

Abstract
Radial glial cells in the cerebral cortex serve as progenitors for neurons and glia and guide the migration of cortical neurons. The integrin α3β1 is thought to mediate interactions of migrating neurons with radial glial cells and to function as a receptor for the reelin signaling molecule. Here, we challenge this view and demonstrate that β1 integrins in migrating neurons are not essential for the formation of cell layers in the cerebral cortex. Cortical cell layers also form normally in mice deficient in the integrin α3β1. However, we provide evidence that β1 integrins in radial glia control the morphological differentiation of both glia and neurons. We conclude that β1 integrins in radial glia are required for the proper development of the cerebral cortex, whereas β1 integrins in migrating neurons are not essential for glial-guided migration and reelin signaling.
Keywords: integrin, cell migration, radial glia, cerebral cortex, nervous system, reelin
Introduction
The mature cerebral cortex consists of cell layers containing distinct neuronal subtypes that are formed by series of well orchestrated cell specification and migration events. Excitatory neurons are generated from precursors in the cortical ventricular zone (VZ) and subventricular zone (SVZ) and migrate along radial glial fibers to populate the cortical wall. Later-born neurons bypass earlier neurons and come to sit in ever-more-superficial layers. Most inhibitory interneurons are born in the ganglionic eminences from where they migrate tangentially and settle into the emerging cortical cell layers (Hatten, 2002; Marin and Rubenstein, 2003; Nadarajah et al., 2003; Kriegstein and Noctor, 2004).
Although the complete composition of the precursor pool in the VZ and SVZ is not known, it is clear that radial glial cells constitute an important part of the pool. Radial glial cells therefore serve a dual purpose as progenitors for neurons and as a scaffold to guide their migration (Campbell and Gotz, 2002; Fishell and Kriegstein, 2003; Kriegstein and Gotz, 2003). Although the intracellular signaling mechanisms controlling neuronal migration are beginning to be defined (such as those involving Lis1, p35, CDK5, doublecortin, and doublecortin-like kinase) (Bielas et al., 2004; Tsai and Gleeson, 2005; Weimer and Anton, 2006; Ayala et al., 2007), the receptors that control the differentiation of radial glial cells and mediate their interactions with neurons are less well understood. Recent studies have provided evidence that notch-delta signaling controls the specification of radial glial cells (Gaiano et al., 2000), whereas neuregulins regulates the formation of radial glial processes (Anton et al., 1997; Schmid et al., 2003). β1 integrins, a family of 12 heterodimeric extracellular matrix (ECM) receptors consisting of a β1 subunit and different α subunits (Hynes, 2002), have also been implicated in controlling the differentiation of radial glial cells. Mice that lack expression of the integrin β1 subunit in radial glial cells (Itgb1-CNSko mice) show perturbations in the formation of cortical cell layers that are associated with defects in the anchorage of radial glial fibers at the meningeal basement membrane (Graus-Porta et al., 2001). Similar defects have been observed in mice with mutations in the genes encoding (1) the integrin α6-subunit or both α3 and α6 (Georges-Labouesse et al., 1998; De Arcangelis et al., 1999); (2) ECM components of the meningeal basement membrane (Miner et al., 1998; Costell et al., 1999; Halfter et al., 2002; Poschl et al., 2004); and (3) the integrin downstream effectors FAK (focal adhesion kinase) (Beggs et al., 2003) and ILK (integrin-linked kinase) (Niewmierzycka et al., 2005). These findings suggest that integrins form a transmembrane signaling complex that regulates glial process outgrowth and endfeet anchorage at the meningeal basement membrane.
Recent studies have shed light on the receptors that mediate interactions between radial glial cells and migrating neurons. The gap junctional proteins connexin 26 and 43 are expressed at the contact points between radial glial fibers and migrating neurons to provide dynamic adhesive contacts (Elias et al., 2007). Members of the β1 integrin family are also thought to control interactions between neurons and radial glial fibers that are critical for glial-guided migration. In particular, it has been described that migrating neurons express the integrin α3 subunit, which heterodimerizes with the integrin β1 subunit to form the functional α3β1 receptor. Function blocking antibodies against α3 perturb neuron–glia interactions in vitro, and mice with a null mutation in the integrin α3 subunit gene (Itga3-null mice) have been reported to have defects in the migration of cortical neurons (Anton et al., 1999; Schmid et al., 2004). In addition, it has been reported that α3β1 is a coreceptor for the reelin signaling molecule (Dulabon et al., 2000). Reelin is strongly expressed in Cajal–Retzius cells in the cortical marginal zone and required for the proper formation of cortical layers. Reelin binds to very low density lipoprotein receptor (VLDLR) and apolipoprotein E receptor 2 (ApoER2), and mutations in the genes for these two receptors lead to cortical layering defects that closely resemble those in reelin-deficient mice. VLDLR and ApoER2 also bind to the cytoplasmic adaptor protein dab-1, which is phosphorylated in a reelin-dependent manner and required for the formation of cortical layers, suggesting that reelin activates a VLDLR/ApoER2-dependent signaling pathway (Rice and Curran, 2001; Herz and Bock, 2002; Tissir and Goffinet, 2003). It has been reported that the integrin α3β1 also binds to reelin and that this interaction leads to the termination of cell migration, possibly in a process involving dab-1 (Dulabon et al., 2000; Sanada et al., 2004; Schmid et al., 2004). However, it is unclear whether reelin serves as a signal that terminates cell migration or whether it acts by a different mechanism (Magdaleno and Curran, 2001; Luque, 2004). In addition, the extent to which the phenotype of Itga3-null mice resembles reeler mice is unclear. In fact, no defects in neuron–glia interaction or direct effect on neuronal migration have been observed in Itgb1-CNSko mice, which lack all β1 integrins (including the integrin α3β1) in radial glia and migrating neurons (Graus-Porta et al., 2001).
To clarify the function of β1 integrins during glial-guided migration and reelin signaling, we have further analyzed the function of β1 integrins by genetic means in mouse model systems. We demonstrate here that β1 integrins in neurons are not essential for the formation of cell layers in the cerebral cortex. Cortical layers also form normally in Itga3-null mice. We conclude that other receptors such as connexin 26 and 43 (Elias et al., 2007) must control neuronal migration during cortical development, either alone or in a redundant manner with β1 integrins. In contrast, integrin expression in glial cells is critical for the formation of cortical layers and the morphological differentiation of both glia and neurons.
Materials and Methods
Animals and antibodies. Itgb1-CNSko
mice and Itgb1-NEXKo mice were generated by crossing Itgb1flox/flox mice (Graus-Porta et al., 2001) with Itgb1 flox/+ Nestin-Cre +/− mice (Tronche et al., 1999) or Itgb1 flox/+ NEX-Cre +/− mice (Wu et al., 2005). Genotyping was performed as described previously (Graus-Porta et al., 2001). Itga3-null mice and CRE activity reporter mice Rosa26lacZ-loxP and Z/EG have been described previously (Kreidberg et al., 1996; Novak et al., 2000).
Histology and immunohistochemistry.
For LacZ staining, embryos at embryonic day 11 (E11) and E12.5 were fixed in 1% paraformaldehyde (PFA) for 20 min and then incubated overnight at 37°C in the following solution: 2 mm MgCl2, 0.02% NP40, 0.01 deoxycholate, 5 mm potassium ferrocyanide, 5 mm potassium ferricyanide, and 0.5 mg/ml X-gal, in PBS. Nissl staining and immunohistochemistry were performed as described previously (Graus-Porta et al., 2001; Belvindrah et al., 2002). Antibodies were as follows: polyclonal anti-GFAP (DAKO, Glostrup, Denmark), polyclonal anti-CRE (Covance, Berkeley,CA), polyclonal anti-calretinin (Chemicon, Temecula, CA), polyclonal anti-CUX1 (kind gift from C. Walsh, Boston, MA), monoclonal anti-Neurofilament (Smi32) (Covance, Berkeley, CA), polyclonal anti-microtubule-associated protein 2 (MAP2) (a kind gift from A. Matus, Basel, Switzerland), polyclonal anti-TBR1 (a kind gift from R. Hevner, Seattle, WA), monoclonal anti-reelin (G10 clone, Abcam, Cambridge, MA), polyclonal GFP (our own antibody raised in rabbits against a GFP-GST fusion protein), polyclonal anti-dcx (guinea pig, Abcam, Cambridge, MA). For detection, we used secondary antibodies coupled to fluorochromes (Molecular Probes, Eugene, OR) or ABC detection kit and DAB substrate (Vector, Burlingame, CA). Nuclei were counterstained with DAPI (1 μg/ml) (Sigma, St Louis, MO). GFP fluorescence and antibody stainings were analyzed by confocal microscopy (Fluoview-LSM, Olympus, Melville, NY) or deconvolution microscopy (Deltavision, Applied Precision). Values are mean ± SEM. A Student's t test was performed.
Flow cytometry.
Cortical hemispheres were dissected into cold HBSS without (w/o) Ca2+ and Mg2+ (Invitrogen, Carlsbad, CA) from E14.5 mice. Cells were dissociated for 6 min at 37°C with 5 mg/ml trypsin (Worthington, Lakewood, NJ) and 0.1% DNase I (Worthington), dissociated mechanically in sorting buffer (HBSS w/o Ca 2+ and Mg2+ and phenol red, 1 mm EDTA, 25 mm HEPES, pH 7.0, 1% FBS) and filtered through a 40 μm nylon cell strainer (BD Biosciences, San Jose, CA). GFP-positive cells were fixed for 10 min at 4°C with 4% PFA and analyzed for β1 integrin expression by flow cytometry as described previously (Czuchra et al., 2006), using anti β1 antibody HMB1–1 (Ebiosciences).
Cortical size and cell numbers in cortical layers.
The size of the cortex was measured using SPOT software (Diagnostic Instruments Inc.). Three sections from three Itgb1-NEXko mice and three control littermates were analyzed at P60, as well as three Itga3-null mice and three control littermates at P0. Longitudinal distances were measured from the meninges to the dorsal limit of the corpus callosum, in five different rostrocaudal levels (see drawing in Fig. 4 e). For cell densities, Nissl+ cells were counted in a 5000 μm2 square in three sections in layers II and III from three Itgb1-NEXko mice and three littermate controls, and in a 1000 μm2 square in three sections in three Itga3-null mice and three littermate controls at P0. Values are mean ± SEM. A Student's t test was performed. To quantify cell numbers in neuronal cell layers, Tbr-1+ (for layers II–IV and VI) and Cux1+ (for layers II–III) cells were counted in a 10,000 μm2 square in three different sections from three Itga3-null mice and three Itgb1-NEXko mice with their respective littermate controls. Values are mean ± SEM. A Student's t test was performed.
Figure 4.

Analysis of the cerebral cortex of adult Itgb1-NEXko mice. a, b, Sagittal section through the cerebral cortex of Itgb1-NEXko mice and control littermates (wt, wild-type) at P60 were stained with Nissl. The overall morphology of the cerebral cortex appeared unaffected in the mutant mice. c, Cell density was determined in layer II/III in three wild-type and three Itgb1-CNSko mice. There was no difference in cell density. d, e, The thickness of the cerebral cortex was determined at five anatomical levels as indicated in the diagram. There was no significant difference between wild-type and Itgb1-CNSko mice. Scale bars: a, b, 400 μm.
Western blots and primary cell culture.
Primary cell cultures were established from the cerebral cortex of newborn mice following published procedures. In short, cortical hemispheres were dissected, the meninges were removed, and single cell suspensions were prepared as described previously (Hatten et al., 1998), except that cells were not separated by Percoll gradient centrifugation. Cells were resuspended in basal medium Eagle containing glutamine, 0.35% glucose, 10% horse serum, 5% FCS, and Pen-Strep and plated at a cell density of 4 × 105 onto 12-well plates coated with poly-d-lysine (PDL) and the indicated purified ECM glycoproteins. Cells were fixed after 1 or 5 d in culture with 2% PFA and processed for immunocytochemistry as described above. The length of GFAP+ processes (glia) and MAP2+ processes (neurites) were measured from the soma to the edge of the growth cone with MetaMorph Software (Universal Imaging, West Chester, PA). For all three genotypes (wild-type, Itgb1-CNSko, and Itgb1-NEXko), values were determined from three different samples, and the mean ± SEM was determined. A Student's t test was performed for statistical evaluation.
Results
Characterization of Nex-CRE mice
We have previously described a mouse line that is suitable for the inactivation of the β1 integrin subunit gene (Itgb1-flox) (Graus-Porta et al., 2001). We have also shown that inactivation of the Itgb1 gene in radial glial cells in the CNS, using a nestin-CRE mouse, leads to severe defects in the formation of cortical layers that can largely be attributed to defects in the cortical marginal zone, such as the anchorage of radial glial processes at the meningeal basement membrane and the formation of the Cajal–Retzius cell layer (Graus-Porta et al., 2001). However, because cortical morphology is severely affected in the mutants (referred to in the following as Itgb1-CNSko mice), we could not exclude entirely that β1 integrins are also essential for migration of cortical neurons.
To address the requirement of β1 integrins for cell migration, we wanted to inactivate the Itgb1 gene specifically in migrating neurons. For this purpose, we used the Nex-CRE mouse line, in which CRE is expressed from the endogenous transcriptional control region of the Nex gene. Using a GFP reporter mouse line, it has been shown that Nex-CRE induces CRE-mediated recombination in transient amplifying cells in the SVZ that give rise to pyramidal neurons (Wu et al., 2005). To characterize the Nex-CRE mouse line further, we crossed them with a reporter mouse line that carries a Rosa26lacZ-loxP gene. In this mouse line, expression of a nuclear localized LacZ is induced by CRE-mediated recombination (Mao et al., 1999). In whole-mount stainings, LacZ could be detected at E11 in early differentiating cranial ganglia (Fig. 1 a, arrow). At E12.5, LacZ expression in cranial ganglia was more pronounced and was also detected at the mid-hindbrain junction, and in the developing telencephalon and spinal cord (Fig. 1 b–d, arrows). Higher-magnification views of the telencephalon showed a gradient of LacZ-positive cells with high expression close to the VZ (Fig. 1 d, arrows), and scattered expression throughout the developing cortical hemispheres. The LacZ expression pattern was consistent with strong expression in precursor cells in the VZ/SVZ, which generate migrating neurons that disperse across the cortical hemispheres giving rise to the scattered appearance of the LacZ signal. In the adult, LacZ activity was evident throughout all cell layers of the cerebral cortex, and in the dentate gyrus and CA1-CA3 regions of the hippocampus (Fig. 1 e,g,h). In addition, a subset of cells in the mitral cell layer and below the glomerular layer of the olfactory bulb was LacZ positive, as well as some cells in the internal granule cell layer of the cerebellum (Fig. 1 e,f,i).
Figure 1.

Analysis of Nex-CRE mice. Nex-CRE mice were crossed with Rosa26lacZ-loxP mice and analyzed for nuclear localized LacZ expression (blue). a, At E11, LacZ was expressed in cranial ganglia (arrow). b, c, At E12.5, LacZ was expressed in basal ganglia, the mid-hindbrain junction, and the telencephalon (arrows in b). Expression was also observed in the spinal cord (arrows in c). d, At E12.5 prominent LacZ expression was observed close to the ventricular zone (arrows) and in scattered cells throughout the cortical wall. e–i, Histological sections of adult mice. e, LacZ was prominently expressed throughout the telencephalon and in the olfactory bulb. f, In the olfactory bulb, LacZ was expressed in the mitral cell layer and in the periglomerular layer. g–i, LacZ was widely expressed throughout the cortical wall (g), in the hippocampus (h) and in a subset of cells in the internal granule cell layer in the cerebellum (i). Scale bars: a–c, 1 mm; d, 360 μm; e, 820 μm; f, 260 μm; g, 215 μm; h, 205 μm; i, 260 μm.
To further define the cells in the cerebral cortex that expressed LacZ, we analyzed histological sections by staining with molecular markers. Costaining with GFAP demonstrated that LacZ-positive cells were negative for GFAP, suggesting that they were not glial cells (Fig. 2 a,b). In contrast, essentially all LacZ-positive cells were positive for the neuronal marker NeuN and vice versa (Fig. 2 d–f). These findings were confirmed with dissociated cells in culture. LacZ-positive cells were interspersed between GFAP-positive glial cells (Fig. 2 c), whereas costaining with antibodies to CRE and the neuronal marker MAP2 confirmed that essentially all neurons expressed CRE (Fig. 2 g).
Figure 2.

Nex-CRE mice induce recombination in cortical neurons but not in glia. a, b, Sagittal sections through the cortex were stained for LacZ (blue) and GFAP (brown). GFAP-positive cells (arrowheads) were LacZ negative. Arrows point to LacZ-positive cells that were interspersed between GFAP-positive cells; cc, corpus callosum. d–f, Sagittal cortical sections were stained for LacZ (blue) and NeuN (brown). LacZ-positive cells were NeuN positive, confirming their identity as neurons. Note that LacZ staining in d–f was for a shorter time than in a and b. Therefore, the signal appeared less strong and less diffuse in d–f, revealing strong staining in one or two dots within nuclei (arrows in f). c, g, Cortical neurons and glial cells from P0 mice were cultured on PDL/LN substrates and stained. c, LacZ-positive cells (blue) were interspersed between GFAP-positive cells (brown). g, MAP2-positive neurons (red), expressed CRE (green) in nuclei. Scale bars: a, 330 μm; b, c, 23 μm; d, 115 μm; e, 30 μm; f, g, 18 μm.
Together, our findings suggest that Nex-CRE induces efficient recombination in progenitor cells in the SVZ that give rise to pyramidal neurons that populate cortical cell layers. Using a CRE reporter line, we demonstrate that Nex-CRE is already active in the telencephalon at or before E12.5 and that neurons throughout all cortical layers undergo CRE-mediated recombination. In addition, we observed CRE-mediated recombination in cranial ganglia and a subset of cells in the olfactory bulb, hippocampus, and cerebellum.
Nex-CRE induces efficient recombination in migrating neurons and leads to the inactivation of β1 expression in Itgb1-NEXko mice
The analysis of the lacZ expression pattern induced by Nex-CRE (Figs. 1, 2) suggested that the vast majority of migrating neurons that leave the SVZ and populate cortical layers undergo CRE-mediated recombination. However, whereas expression of LacZ in individual cells could readily be demonstrated at postnatal ages (Fig. 2), LacZ expression at embryonic ages was more difficult to visualize because of the relatively weak expression of the LacZ transgene from the Rosa26 locus. Therefore, to analyze the extent of CRE-mediated recombination during embryonic ages, we crossed the Nex-CRE mice with the Z/EG reporter mouse line, which expresses high levels of GFP upon CRE-mediated recombination (Novak et al., 2000). Analysis of whole mounts revealed a recombination pattern similar to that observed with the LacZ reporter (Fig. 3 a–c). In vibratome sections, strong GFP fluorescence was detectable throughout the cortical wall (Fig. 3 d). To define more precisely the cells that expressed GFP, we analyzed coronal sections of E14.5 and E16.5 animals (Fig. 3 e–n). GFP expression was absent from the VZ but clearly detectable within the SVZ and throughout the cortical plate (Fig. 3 e–h), consistent with the expression in differentiating and migrating neurons. Costaining with DAPI revealed that the vast majority of cells within the SVZ and CP were GFP positive (Fig. 3 i–k). We next stained sections with antibodies to doublecortin, which stains premigratory neurons in the SVZ and migrating neurons within the cortical wall (des Portes et al., 1998; Francis et al., 1999; Gleeson et al., 1999). Costaining with antibodies to GFP revealed that the GFP-positive cells expressed doublecortin (Fig. 3 l–n), confirming their identity as migrating neurons. Higher-magnification views revealed that essentially all cells in the dorsal most aspect of the SVZ expressed GFP (Fig. 3 l′–n′). We conclude that the Nex-CRE transgene is expressed in most if not all neurons that migrate from the SVZ into the cortical wall.
Figure 3.

Characterization of NEX-CRE mice and analysis of Itgb1 expression by flow cytometry. a–n, Z/EG reporter mice carrying a CRE-inducible GFP transgene were crossed with NEX-CRE mice to analyze the CRE recombination pattern. a–c, GFP fluorescence was evident in the developing cerebral cortex of E12.5–E16.5 embryos by whole-mount analysis. d, GFP fluorescence throughout the cerebral cortex was also evident in vibratome sections. e–h, Coronal sections of mice at E14.5 and E16.5 were stained with antibodies to GFP. GFP expression was evident in the SVZ and cortical plate (CP), but not in the VZ. In e and g, nuclei were counterstained with DAPI (blue). i–k, Higher-magnification views of coronal sections stained with DAPI and antibodies to GFP. The vast majority of cells were GFP positive (arrows). l–n, Sections from E15.5 animals were stained with antibodies to doublecortin (dcx, red) and GFP (green). (l′–n′) Higher-magnification views of the area outlined in l–n. Note overlapping patterns of cytoplasmic staining for doublecortin and GFP. o, The expression levels of β1 integrins were evaluated in GFP-positive cells that had been isolated by FACS sorting from Nex-CRE mice containing a CRE-inducible GFP transgene (control), or from Itgb1-NEXko mice carrying a CRE-inducible transgene (mutant). The green line shows samples incubated with antibody to β1 integrins (β1 Ab); in the blue line samples, the secondary antibody alone was added (second); and in the red line samples, no antibody was added. Scale bar: a, 600 μm; b, c, 3.75 mm; d, 2 mm; e, f, 150 μm; g, h, 300 μm; i–k, 37.5 μm; l–n, 150 μm; l′–n′, 37.5 μm.
We next wanted to inactivate the Itgb1 gene in migrating neurons by crossing Nex-CRE carrying one Itgb1-null allele (Nex-CRE/Itgb1 +/− mice) with Itgb1-flox mice. Two types of crosses were performed. In one set of experiments, Itgb1-flox mice were directly crossed with Nex-CRE/Itgb1 +/− mice to obtain mutant mice that will be referred to as Itgb1-NEXko mice. In a second set of experiments, we wanted to be able to visualize and isolate cells that had undergone CRE-mediated recombination. Therefore, we crossed the CRE-inducible GFP transgene from Z/EG mice first into the Itgb1-flox background to obtain Itgb1/GFP-flox mice. These mice were then crossed with Nex-CRE/Itgb1 +/− mice. Mutant offspring from this second intercross will be referred to as Itgb1-NEXko-GFP mice. Itgb1-NEXko mice and Itgb1-NEXko-GFP mice were obtained in the expected Mendelian frequency and gave similar results in subsequent experiments.
To analyze inactivation of the Itgb1 gene, we took advantage of the GFP reporter in Itgb1-NEXko-GFP. We microdissected the SVZ and cortical plate from E14.5 Itgb1-NEXko-GFP and analyzed GFP-positive cells by flow cytometry for expression of the β1 integrin subunit, using an antibody specific for the extracellular domain of the integrin β1 subunit that is commonly used to evaluate β1 integrin expression levels (Czuchra et al., 2006). The experiments were performed with control mice carrying the wild-type Itgb1 gene and expressing the GFP transgene, and with Itgb1-deficient mice expressing the GFP transgene (Fig. 3 o). Robust expression of the integrin β1 protein could be detected in control mice. In contrast, β1 expression was not detected in the mutant mice, confirming efficient inactivation of the Itgb1-flox gene by Nex-CRE in migrating neurons.
Cortical development proceeds normally in Itgb1-NEXko mice
We next stained cortical sections of P60 mice with Nissl to analyze the organization of the cerebral cortex. Unlike in Itgb1-CNSko mice that lack β1 integrins in glial cells and neurons, cortical layers in Itgb1-NEXko mice appeared normal and neurons did not invade the cortical marginal zone (Fig. 4 a,b). We also quantified the cell density in the cerebral cortex, focusing on layers II and III, where nuclei of individual cells can readily be distinguished. There was no significant difference in cell density between wild-type and Itgb1-NEXko mice (Fig. 4 d). Measurements of cortical thickness at five anatomically distinct levels did not reveal any difference between wild-type and Itgb1-NEXko mice (Fig. 4 e).
We next analyzed the formation of cortical layers in detail using layer-specific molecular markers. These included CUX1, a marker for layer II–IV neurons (Nieto et al., 2004), and nonphosphorylated neurofilament H (clone Smi32), a marker for layer III and V neurons (Campbell and Morrison, 1989). For better comparison, we included not only wild-type and Itgb1-NEXko mice in the analysis, but also Itgb1-CNSko mice that lack β1 integrins in both neurons and glia and have undulating cortical layers (Graus-Porta et al., 2001). CUX1-positive neurons were appropriately localized to layer II–IV in wild-type and Itgb1-NEXko mice (Fig. 5 a,c,d,f). The cells had also reached layer II–IV in Itgb1-CNSko mice, but the cell layer had a wavy appearance and occupied a broader territory, indicative of defects in layer formation (Fig. 5 b,e). Similarly, Smi32-positive neurons had reached layer III and V in wild-type, Itgb1-NEXko, and Itgb1-CNSko mice (Fig. 5 g–i). Although the layers appeared normal in Itgb1-NEXko mice, they were undulating in Itgb1-CNSko mice. Higher resolution pictures revealed that the morphological differentiation of Smi32-positive neurons was not obviously affected in Itgb1-NEXko mice (Fig. 5 j–m). To further define whether the number of cells that populated cortical layers was affected in Itgb1-NEXko mice, we determined the number of CUX1 neurons in cortical layer II–IV. There was no significant difference in cell density or the number of CUX1-positive neurons in wild-type and Itgb1-NEXko mice (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Figure 5.

Cortical layers form normally in Itgb1-NEXko mice. a–f, Sagittal sections through the cerebral cortex of wild-type, Itgb1-CNSko, and Itgb1-NEXko mice at P60 were stained with antibodies to CUX1 (brown), to reveal layer II–IV neurons. CUX1-positive neurons in Itgb1-NEXko mice were confined to cortical layers II–IV. CUX1-positive neurons in Itgb1-CNSko mice were also present in cortical layers II–IV, but the layer had a wavy appearance and appeared to be broader than in wild-type mice. g–m, Sagittal sections through the cerebral cortex were stained with an antibody to a nonphosphorylated form of neurofilament (Smi32) (brown), which specifically stains a subset of layer III and V neurons, as well as a subset of cells in the hippocampus. Neurons in wild-type, Itgb1-CNSko mice, and Itgb1-NEXko mice were appropriately located in layer III and V, but layer in Itgb1-CNSko mice appeared wavy (g–i). Higher-magnification views revealed that dendritic process in layer III (j, k) and the hippocampus (l, m) were not noticeably affected in Itgb1-NEXko mice. Scale bars: a–c, 400 μm; d–f, 125 μm; g–i, 450 μm; j–m, 70 μm.
We have previously shown that the integrity of the Cajal–Retzius cell layer is affected in Itgb1-CNSko mice (Graus-Porta et al., 2001). To evaluate similar defects in Itgb1-NEXko mice, we stained histological sections with antibodies to reelin. The distribution of reelin-positive Cajal–Retzius cells was unaffected in Itgb1-NEXko mice (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Finally, we determined the presence and distribution of GFAP-expressing astrocytes in the cortex of wild-type, Itgb1-CNSko, and Itgb1-NEXko mice (Fig. 6). At P60, we observed a massive accumulation of GFAP-positive astrocytes in Itgb1-CNSko mice, but not in Itgb1-NEXko mice or in wild-type mice (Fig. 6 a–c). To define the time course for the accumulation of GFAP-positive cells, we stained the cerebral cortex at different postnatal ages. At P0, there was no difference between wild-type and Itgb1-CNSko mice. GFAP-positive radial glial fibers were prominent, as well as GFAP-positive astrocytes in the corpus callosum and the cortical marginal zone (Fig. 6 d,g). By P10, cells that were strongly positive for GFAP accumulated in the cortical wall of Itgb1-CNSko mice but were rarely detectable in wild-type mice (Fig. 6 e,h). By 1 year of age, there was a massive accumulation of GFAP-positive cells only in Itgb1-CNSko mice (Fig. 6 f–i), and the cells had the characteristic morphology of astrocytes (Fig. 6 k). The appearance of the GFAP-positive cells at late postnatal ages is consistent with a reactive gliosis indicative of defects in the function of cortical neurons and/or glial cells. No gliosis was observed in Itgb1-NEXko at any age analyzed (Fig. 6 c) (data not shown).
Figure 6.

Reactive gliosis in Itgb1-CNSko mice. a–c, Histological sections were stained at P60 with antibodies to GFAP and processed with secondary antibodies coupled to peroxidase. In Itgb1-CNSko mice, astrocytes that strongly express GFAP (brown) were abundantly present. d–k, GFAP expression was evaluated by immunofluorescence microscopy (green) at different postnatal ages. At P0, GFAP-positive cells were detectable in the meninges and corpus callosum close to the VZ of wild-type (d) and Itgb1-CNSko mice (g). At P10, patches of astrocytes expressing high amounts of GFAP started to be detectable within the cortical wall of Itgb1-CNS knock-out mice (h), but not in wild-type mice (e). GFAP-positive cells were much more abundant in Itgb1-CNSko mice by 1 year of age and distributed throughout the cerebral cortex (f, i). j, k, High-magnification view of the cortex of mice at 1 year of age. Note that the GFAP-positive cells in Itgb1-CNSko mice show the characteristic morphology of astrocytes. Scale bars: a–c, 60 μm; d–i, 200 μm; j, k, 50 μm.
Together, our findings show that when β1 integrins are inactivated in the precursors of neurons and glia (Itgb1-CNSko mice), the formation of cortical cell layers is perturbed. A reactive gliosis develops postnatally, indicative of defects in the function of neurons and/or glia. In contrast, integrin expression in migrating pyramidal neurons (Itgb1-NEXko mice) is not essential for the migration of neurons into cortical cell layers and does not cause gliosis.
Cortical development proceeds normally in Itga3-null mice
Previous studies that were largely based on in vitro data have concluded that the integrin α3β1 in migrating cortical neurons regulates their interactions with radial glial fibers and transmits a reelin signal into migrating neurons (Anton et al., 1999; Dulabon et al., 2000; Schmid et al., 2004). Because we did not observe defects in the formation of cortical layers in Itgb1-NEXko mice, one may argue that although α3β1 may regulate migration and reelin signaling positively, a second β1 integrin heterodimer may counteract migration and reelin signaling. Removal of the antagonistic integrins may lead to no net defect in the formation of cortical cell layers. To resolve these issues, we analyzed in detail integrin Itga3-null mice, which completely lack expression of the Itga3-encoded protein, and therefore the functional α3β1 receptor (Kreidberg et al., 1996). The analysis was performed with P0 animals, because the mutant mice die within the first day of birth, most likely as a consequence of kidney defects (Kreidberg et al., 1996). Staining of sagittal sections did not reveal any obvious defect in the overall organization of the cerebral cortex in Itga3-null mice, and distinct cell layers could be resolved (Fig. 7 a–d; supplemental Fig. 3, available at www.jneurosci.org as supplemental material). In addition, there was no detectable difference in cell density or thickness of the cerebral cortex (Fig. 7 e,f).
Figure 7.

Histological analysis of the cerebral cortex at P0 in Itga3-null mice. a–d, Overall brain morphology and the organization of the cerebral cortex appeared unaltered when analyzed by Nissl staining. e, f, Cell density in layers V/VI and cortical thickness (at similar anatomical levels as shown in Fig. 4 d) was not significantly altered in Itga3-null mice. Scale bars: a, b, 425 μm; c, d, 690 μm.
We next stained sections with a variety of molecular markers such as (1) βIII-tubulin (Tuj1), to reveal the overall appearance of the cortex; (2) calretinin and reelin, to determine the position of Cajal–Retzius cells and interneurons in the cortical marginal zone (Weisenhorn et al., 1994); (3) TBR1, to stain neurons in layers II–IV and VI (Englund et al., 2005); (4) CUX1, to stain neurons in layers II–IV (Nieto et al., 2004); and (5) Smi32, to stain neurons in layers III and V (Campbell and Morrison, 1989) (Figs. 8, 9) (data not shown). Staining with Tuj1 did not reveal any difference between wild-type and mutant mice (Fig. 8 a,b); the positioning of interneurons and Cajal–Retzius cells in the cortical marginal zone was unaffected (Fig. 8 c–h); neuronal subtypes as revealed by staining with antibodies to TBR1 (layers II–IV and VI) and CUX1 (layers II-III) assembled into appropriate layers without any obvious perturbations at any cortical level (Fig. 9). We also counted the number of TBR1-positive cells in layer II–IV and VI, and did not observe any defect in the mutant mice (supplemental Fig. 4, available at www.jneurosci.org as supplemental material), demonstrating that the correct number of cells had reached their appropriate target cell layers. Finally, migrating interneurons invading the cortical wall could be detected in both wild-type and mutant animals (Fig. 8 e,f, arrows). Together, these findings demonstrate that the integrin α3 subunit is not essential for the formation of cell layers in the cerebral cortex. The data also demonstrate that the phenotype of Itga3-null mice is clearly distinct from the phenotype of reeler mice, because no inversion of cell layers or intermingling of neurons from different cell layers was observed. We therefore conclude that the α3β1 integrin is also not essential for reelin signaling.
Figure 8.

The cortical marginal zone is unaffected in Itga3-null mice. Sagittal sections through the cerebral cortex of wild-type and Itga3-null mice at P60 were stained with antibodies to βΙΙΙ-tubulin (Tuj1 clone, green), to reveal overall organization of the cortex (a, b); calretinin (calret, red), to visualize cells in the cortical marginal zone and interneurons that migrate into cortical layers (arrows in e, f) (c–f); reelin (green), to reveal Cajal–Retzius cells (nuclei are stained with DAPI and shown in blue) (g, h). Scale bars: a, b, 70 μm; c, d, g, h, 80 μm; e, f, 40 μm. mz, Marginal zone.
Figure 9.
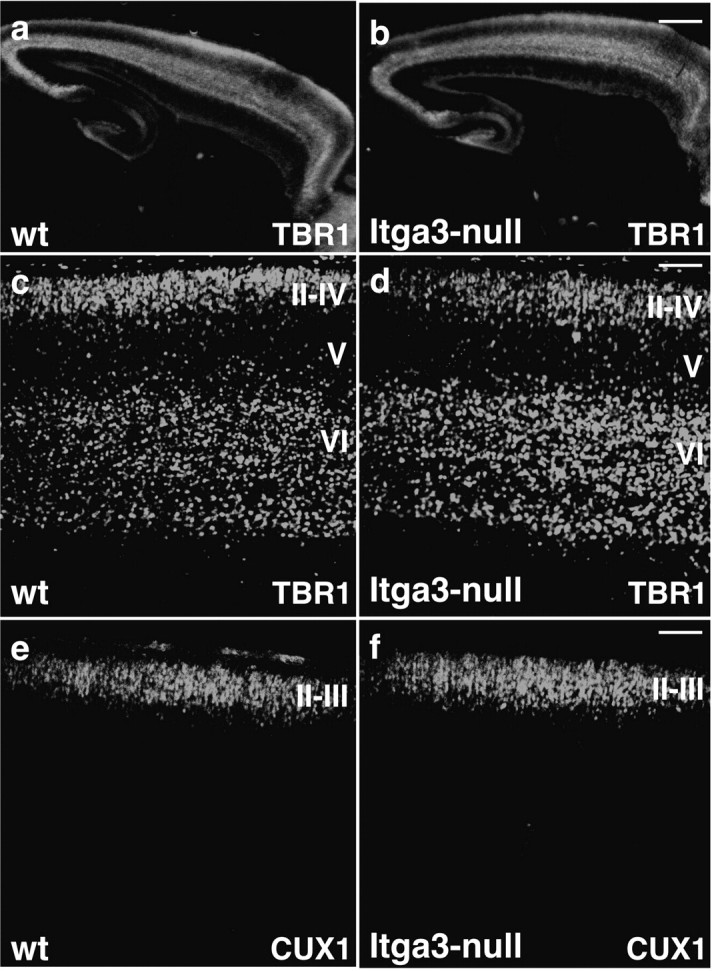
Cortical layers form normally in Itga3-null mice. Sagittal sections through the cerebral cortex of wild-type and Itga3-null mice at P0 were stained with antibodies to TBR1, to reveal neurons in layers II–IV and VI (a–d); and CUX1, to reveal neurons in layers II–IV (e, f). Note that there was no difference in the distribution of different neuronal subtypes in wild-type and Itga3-null mice. Scale bars: a, b, 425 μm; c, d, 80 μm; e, f, 100 μm.
Glial control of neuronal development
An analysis of the morphological differentiation of cortical neurons with MAP2 staining revealed that the dendritic trees of cortical neurons in wild-type mice were aligned perpendicular to the brain surface, whereas those in Itgb1-CNSko mice projected in abnormal angles (Fig. 10 a,b). The defects in Itgb1-CNSko mice were already obvious before birth, but absent in developing and adult Itgb1-NEXko mice (Fig. 10 c). These findings suggest that radial glial cells may control the morphological differentiation of cortical neurons. Alternatively, the disruptions in the cortical marginal zone in Itgb1-CNSko mice (Graus-Porta et al., 2001) may secondarily affect neuronal morphology. Unfortunately, because radial glial cells are the precursors for cortical neurons, it has so far not been possible to generate CRE mice that selectively inactivate integrins in radial glia but leave expression in cortical neurons intact. Therefore, to test more directly for effects of glial cells on the morphological differentiation of cortical neurons, we turned to explant cultures. Cortices were dissected from mice, and the cells were dissociated and maintained for several days in culture (Fig. 10 d–n). The length of the longest processes formed by neurons and glial cells was determined (Fig. 10 m,n). After 1 d in culture, staining of cortical cultures from wild-type mice with antibodies to MAP2 revealed that neurons had formed short neurites (Fig. 10 d). After 5 d in culture, the neurites were substantially longer as expected (Fig. 10 e,m). Staining of the cultures with GFAP revealed that glial cells had also undergone morphological maturation and formed long processes (Fig. 10 f,n). In cultures that were derived from Itgb1-CNSko mice, in which both neurons and glial cells lack the expression of β1 integrins, the formation of cell processes was severely affected in both cell types (Fig. 10 g–i,m,n). In striking contrast, no morphological defects were observed in either glia or neurons derived from Itgb1-NEXko mice. Glial cells formed elaborate projections, and even the β1 integrin-deficient neurons extended long neurites (Fig. 10 j–n). Together, these findings show that the defect in neurite outgrowth of β1 integrin-deficient neurons is rescued when glial cells express β1 integrins and undergo normal morphological differentiation, suggesting that the perturbations in neurite outgrowth are a secondary consequence of defects in glial cells.
Figure 10.

Defects in neuronal and glial process outgrowth. a–c, Sections through the cerebral cortex of P0 animals of the indicated genotype were stained with antibodies to MAP2 (green), to reveal dendrites. Note that dendrite morphology was severely affected in Itgb1-CNSko mice only. d–l, Dissociated cells from the cerebral cortex of P0 animals of the indicated genotype were plated onto PDL/LN substrates and cultured for 1 d (1div) or 5 d (5div). The cultures were stained with antibodies to MAP2 or GFAP (brown), as indicated. d–f, In cultures containing cells from wild-type mice, neurite outgrowth was evident at 1div and more pronounced by 5div. Many glial cells also formed extensive projections (arrows in f). g–i, Neurite outgrowth and the formation of glial processes was severely impaired in primary cultures containing cells from Itgb1-CNSko mice. j–l, Neurite outgrowth and the formation of glial processes were restored in cultures with cells from Itgb1-NEXko mice. m, n, The length of the glial processes and neurites was determined. Values indicated the mean and SD. A Student's t test was performed (***p < 0.01). Scale bars: a–c, 50 μm; d–l, 76 μm.
Discussion
We provide here genetic evidence that β1 integrins, and in particular the integrin α3β1, are not required for glial-guided migration and reelin signaling. This model is supported by the findings that the formation of cell layers is not affected in Itgb1-NEXko mice, which specifically lack β1 integrins in neurons that migrate along radial glial fibers from the VZ into the emerging cortical wall, and in Itga3-null mice, which do not express the integrin α3 subunit (and therefore the α3β1 receptor) in any cell type including the migrating neurons. However, in Itgb1-CNSko mice, which lack β1 integrins in both radial glia and neurons, the formation of cortical cell layers is affected because of defects in the cortical marginal zone that include perturbations in the anchorage of glial endfeet (Graus-Porta et al., 2001). Interestingly, in Itgb1-CNSko mice the morphology of the apical dendrites of pyramidal neurons is also perturbed. This defect is not seen in Itgb1-NEXko mice that lack β1 specifically in neurons, suggesting that the morphological abnormalities are caused, at least in part, by defects in glial cells. Consistent with this model, neurite outgrowth in mixed neuron–glia cultures is not dependent on β1 integrin expression in neurons, but outgrowth is affected when β1 integrins are inactivated in both neurons and glia. Although further studies are necessary to address this point, our findings suggest that interactions between neurons and glia that are mediated by direct cell contact or secreted signaling molecules are critical for proper neuronal differentiation. Overall, our findings suggest that β1 integrins in glial cells but not in pyramidal neurons are essential for the development of properly organized cell layers in the cerebral cortex. Other receptors such as connexins (Elias et al., 2007) control the migratory behavior of cortical neurons, either alone or in combination with β1 integrins.
Integrins and glial-guided migration
The function of β1 integrins in cell migration during cortical development has been controversial. Initially, it was reported that the integrin α3 subunit is expressed in migrating neurons and that function-blocking antibodies against α3 affect neuron–glia interactions in culture (Anton et al., 1999). It has also been reported that the integrin α3β1 is required for the migration of cortical neurons along radial glial fibers. However, neurons are assembled into layers in Itgb1-CNSko mice, which lack all β1 integrins in the developing cerebral cortex, suggesting that β1 integrins (including α3β1) are not required for radial glial-guided migration. The findings that we present here strongly support this latter view. First, we demonstrate that the cortical layers form normally when the Itgb1 gene is inactivated in the precursors of migrating neurons with Nex-CRE. Previous studies (Wu et al., 2005) and our characterization of the Nex-CRE mice show that CRE is expressed efficiently in the SVZ in transient amplifying cells and premigratory neurons that give rise to pyramidal neurons. CRE activity is prominent in the SVZ by E12.5, suggesting that there is sufficient time for inactivating expression of genes such as β1 integrins in the period of cortical development that stretches until postnatal ages. Consistent with this interpretation, we show β1 protein expression is abolished in the migrating CRE-expressing cells in Itgb1-NEXko-GFP mice. In addition, we have analyzed the cortex of Itga3-null mice, which completely lack expression of the α3 protein, and therefore the functional α3β1 receptor, in all cell types. We have studied the mice with a panel of molecular markers that have allowed us to determine the identity of neurons in different cortical layers, as well as cell numbers. These markers were largely not available when the Itga3-null mice were initially described. We also made sure that we study mice from the same source as used in previous experiments (Kreidberg et al., 1996) to exclude allele-specific variations in phenotype. Stainings with antibodies to βΙΙΙ-tubulin, calretinin, TBR1, CUX-1, and reelin did not reveal any defects in the position of cortical neurons or in the organization of cortical layers at any anatomical level. Neuronal cell density and cortical size were also unaffected in the mutants. How can these data be reconciled with earlier published data? As one possibility, there may be minor defects in the motility of neurons in Itga3-null mice that may have been detected in earlier in vitro studies, but that are not important for the development of cortical cell layers in vivo. Interestingly, culture experiments have been performed to study the function not only of the integrin α3 subunit but also of the αυ subunit in cortical development (Anton et al., 1999). Although these in vitro studies have led to the conclusion that both the integrin α3 and αυ subunits control neuron–glia interactions, genetic studies now demonstrate that neurons still migrate normally in the cortex of mice with a CNS restricted knock-out of the Itgav subunit gene (McCarty et al., 2005). β1 and αυ integrins may possibly control neuron–glia interaction in collaboration with other yet to be defined receptors. Neurons in the in vitro setting may rely more strongly on integrins, whereas a second class of receptors may compensate in vivo for the loss of integrin expression. Similar observations have been made for integrin functions in other settings. For example, β1 integrins control motor neuron outgrowth in vitro on purified extracellular matrix substrates but are not essential for axon outgrowth by motor neurons in vivo (Schwander et al., 2004).
Integrins and reelin signaling
The integrin α3β1 has also been reported to bind to reelin and to transmit a reelin detachment/stop signal into migrating neurons (Anton et al., 1999; Dulabon et al., 2000; Schmid et al., 2004). Although we cannot exclude that α3β1 binds reelin, recent studies by others and our findings presented here provide strong evidence that this integrin is not an essential component of the reelin signaling pathway: (1) our staining with a panel of layer-specific molecular markers shows that the inside-out sequence of cortical layers is not obviously altered in Itga3-null mice, Itgb1-CNSko mice, and Itgb1-NEXko mice, and therefore clearly different from the phenotype in reeler mice; (2) although dab-1 phosphorylation is instrumental in reeling signaling (Howell et al., 2000), α3β1 integrins does not control dab-1 phosphorylation (Dulabon et al., 2000); (3) recent genetic evidence suggests a promigratory function for reelin molecule from deeper cortical layers (IV–VI) (Magdaleno et al., 2002; Meyer et al., 2004; Yoshida et al., 2006) that would be incompatible with a mechanism by which binding of reelin to α3β1 inhibits migration; (4) mice with mutations in the reelin receptors VLDLR−/− and ApoERII−/− have a reeler-like phenotype (Trommsdorff et al., 1999). In summary, these studies suggest that reelin signaling is mediated by the VLDLR and ApoERII receptors independently of any β1 integrin (Magdaleno and Curran, 2001; Luque, 2004).
Integrins and the development of the cerebral cortex
Although our findings show that β1 integrins in cortical neurons are not essential for the formation of cortical layers, β1 integrins have important functions for corticogenesis. Studies by several laboratories have demonstrated that β1 integrins and their ligands and signaling effectors are required for the outgrowth of radial glial fibers and their attachment at the meningeal basement membrane. In the absence of integrin signaling, radial glial processes show defects in morphology, and the meningeal basement membrane is not properly remodeled/maintained (Miner et al., 1998; Costell et al., 1999; Graus-Porta et al., 2001; Halfter et al., 2002; Beggs et al., 2003; Poschl et al., 2004; Niewmierzycka et al., 2005). In addition, we show here that the defects in radial glial cells affect the morphological differentiation of pyramidal neurons. Accordingly, the projection pattern of dendrites is perturbed in the cortex of Itgb1-CNSko mice but not in Itgb1-NEXko mice. Because the Itgb1 subunit gene is inactivated in cortical neurons in Itgb1-NEXko mice, whereas recombination occurs in both neurons and glia in Itgb1-CNSko mice, defects in neurons are likely a secondary consequence of defects in glial cells. In agreement with this hypothesis, in primary cells in culture, defects in neurite outgrowth are observed only in coculture of β1-deficient neurons and glia, but not in coculture of β1-deficient neurons with β1-expressing glia. As one possibility, direct contact between neurons and glia may be important to control neuronal development. Alternatively, glial cells may secrete proteins that control the development of cortical neurons. Interesting candidates are members of the thrombospondin family, which have been identified as glial-derived proteins that control synapse formation (Christopherson et al., 2005).
In summary, the findings presented here suggest that β1 integrins in radial glial cells are critical for the formation of cell layers in the cerebral cortex. In contrast, β1 integrin expression in neurons is not essential for glial-guided migration and reelin signaling.
Footnotes
This work was supported by National Institutes of Health Grants R01 NS046456 and U01 MH078833 (U.M.). We thank members of the Müller laboratory for helpful discussion; C. Walsh, A. Matus, and R. Hevner for antibodies; J. Kreidberg for Itgα3-null mice; and Jerold Chun for comments on this manuscript.
References
- Anton ES, Marchionni MA, Lee KF, Rakic P. Role of GGF/neuregulin signaling in interactions between migrating neurons and radial glia in the developing cerebral cortex. Development. 1997;124:3501–3510. doi: 10.1242/dev.124.18.3501. [DOI] [PubMed] [Google Scholar]
- Anton ES, Kreidberg JA, Rakic P. Distinct functions of alpha3 and alpha(v) integrin receptors in neuronal migration and laminar organization of the cerebral cortex. Neuron. 1999;22:277–289. doi: 10.1016/s0896-6273(00)81089-2. [DOI] [PubMed] [Google Scholar]
- Ayala R, Shu T, Tsai LH. Trekking across the brain: the journey of neuronal migration. Cell. 2007;128:29–43. doi: 10.1016/j.cell.2006.12.021. [DOI] [PubMed] [Google Scholar]
- Beggs HE, Schahin-Reed D, Zang K, Goebbels S, Nave KA, Gorski J, Jones KR, Sretavan D, Reichardt LF. FAK deficiency in cells contributing to the basal lamina results in cortical abnormalities resembling congenital muscular dystrophies. Neuron. 2003;40:501–514. doi: 10.1016/s0896-6273(03)00666-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belvindrah R, Rougon G, Chazal G. Increased neurogenesis in adult mCD24-deficient mice. J Neurosci. 2002;22:3594–3607. doi: 10.1523/JNEUROSCI.22-09-03594.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielas S, Higginbotham H, Koizumi H, Tanaka T, Gleeson JG. Cortical neuronal migration mutants suggest separate but intersecting pathways. Annu Rev Cell Dev Biol. 2004;20:593–618. doi: 10.1146/annurev.cellbio.20.082503.103047. [DOI] [PubMed] [Google Scholar]
- Campbell K, Gotz M. Radial glia: multi-purpose cells for vertebrate brain development. Trends Neurosci. 2002;25:235–238. doi: 10.1016/s0166-2236(02)02156-2. [DOI] [PubMed] [Google Scholar]
- Campbell MJ, Morrison JH. Monoclonal antibody to neurofilament protein (SMI-32) labels a subpopulation of pyramidal neurons in the human and monkey neocortex. J Comp Neurol. 1989;282:191–205. doi: 10.1002/cne.902820204. [DOI] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 2005;120:421–433. doi: 10.1016/j.cell.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Costell M, Gustafsson E, Aszodi A, Morgelin M, Bloch W, Hunziker E, Addicks K, Timpl R, Fassler R. Perlecan maintains the integrity of cartilage and some basement membranes. J Cell Biol. 1999;147:1109–1122. doi: 10.1083/jcb.147.5.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czuchra A, Meyer H, Legate KR, Brakebusch C, Fassler R. Genetic analysis of beta1 integrin “activation motifs” in mice. J Cell Biol. 2006;174:889–899. doi: 10.1083/jcb.200604060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Arcangelis A, Mark M, Kreidberg J, Sorokin L, Georges-Labouesse E. Synergistic activities of alpha3 and alpha6 integrins are required during apical ectodermal ridge formation and organogenesis in the mouse. Development. 1999;126:3957–3968. doi: 10.1242/dev.126.17.3957. [DOI] [PubMed] [Google Scholar]
- des Portes V, Pinard JM, Billuart P, Vinet MC, Koulakoff A, Carrie A, Gelot A, Dupuis E, Motte J, Berwald-Netter Y, Catala M, Kahn A, Beldjord C, Chelly J. A novel CNS gene required for neuronal migration and involved in X-linked subcortical laminar heterotopia and lissencephaly syndrome. Cell. 1998;92:51–61. doi: 10.1016/s0092-8674(00)80898-3. [DOI] [PubMed] [Google Scholar]
- Dulabon L, Olson EC, Taglienti MG, Eisenhuth S, McGrath B, Walsh CA, Kreidberg JA, Anton ES. Reelin binds alpha3beta1 integrin and inhibits neuronal migration. Neuron. 2000;27:33–44. doi: 10.1016/s0896-6273(00)00007-6. [DOI] [PubMed] [Google Scholar]
- Elias LA, Wang DD, Kriegstein AR. Gap junction adhesion is necessary for radial migration in the neocortex. Nature. 2007;448:901–907. doi: 10.1038/nature06063. [DOI] [PubMed] [Google Scholar]
- Englund C, Fink A, Lau C, Pham D, Daza RA, Bulfone A, Kowalczyk T, Hevner RF. Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J Neurosci. 2005;25:247–251. doi: 10.1523/JNEUROSCI.2899-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishell G, Kriegstein AR. Neurons from radial glia: the consequences of asymmetric inheritance. Curr Opin Neurobiol. 2003;13:34–41. doi: 10.1016/s0959-4388(03)00013-8. [DOI] [PubMed] [Google Scholar]
- Francis F, Koulakoff A, Boucher D, Chafey P, Schaar B, Vinet MC, Friocourt G, McDonnell N, Reiner O, Kahn A, McConnell SK, Berwald-Netter Y, Denoulet P, Chelly J. Doublecortin is a developmentally regulated, microtubule-associated protein expressed in migrating and differentiating neurons. Neuron. 1999;23:247–256. doi: 10.1016/s0896-6273(00)80777-1. [DOI] [PubMed] [Google Scholar]
- Gaiano N, Nye JS, Fishell G. Radial glial identity is promoted by Notch1 signaling in the murine forebrain. Neuron. 2000;26:395–404. doi: 10.1016/s0896-6273(00)81172-1. [DOI] [PubMed] [Google Scholar]
- Georges-Labouesse E, Mark M, Messaddeq N, Gansmuller A. Essential role of alpha 6 integrins in cortical and retinal lamination. Curr Biol. 1998;8:983–986. doi: 10.1016/s0960-9822(98)70402-6. [DOI] [PubMed] [Google Scholar]
- Gleeson JG, Lin PT, Flanagan LA, Walsh CA. Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron. 1999;23:257–271. doi: 10.1016/s0896-6273(00)80778-3. [DOI] [PubMed] [Google Scholar]
- Graus-Porta D, Blaess S, Senften M, Littlewood-Evans A, Damsky C, Huang Z, Orban P, Klein R, Schittny JC, Muller U. Beta1-class integrins regulate the development of laminae and folia in the cerebral and cerebellar cortex. Neuron. 2001;31:367–379. doi: 10.1016/s0896-6273(01)00374-9. [DOI] [PubMed] [Google Scholar]
- Halfter W, Dong S, Yip YP, Willem M, Mayer U. A critical function of the pial basement membrane in cortical histogenesis. J Neurosci. 2002;22:6029–6040. doi: 10.1523/JNEUROSCI.22-14-06029.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatten ME. New directions in neuronal migration. Science. 2002;297:1660–1663. doi: 10.1126/science.1074572. [DOI] [PubMed] [Google Scholar]
- Hatten ME, Gao WQ, Morrison ME, Mason CA. The cerebellum: purification and coculture of identified cell populations. In: Banker G, Goslin K, editors. Culturing nerve cells. Cambridge, MA: MIT; 1998. [Google Scholar]
- Herz J, Bock HH. Lipoprotein receptors in the nervous system. Annu Rev Biochem. 2002;71:405–434. doi: 10.1146/annurev.biochem.71.110601.135342. [DOI] [PubMed] [Google Scholar]
- Howell BW, Herrick TM, Hildebrand JD, Zhang Y, Cooper JA. Dab1 tyrosine phosphorylation sites relay positional signals during mouse brain development. Curr Biol. 2000;10:877–885. doi: 10.1016/s0960-9822(00)00608-4. [DOI] [PubMed] [Google Scholar]
- Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- Kreidberg JA, Donovan MJ, Goldstein SL, Rennke H, Shepherd K, Jones RC, Jaenisch R. Alpha 3 beta 1 integrin has a crucial role in kidney and lung organogenesis. Development. 1996;122:3537–3547. doi: 10.1242/dev.122.11.3537. [DOI] [PubMed] [Google Scholar]
- Kriegstein AR, Gotz M. Radial glia diversity: a matter of cell fate. Glia. 2003;43:37–43. doi: 10.1002/glia.10250. [DOI] [PubMed] [Google Scholar]
- Kriegstein AR, Noctor SC. Patterns of neuronal migration in the embryonic cortex. Trends Neurosci. 2004;27:392–399. doi: 10.1016/j.tins.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Luque JM. Integrin and the Reelin-Dab1 pathway: a sticky affair? Brain Res Dev Brain Res. 2004;152:269–271. doi: 10.1016/j.devbrainres.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Magdaleno S, Keshvara L, Curran T. Rescue of ataxia and preplate splitting by ectopic expression of Reelin in reeler mice. Neuron. 2002;33:573–586. doi: 10.1016/s0896-6273(02)00582-2. [DOI] [PubMed] [Google Scholar]
- Magdaleno SM, Curran T. Brain development: integrins and the Reelin pathway. Curr Biol. 2001;11:R1032–R1035. doi: 10.1016/s0960-9822(01)00618-2. [DOI] [PubMed] [Google Scholar]
- Mao X, Fujiwara Y, Orkin SH. Improved reporter strain for monitoring Cre recombinase-mediated DNA excisions in mice. Proc Natl Acad Sci USA. 1999;96:5037–5042. doi: 10.1073/pnas.96.9.5037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin O, Rubenstein JL. Cell migration in the forebrain. Annu Rev Neurosci. 2003;26:441–483. doi: 10.1146/annurev.neuro.26.041002.131058. [DOI] [PubMed] [Google Scholar]
- McCarty JH, Lacy-Hulbert A, Charest A, Bronson RT, Crowley D, Housman D, Savill J, Roes J, Hynes RO. Selective ablation of {alpha}v integrins in the central nervous system leads to cerebral hemorrhage, seizures, axonal degeneration and premature death. Development. 2005;132:165–176. doi: 10.1242/dev.01551. [DOI] [PubMed] [Google Scholar]
- Meyer G, Cabrera Socorro A, Perez Garcia CG, Martinez Millan L, Walker N, Caput D. Developmental roles of p73 in Cajal-Retzius cells and cortical patterning. J Neurosci. 2004;24:9878–9887. doi: 10.1523/JNEUROSCI.3060-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miner JH, Cunningham J, Sanes JR. Roles for laminin in embryogenesis: exencephaly, syndactyly, and placentopathy in mice lacking the laminin alpha5 chain. J Cell Biol. 1998;143:1713–1723. doi: 10.1083/jcb.143.6.1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadarajah B, Alifragis P, Wong RO, Parnavelas JG. Neuronal migration in the developing cerebral cortex: observations based on real-time imaging. Cereb Cortex. 2003;13:607–611. doi: 10.1093/cercor/13.6.607. [DOI] [PubMed] [Google Scholar]
- Nieto M, Monuki ES, Tang H, Imitola J, Haubst N, Khoury SJ, Cunningham J, Gotz M, Walsh CA. Expression of Cux-1 and Cux-2 in the subventricular zone and upper layers II–IV of the cerebral cortex. J Comp Neurol. 2004;479:168–180. doi: 10.1002/cne.20322. [DOI] [PubMed] [Google Scholar]
- Niewmierzycka A, Mills J, St-Arnaud R, Dedhar S, Reichardt LF. Integrin-linked kinase deletion from mouse cortex results in cortical lamination defects resembling cobblestone lissencephaly. J Neurosci. 2005;25:7022–7031. doi: 10.1523/JNEUROSCI.1695-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak A, Guo C, Yang W, Nagy A, Lobe CG. Z/EG, a double reporter mouse line that expresses enhanced green fluorescent protein upon Cre-mediated excision. Genesis. 2000;28:147–155. [PubMed] [Google Scholar]
- Poschl E, Schlotzer-Schrehardt U, Brachvogel B, Saito K, Ninomiya Y, Mayer U. Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development. 2004;131:1619–1628. doi: 10.1242/dev.01037. [DOI] [PubMed] [Google Scholar]
- Rice DS, Curran T. Role of the reelin signaling pathway in central nervous system development. Annu Rev Neurosci. 2001;24:1005–1039. doi: 10.1146/annurev.neuro.24.1.1005. [DOI] [PubMed] [Google Scholar]
- Sanada K, Gupta A, Tsai LH. Disabled-1-regulated adhesion of migrating neurons to radial glial fiber contributes to neuronal positioning during early corticogenesis. Neuron. 2004;42:197–211. doi: 10.1016/s0896-6273(04)00222-3. [DOI] [PubMed] [Google Scholar]
- Schmid RS, Shelton S, Stanco A, Yokota Y, Kreidberg JA, Anton ES. {alpha}3{beta}1 integrin modulates neuronal migration and placement during early stages of cerebral cortical development. Development. 2004;131:6023–6031. doi: 10.1242/dev.01532. [DOI] [PubMed] [Google Scholar]
- Schmid RS, McGrath B, Berechid BE, Boyles B, Marchionni M, Sestan N, Anton ES. Neuregulin 1-erbB2 signaling is required for the establishment of radial glia and their transformation into astrocytes in cerebral cortex. Proc Natl Acad Sci USA. 2003;100:4251–4256. doi: 10.1073/pnas.0630496100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwander M, Shirasaki R, Pfaff SL, Muller U. β1 integrins in muscle, but not in motor neurons, are required for skeletal muscle innervation. J Neurosci. 2004;24:8181–8191. doi: 10.1523/JNEUROSCI.1345-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tissir F, Goffinet AM. Reelin and brain development. Nat Rev Neurosci. 2003;4:496–505. doi: 10.1038/nrn1113. [DOI] [PubMed] [Google Scholar]
- Trommsdorff M, Gotthardt M, Hiesberger T, Shelton J, Stockinger W, Nimpf J, Hammer RE, Richardson JA, Herz J. Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell. 1999;97:689–701. doi: 10.1016/s0092-8674(00)80782-5. [DOI] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schutz G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet. 1999;23:99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- Tsai LH, Gleeson JG. Nucleokinesis in neuronal migration. Neuron. 2005;46:383–388. doi: 10.1016/j.neuron.2005.04.013. [DOI] [PubMed] [Google Scholar]
- Weimer JM, Anton ES. Doubling up on microtubule stabilizers: synergistic functions of doublecortin-like kinase and doublecortin in the developing cerebral cortex. Neuron. 2006;49:3–4. doi: 10.1016/j.neuron.2005.12.016. [DOI] [PubMed] [Google Scholar]
- Weisenhorn DM, Prieto EW, Celio MR. Localization of calretinin in cells of layer I (Cajal-Retzius cells) of the developing cortex of the rat. Brain Res Dev Brain Res. 1994;82:293–297. doi: 10.1016/0165-3806(94)90171-6. [DOI] [PubMed] [Google Scholar]
- Wu SX, Goebbels S, Nakamura K, Kometani K, Minato N, Kaneko T, Nave KA, Tamamaki N. Pyramidal neurons of upper cortical layers generated by NEX-positive progenitor cells in the subventricular zone. Proc Natl Acad Sci USA. 2005;102:17172–17177. doi: 10.1073/pnas.0508560102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida M, Assimacopoulos S, Jones KR, Grove EA. Massive loss of Cajal-Retzius cells does not disrupt neocortical layer order. Development. 2006;133:537–545. doi: 10.1242/dev.02209. [DOI] [PubMed] [Google Scholar]