

Abstract
Whether the subunit composition of NMDA receptors (NMDARs) controls the direction of long-term plasticity is currently disputed. In the visual layers of NR2A−/− juvenile superior colliculus (SC), synapses lose miniature NMDAR currents, leaving NR2B-rich receptors in extrasynaptic regions. Compared with wild type (WT), evoked NMDAR currents in mutant neurons have slower rise and decay times and lower NMDAR/AMPAR current ratios. Moreover, NMDAR and L-type Ca2+ channel-dependent SC long-term potentiation (LTP) is absent in NR2A−/− cells, whereas both WT and mutant neurons show long-duration, low-frequency-induced, long-term depression (LLF-LTD) that is blocked by either AP-5, nimodipine, or Ro 25-6981 [R-(R,S)-α-(4-hydroxyphenyl)-β-methyl-4-(phenylmethyl)-1-piperidine propranol]. Thus, NMDAR currents or signaling localized at the postsynaptic density are essential to SC NMDAR-dependent LTP, whereas extrasynaptic or NR2B-rich NMDARs are necessary for LLF-LTD. However, synaptic NMDARs as well as the NR2A subunit are missing in NR2A−/− mice. Therefore, NR2 subunit-specific ligand binding/channel properties and/or separate signaling pathways interacting with NMDARs at synaptic versus extrasynaptic receptors could underlie these results.
Keywords: NR2A−/−, synaptic NMDAR, extrasynaptic NMDAR, LTP, LTD, L-type Ca2+ channel
Introduction
NMDA receptor (NMDAR) activity can strengthen or weaken brain synapses, but whether current through NR2A- or NR2B-subunit-containing NMDARs dictates long-term potentiation (LTP) versus depression (LTD) is subject to much debate (Kohr, 2006). NR2A−/− mice are reported to show reduced LTP in the hippocampus (Sakimura et al., 1995; Ito et al., 1996; Kiyama et al., 1998) but not in barrel cortex or the nucleus of the stria terminals (Lu et al., 2001a; Weitlauf et al., 2005). The visual cortex of NR2A−/− mice shows LTP of field EPSPs but lacks both metaplasticity (Philpot et al., 2007) and orientation tuning (Fagiolini et al., 2003). In hippocampal CA1, the NR2A antagonist NVP-AAM077 ([(R)-[(S)-1-(4-bromo-phenyl)-ethylamino]-(2,3-dioxo-1,2,3,4-tetrahydroquinoxalin-5-yl)-methyl]-phosphonic acid) appears to block LTP, whereas NR2B blockade disrupts LTD (Liu et al., 2004). However, in this same region, calcium/calmodulin-dependent kinase II binding to the NR2B tail appears critical to LTP (Barria and Malinow, 2005).
The distribution of NMDARs is also important to their ability to induce LTP versus LTD. NR2A-rich receptors concentrate at subsynaptic sites, whereas NR2B-rich receptors are prominent in perisynaptic or extrasynaptic regions (Tovar and Westbrook, 1999; van Zundert et al., 2004; Thomas et al., 2006). Activation of subsynaptic NMDARs in hippocampal cultures induces LTP, whereas activation of extrasynaptic NMDARs produces LTD (Lu et al., 2001b). Pairing low-frequency stimulation with postsynaptic depolarization produces hippocampal LTP regardless of NR2A or NR2B activation (Berberich et al., 2005). However, pairing synaptic stimulation with pulses of depolarizing current is also reported to produce LTP even when NMDARs are blocked with AP-5 (Kullman et al., 1992).
In visual superior colliculus (SC) synapses of the NR2−/− mouse, miniature NMDAR (mNMDAR) currents disappear before eye opening [postnatal day 13 (P13)], whereas extrasynaptic NR2B-NMDARs remain and mediate evoked NMDAR currents with prolonged rise times (Townsend et al., 2003). Mutant mice lacking the NR2A cytoplasmic domain show a similar decrease in synaptic NMDARs in hippocampal CA1 (Steigerwald et al., 2000). The NR2A cytoplasmic domain therefore appears necessary to bind NMDARs to the synapse, and their loss from that position in NR2A−/− mutants is consistent with the relatively poor binding of the remaining NR2B cytoplasmic tail to the mature scaffold postsynaptic density-95 (PSD-95) (Townsend et al., 2003). In rat SC, L-type Ca2+ channels and NMDARs act synergistically to induce LTP (Zhao et al., 2006). Here we show that juvenile NR2A−/− mice (P15–P17) lack synaptic NMDARs and show no SC LTP but retain NR2B-NMDAR- and L-type Ca2+ channel-dependent long-duration, low-frequency-induced, long-term depression (LLF-LTD). Blockade of NR2B-rich NMDARs eliminates LLF-LTP in both WT and NR2A−/− neurons. The data suggest that synaptic NMDARs are critical to SC LTP, whereas extrasynaptic NMDARs are critical to LLF-LTD but they cannot fully address the question of NMDAR subunit specificity in these responses.
Materials and Methods
Slice preparation.
Wild-type (WT) C57BL/6 mice and NR2A−/− mice (Sakimura et al., 1995) (a gift from M. Mishina, University of Tokyo School of Medicine, Tokyo, Japan) with a C57BL/6 background were bred and maintained in Massachusetts Institute of Technology facilities. All animal procedures were in accord with approved Massachusetts Institute of Technology Animal Care and Use Committee protocols. Parasagittal SC slices were prepared from these mice at P15–P17 as described previously (Zhao et al., 2006).
Electrophysiology.
All recordings used whole-cell patch clamping from narrow field vertical neurons in the stratum griseum superficale of the SC. When plasticity was examined the artificial CSF (ACSF) and pipette solutions were as in the study of Zhao et al. (2006). For miniature and evoked current recording the pipette solution contained the following (in mm): 122.5 Cs-gluconate, 17.5 CsCl, 10 HEPES, 0.2 NaEGTA, 4 ATP-Mg, 0.4 GTP-Na, and 8 NaCl, pH adjusted to 7.3 with CsOH.
Miniature EPSCs (mEPSCs) were recorded at −70 mV in the presence of 1 μm tetrodotoxin (Sigma, St. Louis, MO), 10 μm bicuculline methiodide (BMI) (GABAA receptor antagonist; Sigma), 5 μm glycine, and 0 mm Mg2+. All mEPSCs more than two times baseline noise (2.5 pA) in a 1–3 min period (≥360 events) were averaged.
Evoked AMPAR and NMDAR currents (eAMPARcs and eNMDARcs) were obtained at −70 and +40 mV, respectively, with 20 μm BMI. eNMDARcs were recorded after eAMPARcs with 20 μm 2,3-dihydroxy-6-nitro-7-sulfonyl-benzo[f]quinoxaline (NBQX) (selective AMPA/kainate receptor antagonist; Tocris Bioscience, Ballwin, MO). Constant current stimuli (0.05 Hz, 0.2 ms duration) were delivered through a bipolar tungsten electrode in the stratum opticum of SC slices.
Decay time (90–37% peak amplitude for minis and 0.37 peak amplitude for evoked events), rise time (10–90% peak amplitude), and amplitude of averaged mEPSCs, eAMPARcs, and eNMDARcs were measured with Mini Analysis (Synaptosoft, Decatur, GA). Stimulation intensity for evoked currents was defined relative to the threshold for inducing a response. Paired-pulse ratios (PPRs) used 100 ms separation and were calculated as second eAMPARc amplitude/first eAMPARc amplitude.
LTP induction was as described previously (Zhao et al., 2006). BMI (12 μm) was present in all EPSP recording. AP-5 (NMDAR antagonist, 100 μm; Sigma), nimodipine (Nim) (L-type Ca2+ channel antagonist, 20 μm; Tocris Bioscience), and Ro 25-6981 [R-(R,S)-α-(4-hydroxyphenyl)-β-methyl-4-(phenylmethyl)-1-piperidine propranol] (Ro 25) (NR2B NMDAR antagonist, 5 μm; Sigma) were used as noted. In pilot studies, Ro 25 strongly reduced (∼80%) NR2B-NMDARS in NR2A−/− SC neurons. In all experiments, Ro 25 was added to the ACSF 15 min before whole-cell configuration was achieved. LTP and LTD were induced and assayed as described in the figure legends.
One cell per slice was used for evoked response recordings, one to two cells per slice for mEPSC recordings, and n is given as the number of experiments/the number of animals. Data are expressed as mean ± SEM. Statistical significance (*p < 0.05 and **p < 0.01) was determined using Student's t and ANOVA tests as stated in the figure legends.
Results
Juvenile NR2A−/− SC neurons lose mNMDARcs
Previous work documented loss of mNMDARcs in the NR2A−/− SC by P13 (Townsend et al., 2003). This difference from WT was retained in the P15–P17 pups used here. We recorded mEPSCs from WT and NR2A−/− neurons in the absence and presence of 50 μm AP-5 (Fig. 1A). In WT, the ratios of decay time with AP-5/without AP-5 were significantly lower than in NR2A−/− (Fig. 1B), indicating a significant contribution of mNMDARcs in WT compared with NR2A−/−. Neither mEPSC rise time (Fig. 1C) nor amplitude ratios (Fig. 1D) differed between the two strains.
Figure 1.
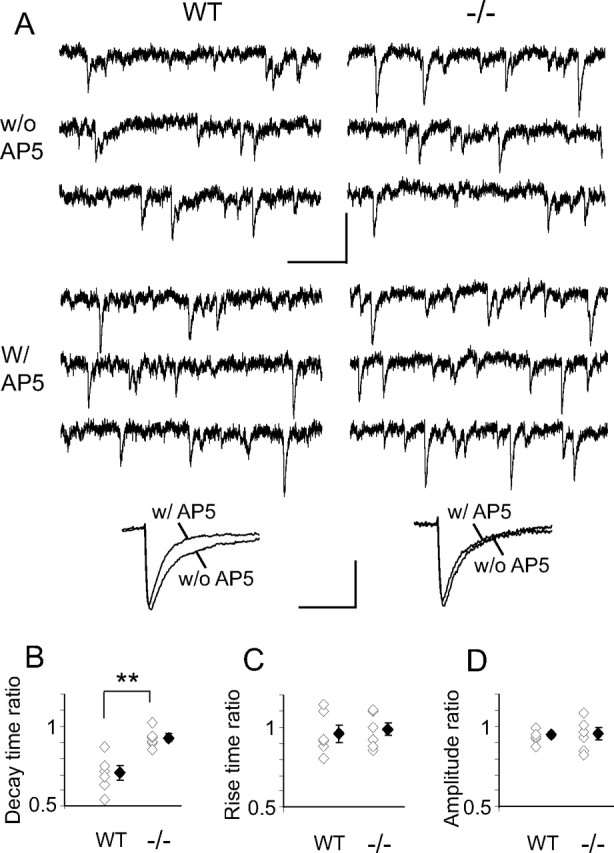
AP-5 shortens mEPSC decay time in WT but not in NR2A−/− SC neurons. A, Top, Sample traces of mEPSCs recorded in Mg2+-free ACSF at −70 mV from WT and NR2A−/− SC in the absence (w/o AP5) and presence (w/AP5) of AP-5. Calibration: 20 pA, 100 ms. Bottom, Representative averaged traces of mEPSCs before and 5 min after AP-5 application in a WT (538 and 453 events, respectively) and a NR2A−/− (627 and 725 events, respectively) neuron. Calibration: 5 pA, 13 ms. B–D, Summary plots showing ratios for each cell alongside averaged mEPSC decay time, rise time, and amplitude ratios (averaged mEPSC with AP-5, averaged mEPSC without AP-5; n = 6 and 2 for each genotype). White diamonds represent individual experiments, and black diamonds are means of all the experiments in that group. B, In WT cells, the decay time ratio is significantly lower than that in NR2A−/− cells (WT, 0.71 ± 0.05; NR2A−/−, 0.93 ± 0.02; p = 0.001). There is no difference between WT and NR2A−/− neurons in rise time ratio (C) (WT, 0.96 ± 0.06; NR2A−/−, 0.98 ± 0.04; p = 0.69) or amplitude ratio (D) (WT, 0.94 ± 0.02; NR2A−/−, 0.95 ± 0.04; p = 0.82). Unpaired two-tail Student's t test.
Different evoked NMDAR/AMPAR current ratios between NR2A−/− and WT neurons
Townsend et al. (2003) also demonstrated at P11–P13 slower rise and decay time eNMDARcs in NR2A−/− SC neurons compared with WT, with no difference in AMPARc rise or decay times. We corroborated this after eye opening [eNMDARc rise times in NR2A knock-out NR2AKO−/− slower than eNMDARc rise times in WT, p < 0.001; eAMPARc rise times in NR2A−/− not different from eAMPARcs in WT, p = 0.66; recorded at +40 mV with NBQX and at −70 mV, respectively]. eNMDARc/eAMPARc ratios for both WT and mutant neurons were then compared at multiple stimulation intensities (Fig. 2A). Evoked NMDARc/AMPARc rise time and decay time ratios were significantly higher in NR2A−/− than that in WT neurons, confirming respectively slower rise time eNMDARcs and more NR2B-rich NMDARs in NR2A−/− neurons. Amplitude ratios were significantly lower in NR2A−/− SC, indicating fewer total NMDARs in the mutant. The ratios remained constant for both strains over the full range of stimulating intensities. Differences between WT and NR2A−/− NMDAR currents were not caused by altered presynaptic release because PPRs did not differ between genotypes (Fig. 2B).
Figure 2.
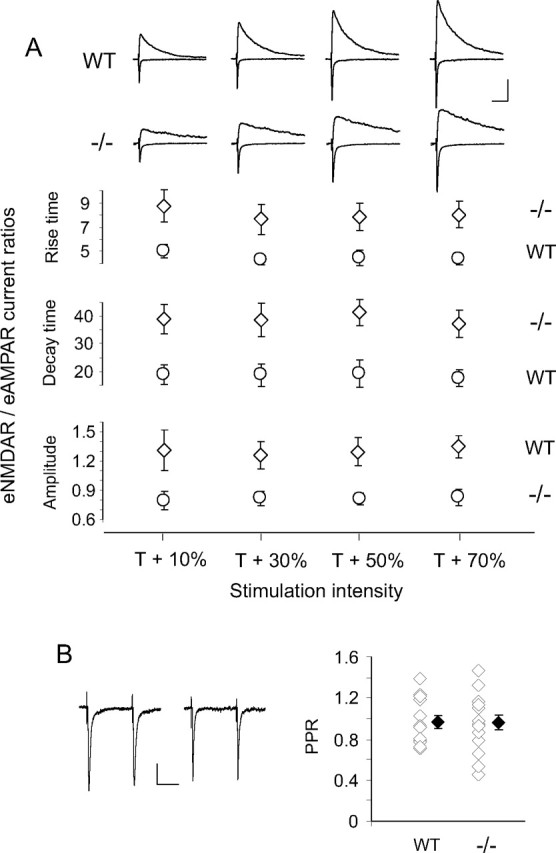
WT and NR2A−/− SC neurons show different NMDAR/AMPAR evoked current ratios but similar presynaptic release probabilities. A, Top, Sample average recordings of eAMPARcs at −70 mV and eNMDARcs at +40 mV in WT and NR2A−/− SC neurons over a range of stimulation intensities. Each average is of 10 evoked currents. Calibration: 50 pA, 100 ms. Bottom, Summary plots showing averaged eNMDARc/eAMPARc ratios for rise time, decay time, and amplitude (n = 6 and 2 for each genotype). Genotype had a significant effect on rise time ratio (F = 3.7, df = 7, p < 0.0001), decay time ratio (F = 5.7, df = 7, p < 0.0001), and amplitude ratio (F = 4.9, df = 7, p < 0.0001). NR2A−/− neurons had higher rise time and decay time ratios and lower amplitude ratios than WT neurons. Stimulus intensity had no effect (p > 0.05) on any ratio, and there was no significant interaction between stimulus intensity and genotype (p > 0.05) (two-factor ANOVA). B, PPR sample traces (left; calibration: 20 pA, 50 ms) and summary graph (right) showing no difference between WT and NR2A−/− neurons (WT, 0.95 ± 0.06, n = 14 and 3; NR2A−/−, 0.95 ± 0.07, n = 14 and 3; p = 0.97). Each sample trace is the average of 10 paired-pulse responses. T, Threshold. White diamonds represent individual experiments, and black diamonds are means of all the experiments in that group.
NR2A−/− mice lack SC LTP
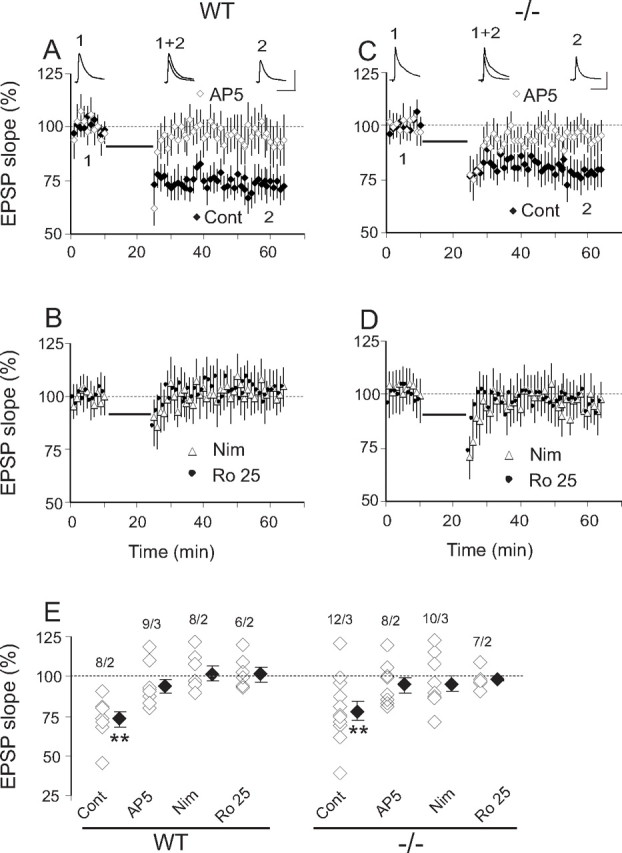
As in rat SC neurons, LTP could be induced with 20 Hz stimulation for 20 s at intensity of 30–50% spike threshold (ST) in WT but not in NR2A−/− neurons (Fig. 3A,B,E). Increasing induction intensity produced a small but significant depression in WT but not in NR2A−/− neurons (Fig. 3A,B,E). Stimulating frequencies of 10 Hz at 30–50% ST produced no change in both WT and NR2A−/− neurons (Fig. 3E), whereas 50 Hz stimulation produced a significant LTD in both WT and NR2A−/− neurons (Fig. 3A,B,E). SC LTP shows similar frequency sensitivity in rats (Zhao et al., 2006). Using the effective 20 Hz, 30–50% ST protocol in WT neurons, bath application of AP-5 or AP-5 plus Nim prevented plasticity, Nim or Nim plus Ro 25 resulted in LTD (Fig. 3C,E), Nim applied after stimulation blocked change in EPSP slopes (Fig. 3D,E), and LTP switched to LTD with Nim during preinduction (Fig. 3D,E). Therefore, L-type Ca2+ channel activity is necessary for both the induction and maintenance of SC LTP in WT. In NR2A−/− cells, 20 Hz, 30–50% ST did not change EPSP slopes in the presence of Nim (Fig. 3E). Nim alone did not change the baseline activation in either genotype (data not shown). The mechanisms of the LTD induced by high-frequency stimuli or L-type Ca2+ channel blockade remain unknown. As suggested by Zhao et al. (2006), they could involve fatigue of presynaptic terminals and postsynaptic conductance that change excitability or entrainment of metabotropic glutamate receptor-mediated LTP. Nim versus Nim plus Ro 25 in WT suggest that NR2A-containing receptors are involved in the LTD induced by 20 Hz. Consistently, no LTD is induced in NR2A−/− mice in the presence of Nim. In contrast, LTD induced by 50 Hz appears to be NR2A independent.
Figure 3.

NMDAR and L-type Ca2+ channel-dependent LTP is present in WT but not in NR2A−/− SC neurons. A, E, In WT SC, only 20 Hz at 30–50% ST induced LTP (126.5 ± 6.9, n = 10 and 3, p = 0.004), 20 Hz at 80–90% ST produced a small but significant depression (92.2 ± 2.1, n = 6 and 2, p = 0.01), 10 Hz at 30–50% ST produced no change from baseline (95.4 ± 3.1, n = 5 and 2, p = 0.2), and 50 Hz at 30–50% produced a significant LTD (82.3 ± 5.6, n = 9 and 3, p = 0.02). Sample traces are averages of 30 evoked EPSPs obtained during the 10 min of baseline (1) and 30–40 min after induction (2) with 20 Hz at 30–50% ST (calibration: 10 mV, 50 ms). For all parts of Figure 3, stimulus intensity is given as percentage of ST identified as percentage alone, and all stimuli are 20 s in duration. EPSP slope is given as percentage change from baseline (average evoked EPSC during 10 min baseline over average EPSP slope 30–40 min after induction). B, E, In NR2A−/− SC, four of five stimulation protocols produced no significant change from baseline (20 Hz at 30–50% ST, 102.7 ± 5.9, n = 9 and 3, p = 0.68; 20 Hz at 80–90% ST, 90.1 ± 6.7, n = 6 and 2, p = 0.22; 10 Hz at 30–50% ST, 94.7 ± 4.5, n = 5 and 2, p = 0.35; and 20 Hz 30–50% ST in the presence of Nim, 97 ± 4.6, n = 8 and 2, p = 0.51). The exception was 50 Hz at 30–50% ST, which produced an LTD (81.1 ± 6.1, n = 8 and 3, p = 0.02) similar to that seen with 50 Hz stimulation in WT. Sample traces are averages of 30 evoked EPSPs obtained during the 10 min of baseline (1) and 30–40 min after induction (2) with 20 Hz at 30–50% ST (calibration bar: 10 mV, 50 ms). C, E, In WT neurons, LTP induction with 20 Hz at 30–50% was prevented by AP-5 alone (94.9 ± 4.8, n = 7 and 2, p = 0.36) or AP-5 plus Nim (98.9 ± 4.8, n = 8 and 2, p = 0.81) and reversed to LTD by Nim alone (78.7 ± 5, n = 8 and 3, p < 0.01) or Nim plus Ro 25 (74.4 ± 5.6, n = 9 and 3, p < 0.01). D, Top, In WT neurons, LTP induction with 20 Hz at 30–50% ST could be reversed to LTD by Nim use before induction (85.7 ± 4.1, n = 6 and 2, p = 0.02). Bottom, All plasticity was prevented by Nim application after induction (92.1 ± 4.6, n = 6 and 2, p = 0.18). E, Summary of mean EPSP slope changes from baseline at 30–40 min after induction under the different conditions in WT and NR2A−/− SC neurons. Each open diamond represents an individual experiment, and filled diamonds are the means of all the experiments in that group. All stimuli are at 30–50% ST except when noted. All stimulation frequencies are 20 Hz unless otherwise stated, and all stimulation durations are 20 s. Pre-Nim and Post-Nim, Before and after induction application of Nimodipine. Paired-sample, two-tailed Student's t test.
LLF-LTD is unaltered in WT and NR2A−/− SC neurons
Significant LTD was induced with 900 stimuli at 30–50% ST presented at 1 Hz (LLF stimulation) in WT (Fig. 4A,E) and NR2A−/− (Fig. 4C,E) neurons. In both mouse strains, application of AP-5 (Fig. 4A,C,E), or Nim (Fig. 4B,D,E) or Ro 25 (Fig. 4B,D,E) alone eliminated this depression. Therefore, blockade of either NR2B-NMDARs or L-type Ca2+ channels is sufficient to eliminate SC LTD whether or not NR2A subunits are present.
Figure 4.
NMDAR and L-type Ca2+ channel-dependent LTD (LLF-LTD) is present in both WT and NR2A−/− mouse SC neurons and requires NR2B. A, B, In WT SC, 900 stimuli at 1 Hz (LLF stimulation, thick line) induced significant LLF-LTD (Cont; 72.9 ± 4.9, n = 8 and 2, p < 0.01) that was blocked by AP-5, (94.2 ± 4.4, n = 9 and 3, p = 0.26), Nim (101.2 ± 5.1, n = 8 and 2, p = 0.71), or Ro 25 (102.8 ± 4.5, n = 6 and 2, p = 0.53). C, D, NR2A−/− SC neurons show responses similar to WT with LLF stimulation. LLF-LTD (Cont; 77.9 ± 6, n = 12 and 3, p < 0.01) was blocked by AP-5 (94.4 ± 4.7, n = 8 and 2, p = 0.34), Nim (95.2 ± 4.6, n = 10 and 3, p = 0.35), or Ro 25 (97.3 ± 2, n = 7 and 2, p = 0.28). WT versus NR2A−/− LLF-LTD was not significantly different (p = 0.58). Sample traces represent averages of 30 EPSPs obtained during the 10 min of base line (1) and 30–40 min after 900 stimuli at 1 Hz induction (2) in WT and NR2A−/− neurons (calibration: 10 mV, 50 ms). E, Summary of mean EPSP slope changes in WT and NR2A−/− neurons under the different pharmacological conditions. Format as in Figure 3E (paired-sample, two-tailed Student's t test). White diamonds represent individual experiments, and black diamonds are means of all the experiments in that group.
Discussion
Evoked neurotransmitter release can activate extrasynaptic NMDARs even in neurons that show no evidence of mNMDAR currents (Chen and Diamond, 2002; Clark and Cull-Candy, 2002). Thus, the presence or absence of synaptic NMDARs can be determined only when miniature currents are recorded. Steigerwald et al. (2000), Townsend et al. (2003), and Fu et al. (2005) all report the importance of the NR2A subunit or its cytoplasmic domain for maintaining mNMDAR currents as the brain matures. In all reports, AMPAR-mediated miniature and evoked currents remain. Nevertheless, adult NR2A−/− mice or mice with truncated NR2A lack field potential LTP in CA1 in response to a single tetanization but can show an NR2B-dependent LTP of comparable magnitude with WT LTP when induction consists of multiple tetanic stimuli (Kiyama et al., 1998; Kohr et al., 2003). However, the contribution of L-type Ca2+ channels to this high-threshold LTP remains unknown, although L-type Ca2+ channels have been reported to contribute to CA1 LTP particularly in response to intense stimulation (Cavus and Teyler, 1996).
The present results are consistent with previous work in several preparations (Liu et al., 2004; Massey et al., 2004; Lu et al., 2001b): Namely, synaptic, NR2A-rich NMDARs are obligatory for the induction of SC LTP, and extrasynaptic NR2B-rich NMDARs are critical to SC LTD induced by long, low-frequency stimulation. However, three questions critical to the NR2A/NR2B debate remain unanswered. Are there unique attributes of NR2A subunits that require a concentration of this protein at synapses to initiate NMDAR-dependent LTP? At P15–P17, PSD-95 is the dominant scaffold for synaptic SC NMDARs (van Zundert et al., 2004). Are the cytoplasmic signaling molecules specifically associated with PSD-95 critical to LTP? Do NR2B-rich NMDARs selectively drive depression or is their requirement in SC LLF-LTD attributable to a different signaling complex that they specifically associate with as a result of being predominantly extrasynaptic? Answers to these questions are important. They should reveal how one receptor produces two diametrically opposite effects on synaptic plasticity and brain function.
Footnotes
This work was supported by National Institutes of Health Grant EY014074 (M.C.-P.). We thank Marnie Phillips and H. R. Horvitz for helpful comments on this manuscript.
References
- Barria A, Malinow R. NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron. 2005;48:289–301. doi: 10.1016/j.neuron.2005.08.034. [DOI] [PubMed] [Google Scholar]
- Berberich S, Punnakkal P, Jensen V, Pawlak V, Seeburg PH, Hvalby O, Kohr G. Lack of NMDA receptor subtype selectivity for hippocampal long-term potentiation. J Neurosci. 2005;25:6907–6910. doi: 10.1523/JNEUROSCI.1905-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavus I, Teyler T. Two forms of long-term potentiation in area CA1 activate different signal transduction cascades. J Neurophysiol. 1996;76:3038–3047. doi: 10.1152/jn.1996.76.5.3038. [DOI] [PubMed] [Google Scholar]
- Chen S, Diamond JS. Synaptically released glutamate activates extrasynaptic NMDA receptors on cells in the ganglion cell layer of rat retina. J Neurosci. 2002;22:2165–2173. doi: 10.1523/JNEUROSCI.22-06-02165.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark BA, Cull-Candy SG. Activity-dependent recruitment of extrasynaptic NMDA receptor activation at an AMPA receptor-only synapse. J Neurosci. 2002;22:4428–4436. doi: 10.1523/JNEUROSCI.22-11-04428.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagiolini M, Katagiri H, Miyamoto H, Mori H, Grant SG, Mishina M, Hensch TK. Separable features of visual cortical plasticity revealed by N-methyl-d-aspartate receptor 2A signaling. Proc Natl Acad Sci USA. 2003;100:2854–2859. doi: 10.1073/pnas.0536089100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Z, Logan SM, Vicini S. Deletion of the NR2A subunit prevents developmental changes of NMDA-mEPSCs in cultured mouse cerebellar granule neurones. J Physiol (Lond) 2005;563:867–881. doi: 10.1113/jphysiol.2004.079467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito I, Sakimura K, Mishina M, Sugiyama H. Age-dependent reduction of hippocampal LTP in mice lacking N-methyl-d-aspartate receptor epsilon 1 subunit. Neurosci Lett. 1996;203:69–71. doi: 10.1016/0304-3940(95)12258-3. [DOI] [PubMed] [Google Scholar]
- Kiyama Y, Manabe T, Sakimura K, Kawakami F, Mori H, Mishina M. Increased thresholds for long-term potentiation and contextual learning in mice lacking the NMDA-type glutamate receptor epsilon1 subunit. J Neurosci. 1998;18:6704–6712. doi: 10.1523/JNEUROSCI.18-17-06704.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohr G. NMDA receptor function: subunit composition versus spatial distribution. Cell Tissue Res. 2006;326:439–446. doi: 10.1007/s00441-006-0273-6. [DOI] [PubMed] [Google Scholar]
- Kohr G, Jensen V, Koester HJ, Mihaljevic AL, Utvik JK, Kvello A, Ottersen OP, Seeburg PH, Sprengel R, Hvalby O. Intracellular domains of NMDA receptor subtypes are determinants for long-term potentiation induction. J Neurosci. 2003;23:10791–10799. doi: 10.1523/JNEUROSCI.23-34-10791.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullman DM, Perkel DJ, Manabe T, Nicoll RA. Ca2+ entry via postsynaptic voltage-sensitive Ca2+ channels can transiently potentiate synaptic transmission in the hippocampus. Neuron. 1992;9:1175–1183. doi: 10.1016/0896-6273(92)90075-o. [DOI] [PubMed] [Google Scholar]
- Liu L, Wong TP, Pozza MF, Lingenhoehl K, Wang Y, Sheng M, Auberson YP, Wang YT. Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science. 2004;304:1021–1024. doi: 10.1126/science.1096615. [DOI] [PubMed] [Google Scholar]
- Lu HC, Gonzalez E, Crair MC. Barrel cortex critical period plasticity is independent of changes in NMDA receptor subunit composition. Neuron. 2001a;32:619–634. doi: 10.1016/s0896-6273(01)00501-3. [DOI] [PubMed] [Google Scholar]
- Lu W, Man H, Ju W, Trimble WS, MacDonald JF, Wang YT. Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron. 2001b;29:243–254. doi: 10.1016/s0896-6273(01)00194-5. [DOI] [PubMed] [Google Scholar]
- Massey PV, Johnson BE, Moult PR, Auberson YP, Brown MW, Molnar E, Collingridge GL, Bashir ZI. Differential roles of NR2A and NR2B-containing NMDA receptors in cortical long-term potentiation and long-term depression. J Neurosci. 2004;24:7821–7828. doi: 10.1523/JNEUROSCI.1697-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philpot BD, Cho KK, Bear MF. Obligatory role of NR2A for metaplasticity in visual cortex. Neuron. 2007;53:495–502. doi: 10.1016/j.neuron.2007.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakimura K, Kutsuwada T, Ito I, Manabe T, Takayama C, Kushiya E, Yagi T, Aizawa S, Inoue Y, Sugiyama H, Mishina M. Reduced hippocampal LTP and spatial learning in mice lacking NMDA receptor epsilon 1 subunit. Nature. 1995;373:151–155. doi: 10.1038/373151a0. [DOI] [PubMed] [Google Scholar]
- Steigerwald F, Schulz TW, Schenker LT, Kennedy MB, Seeburg PH, Kohr G. C-Terminal truncation of NR2A subunits impairs synaptic but not extrasynaptic localization of NMDA receptors. J Neurosci. 2000;20:4573–4581. doi: 10.1523/JNEUROSCI.20-12-04573.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas CG, Miller AJ, Westbrook GL. Synaptic and extrasynaptic NMDA receptor NR2 subunits in cultured hippocampal neurons. J Neurophysiol. 2006;95:1727–1734. doi: 10.1152/jn.00771.2005. [DOI] [PubMed] [Google Scholar]
- Townsend M, Yoshii A, Mishina M, Constantine-Paton M. Developmental loss of miniature N-methyl-d-aspartate receptor currents in NR2A knockout mice. Proc Natl Acad Sci USA. 2003;100:1340–1345. doi: 10.1073/pnas.0335786100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL. The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J Neurosci. 1999;19:4180–4188. doi: 10.1523/JNEUROSCI.19-10-04180.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Zundert B, Yoshii A, Constantine-Paton M. Receptor compartmentalization and trafficking at glutamate synapses: a developmental proposal. Trends Neurosci. 2004;27:428–437. doi: 10.1016/j.tins.2004.05.010. [DOI] [PubMed] [Google Scholar]
- Weitlauf C, Honse Y, Auberson YP, Mishina M, Lovinger DM, Winder DG. Activation of NR2A-containing NMDA receptors is not obligatory for NMDA receptor-dependent long-term potentiation. J Neurosci. 2005;25:8386–8390. doi: 10.1523/JNEUROSCI.2388-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao JP, Phillips MA, Constantine-Paton M. Long-term potentiation in the juvenile superior colliculus requires simultaneous activation of NMDA receptors and L-type Ca2+ channels and reflects addition of newly functional synapses. J Neurosci. 2006;26:12647–12655. doi: 10.1523/JNEUROSCI.3678-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]