP3 dependence of the InsP3R. The open probability of the InsP3R-I inserted into lipid bilayers was increased threefold when O-GlcNAcase was added to remove GlcNAc from the channel. The RyR is also O-GlcNAcylated, but removal of O-GlcNAc does not change its open probability. We also studied the effect of O-GlcNAc modification on the ability of cerebellar interneurons to produce InsP3-dependent Ca2+ transients. We found that addition of UDP-GlcNAc decreased the ability of these cells to produce a Ca2+ transient when InsP3 was elevated, as predicted from the results obtained at the single-channel level. These results uncover a new mechanism of regulation of the InsP3R-I channel that is specific to this family of intracellular Ca2+ release channel.
InsP3R-I from several sources (mouse cerebella, SH-SY5Y cells, and DT-40 cells) were modified by GlcNAc. Both addition and removal of O-GlcNAc could be accomplished with the enzymes known to be present in the cytoplasm and nucleus (Kreppel et al., 1997; Gao et al., 2001). The reactions are specific to O-linked sugars: O-GlcNAc immunoreactivity was removed when the InsP3R-I was treated with O-GlcNAcase but not when it was treated with enzymes that remove oligosaccharides from N-linked glycoproteins (Fig. 1B). Additional support for the specificity is that neither addition of boiled O-GlcNAcase nor of UDP-GlcNAc alone altered channel activity.
Other studies have demonstrated that O-GlcNAcylation regulates protein function either by altering enzyme activity, protein–protein interactions, subcellular localization, or protein phosphorylation. O-GlcNAc modification of glycogen synthase results in the retention of the enzyme in a glucose 6-phosphate-dependent state and contributes to the reduced activation of the enzyme in insulin resistance (Parker et al., 2003). O-GlcNAcylation of the activation domain of Sp1 blocks its in vitro and in vivo interactions with other Sp1 molecules and its transcriptional capability (Yang et al., 2001). Microtubule-associated Tau proteins are also modified by phosphorylation and O-GlcNAcylation. An increase in Tau protein phosphorylation level correlates with a decrease in O-GlcNAcylation level and with a reduced transfer of Tau into the nucleus (Lefebvre et al., 2003).
To survive environmental, physiological, or chemical stress, cells must initiate signal transduction events that activate mechanisms that counteract apoptosis. Recent studies have found that conditions of stress generate a global increase in O-GlcNAc protein modification and that increasing O-GlcNAc levels protects cells (Zachara et al., 2004; Zachara and Hart, 2006). It was also observed that O-GlcNAc regulates both the rate and extent of the stress-induced induction of heat shock proteins, providing a molecular basis to the cellular protection produced by O-GlcNAc (Zachara et al., 2004). Ca2+-sensitive activation of caspases is an important pathway for the induction of apoptosis, and InsP3R-I-sensitive Ca2+ stores have been shown to play a role (Hajnoczky et al., 2000). Our observation that O-GlcNAcylated InsP3R-I channels are low activity channels provides another possible explanation for how increased O-GlcNAcylation results in cellular protection against apoptosis.
Interestingly, an increase in O-GlcNAc level in myocardial cells has been associated with protection against stresses, such as ischemia and reperfusion, which lead to Ca2+ overload. In a study of whole heart models, GlcNAc pretreatment elevated protein O-GlcNAcylation resulting in a marked increase in tolerance to injury caused by ischemia/reperfusion, characterized by a decreased release of lactate dehydrogenase (a marker of tissue injury) and improved contractile function (Liu et al., 2006). The recent finding that increases in cytosolic Ca2+ produced by InsP3 generating agonists in cardiomyocytes are inhibited by increased O-GlcNAc levels (Nagy et al., 2006) is consistent with our observation that O-GlcNAcylated InsP3R-I channels are low activity channels. We also showed that O-GlcNAcylation of RyR does not regulate channel activity. In the failing human heart, expression of these two intracellular Ca2+ release channels is regulated in opposite directions, where RyR is downregulated and InsP3R-I is upregulated (Go et al., 1995). Together, these results indicate that Ca2+ release through the InsP3R-I plays an important role in the cardioprotection provided by GlcNAc against myocardial Ca2+ overload.
Diabetic cardiomyopathy is characterized by poor cardiac contractility linked to impaired sarcoplasmic reticulum Ca2+ cycling, including reduced sarcoplasmic reticulum Ca2+ content, decreased diastolic Ca2+ uptake and systolic Ca2+ release. Two recent papers have documented these alterations of Ca2+ signaling in the diabetic cardiomyocyte and have linked them to increased levels of protein O-GlcNAcylation (Clark et al., 2003; Hu et al., 2005). It was shown in diabetic heart models that protein O-GlcNAcylation is increased in both type I and type II diabetes (Hu et al., 2005). Animals with streptozotocin-induced diabetes and hyperglycemia show a greatly increased concentration of the hexosamine metabolite UDP-GlcNAc (Robinson et al., 1995). Thus, under diabetic conditions, high UDP-GlcNAc leads to an increase in protein O-GlcNAcylation (Akimoto et al., 2000; Clark et al., 2003). Adenovirus-mediated overexpression of O-GlcNAcase was shown to reduce cellular O-GlcNAcylation, enhance the diabetic cardiomyocyte transient, the sarcoplasmic reticulum Ca2+ loading, and improve contractile function (Hu et al., 2005). Our finding that increased O-GlcNAcylation of the InsP3R-I results in decreased Ca2+ release is consistent with the diabetes-induced reduction in systolic Ca2+ release and the enhancement of the diabetic cardiomyocyte transient resulting from O-GlcNAcase overexpression.
Brain damage is a recognized complication of diabetes. Data from epidemiological studies suggest that diabetes is a risk factor for neurocognitive dysfunction (Launer, 2005). The pathophysiological basis of the dysfunction remains controversial with some studies identifying recurrent hypoglycemia as the primary cause of neuronal damage, whereas others associate these effects to chronic hyperglycemia (McNay et al., 2006; Wessels et al., 2006). To further complicate the issue, recent reports indicate that, although brain dysfunction has been consistently reported in many diabetic patients, the literature fails to find serious and progressive global cognitive impairment (Ryan, 2006; Wessels et al., 2006). In contrast, a recent study of the effect of recurrent hypoglycemia in cognitive function of a diabetic rat model concludes that recurrent hypoglycemia actually prevents the age-related decline in hippocampus-associated cognitive function (McNay et al., 2006). The remarkable implication of these data is that diabetic-associated changes in brain structure and function exist and can be measured, although only subtle neurocognitive dysfunction has been detected. A neuronal protective process must contribute to the relative preservation of neurocognitive function in the presence of damaged tissue. The brain is one of the tissues with the highest expression levels of OGT and O-GlcNAcase (Haltiwanger et al., 1992; Gao et al., 2001). Protein O-GlcNAcylation is increased in both type I and type II diabetes (Hu et al., 2005). Diabetes causes a reduction in the amplitude of InsP3-induced Ca2+ release in primary and secondary nociceptive neurons (Clark et al., 2003; Kruglikov et al., 2004). Because excessive cytosolic Ca2+ is a well known mediator of neuronal death, increased O-GlcNAcylation of InsP3R-I leading to low channel activity would provide protection against apoptosis and associated changes in brain structure and function.
Growing evidence implicates Ca2+ signaling disruptions in the etiology of neurodegenerative disorders like Alzheimer's disease (AD). Most cases of AD are sporadic, but ∼10% are inherited (Thinakaran and Sisodia, 2006). Mutations in presenilins 1 and 2 account for ∼20–50% of these forms of familial AD (FAD) (Tandon and Fraser, 2002). Impaired Ca2+ homeostasis and exaggerated InsP3-mediated Ca2+ signals are observed in mice, Xenopus oocytes, and PC12 cells expressing presenilin mutations (Guo et al., 1996; Leissring et al., 1999; Stutzmann et al., 2004). In a compelling study, Tu et al. (2006) show that wild-type, but not mutant presenilins, form low-conductance divalent-cation-permeable ion channels in planar lipid bilayers. The authors suggest that expression of mutant presenilins leads to loss of the normal endoplasmic reticulum Ca2+ leak current and therefore cause an increased loading of the Ca2+ store, which would explain the exaggerated InsP3-mediated Ca2+ signals observed by previous studies with expression of presenilin mutations. Recently, it has also been observed that, in animal models of starved mice used to mimic the low glucose uptake/metabolism observed in the brains of individuals with AD, reduced glucose uptake resulted in decreased protein O-GlcNAcylation (Liu et al., 2004). Therefore, another contributor to the enhancement of InsP3-evoked Ca2+ release observed in FAD can be explained with our finding that decreased O-GlcNAcylation of the InsP3R-I increases Ca2+ release. This new mechanism of regulation of the InsP3R-I may also prove relevant for understanding the potential role of Ca2+ signaling disruptions in sporadic AD.
Under physiological conditions, the InsP3R-I channel couples activation of cell surface receptors to intracellular Ca2+ release. Regulation of the InsP3R-I channel is thought to be a major component in the determination of the spatiotemporal characteristics of agonist-evoked Ca2+ signals (Berridge, 1993). Previous studies have shown that O-GlcNAcylation is a dynamic and inducible modification (for review, see Wells et al., 2001). We have shown that O-GlcNAcylation of the InsP3R-I can be modulated both in vitro and in vivo. These modifications present a new mechanism of regulation of the InsP3R-I channel, which may emerge as a major player in the control of fundamental cellular processes regulated by changes in the cytosolic Ca2+ concentration.
Footnotes
This work was supported by grants from the Diabetes Endocrinology Research Center and the National Institutes of Health (B.E.E.), Centre National de la Recherche Scientifique (T.C., B.E.E.), a Howard Hughes Medical Institute predoctoral fellowship (C.J.G.), and a German National Merit Foundation scholarship (E.W.). We thank Natasha Zachara and Gerald Hart for teaching us about the GlcNAC pathway and its importance in cellular regulation. We also thank Isabel Llano, Alain Marty, Benedicte Rossi, and Brenda DeGray for thoughtful discussions about the experiments and comments on this manuscript.
References
- Akimoto Y, Kreppel LK, Hirano H, Hart GW. Increased O-GlcNAc transferase in pancreas of rats with streptozotocin-induced diabetes. Diabetologia. 2000;43:1239–1247. doi: 10.1007/s001250051519. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. Inositol trisphosphate and calcium signalling. Nature. 1993;361:315–325. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I. The inositol 1,4,5-trisphosphate receptors. Cell Calcium. 2005;38:261–272. doi: 10.1016/j.ceca.2005.06.030. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I, Watras J, Ehrlich BE. Bell-shaped calcium-response curves of Ins(1,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature. 1991;351:751–754. doi: 10.1038/351751a0. [DOI] [PubMed] [Google Scholar]
- Choe CU, Ehrlich BE. The inositol 1,4,5-trisphosphate receptor (IP3R) and its regulators: sometimes good and sometimes bad teamwork. Sci STKE. 2006;2006:re15. doi: 10.1126/stke.3632006re15. [DOI] [PubMed] [Google Scholar]
- Clark RJ, McDonough PM, Swanson E, Trost SU, Suzuki M, Fukuda M, Dillmann WH. Diabetes and the accompanying hyperglycemia impairs cardiomyocyte calcium cycling through increased nuclear O-GlcNAcylation. J Biol Chem. 2003;278:44230–44237. doi: 10.1074/jbc.M303810200. [DOI] [PubMed] [Google Scholar]
- Comer FI, Vosseller K, Wells L, Accavitti MA, Hart GW. Characterization of a mouse monoclonal antibody specific for O-linked N-acetylglucosamine. Anal Biochem. 2001;293:169–177. doi: 10.1006/abio.2001.5132. [DOI] [PubMed] [Google Scholar]
- Ehrlich BE, Kaftan E, Bezprozvannaya S, Bezprozvanny I. The pharmacology of intracellular Ca2+-release channels. TiPs. 1994;15:145–149. doi: 10.1016/0165-6147(94)90074-4. [DOI] [PubMed] [Google Scholar]
- Gao Y, Wells L, Comer FI, Parker GJ, Hart GW. Dynamic O-glycosylation of nuclear and cytosolic proteins: cloning and characterization of a neutral, cytosolic beta-N-acetylglucosaminidase from human brain. J Biol Chem. 2001;276:9838–9845. doi: 10.1074/jbc.M010420200. [DOI] [PubMed] [Google Scholar]
- Go LO, Moschella MC, Watras J, Handa KK, Fyfe BS, Marks AR. Differential regulation of two types of intracellular calcium release channels during end-stage human heart failure. J Clin Invest. 1995;95:888–894. doi: 10.1172/JCI117739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Q, Furukawa K, Sopher BL, Pham DG, Xie J, Robinson N, Martin GM, Mattson MP. Alzheimer's PS-1 mutation perturbs calcium homeostasis and sensitizes PC12 cells to death induced by amyloid beta-peptide. NeuroReport. 1996;8:379–383. doi: 10.1097/00001756-199612200-00074. [DOI] [PubMed] [Google Scholar]
- Hajnoczky G, Csordas G, Madesh M, Pacher P. Control of apoptosis by IP(3) and ryanodine receptor driven calcium signals. Cell Calcium. 2000;28:349–363. doi: 10.1054/ceca.2000.0169. [DOI] [PubMed] [Google Scholar]
- Haltiwanger RS, Blomberg MA, Hart GW. Glycosylation of nuclear and cytoplasmic proteins. Purification and characterization of a uridine diphospho-N-acetylglucosamine:polypeptide beta-N-acetylglucosaminyltransferase. J Biol Chem. 1992;267:9005–9013. [PubMed] [Google Scholar]
- Haltiwanger RS, Grove K, Philipsberg GA. Modulation of O-linked N-acetylglucosamine levels on nuclear and cytoplasmic proteins in vivo using the peptide O-GlcNAc-beta-N-acetylglucosaminidase inhibitor O-(2-acetamido-2-deoxy-d-glucopyranosylidene)amino-N-phenylcarbamate. J Biol Chem. 1998;273:3611–3617. doi: 10.1074/jbc.273.6.3611. [DOI] [PubMed] [Google Scholar]
- Helenius A, Aebi M. Roles of N-linked glycans in the endoplasmic reticulum. Annu Rev Biochem. 2004;73:1019–1049. doi: 10.1146/annurev.biochem.73.011303.073752. [DOI] [PubMed] [Google Scholar]
- Hu Y, Belke D, Suarez J, Swanson E, Clark R, Hoshijima M, Dillmann WH. Adenovirus-mediated overexpression of O-GlcNAcase improves contractile function in the diabetic heart. Circ Res. 2005;96:1006–1013. doi: 10.1161/01.RES.0000165478.06813.58. [DOI] [PubMed] [Google Scholar]
- Jayaraman T, Ondrias K, Ondriasova E, Marks AR. Regulation of the inositol 1,4,5-trisphosphate receptor by tyrosine phosphorylation. Science. 1996;272:1492–1494. doi: 10.1126/science.272.5267.1492. [DOI] [PubMed] [Google Scholar]
- Johenning FW, Zochowski M, Conway SJ, Holmes AB, Koulen P, Ehrlich BE. Distinct intracellular calcium transients in neurites and somata integrate neuronal signals. J Neurosci. 2002;22:5344–5353. doi: 10.1523/JNEUROSCI.22-13-05344.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreppel LK, Blomberg MA, Hart GW. Dynamic glycosylation of nuclear and cytosolic proteins. Cloning and characterization of a unique O-GlcNAc transferase with multiple tetratricopeptide repeats. J Biol Chem. 1997;272:9308–9315. doi: 10.1074/jbc.272.14.9308. [DOI] [PubMed] [Google Scholar]
- Kruglikov I, Gryshchenko O, Shutov L, Kostyuk E, Kostyuk P, Voitenko N. Diabetes-induced abnormalities in ER calcium mobilization in primary and secondary nociceptive neurons. Pflügers Arch. 2004;448:395–401. doi: 10.1007/s00424-004-1263-8. [DOI] [PubMed] [Google Scholar]
- Launer LJ. Diabetes and brain aging: epidemiologic evidence. Curr Diab Rep. 2005;5:59–63. doi: 10.1007/s11892-005-0069-1. [DOI] [PubMed] [Google Scholar]
- Lefebvre T, Ferreira S, Dupont-Wallois L, Bussiere T, Dupire MJ, Delacourte A, Michalski JC, Caillet-Boudin ML. Evidence of a balance between phosphorylation and O-GlcNAc glycosylation of Tau proteins—a role in nuclear localization. Biochim Biophys Acta. 2003;1619:167–176. doi: 10.1016/s0304-4165(02)00477-4. [DOI] [PubMed] [Google Scholar]
- Leissring MA, Paul BA, Parker I, Cotman CW, LaFerla FM. Alzheimer's presenilin-1 mutation potentiates inositol 1,4,5-trisphosphate-mediated calcium signaling in Xenopus oocytes. J Neurochem. 1999;72:1061–1068. doi: 10.1046/j.1471-4159.1999.0721061.x. [DOI] [PubMed] [Google Scholar]
- Liu F, Iqbal K, Grundke-Iqbal I, Hart GW, Gong CX. O-GlcNAcylation regulates phosphorylation of tau: a mechanism involved in Alzheimer's disease. Proc Natl Acad Sci USA. 2004;101:10804–10809. doi: 10.1073/pnas.0400348101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Pang Y, Chang T, Bounelis P, Chatham JC, Marchase RB. Increased hexosamine biosynthesis and protein O-GlcNAc levels associated with myocardial protection against calcium paradox and ischemia. J Mol Cell Cardiol. 2006;40:303–312. doi: 10.1016/j.yjmcc.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Llano I, Gerschenfeld HM. Inhibitory synaptic currents in stellate cells of rat cerebellar slices. J Physiol. 1993;468:177–200. doi: 10.1113/jphysiol.1993.sp019766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llano I, Tan YP, Caputo C. Spatial heterogeneity of intracellular Ca2+ signals in axons of basket cells from rat cerebellar slices. J Physiol. 1997;502:509–519. doi: 10.1111/j.1469-7793.1997.509bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubas WA, Hanover JA. Functional expression of O-linked GlcNAc transferase Domain structure and substrate specificity. J Biol Chem. 2000;275:10983–10988. doi: 10.1074/jbc.275.15.10983. [DOI] [PubMed] [Google Scholar]
- Marshall S, Duong T, Orbus RJ, Rumberger JM, Okuyama R. Measurement of UDP-N-acetylglucosaminyl transferase (OGT) in brain cytosol and characterization of anti-OGT antibodies. Anal Biochem. 2003;314:169–179. doi: 10.1016/s0003-2697(02)00686-3. [DOI] [PubMed] [Google Scholar]
- McNay EC, Williamson A, McCrimmon RJ, Sherwin RS. Cognitive and neural hippocampal effects of long-term moderate recurrent hypoglycemia. Diabetes. 2006;55:1088–1095. doi: 10.2337/diabetes.55.04.06.db05-1314. [DOI] [PubMed] [Google Scholar]
- Michikawa T, Hamanaka H, Otsu H, Yamamoto A, Miyawaki A, Furuichi T, Tashiro Y, Mikoshiba K. Transmembrane topology and sites of N-glycosylation of inositol 1,4,5-trisphosphate receptor. J Biol Chem. 1994;269:9184–9189. [PubMed] [Google Scholar]
- Nagase T, Ito KI, Kato K, Kaneko K, Kohda K, Matsumoto M, Hoshino A, Inoue T, Fujii S, Kato H, Mikoshiba K. Long-term potentiation and long-term depression in hippocampal CA1 neurons of mice lacking the IP(3) type 1 receptor. Neuroscience. 2003;117:821–830. doi: 10.1016/s0306-4522(02)00803-5. [DOI] [PubMed] [Google Scholar]
- Nagy T, Champattanachai V, Marchase RB, Chatham JC. Glucosamine inhibits angiotensin II-induced cytoplasmic Ca2+ elevation in neonatal cardiomyocytes via protein-associated O-linked N-acetylglucosamine. Am J Physiol. 2006;290:C57–C65. doi: 10.1152/ajpcell.00263.2005. [DOI] [PubMed] [Google Scholar]
- Parker GJ, Lund KC, Taylor RP, McClain DA. Insulin resistance of glycogen synthase mediated by O-linked N-acetylglucosamine. J Biol Chem. 2003;278:10022–10027. doi: 10.1074/jbc.M207787200. [DOI] [PubMed] [Google Scholar]
- Robinson KA, Weinstein ML, Lindenmayer GE, Buse MG. Effects of diabetes and hyperglycemia on the hexosamine synthesis pathway in rat muscle and liver. Diabetes. 1995;44:1438–1446. doi: 10.2337/diab.44.12.1438. [DOI] [PubMed] [Google Scholar]
- Ryan CM. Diabetes and brain damage: more (or less) than meets the eye? Diabetologia. 2006;49:2229–2233. doi: 10.1007/s00125-006-0392-3. [DOI] [PubMed] [Google Scholar]
- Snow CM, Senior A, Gerace L. Monoclonal antibodies identify a group of nuclear pore complex glycoproteins. J Cell Biol. 1987;104:1143–1156. doi: 10.1083/jcb.104.5.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutzmann GE, Caccamo A, LaFerla FM, Parker I. Dysregulated IP3 signaling in cortical neurons of knock-in mice expressing an Alzheimer's-linked mutation in presenilin1 results in exaggerated Ca2+ signals and altered membrane excitability. J Neurosci. 2004;24:508–513. doi: 10.1523/JNEUROSCI.4386-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tandon A, Fraser P. The presenilins. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-11-reviews3014. reviews3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang TS, Tu H, Wang Z, Bezprozvanny I. Modulation of type 1 inositol (1,4,5)-trisphosphate receptor function by protein kinase A and protein phosphatase 1α. J Neurosci. 2003;23:403–415. doi: 10.1523/JNEUROSCI.23-02-00403.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thinakaran G, Sisodia SS. Presenilins and Alzheimer disease: the calcium conspiracy. Nat Neurosci. 2006;9:1354–1355. doi: 10.1038/nn1106-1354. [DOI] [PubMed] [Google Scholar]
- Thrower EC, Choe CU, So SH, Jeon SH, Ehrlich BE, Yoo SH. A functional interaction between chromogranin B and the inositol 1,4,5-trisphosphate receptor/Ca2+ channel. J Biol Chem. 2003;278:49699–49706. doi: 10.1074/jbc.M309307200. [DOI] [PubMed] [Google Scholar]
- Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee SF, Hao YH, Serneels L, De Strooper B, Yu G, Bezprozvanny I. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer's disease-linked mutations. Cell. 2006;126:981–993. doi: 10.1016/j.cell.2006.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells L, Hart GW. O-GlcNAc turns twenty: functional implications for post-translational modification of nuclear and cytosolic proteins with a sugar. FEBS Lett. 2003;546:154–158. doi: 10.1016/s0014-5793(03)00641-0. [DOI] [PubMed] [Google Scholar]
- Wells L, Vosseller K, Hart GW. Glycosylation of nucleocytoplasmic proteins: signal transduction and O-GlcNAc. Science. 2001;291:2376–2378. doi: 10.1126/science.1058714. [DOI] [PubMed] [Google Scholar]
- Wessels AM, Simsek S, Remijnse PL, Veltman DJ, Biessels GJ, Barkhof F, Scheltens P, Snoek FJ, Heine RJ, Rombouts SA. Voxel-based morphometry demonstrates reduced grey matter density on brain MRI in patients with diabetic retinopathy. Diabetologia. 2006;49:2474–2480. doi: 10.1007/s00125-006-0283-7. [DOI] [PubMed] [Google Scholar]
- Wojcikiewicz RJ, Luo SG. Phosphorylation of inositol 1,4,5-trisphosphate receptors by cAMP-dependent protein kinase. Type I, II, and III receptors are differentially susceptible to phosphorylation and are phosphorylated in intact cells. J Biol Chem. 1998;273:5670–5677. doi: 10.1074/jbc.273.10.5670. [DOI] [PubMed] [Google Scholar]
- Yang X, Su K, Roos MD, Chang Q, Paterson AJ, Kudlow JE. O-Linkage of N-acetylglucosamine to Sp1 activation domain inhibits its transcriptional capability. Proc Natl Acad Sci USA. 2001;98:6611–6616. doi: 10.1073/pnas.111099998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachara NE, Hart GW. Cell signaling, the essential role of O-GlcNAc! Biochim Biophys Acta. 2006;1761:599–617. doi: 10.1016/j.bbalip.2006.04.007. [DOI] [PubMed] [Google Scholar]
- Zachara NE, O'Donnell N, Cheung WD, Mercer JJ, Marth JD, Hart GW. Dynamic O-GlcNAc modification of nucleocytoplasmic proteins in response to stress. A survival response of mammalian cells. J Biol Chem. 2004;279:30133–30142. doi: 10.1074/jbc.M403773200. [DOI] [PubMed] [Google Scholar]