

Abstract
Pain hypersensitivity is a cardinal sign of tissue damage, but how molecules from peripheral tissues affect sensory neuron physiology is incompletely understood. Previous studies have shown that activin A increases after peripheral injury and is sufficient to induce acute nociceptive behavior and increase pain peptides in sensory ganglia. This study was designed to test the possibility that the enhanced nociceptive responsiveness associated with activin involved sensitization of transient receptor potential vanilloid I (TRPV1) in primary sensory neurons. Activin receptors were found widely distributed among adult sensory neurons, including those that also express the capsaicin receptor. Whole-cell patch-clamp recording from sensory neurons showed that activin acutely sensitized capsaicin responses and depended on activin receptor kinase activity. Pharmacological studies revealed that the activin sensitization of capsaicin responses required PKCε signaling, but not PI3K (phosphoinositide 3-kinase), ERK (extracellular signal-regulated protein kinase), PKA, PKCα/β, or Src. Furthermore, activin administration caused acute thermal hyperalgesia in wild-type mice, but not in TRPV1-null mice. These data suggest that activin signals through its own receptor, involves PKCε signaling to sensitize the TRPV1 channel, and contributes to acute thermal hyperalgesia.
Keywords: activin, capsaicin, nociceptor, TRPV1, hyperalgesia, pain
Introduction
A well recognized adjunct to tissue damage and repair is the somatosensory tenderness around a wound that triggers nocifensive behaviors (e.g., withdrawal) that serve to guard against additional damage during the healing process. Recent evidence suggests that activins, members of the transforming growth factor β (TGFβ) superfamily, play an important role in this response to tissue damage.
Activin ligands act primarily as homodimers of βAβA (activin A) or βBβB (activin B) binding to heteromeric transmembrane receptors, which signal through either canonical (e.g., Smad) or atypical [e.g., Erk (extracellular signal-regulated protein kinase)] pathways to affect cell function (Pangas and Woodruff, 2000). Recent attention has focused on the role of activins in tissue repair and wound healing. Skin damage, systemic inflammation, and arthropathies result in dramatic elevations in activin expression (Hubner et al., 1996, 1997; Munz et al., 1999a; Jones et al., 2000, 2004; Phillips et al., 2001; Cruise et al., 2004). Transgenic overexpression of activin A (Munz et al., 1999b) or of the activin-binding protein, follistatin (Wankell et al., 2001), in mice results in accelerated or retarded wound healing, respectively.
Recent evidence suggests that activin itself causes both acute and prolonged tactile allodynia in vivo (Xu et al., 2005). Activin has been shown to increase expression of calcitonin-gene related peptide (CGRP) (Ai et al., 1999; Cruise et al., 2004), a peptide marker of the major subset of peripheral nociceptors that may contribute to the prolonged phase of hyperalgesia (Urban et al., 1994). However, because sensitization of nociception by activin occurs within 60 min of administration, the underlying mechanisms of this acute response are unlikely to be transcriptional; rather, it is more likely that posttranslational events underlie the phenomenon. Increasing evidence has linked many inflammatory mediators elevated in both acute and chronic pain conditions to sensitization of the capsaicin receptor ion channel [transient receptor potential vanilloid I (TRPV1)] through a variety of posttranslational mechanisms (Premkumar and Ahern, 2000; Bhave et al., 2002, 2003; Hu et al., 2002; Numazaki et al., 2002; Mohapatra and Nau, 2003). For example, nerve growth factor (NGF), prostaglandins (PGE2), protons, bradykinin (BK), and serotonin (5-HT) are known to sensitize TRPV1 through both common and distinct signaling pathways (Lopshire and Nicol, 1998; Shu and Mendell, 2001; Bonnington and McNaughton, 2003; Sugiuar et al., 2004; Zhuang et al., 2004; Ahern et al., 2005; Zhang et al., 2005; Stein et al., 2006; Zhu and Oxford, 2007). Furthermore, TRPV1-null mice exhibit largely normal acute pain behaviors, but not enhanced pain behaviors in response to inflammatory challenges (Caterina et al., 2000; Davis et al., 2000).
We sought to examine the possibility that the enhanced pain responsiveness associated with direct activin administration or its elevation during injury might involve sensitization of TRPV1 in primary sensory neurons. Using electrophysiological, pharmacological, biochemical, and behavioral approaches, we have confirmed such a link and have identified the signal transduction events connecting activin receptor activation to enhanced TRPV1 activity.
Materials and Methods
Reagents
Activin A was purchased from R&D Systems (Minneapolis, MN) and reconstituted in sterile 0.1% BSA saline at 10 μg/ml as stock solution. The activin receptor-like kinase 4 (ALK4) inhibitor SB431542 was purchased from Tocris (Ellisville, MO) and dissolved in dimethylsulfoxide (DMSO) at 50 mm stock solution. The mitogen-activated protein (MAP) kinase kinase (MEK) inhibitors PD98059 and U0126, phosphoinositide 3-kinase (PI3K) inhibitor LY294002, c-Src tyrosine kinase inhibitor PP2, protein kinase A (PKA) inhibitor H89, protein kinase C (PKC) inhibitor bisindolylmaleimide I (BIM), PKC inhibitory peptide (PKC 19–31), PKCε translocation inhibitor peptide (EAVSLKPT) (PKCε TIP), negative control scrambled peptide (LSETKPAV) (PKCε NCP), and the PKCα/β inhibitor Gö6976 were all purchased from Calbiochem (San Diego, CA) and dissolved in DMSO or H2O as stock solutions. All stock solutions were aliquoted and stored at −80°C until use. ActRIB monoclonal antibody (ACVR1B, M09) was purchased from Abnova (Taipei City, Taiwan), ActRII monoclonal antibody was purchased from GeneTex (San Antonio, TX). ActRIB, ActRII, and ActRIIB polyclonal antibodies were generous gifts from Dr. Wylie Vale (Salk Institute, San Diego, CA). Primary goat polyclonal antibodies to activin receptors were also purchased from R&D Systems (RIA catalog #AF637; RIB catalog # AF222; RIIB catalog #AF339; RIIA catalog #AF340). PKCε polyclonal antibody was purchased from Assay Designs (Ann Arbor, MI). Rat TRPV1 C-terminal antibody was raised in a rabbit against amino acid residues 816–838 conjugated to keyhole limpet hemocyanin using a commercial service (Covance, Princeton, NJ). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and actin monoclonal antibodies were purchased from Millipore (Billerica, MA). All other reagents were purchased from Sigma (St. Louis, MO).
Adult dorsal root ganglion cell culture
Sprague Dawley rats (Harlan, Indianapolis, IN) were maintained at the Laboratory Animal Resource Center of Indiana University School of Medicine. Animals were anesthetized with isoflurane and then decapitated, which is consistent with the recommendations of the Panel on Euthanasia of the American Veterinary Medical Association. Dorsal root ganglion (DRG) neurons from young adult Sprague Dawley rats (150–200 g) were dissociated and cultured as described previously (Koplas et al., 1997; Shu and Mendell, 1999). Dissociated cells were plated on poly-d-lysine-coated glass coverslips and maintained in DMEM (Invitrogen, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS; Hyclone, Logan, UT) and 100 U/ml penicillin and 100 mg/ml streptomycin for 16–18 h at 37°C under 5% CO2.
Electrophysiology
Currents were recorded under voltage clamp by whole-cell patch recording methods. In all experiments, the holding potential was −60 mV. Electrodes (N51A borosilicate glass) exhibited resistances of 2–5 MΩ. The standard external solution (SES) contained (in mm; all from Sigma): 145 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, and 10 glucose, pH 7.3. The internal solution consisted of (in mm): 130 K-gluconate, 10 EGTA, 1 MgCl2, 1 CaCl2, 10 HEPES, and 2 Mg-ATP, pH 7.4. Data were collected using an Axopatch 200A patch-clamp amplifier, Digidata 1200 interface, and pClamp 7.0 software (Molecular Devices, Foster City, CA), filtered at 1 kHz, and sampled at 5 kHz.
Drug treatment
Two capsaicin challenges (50 nm, 40 s) were delivered to cells from a quartz capillary (250 μm inner diameter; Polymicro Technologies, Phoenix, AZ) with a 10 min interval between challenges. External solutions with 0.1% BSA saline solution (control), 10 ng/ml activin A (experiment), or 10 ng/ml activin A plus various pharmacological reagents (interference) were superfused during the 10 min interval. For experiments assessing pharmacological interference with the activin effect, a 10 min pretreatment with the relevant inhibiting reagent preceded recording.
Immunocytochemistry
Isolated DRG neurons
DRG neurons were cultured overnight, rinsed with HBSS, and fixed with 4% paraformaldehyde for 20 min at room temperature. After three PBS rinses, neurons were permeabilized with 0.1% Triton X-100 plus 5% goat serum in PBS for 30 min at 37°C. Primary antibody (1:5000 polyclonal TRPV1; 1:200 monoclonal ActRIB or ActRII antibodies; or 1:1000 to 1:500 polyclonal ActRIB, ActRII, and ActRIIB) were applied overnight at 4°C. After washing, secondary antibody (1:20,000 goat anti-rabbit Alexa 594 or 1:2000 goat anti-mouse Alexa 488) was applied for 1.5 h at 37°C, and cells were rinsed well and mounted in 4′,6′-diamidino-2-phenylindole (DAPI)-containing Vectashield.
Adult DRG sections
Adult rats were perfused with 4% paraformaldehyde/0.1 m P04, pH 7.4, and lumbar DRGs were dissected and postfixed for 1 h at room temperature. Ganglia were transferred to 30% sucrose at 4°C overnight to cryoprotect the tissue. After embedding in OCT (optimal cutting temperature medium), 10 μm sections were collected on gelatin-coated slides. After washing in PBS, slides were incubated in dilution buffer for 1 h containing Triton X-100 as above. Primary goat polyclonal antibodies to activin receptors obtained from R&D Systems (RIA catalog #AF637; RIB catalog # AF222; RIIB catalog # AF339; RIIA catalog #AF340) were used at 5–10 μg/ml in dilution buffer and applied to slides overnight in a humid chamber. After washes, slides were incubated in biotinylated donkey anti-goat IgG at 1:300 in dilution buffer for 2 h at room temperature, washed, and developed with a diaminobenzidine substrate after avidin–biotin amplification (Vectastain kit). No reactivity was observed in control studies in which primary antibody was omitted.
PKCε translocation
DRG neurons were cultured overnight and stimulated by vehicle, 300 nm phorbol-12-myristate-13-acetate (PMA) for 120 s (positive control), 1 μm BK for 30 s, or 10 ng/ml activin A for 30, 300, or 600 s. In some cases, neurons were pretreated with the activin receptor ActRIB antagonist SB431542 (20 μm) for 10 min. Neurons on coverslips were fixed with 4% paraformaldehyde (w/v) plus 4% sucrose (w/v) in 50% PBS (Invitrogen) for 20 min at 37°C. After rinses in PBS containing 0.2% fish skin gelatin (w/v; Sigma) (PBS/FSG), neurons were permeabilized with 0.1% Triton X-100 in PBS for 30 min at 37°C. Fixed neurons were then exposed to primary polyclonal anti-PKCε (Assay Designs) overnight at 4°C before three times rinse with PBS/FSG and labeling with 1:20,000 goat anti-rabbit Alexa 594 (Invitrogen) secondary antibody. Vectashield containing DAPI (Vector Laboratories, Burlingame, CA) was used as mounting medium.
Reverse transcriptase-PCR
Total RNA was extracted using RNAEasy kits from Qiagen (Valencia, CA) according to the manufacturer's instructions. The resulting RNA samples were digested with RNase-free DNase at 37°C for 1 h to remove the trace genomic DNA contamination and followed by phenol-chloroform extraction and alcohol precipitation. The quality and integrity of resulting RNA samples were checked by gel electrophoresis and spectrophotometry. Two micrograms of RNA were denatured with 1 μm oligo dT (dT 15) at 70°C for 5 min, followed by reaction with MMLV reverse transcriptase at 42°C for 1 h to synthesize cDNA. cDNA samples (1 μl) were used as template for the PCR by the following protocol: 94°C for 5 min, then 35 cycles of 94°C for 30 s, 60°C for 30 s, and 72°C for 1 min, followed by 72°C for 7 min. The primers for amplification were as follows: ActRI (sense), 5′-TCTGTGCTAATGATGATGGCTCTCC-3′; ActRI (antisense), 5′-TTCTGCGATCCAGGGAAGGATTTC-3′; ActRIB (sense), 5′-TTGAGGAAATGCGAAAGGTCG-3′; ActRIB (antisense), 5′-GGGACAAAGTCTTCTTGATGC-3′; ActRII (sense), 5′-CCATCTCTTGAAGATATGCAGGA-3′; ActRII (antisense), 5′-GTGACCACTGTTACAATGTCCTC-3′; ActRIIB (sense), 5′-CTTTGTGGCTGTGAAGATCTTC-3′; and ActRIIB (antisense), 5′-CATTCTTGCTTTTGAACTCCCTG-3′ (Ai. et al., 1999). The resulting PCR products were separated in a 2% agarose gel.
Western blotting
Thirty thousand neurons in each well of a 12-well dish were maintained overnight in growth medium described above. After rinsing once with PBS, new medium without FBS was added to the neurons for 4 h, followed by control vehicle, NGF (100 ng/ml), or activin A (1.0, 2.5, 5.0, or 10.0 ng/ml) for 10 min. After two rinses with PBS, neurons were lysed with buffer (30 mm Tris, pH 7.4, 150 mm NaCl, 1% Triton X-100, 0.1% SDS, 1 mm PMSF, 10 mm EDTA, 1 mm Na2VO3, 160 mm NaF). The protein concentrations of the lysates were determined using a BCA Protein Assay Kit (Pierce, Rockford, IL). Protein (30–50 μg) from DRG neurons was separated by approximately 6–10% SDS-PAGE and transferred to PVDF membrane. After a 30 min incubation with blocking buffer (Licor, Lincoln, NE), membranes were incubated with primary antibody (1:1000 for rabbit ERK, pERK, Akt, pAkt; 1:500 for rabbit TRPV1 C-terminal antibody; 1:2000 for mouse GAPDH or actin antibody; 1:200 for mouse anti-ActRIB or -ActRII antibody; 1:1000 for rabbit anti-ActRIB, -ActRII, and -ActRIIB antibody) for 1.5–2 h at room temperature. After three TBS-T washes, the membranes were incubated with 1:20,000 goat anti-rabbit antibody conjugated with AlexaFluor 680 or 1:10,000 goat anti-mouse antibody conjugated with infrared dye 800 (Invitrogen) plus 0.5% SDS for 1.5 h at room temperature. After three TBS-T washes, the membranes were scanned by Odyssey (Licor) at channel 700 or 800.
Behavioral assays of thermal hyperalgesia and tactile allodynia in wild-type or TRPV1-null mice
C57BL/6J and TRPV1-null mice (B6.129X1-Trpv1tm1Jul/J, backcrossed to C57BL/6J for 10 generations; The Jackson Laboratory, Bar Harbor, ME) 8 weeks of age were used for behavioral studies, and animal protocols were approved by the Institutional Animal Care and Use Committee at Case Western Reserve University School of Medicine. Mice were habituated to plastic chambers for 1 h per time, twice a day and 3 continuous days. Before each test, the mice were habituated for 1 h. In all experiments, the experimenter was blinded to the genotypes of the mice being analyzed. Mice were anesthetized with 3% isoflurane inhalation anesthetic, both legs were shaved, and 5 μl of saline or 250 ng/5 μl of carrier-free activin A (R&D Systems) was injected to the lateral ankle skin with a 26 gauge needle connected to a 25 μl Hamilton syringe. Basal nociceptive responses were collected at 1, 24, and 48 h. Thermal hyperalgesia was detected by a Hargreaves test (Hargreaves et al., 1988) (Ugo Basile plantar analgesia instrument; Ugo Basile, Comerio, Italy) at 1, 24, and 48 h. A mobile radiant heat source was located under the glass table and focused onto the plantar area of the hindpaw. Paw withdrawal latencies (PWLs) were recorded automatically. The intensity of the radiant heat gave a basal latency of 4–5 s on an intact normal mouse. Each trial was repeated five times at 4 min intervals, and the PWL is the average of the five responses. Responses to mechanical stimulation in mice were assessed using calibrated von Frey filaments using the up–down paradigm as previously performed for assessment of tactile allodynia in rats (Xu et al., 2005).
Data analysis
A one-way repeated-measure ANOVA was used to analyze the majority of data. When p < 0.05 a Newman–Keul's post hoc test was also used to make individual comparisons between groups. A Student's t test was also used for data analysis when only two conditions are being compared and significance set at p < 0.05.
Results
Activin receptors are expressed in intact and cultured adult rat DRG neurons
Like other members of the TGFβ superfamily, activin initiates signaling and cellular responses through forming a heteromeric complex of two types of transmembrane receptor serine/threonine kinases classified as type II (ActRII or ActRIIB) and type I (ActRI/ALK2 or ActRIB/ALK4) (Pangas and Woodruff, 2000). Although originally thought to be an activin receptor, ActRI/ALK2 has more recently been classified as a receptor for bone morphogenetic proteins (BMPs) and does not bind activin (Macias-Silva et al., 1998). We evaluated the expression of these activin receptors in cultured adult rat DRG neurons. Reverse transcriptase-PCR revealed that messenger RNA for ActRI (BMP receptor), ActRIB, ActRII, and ActRIIB were all expressed in adult rat DRG neurons (Fig. 1A), consistent with other reports (Ai et al., 1999; Kos et al., 2001), although expression of ActRIIB was relatively low. Similarly, Western blotting revealed an ∼52 kDa ActRIB immunoreactive band, an ∼70 kDa immunoreactive band for unglycosylated ActRII, and two 85–90 kDa glycosylated ActRII bands as well as an ∼70 kDa immunoreactive band for ActRIIB in the rat DRG neuronal culture (Fig. 1B), which is consistent with other reports (Mathews and Vale, 1993; Funaba et al., 2001). Furthermore, ActRIB, ActRII, and ActRIIB immunoreactivities were detected in all cultured DRG neurons (Fig. 2B,C), including TRPV1-expressing small or medium size DRG neurons (Fig. 2C). Indeed, TRPV1-expressing neurons comprised ∼51.8 ± 1.2% (n = 8 coverslips, 2327 neurons counted) of the neurons and represented mainly small- or medium-diameter neurons (20–30 μm), which is consistent with other reports (Guo et al., 1999). Furthermore, consistent with this pattern, activin receptors were detected in virtually all neurons by immunocytochemistry in thin sections of adult DRG (Fig. 2A), suggesting that dissociation and short-term culture of neurons did not artifactually induce receptor expression. Thus, activin receptors were broadly expressed in adult DRG neurons.
Figure 1.
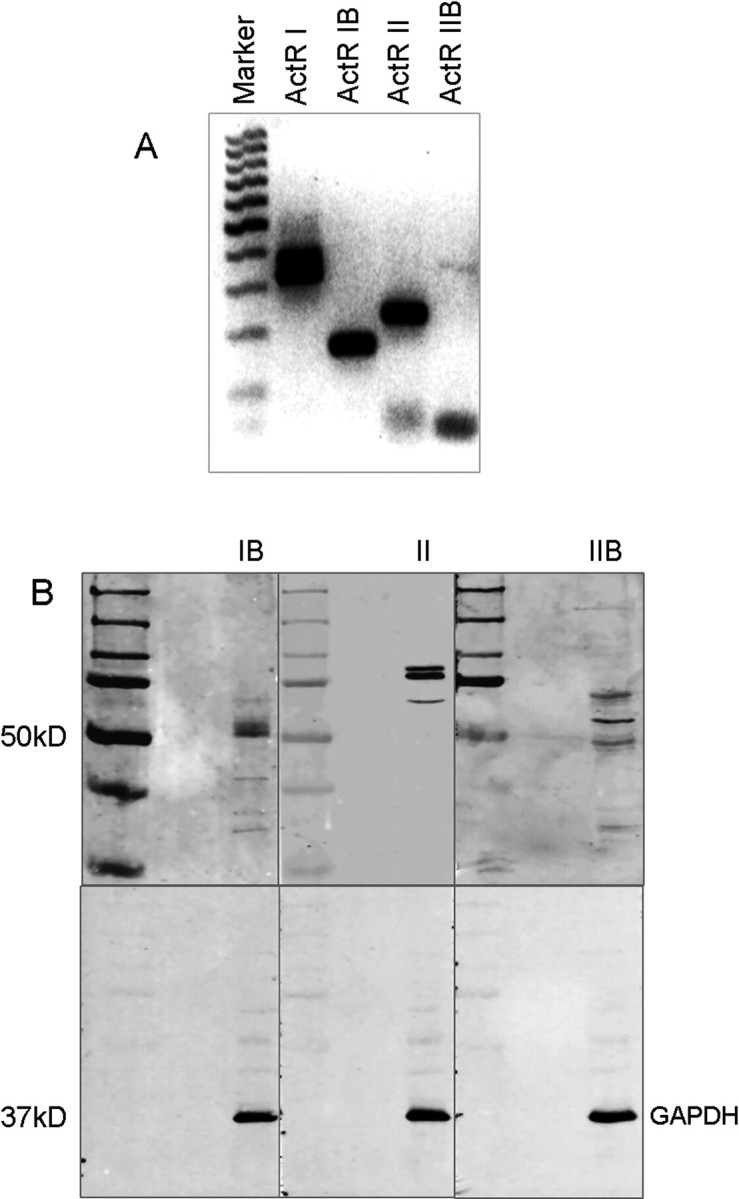
Expression of activin receptor mRNA and protein in adult rat DRG neurons. A, Reverse transcriptase-PCR revealed strong expression of mRNAs for ActRI, ActRIB, and ActRII and weak expression of ActRIIB mRNA in the rat DRG neurons. B, ActRIB, ActRII, and ActRIIB immunoreactivity were detected by Western blotting of proteins from cultured rat DRG neurons. Western blotting revealed an ∼52 kDa ActRIB immunoreactive band, an ∼70 kDa immunoreactive band for unglycosylated ActRII, and 2 85–90 kDa bands for glycosylated ActRII as well as an ∼70 kDa immunoreactive band for ActRIIB.
Figure 2.
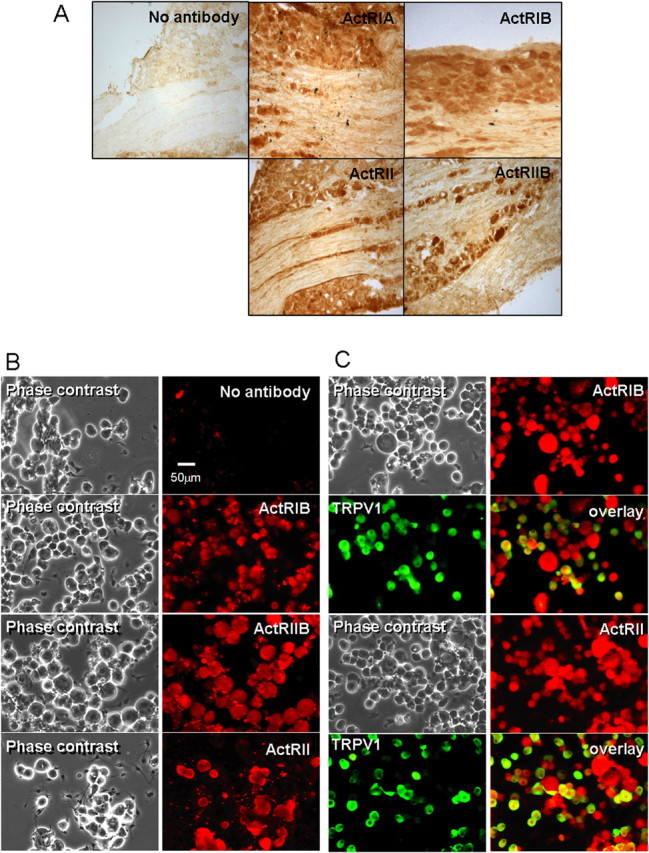
Activin receptors are expressed in intact as well as cultured DRG neurons. A, Immunohistochemical detection of ActRI, ActRIB, ActRII, and ActRIIB in sections of adult DRG as indicated by brown DAB reaction product. It is apparent that receptors are expressed in most neurons and nerve fibers within the ganglion. No reactivity was observed in sections in which primary antibody was omitted. B, C, Phase and pseudocolor fluorescence images of cultured DRG neurons. Primary antibody conditions are labeled on each fluorescence panel. ActRIB, ActRII, and ActRIIB immunoreactivity was detected by immunocytochemistry in all identified neurons, including TRPV1-expressing small and medium-size neurons.
Activin A acutely sensitizes capsaicin currents in cultured DRG neurons
Whole-cell currents were recorded from cultured DRG neurons under voltage-clamp conditions (−60 mV). Each of two 50 nm capsaicin administrations separated by a 10 min exposure to SES containing 0.1% BSA induced inward currents, the second being consistently smaller than the first because of calcium-dependent desensitization (Docherty et al., 1996; Koplas et al., 1997; Shu and Mendell, 1999, 2001; Zhu et al., 2004; Zhuang et al., 2004; Zhu and Oxford, 2007) (Fig. 3A). In contrast, when 10 ng/ml activin A was perfused during the 10 min interval, the second current response was augmented (Fig. 3A). Although stable longer recordings in individual neurons are a challenge, we have occasionally observed this sensitization to last for >20 min. Activin A increased the ratio of the second capsaicin response to the first response by 8.44 ± 1.90-fold (n = 49), whereas the ratio declined to 0.51 ± 0.06 (n = 18) for the vehicle control (Fig. 3B) (p < 0.05). Pretreatment with ALK4 inhibitor SB431542 (20 μm) (Inman et al., 2002) for 10 min plus exposure to both activin A and SB431542 during the interval between capsaicin challenges prevented the augmentation (Fig. 3A). Under this condition, the second-to-first response ratio was not different from that observed in vehicle controls (0.43 ± 0.09; n = 13) (Fig. 3B), indicating that activin A sensitizes capsaicin current through its receptor ALK4.
Figure 3.
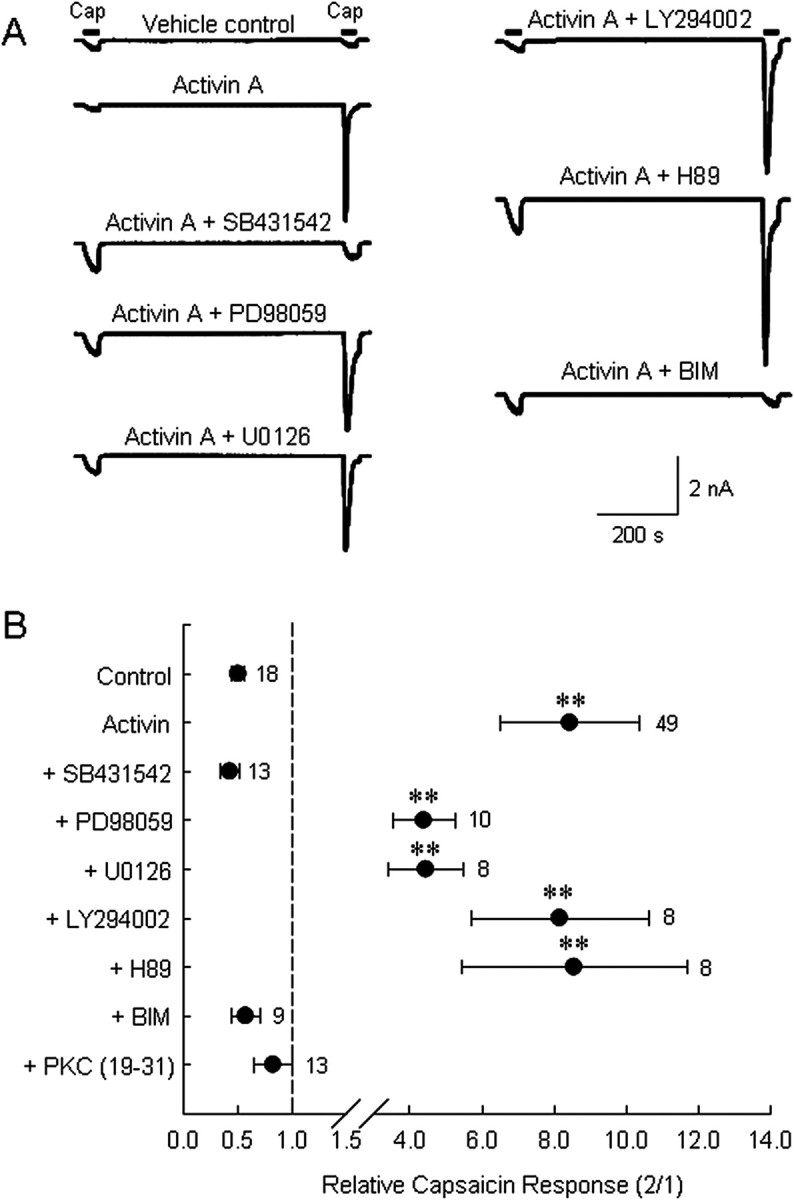
Activin A acutely sensitizes the capsaicin-induced current in the cultured DRG neurons through the activin type I receptor ALK4 and PKC activation. A, Representative current traces from individual DRG neurons challenged by two 50 nm capsaicin (Cap) administrations separated by a 10 min treatment with vehicle, 10 ng/ml activin A, or 10 ng/ml activin plus a test reagent (additional 10 min pretreatment). The reagents tested were 20 μm SB431542 (ALK4 inhibitor), 10 μm PD98059 (MEK inhibitor), 5 μm U0126 (MEK inhibitor), 10 μm LY294002 (PI3K inhibitor), 5 μm H89 (PKA inhibitor), or 1 μm BIM (PKC inhibitor). B, Summary data (mean ± SEM) of normalized capsaicin responses (second peak current/first peak current; note change in scale after axis break) indicate that activin acts to sensitize capsaicin currents in DRG neurons through ALK4 and involves protein kinase C, but not Erk1/2, PI3K, or PKA. Additional data represent tests of activin-induced sensitization with the pseudosubstrate peptide inhibitor of PKC included in the recording pipette solution (2 μm). **p < 0.01 compared with control values. Number of observations are indicated next to each data point.
Several major kinase signaling pathways do not mediate activin A-induced TRPV1 sensitization
Because activin A causes potentiation of capsaicin currents within minutes, it is unlikely that transcriptional effects through Smad signaling are involved. In this regard, several Smad-independent signaling pathways can be initiated by activin (Moustakas and Heldin, 2005). Activation of ERK, PI3K, src tyrosine kinase, PKA, and PKC have been implicated in the acute sensitization of TRPV1 by a variety of agents, including NGF, BK, and 5-HT (Lopshire and Nicol, 1998; Premkumar and Ahern, 2000; Bhave et al., 2002, 2003; Hu et al., 2002; Bonnington and McNaughton, 2003; Mohapatra and Nau, 2003; Sugiuar et al., 2004; Zhuang et al., 2004; Zhang et al., 2005; Stein et al., 2006; Zhu and Oxford, 2007). Pharmacological interference was used to test the involvement of each of these pathways in activin-induced TRPV1 potentiation.
We first examined the role of ERK and PI3K activation. After a 10 min pretreatment of neurons with the specific MEK inhibitors PD98059 (10 μm) (Pang et al., 1995) or U0126 (5 μm) (Favata et al., 1998), two capsaicin responses were again elicited separated by a 10 min exposure to 10 ng/ml activin A either with or without PD98059 or U0126. Neither PD98059 nor U0126 treatment significantly changed the activin-induced potentiation of capsaicin current (Fig. 3A). The second-to-first response ratio was 4.39 ± 0.87 (n = 10) for the PD98059 treatment group and 4.45 ± 1.03 (n = 8) for the U0126 treatment group (Fig. 3B), neither of which is significantly different from the activin A-only group. This result suggests that ERK signaling cascades are unlikely to be involved in acute sensitization of capsaicin current by activin.
To test the involvement of the PI3K signaling pathway in activin-induced TRPV1 sensitization, neurons were treated before (10 min) and during capsaicin challenges with the specific PI3K inhibitor LY294002 (10 μm) (Vlahos et al., 1994). As seen for ERK inhibitors, PI3K inhibition did not prevent activin-induced capsaicin current potentiation (Fig. 3A). The average second-to-first response ratio for the LY294002 treatment group (Fig. 3B) was 8.16 ± 2.47 (n = 8), which was not significantly different from the activin A-only group, 8.83 ± 2.03 (n = 14). We conclude that PI3K signaling is not involved in activin enhancement of capsaicin responses.
We also examined whether activin A administration led to acute increases in phosphorylation of ERK or Akt in DRG neurons as indicators of stimulation of ERK and PI3K pathways, respectively. Previously, we have shown that NGF, which potentiates TRPV1 responsiveness, also augments p-ERK and p-Akt in cultured DRG neurons within 10 min (Zhu and Oxford, 2007). Therefore, different doses of activin and 100 ng/ml NGF (a positive control) were applied to cultured DRG neurons for 10 min, and p-ERK and p-Akt levels were examined by Western blotting. Whereas NGF significantly increased p-ERK and p-Akt as expected, activin failed to augment either p-ERK or p-Akt stimulation (Fig. 4A,B). Together, these data suggest that MAP kinase and PI3 kinase activities are not required to mediate activin-induced TRPV1 sensitization.
Figure 4.

Activin A does not promote phosphorylation of ERK or Akt in cultured DRG neurons. A, Representative Western blots of p-ERK and p-Akt production in rat DRG neurons after 10 min of exposure to control vehicle, 1.0, 2.5, 5.0, and 10 ng/ml activin A (A) as well as 100 ng/ml NGF (N), respectively. B, Normalized density of bands [(phospho-protein/corresponding total protein)/internal control actin] for the indicated treatments. Each bar represents mean ± SEM of six (Akt) or seven (ERK) density measurements. *p < 0.01 and **p < 0.0001 compared with respective controls.
Inhibition of c-src kinase does not abolish activin A-induced TRPV1 sensitization
Recently, phosphorylation of a specific tyrosine residue on TRPV1 by c-src kinase has been proposed to be important in NGF-induced sensitization by promoting insertion of TRPV1 channels into the plasma membrane (Zhang et al., 2005). We therefore examined a possible role for src kinase in activin-induced TRPV1 acute sensitization. A c-src kinase-specific inhibitor PP2 (10 μm) (Hanke et al., 1996) was added to the recording pipette during whole-cell recording and capsaicin challenges begun only after 10 min of intracellular access of the inhibitor. Under these conditions, the activin-induced TRPV1 current sensitization was not significantly different from control recordings without PP2. The second-to-first capsaicin response ratio was 0.54 ± 0.09 (n = 8) for the control group, increasing to 3.07 ± 0.46 (n = 11) for neurons exposed to activin A, and 2.52 ± 0.33 (n = 15) for those neurons exposed to activin A and internally dialyzed with PP2 (Fig. 5). This result suggests that c-src kinase is unlikely to play a role in activin-induced acute TRPV1 sensitization.
Figure 5.

Inhibition of c-src tyrosine kinase does not prevent activin A sensitization of TRPV1. Bars represent mean ± SEM of normalized capsaicin responses for the indicated conditions of activin A treatment (10 ng/ml) and PP2 treatment (10 μm). **p < 0.05 compared with control; #p > 0.05 compared with activin A only.
Inhibition of PKC, but not PKA, abrogates activin A-induced potentiation of TRPV1 in DRG neurons
TRPV1 sensitivity is known to be enhanced by phosphorylation of specific amino acid residues by both PKA and PKC (Lopshire and Nicol, 1998; Premkumar and Ahern, 2000; Bhave et al., 2002, 2003; Hu et al., 2002; Numazaki et al., 2002; Mohapatra and Nau, 2003). We thus sought to determine whether PKA or PKC might be involved in activin-induced sensitization of TRPV1. Using the same experimental protocol, the PKA inhibitor H89 (5 μm) (Chijiwa et al., 1990) or the PKC inhibitor BIM (1 μm) (Wilkinson et al., 1993) was applied to single neurons (10 min pretreatment and 10 min coperfusion with 10 ng/ml activin A) challenged by capsaicin. The PKA inhibitor H89 was without impact on acute potentiation of capsaicin current induced by activin A (Fig. 3A). The second-to-first capsaicin response ratio was 8.56 ± 3.12 (n = 8) for the H89 treatment group, similar to the activin A-only group (Fig. 3B). In contrast, pretreatment and coperfusion of 1 μm BIM with 10 ng/ml activin A completely abolished the sensitizing effect of activin A on capsaicin current (Fig. 3A) with a response ratio of only 0.58 ± 0.13 (n = 9), which is significantly different from the activin A-only group (p < 0.01), but not the vehicle control group (p > 0.5) (Fig. 3B). This result was confirmed in other neurons by adding 2 μm of the PKC pseudosubstrate inhibitory peptide (PKC 19–31) to the recording pipette before the experiment. Under these conditions the second-to-first response ratio declined to 0.83 ± 0.18 (n = 13), which is significantly different from the activin A group (p < 0.01) but not from vehicle controls (p > 0.1) (Fig. 3B). These results suggest that PKC, but not PKA, is involved in sensitization of TRPV1 by activin.
Activin A sensitizes TRPV1 through activation of the PKCε isoform
Several PKC isoforms (βI, βII, δ, ε, and ζ) are present in rat DRG neurons (Cesare et al., 1999) that might mediate TRPV sensitization by activin. Activation of several of these isoforms requires their translocation from the cytosol to the plasma membrane for interaction with membrane phospholipids. In particular, activation of PKCε translocation can be triggered by PMA and is believed to underlie the sensitization of TRPV1 by bradykinin (Cesare and McNaughton, 1996; Cesare et al., 1999). To examine the possible involvement of such translocation in activin sensitization of TRPV1, PKCε translocation was assayed by confocal microscopy in adult DRG neurons. Under vehicle control conditions, immunoreactivity of PKCε was evenly distributed in the cytoplasm of adult rat DRG neurons (Fig. 6A). Application of the potent PKC activator PMA (300 nm, 2 min) caused a strong membrane association of PKCε immunoreactivity in almost all of DRG neurons (Fig. 6A,B). Bradykinin (1 μm, 30 s) also caused translocation of PKCε to the plasma membrane in 39.3 ± 2.5% (n = 3 coverslips) of DRG neurons (Fig. 6A,B), which is quantitatively consistent with previous observations (Cesare et al., 1999). Activin A (10 ng/ml) also caused rapid translocation of PKCε immunoreactivity to the plasma membrane in most DRG neurons (Fig. 6A,B). The translocation of PKCε immunoreactivity to the plasma membrane increased during prolonged activin A exposure, as evidenced by the progressive increase in the number of neurons exhibiting such localization (Fig. 6B). After a 30 min exposure to 10 ng/ml activin A, 85.7 ± 1.1% of neurons exhibited PKCε membrane localization. Interestingly, at later exposure times, PKCε immunoreactivity showed an uneven punctate membrane distribution, and after 60 min of exposure, a distribution similar to control conditions was observed in nearly all cells. The translocation of PKCε to the plasma membrane by activin A exposure was mediated through the activin A receptor complex, because pretreatment with the ALK4-specific inhibitor SB431542 dramatically decreased this response (Fig. 6B).
Figure 6.

Activin A causes translocation of PKCε to the cell membrane of rat DRG neurons. A, Distribution of PKCε immunoreactivity of rat DRG neurons after different treatment. Top, Confocal slice images of PKCε immunoreactivity under the indicated conditions. Bottom, Corresponding fluorescence intensity profile along a 40 μm line crossing each neuron. A uniform distribution of PKCε immunoreactivity was seen in the cytoplasm under control conditions, whereas a prominent relocation to the plasma membrane was seen for the conditions of PMA (300 nm, 2 min), BK (1 μm, 30 s), and activin (10 ng/ml, 30 s and 30 min). B, Time course of PKCε membrane translocation quantified as the percentage of neurons showing positive translocation. A positive neuron was taken as any neuron where the peak (membrane) fluorescence was ≥120% of the mean of 8 pixels in the center of the scan. At control time (no activin exposure), ∼3% of neurons showed PKCε membrane translocation, whereas at 30 s of activin exposure, ∼37% of neurons demonstrated PKCε membrane translocation, which peaked at ∼85% after 30 min. SB 431542 dramatically decreased PKCε membrane translocation. The potent PKC activator PMA (300 nm, 2 min) caused ∼91% of neurons to demonstrate PKCε membrane translocation, whereas BK (1 μm, 30 s) treatment triggered PKCε membrane translocation in ∼39% of neurons. Numbers of neurons measured are indicated for each data point.
To determine whether translocation and activation of the PKCε isoform contributed functionally and specifically to TRPV1 sensitization, we attempted to interfere with this process and assess activin A-induced sensitization. We first examined the effect of a PKCε translocation inhibitor peptide (PKCε TIP) (Yedovitzky et al., 1997) on activin-induced TRPV1 sensitization in individual DRG neurons. Addition of 200 μm PKCε TIP to the recording pipette completely abolished the activin A-induced TRPV1 sensitization, whereas the addition of 200 μm of a scrambled control peptide did not block TRPV1 sensitization by activin A. The second-to-first capsaicin response ratio in activin A with PKCε TIP in the pipette was 0.71 ± 0.17 (n = 12), which is significantly different from the activin A-only group (8.44 ± 1.90, n = 49, p < 0.0001) or the activin A plus scrambled control peptide group (9.05 ± 2.82, n = 14, p < 0.01) (Fig. 7). This result strongly suggests that activin-induced sensitization of TRPV1 requires activation of PKCε.
Figure 7.

Inhibition of PKCε translocation, but not PKCα activation, blocks activin A-induced TRPV1 sensitization. PKCε TIP (200 μm) added into recording pipette inhibited TRPV1 sensitization by activin, whereas PKCε NCP had no effect. Preincubation and coincubation of neurons with the PKCα/β inhibitor Gö6976 (10 nm) neither blocked activin action nor mimicked it in the absence of activin. Symbols represent mean ± SEM of the second-to-first capsaicin response ratio. **p < 0.01 relative to control; *p < 0.05 relative to activin. Numbers of observations for each condition are indicated next to each symbol.
To rule out the possibility that PKCα or PKCβ isoforms, previously implicated in sensitization of nociceptive neurons, might contribute to sensitization of TRPV1 by activin A, we tested whether Gö6976, a PKCα/β-selective inhibitor (Martiny-Baron et al., 1993), would alter this response. Pretreatment and cotreatment of neurons with 10–100 nm Gö6976 neither sensitized TRPV1 itself nor blocked the sensitization of capsaicin-induced currents by activin A (Fig. 7) suggesting that PKCα and PKCβ isoforms do not mediate the activin A response.
Activin induced thermal hyperalgesia in mice requires the TRPV1 channel
Because our electrophysiological studies reveal a strong sensitization of TRPV1 by activin and TRPV1 has been implicated by many laboratories as a critical molecule for the expression of acute inflammatory hyperalgesia, we hypothesized that TRPV1 may be required for hyperalgesia associated with elevated levels of activin. To test this hypothesis, the effect of activin administration on thermal pain responses was evaluated in TRPV1-null and wild-type (WT) mice. Activin or saline was injected subcutaneously into one ankle, and thermal hyperalgesia was assayed over time in operator-blinded studies. WT mice injected with saline demonstrated thermal pain-induced paw withdrawal within ∼5 s. Activin administration in WT mice produced more rapid paw withdrawal, as expected, because TRPV1 channels were intact in these animals. This behavioral thermal hyperalgesia induced by activin was significant when assessed 1 h after activin injection (average withdrawal latency = 2.9 ± 0.3 s), but was absent 24 and 48 h after injection (Fig. 8A). In TRPV1−/− mice, withdrawal latencies under control saline injections were greater than in WT mice, consistent with previous observations (Keeble et al., 2005; Pogatzki-Zahn et al., 2005). In contrast to WT mice, activin injection into TRPV1−/− null mice produced withdrawal latencies comparable with saline injection, and these latencies were not different from saline injections at any time examined. These studies indicate that TRPV1 is required for the induction of thermal hyperalgesia by activin. Furthermore, they indicate that in mice after an acute elevation of activin, enhanced thermal sensitivity is not maintained.
Figure 8.

Activin injection induces thermal hyperalgesia in wild-type but not TRPV1-null mice. A, Activin (250 ng/5 μl) injection induces thermal hyperalgesia in wild-type mice (gray bars, n = 19) but not TRPV1-null mice (black bars, n = 16). Thermal hyperalgesia was measured by paw withdrawal latency (in seconds) with plantar heating at 1–48 h after injection. Data groupings represent assessments done at the indicated times after activin injection. Wild-type mice show 4–5 s withdrawal latency in the absence of activin (white bars), whereas TRPV1-null mice exhibit longer latencies (hatched bar, n = 8). *p < 0.05 relative to wild-type responses in activin. B, Activin (250 ng/5 μl) injection induces mild tactile allodynia at 1 h in wild-type mice (gray bar, n = 8) but not TRPV1-null mice (black bar, n = 8). Wild-type saline injected mice show 1 g withdrawal threshold (white bar, n = 4). *p < 0.05 denotes the significance level.
Because our previous observations in rats indicated that activin induces tactile allodynia (Xu et al., 2005), we examined this possibility in mice. Although not statistically significant, there was a slight enhancement of the withdrawal reflex evoked by mechanical stimuli in wild-type mice 1 h after an ankle injection with activin (Fig. 8B), whereas the withdrawal was significantly blunted 1 h after activin injection in TRPV1−/− mice.
Discussion
Effective repair of damage associated with wounds, inflammation, or other injuries involves at least three important processes. The first is to heighten pain sensation to promote nocifensive behaviors that avoid further damage. The second is the delivery of sufficient nutrients to the damage site through increased circulation. The third is promoting reconstruction through cell proliferation, migration, and differentiation. To orchestrate these processes, a variety of proinflammatory factors, including cytokines, bradykinin, NGF, and PGE2, are released or upregulated around the site of damage, including the dermis. Among these factors, activin, a proinflammatory cytokine, is also elevated and plays an important role in wound repair, fibrosis, and inflammation (Werner and Alzheimer, 2006). In recent years, research into the role of activin in wound repair has focused on the last two processes: facilitating the repair process and modulating scar formation (Munz et al., 1999b; Wankell et al., 2001), and enhancing CGRP expression in the innervating DRG neurons and spinal cord (Cruise et al., 2004; Xu et al., 2005). CGRP, a strong vasodilator, is released in the periphery, where it enhances blood flow in support of repair. Although the role of activin in enhancing pain sensation has not been as intensively studied, several recent observations support its involvement. Activin injection directly into the skin of a rat produces tactile allodynia (Xu et al., 2005) and increases the neuronal expression of CGRP (Cruise et al., 2004), a peptide implicated in nociception as well as vasodilation. In the present study, we have extended these observations to reveal a fundamental mechanism underlying activin-induced hyperalgesia supported by three novel findings: (1) activin A potently sensitizes TRPV1 through activation of PKCε in adult DRG neurons, (2) activin A activates PKCε through its type I receptor serine/threonine kinase ALK4, and (3) thermal hyperalgesia induced by activin A in vivo requires TRPV1.
Activin sensitizes TRPV1 to mediate acute thermal hyperalgesia. TRPV1 is a multimodal transducer of noxious stimuli, including chemicals such as capsaicin and protons and physical changes such as heat. These responses were initially taken as evidence for a role of TRPV1 in normal nociception (Tominaga et al., 1998). Recently, however, evidence from physiological and behavioral assessments of TRPV1-null mice has indicated that TRPV1 is not required for detecting noxious heat (Woodbury et al., 2004), but plays a significant role in pathological nociception (e.g., hyperalgesia) associated with tissue injury and inflammation (Caterina et al., 2000; Davis et al., 2000; Bolcskei et al., 2005; Pogatzki-Zahn et al., 2005). This is believed to occur through posttranslational changes in the sensitivity and membrane density of TRPV1 as well as transcriptional changes in TRPV1 expression in response to a variety of molecules released during injury or inflammation including NGF, bradykinin, and PGE2 (Lopshire and Nicol, 1998; Shu and Mendell, 2001; Bonnington and McNaughton, 2003; Sugiuar et al., 2004; Zhuang et al., 2004; Zhang et al., 2005; Stein et al., 2006; Zhu and Oxford, 2007). We have now shown that activin can also sensitize TRPV1 within minutes of its application, consistent with its ability to induce hyperalgesia after injection into the skin. Furthermore, hyperalgesia in response to activin injection is absent in TRPV1-null mice, suggesting that sensitization of TRPV1 is both necessary and sufficient for activin-induced acute pain behavior.
Activin A binds to either the constitutively active Ser/Thr kinase activin type II receptor (ActRII) or to one of four isoforms of activin type IIB receptor (ActRIIB) to mediate its biological effects. This binding recruits and transphosphorylates an activin type I receptor (ALK4), which enables subsequent phosphorylation of downstream substrates (Massague, 1998; de Caestecker, 2004). Activin receptor subunits are present in adult DRG neurons at both the mRNA and protein levels (Fig. 1A,B), and both type I and type II receptors are coexpressed with TRPV1 (Fig. 2C). Activin receptors have been reported in embryonic sensory neurons (Ai et al., 1999; Kos et al., 2001), where they are likely to play a role in phenotypic differentiation (Iwahori et al., 1997). In adult DRG neurons, activin sensitization of TRPV1 is blocked by a selective ActRI (ALK4) receptor antagonist (Fig. 3), suggesting that this receptor kinase is required for activin-induced sensitization of TRPV1 in DRG neurons.
Both canonical and noncanonical signaling pathways are activated by these receptors. Canonical signaling involves activation of the Smad family of transcription factors by phosphorylation and translocation to the nucleus to activate target gene transcription. More recently, several noncanonical pathways, including activation of ERK, JNK, and p38 MAP kinases, PI3K, PKC, and PKA have been reported in different cell types (Moustakas and Heldin, 2005). These signals may be required to mediate and/or augment maximal Smad-dependent responses, or they may act on distinct downstream effectors in different cell types. The precise mechanisms bridging these noncanonical signaling pathways with the activated receptor complexes are not fully understood. Given that both the activin-induced hyperalgesia and TRPV1 sensitization are rapid responses, these latter signaling pathways not involving transcriptional changes are strongly implicated.
Many laboratories, including our own, have explored the signaling pathways linking inflammatory agents to the acute sensitization of TRPV1. Although still controversial (Zhang and McNaughton, 2006), the links between activation of the NGF receptor trkA and TRPV1 are the most comprehensively studied, and consensus supports the involvement of both PI3K and src-kinase, and to a lesser extent p42/p44 Erk phosphorylation, in this coupling (Zhuang et al., 2004; Zhang et al., 2005; Stein et al., 2006; Zhu and Oxford, 2007). In contrast to our observations with NGF-induced sensitization, neither inhibition of MEK (upstream of Erk) nor inhibition of PI3K reduced the sensitization of TRPV1 by activin. Furthermore, activin challenges to DRG neurons failed to increase either phosphorylation of Erk or Akt determined biochemically, as would be expected if these pathways were involved and as has been confirmed for NGF (Zhu and Oxford, 2007). Likewise, inhibition of neither src-kinase nor PKA blunted enhancement of TRPV1 responses by activin, suggesting that neither kinase is involved in the sensitization. Phosphorylation of TRPV1 by PKC has been implicated in sensitization by both NGF (Bonnington and McNaughton, 2003; Zhu and Oxford, 2007) and BK (Cesare et al., 1999). With respect to the present study, our pharmacological and translocation data strongly implicate activation of PKC, specifically the PKCε isoform, as also critical to activin-induced sensitization of TRPV1.
Acute sensitization of TRPV1 is becoming appreciated as a common mechanism underlying many injury-induced hyperalgesias. In the case of inflammation or acute wound injury, a variety of released molecules have been shown to sensitize TRPV1, including NGF, BK, prostaglandins, and now activin. Furthermore, phosphorylation by PKC appears to be a common nexus in the signaling pathways linking trkA, B2, and activin receptors to increased TRPV1 sensitivity. Activation of each of these distinct receptor complexes, however, converges on this common effector through different signaling pathways, as revealed through pharmacological interventions. NGF activation of trkA invokes both Erk and PI3K activation, BK activation of B2 receptors invokes PLCβ activation and subsequent PIP2 hydrolysis with concomitant diacylglycerol production, and activin activation of ActII-ALK4 receptors is linked to PKC through an as yet undetermined pathway. In this regard, TGFβ has been shown to activate PKCα in both human keratinocytes and hepatoma cells (Miyazaki et al., 2004; Sakaguchi et al., 2004), although PKCα appears to be absent in sensory neurons of dorsal root ganglia (Cesare et al., 1999). As pharmacological or molecular interference with each of these components abrogates sensitization of TRPV1 from only a single receptor type (i.e., B2 vs trkA vs ALK4), significant cross talk among these receptor linked pathways upstream of PKC activation appears unlikely. This raises the question of why multiple ligands might converge in such a manner rather than use identical or similar signaling pathways (Hucho and Levine, 2007). Given the importance of sensing pathological pain associated with injury and inflammation, perhaps such an arrangement serves to ensure that sensitization invoked by several ligands released during injury will occur without saturation of earlier upstream components of a given pathway that most certainly subserves other cell functions. Without knowledge of the stoichiometry among the different components, it is difficult to test this speculation. It will also be interesting to learn whether activin has effects on other TRP channels involved in nociception (e.g., TRPA1), or indeed, on sodium channel isoforms that contribute to inflammatory pain hypersensitivity (Amaya et al., 2006).
This study reveals activin-mediated sensitization of TRPV1 to be an important component of injury-induced hyperalgesia. Furthermore, activation of PKCε is critical to this sensitization. As discussed, however, the hyperalgesia and associated nocifensive behaviors are only one component of the process of wound healing, and activin plays other important roles in tissue repair and nutrient delivery. Thus, targeting activin receptors per se as substrates for analgesic drug development would appear unwise, because such compounds would likely compromise these other components of recovery. Instead, understanding the distinct postreceptor pathways underlying activin receptor involvement in each of the three processes might afford approaches to interfere in activin-induced hyperalgesia alone.
Footnotes
This work was supported by National Institutes of Health Grants NS39420 (G.S.O.) and NS39316 (A.K.H.). We thank Drs. Michael Vasko, Ted Cummins, and Grant Nicol for constructive discussions regarding this study, Alice Russell for assistance with culture of DRG neurons, and Dr. Wylie Vale for providing the antibodies against ActRIB, ActRII, and ActRIIB receptors.
References
- Ahern GP, Brooks IM, Miyares RL, Wang X. Extracellular cations sensitize and gate capsaicin receptor TRPV1 modulating pain signaling. J Neurosci. 2005;25:5109–5116. doi: 10.1523/JNEUROSCI.0237-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ai X, Cappuzzello J, Hall AK. Activin and bone morphogenetic proteins induce calcitonin gene-related peptide in embryonic sensory neurons in vitro. Mol Cell Neurosci. 1999;14:506–518. doi: 10.1006/mcne.1999.0798. [DOI] [PubMed] [Google Scholar]
- Amaya F, Wang H, Costigan M, Allchorne AJ, Hatcher JP, Egerton J, Stean T, Morisset V, Grose D, Gunthorpe MJ, Chessell IP, Tate S, Green PJ, Woolf CJ. The voltage-gated sodium channel Na(v) 1.9 is an effector of peripheral inflammatory pain hypersensitivity. J Neurosci. 2006;26:12852–12860. doi: 10.1523/JNEUROSCI.4015-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhave G, Zhu W, Wang H, Brasier DJ, Oxford GS, Gereau RW., IV cAMP dependent protein kinase regulates desensitization of the capsaicin receptor (VR1) by direct phosphorylation. Neuron. 2002;35:721–731. doi: 10.1016/s0896-6273(02)00802-4. [DOI] [PubMed] [Google Scholar]
- Bhave G, Hu HJ, Glauner KS, Zhu W, Wang H, Brasier DJ, Oxford GS, Gereau RW., IV Protein kinase C phosphorylation sensitizes but does not activate the capsaicin receptor transient receptor potential vanilloid 1 (TRPV1) Proc Natl Acad Sci USA. 2003;100:12480–12485. doi: 10.1073/pnas.2032100100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolcskei K, Helyes Z, Scabo A, Sandor K, Elekes K, Nemeth J, Almasi R, Pinter E, Petho G, Szolcsanyi J. Investigation of the role of TRPV1 receptors in acute and chronic nociceptive processes using gene-deficient mice. Pain. 2005;117:368–376. doi: 10.1016/j.pain.2005.06.024. [DOI] [PubMed] [Google Scholar]
- Bonnington JK, McNaughton PA. Signaling pathways involved in the sensitisation of mouse nociceptive neurones by nerve growth factor. J Physiol. 2003;551:433–446. doi: 10.1113/jphysiol.2003.039990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, Koltzenburg M, Basbaum AI, Julius D. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288:306–313. doi: 10.1126/science.288.5464.306. [DOI] [PubMed] [Google Scholar]
- Cesare P, McNaughton PA. A novel heat-activated current in nociceptive neurons and its sensitization by bradykinin. Proc Natl Acad Sci USA. 1996;93:15435–15439. doi: 10.1073/pnas.93.26.15435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesare P, Dekker LV, Sardini A, Parker PJ, McNaughton PA. Specific involvement of PKC-ε in sensitization of the neuronal response to painful heat. Neuron. 1999;23:617–624. doi: 10.1016/s0896-6273(00)80813-2. [DOI] [PubMed] [Google Scholar]
- Chijiwa T, Mishima A, Hagiwara M, Sano M, Hayashi K, Inoue T, Naito K, Toshioka T, Hidaka H. Inhibition of forskolin-induced neurite outgrowth and protein phosphorylation by a newly synthesized selective inhibitor of cyclic AMP-dependent protein kinase, N-[2-(p-bromocinnamylamino)ethyl]-5- isoquinolinesulfonamide (H-89), of PC12D pheochromocytoma cells. J Biol Chem. 1990;265:5267–5272. [PubMed] [Google Scholar]
- Cruise BA, Xu P, Hall AK. Wounds increase activin in skin and a vasoactive neuropeptide in sensory ganglia. Dev Biol. 2004;271:1–10. doi: 10.1016/j.ydbio.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Davis JB, Gray J, Gunthorpe MJ, Hatcher JP, Davey PT, Overend P, Harries MH, Latcham J, Clapham C, Atkinson K, Hughes SA, Rance K, Grau E, Harper AJ, Pugh PL, Rogers DC, Bingham S, Randall A, Sheardown SA. Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature. 2000;405:183–187. doi: 10.1038/35012076. [DOI] [PubMed] [Google Scholar]
- de Caestecker M. The transforming growth factor-beta superfamily of receptors. Cytokine Growth Factor Rev. 2004;15:1–11. doi: 10.1016/j.cytogfr.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Docherty RJ, Yeats JC, Bevan S, Boddeke HW. Inhibition of calcineurin inhibits the desensitization of capsaicin-evoked currents in cultured dorsal root ganglion neurones from adult rats. Pflugers Arch. 1996;431:828–837. doi: 10.1007/s004240050074. [DOI] [PubMed] [Google Scholar]
- Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, VanDyk DE, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL, Scherle PA, Trzaskos JM. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- Funaba M, Ogawa K, Abe M. Expression and localization of activin receptors during endochondral bone development. Eur J Endocrinol. 2001;144:63–71. doi: 10.1530/eje.0.1440063. [DOI] [PubMed] [Google Scholar]
- Guo A, Vulchanova L, Wang J, Li X, Elde R. Immunocytochemical localization of the vanilloid receptor 1 (VR1): relationship to neuropeptides, the P2X3 purinoceptor and IB4 binding sites. Eur J Neurosci. 1999;11:946–958. doi: 10.1046/j.1460-9568.1999.00503.x. [DOI] [PubMed] [Google Scholar]
- Hanke JH, Gardner JP, Dow RL, Cangelian PS, Brissette WH, Weringer EJ, Pollok BA, Connelly PA. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. J Biol Chem. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Hu HJ, Bhave G, Gereau RW., IV Prostaglandin and protein kinase A-dependent modulation of vanilloid receptor function by metabotropic glutamate receptor 5: potential mechanism for thermal hyperalgesia. J Neurosci. 2002;22:7444–7452. doi: 10.1523/JNEUROSCI.22-17-07444.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubner G, Hu Q, Smola H, Werner S. Strong induction of activin expression after injury suggests an important role of activin in wound repair. Dev Biol. 1996;173:490–498. doi: 10.1006/dbio.1996.0042. [DOI] [PubMed] [Google Scholar]
- Hubner G, Brauchle M, Gregor M, Werner S. Activin A: a novel player and inflammatory marker in inflammatory bowel disease? Lab Invest. 1997;77:311–318. [PubMed] [Google Scholar]
- Hucho T, Levine JD. Signaling pathways in sensitization: toward a nociceptor cell biology. Neuron. 2007;55:365–376. doi: 10.1016/j.neuron.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Inman GJ, Nocolas FJ, Callahan JF, Harling JD, Gaster LM, Reith AD, Laping NJ, Hill CS. SB-431542 is a potent and specific inhibitor of transforming growth factor-β superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol Pharmacol. 2002;62:65–74. doi: 10.1124/mol.62.1.65. [DOI] [PubMed] [Google Scholar]
- Iwahori Y, Saito H, Torii K, Nishiyama N. Activin exerts a neurotrophic effect on cultured hippocampal neurons. Brain Res. 1997;760:52–58. doi: 10.1016/s0006-8993(97)00275-8. [DOI] [PubMed] [Google Scholar]
- Jones KL, Brauman JN, Groome NP, de Kretser DM, Phillips DJ. Activin A release into the circulation is an early event in systemic inflammation and precedes the release of pollistatin. Endocrinology. 2000;144:1905–1908. doi: 10.1210/endo.141.5.7531. [DOI] [PubMed] [Google Scholar]
- Jones KL, de Kretser DM, Patella S, Phillips DJ. Activin A and follistatin in systemic inflammation. Mol Cell Endocrinol. 2004;225:119–125. doi: 10.1016/j.mce.2004.07.010. [DOI] [PubMed] [Google Scholar]
- Keeble J, Russell F, Curtis B, Starr A, Pinter E, Brain SD. Involvement of transient receptor potential vanilloid 1 in the vascular and hyperalgesic components of joint inflammation. Arthritis Rheum. 2005;52:3248–3256. doi: 10.1002/art.21297. [DOI] [PubMed] [Google Scholar]
- Koplas PA, Rosenberg RL, Oxford GS. The role of calcium in the desensitization of capsaicin responses in rat dorsal root ganglion neurons. J Neurosci. 1997;17:3525–3537. doi: 10.1523/JNEUROSCI.17-10-03525.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kos K, Fine L, Coulombe JN. Activin type II receptors in embryonic dorsal root ganglion neurons of the chicken. J Neurobiol. 2001;47:93–108. doi: 10.1002/neu.1019. [DOI] [PubMed] [Google Scholar]
- Lopshire JC, Nicol GD. The cAMP transduction cascade mediates the prostaglandin E2 enhancement of the capsaicin-elicited current in rat sensory neurons: whole-cell and single-channel studies. J Neurosci. 1998;18:6081–6092. doi: 10.1523/JNEUROSCI.18-16-06081.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macias-Silva M, Hoodless PA, Tang SJ, Buchwald M, Wrana JL. Specific activation of Smad1 signaling pathways by the BMP7 Type I receptor, ALK2. J Biol Chem. 1998;273:25628–25636. doi: 10.1074/jbc.273.40.25628. [DOI] [PubMed] [Google Scholar]
- Martiny-Baron G, Kazanietz MG, Mischak H, Blumberg P, Kochs G, Hug H, Marme D, Schächtele C. Selective inhibition of protein kinase C isozymes by the indolocarbazole Gö 6976. J Biol Chem. 1993;268:9194–9197. [PubMed] [Google Scholar]
- Massague J. TGF-β signal transduction. Annu Rev Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- Mathews LS, Vale WW. Characterization of type II activin receptors. Binding, processing, and phosphorylation. J Biol Chem. 1993;268:19013–19018. [PubMed] [Google Scholar]
- Miyazaki M, Sakaguchi M, Akiyama I, Sakaguchi Y, Nagamori S, Huh NH. Involvement of interferon regulatory factor 1 and S100C/A11 in growth inhibition by transforming growth factor β 1 in human hepatocellular carcinoma cells. Cancer Res. 2004;64:4155–4161. doi: 10.1158/0008-5472.CAN-03-2750. [DOI] [PubMed] [Google Scholar]
- Mohapatra DP, Nau C. Desensitization of capsaicin-activated currents in the vanilloid receptor TRPV1 is decreased by the cyclic AMP-dependent protein kinase pathway. J Biol Chem. 2003;278:50080–50090. doi: 10.1074/jbc.M306619200. [DOI] [PubMed] [Google Scholar]
- Moustakas A, Heldin CH. Non-Smad TGF-beta signals. J Cell Sci. 2005;118:3573–3584. doi: 10.1242/jcs.02554. [DOI] [PubMed] [Google Scholar]
- Munz B, Hubner G, Tretter Y, Alzheimer C, Werner S. A novel role of activin in inflammation and repair. J Endocrinol. 1999a;161:187–193. doi: 10.1677/joe.0.1610187. [DOI] [PubMed] [Google Scholar]
- Munz B, Smola H, Engelhardt F, Bleuel K, Brauchle M, Lein I, Evans LW, Huylebroeck D, Balling R, Werner S. Overexpression of activin A in the skin of transgenic mice reveals new activities of activin in epidermal morphogenesis, dermal fibrosis and wound repair. EMBO J. 1999b;18:5205–5215. doi: 10.1093/emboj/18.19.5205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numazaki M, Tominaga T, Toyooka H, Tominaga M. Direct phosphorylation of capsaicin receptor VR1 by protein kinase C epsilon and identification of two target serine residues. J Biol Chem. 2002;277:13375–13378. doi: 10.1074/jbc.C200104200. [DOI] [PubMed] [Google Scholar]
- Pang L, Sawada T, Decker SJ, Saltiel AR. Inhibition of MAP kinase kinase blocks the differentiation of PC-12 cells induced by nerve growth factor. J Biol Chem. 1995;270:13585–13588. doi: 10.1074/jbc.270.23.13585. [DOI] [PubMed] [Google Scholar]
- Pangas SA, Woodruff TK. Activin signal transduction pathways. Trends Endocrinol Metab. 2000;11:309–314. doi: 10.1016/s1043-2760(00)00294-0. [DOI] [PubMed] [Google Scholar]
- Phillips DJ, Jones KL, Scheerlinck JY, Hedger MP, de Kretser DM. Evidence for activin A and follistatin involvement in the systemic inflammatory response. Mol Cell Endocrinol. 2001;180:155–162. doi: 10.1016/s0303-7207(01)00516-0. [DOI] [PubMed] [Google Scholar]
- Pogatzki-Zahn EM, Shimizu K, Caterina M, Raja SN. Heat hyperalgesia after incision requires TRPV1 and is distinct from pure inflammatory pain. Pain. 2005;115:296–307. doi: 10.1016/j.pain.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Premkumar LS, Ahern GP. Induction of vanilloid receptor channel activity by protein kinase C. Nature. 2000;408:985–990. doi: 10.1038/35050121. [DOI] [PubMed] [Google Scholar]
- Sakaguchi M, Miyazaki M, Sonegawa H, Kashiwagi M, Ohba M, Kuroki T, Namba M, Huh NH. PKCα mediates TGFβ-induced growth inhibition of human keratinocytes via phosphorylation of S100C/A11. J Cell Biol. 2004;164:979–984. doi: 10.1083/jcb.200312041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu XQ, Mendell LM. Nerve growth factor acutely sensitizes the response of adult rat sensory neurons to capsaicin. Neurosci Lett. 1999;274:159–162. doi: 10.1016/s0304-3940(99)00701-6. [DOI] [PubMed] [Google Scholar]
- Shu XQ, Mendell LM. Acutely sensitization by NGF of the response of small-diameter sensory neurons to capsaicin. J Neurophysiol. 2001;86:2931–2938. doi: 10.1152/jn.2001.86.6.2931. [DOI] [PubMed] [Google Scholar]
- Stein AT, Ufret-Vincenty CA, Hua L, Santana LF, Gordon SE. Phosphoinositide 3-kinase binds to TRPV1 and mediates NGF-stimulated TRPV1 trafficking to the plasma membrane. J Gen Physiol. 2006;128:509–522. doi: 10.1085/jgp.200609576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiuar T, Bielefeldt K, Gebhart GF. TRPV1 function in mouse colon sensory neurons is enhanced by metabotropic 5-hydroxytryptamine receptor activation. J Neurosci. 2004;24:9521–9530. doi: 10.1523/JNEUROSCI.2639-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, Skinner K, Raumann BE, Basbaum AI, Julius D. The cloned capsaicin receptor integrates multiple pain producing stimuli. Neuron. 1998;21:531–543. doi: 10.1016/s0896-6273(00)80564-4. [DOI] [PubMed] [Google Scholar]
- Urban L, Thompson SW, Dray A. Modulation of spinal excitability: co-operation between neurokinin and excitatory amino acid neurotransmitters. Trends Neurosci. 1994;17:432–438. doi: 10.1016/0166-2236(94)90018-3. [DOI] [PubMed] [Google Scholar]
- Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002) J Biol Chem. 1994;269:5241–5248. [PubMed] [Google Scholar]
- Wilkinson SE, Parker PJ, Nixon JS. Isoenzyme specificity of bisindolylmaleimides, selective inhibitors of protein kinase C. Biochem J. 1993;294:335–337. doi: 10.1042/bj2940335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodbury CJ, Zwick M, Wang S, Lawson JJ, Caterina MJ, Koltzenburg M, Albers KM, Koerber HR, Davis BM. Nociceptors lacking TRPV1 and TRPV2 have normal heat responses. J Neurosci. 2004;24:6410–6415. doi: 10.1523/JNEUROSCI.1421-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Van Slambrouck C, Berti-Mattera L, Hall AK. Activin induces tactile allodynia and increases calcitonin gene-related peptide after peripheral inflammation. J Neurosci. 2005;25:9227–9235. doi: 10.1523/JNEUROSCI.3051-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wankell M, Munz B, Hubner G, Hans W, Wolf E, Goppelt A, Werner S. Impaired wound healing in transgenic mice overexpressing the activin antagonist follistatin in the epidermis. EMBO J. 2001;20:5361–5372. doi: 10.1093/emboj/20.19.5361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner S, Alzheimer C. Roles of activin in tissue repair, fibrosis, and inflammatory disease. Cytokine Growth Factor Rev. 2006;17:157–171. doi: 10.1016/j.cytogfr.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Yedovitzky M, Mochly-Rosen D, Johnson JA, Gray MO, Ron D, Abramovitch E, Cerasi E, Hesher R. Translocation inhibitors define specificity of protein kinase C isoenzymes in pancreatic β-cells. J Biol Chem. 1997;272:1417–1420. doi: 10.1074/jbc.272.3.1417. [DOI] [PubMed] [Google Scholar]
- Zhang X, McNaughton PA. Why pain gets worse: the mechanism of heat hyperalgesia. J. Gen Physiol. 2006;128:491–493. doi: 10.1085/jgp.200609676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Huang J, McNaughton PA. NGF rapidly increases membrane expression of TRPV1 heat-gated ion channels. EMBO J. 2005;24:4211–4223. doi: 10.1038/sj.emboj.7600893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Oxford GS. Phosphoinositide-3-kinase and mitogen activated protein kinase signaling pathways mediate acute NGF sensitization of TRPV1. Mol Cell Neurosci. 2007;34:689–700. doi: 10.1016/j.mcn.2007.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Galoyan SM, Petruska JC, Oxford GS, Mendell LM. A developmental switch in acute sensitization of small dorsal root ganglion (DRG) neurons to capsaicin or noxious heating by NGF. J Neurophysiol. 2004;92:3148–3152. doi: 10.1152/jn.00356.2004. [DOI] [PubMed] [Google Scholar]
- Zhuang ZY, Xu HX, Clapham DE, Ji RR. Phosphatidylinositol 3-kinase activates ERK in primary sensory neurons and mediates inflammatory heat hyperalgesia through TRPV1 sensitization. J Neurosci. 2004;24:8300–8309. doi: 10.1523/JNEUROSCI.2893-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]