

Abstract
To investigate the role of CREB-mediated gene expression on the excitability of CA1 pyramidal neurons, we obtained intracellular recordings from pyramidal neurons of transgenic mice expressing a constitutively active form of CREB, VP16–CREB, in a regulated and restricted manner. We found that transgene expression increased the neuronal excitability and inhibited the slow and medium afterhyperpolarization currents. These changes may contribute to the reduced threshold for LTP observed in these mice. When strong transgene expression was turned on for prolonged period of time, these mice also showed a significant loss of hippocampal neurons and sporadic epileptic seizures. These deleterious effects were dose dependent and could be halted, but not reversed by turning off transgene expression. Our experiments reveal a new role for hippocampal CREB-mediated gene expression, identify the slow afterhyperpolarization as a primary target of CREB action, provide a new mouse model to investigate temporal lobe epilepsy and associated neurodegeneration, and illustrate the risks of cell death associated to a sustained manipulation of this pathway. As a result, our study has important implications for both the understanding of the cellular bases of learning and memory and the consideration of therapies targeted to the CREB pathway.
Keywords: AHP, CREB, excitability, excitotoxicity, learning and memory, synaptic plasticity
Introduction
Studies in different model systems have established a critical role for the cAMP signaling pathway and cAMP-responsive element binding protein (CREB)-mediated gene expression in different forms of synaptic plasticity related to learning (Lonze and Ginty, 2002; Barco et al., 2003; Josselyn and Nguyen, 2005). In particular, it has been found that the late phase of long-term potentiation (LTP) in the Schaffer collateral pathway correlates with the phosphorylation of CREB in CA1 pyramidal neurons and the induction of CRE-driven gene expression (Bito et al., 1996; Deisseroth et al., 1996; Impey et al., 1996; Lu et al., 1999). Furthermore, the enhanced expression of CRE-driven genes favors the formation and stability of LTP in this pathway (Barco et al., 2002, 2005; Marie et al., 2005), whereas the inhibition of CRE-driven expression by deleting specific CREB isoforms (Bourtchuladze et al., 1994) or by overexpressing a dominant negative form of CREB caused deficits in some forms of LTP (Pittenger et al., 2002; Huang et al., 2004). Knocking out a specific CREB isoform, however, may cause the overexpression of other CRE-binding proteins (Hummler et al., 1994; Blendy et al., 1996), which may compensate the deficiency in CREB activity and reduce the impact of the mutation in the phenotype (Gass et al., 1998; Balschun et al., 2003).
Recent studies have demonstrated that CREB activity can also directly control neuronal excitability. The expression of the constitutively active CREB variant, VP16–CREB, using viral vectors increased the firing frequency and reduced the resting potential of neurons in the locus ceruleus (Han et al., 2006) and the excitability of spiny neurons in the nucleus accumbens (Dong et al., 2006). The long-term effects of these modifications in neuronal physiology and survival have not been investigated. Neither is it known what the relationship is between the regulation of long-term forms of synaptic plasticity by CREB and the changes in neuronal excitability.
To explore the role of CREB-mediated gene expression in controlling the excitability of CA1 pyramidal neurons, we performed intracellular recordings in mice that express VP16–CREB in a regulated manner. Here we report that transgene expression caused a rapid and reversible change of the firing properties of CA1 pyramidal neurons by reducing the slow (sAHP) and medium (mAHP) afterhyperpolarization. Furthermore, we found that the sustained activation of CREB-mediated activity can trigger epileptic seizures once a given threshold is reached, and cause the excitotoxic cell death of pyramidal and granular neurons in the hippocampus. The comparison of transgenic strains with low and high levels of constitutive CREB activity allowed us to separate these two processes.
Materials and Methods
Generation and maintenance of transgenic mice.
Several lines of transgenic mice expressing VP16–CREB under the control of tetO promoter were generated by microinjection of the linear construct as previously described (Barco et al., 2002). The founder mice were backcrossed to C57BL6 F1/J mice more than 15 times to generate the transgenic lines used in our study. We have previously described in detail the mice now referred as VP16–CREBhigh (Barco et al., 2002, 2005). Those bitransgenic animals resulted of the crossing of pCaMKII-tTA mice (line B) (Mayford et al., 1996) and tetO–VP16–CREB line VC27, the line that provided highest level of expression in the hippocampus. In this study we also investigate the phenotype of bitransgenic animals resulted of the crossing of pCaMKII-tTA mice (line B) and tetO–VP16–CREB line VC19, which express the transgene at significantly lower level. We refer to this bitransgenic strain as VP16–CREBlow mice. In all our experiments, we used as control littermate mice carrying either pCaMKII-tTA, tetO–VP16–CREB or none transgene. For VP16–CREBhigh and control littermates, dox was administrated at 40 mg/kg food and removed or added at the indicated times before experimentation. VP16–CREBlow mice were raised without dox. Experiments were performed in adult mice (2-4 month old), at least otherwise indicated in figure or table legends. All mice were maintained and bred under standard conditions, consistent with national guidelines and approved by the Institutional Animal Care and Use Committees.
Electrophysiology.
Whole cell recordings were performed in acute slices maintained in vitro. Mice of either sex were anesthetized with isoflurane, decapitated, and coronal slices that included the dorsal hippocampus (300 μm) were cut in oxygenated (95% O2/5% CO2) ice-cold artificial CSF (ACSF) containing (in mm): NaCl 118, KCl 2.5, NaHCO3 25, NaH2PO4 1.2, MgCl2, 1.3, CaCl2 2.5 and glucose 10. After a recovery period of 30 min at 32°C, slices were maintained at room temperature in ACSF. For recordings, slices were transferred to the recording chamber and superfused with ACSF at 32−34°C. Whole cell recordings were made from CA1 pyramidal neurons using infrared differential interference video microscopy (E600FN, Nikon, Tokyo, Japan). Patch-clamp pipettes were filled with intracellular solution containing (in mm): KMeSO4 135, NaCl 8, HEPES 10, Mg2ATP 2, Na3GTP 0.3 (pH 7.2 osmolarity 290 mOsm/kg). Access resistance was 7-20 MΩ and was monitored throughout the experiment. Experiments were discarded if the series resistance changed by more than 10% during the course of the experiment. Signals were recorded using a MultiClamp 700B amplifier (Molecular Devices, Sunnyvale, CA, USA) filtered at 10 kHz and digitized at 20 kHz (Digidata 1320A, Molecular Devices). All cells described in this study had a membrane potential more negative than −50 mV. Electrical activity was recorded in current and voltage clamp and analyzed using AxoGraph X 1.0 (AxoGraph Scientific, Sydney, Australia). No series resistance compensation was used and membrane potentials were corrected for junction potentials (-4 mV). To investigate the firing properties of neurons, 15 current injection steps (600 ms) were applied from −200 to + 500 pA in 50 pA increments from a holding potential of −70 mV. Passive membrane properties were measured at resting membrane potential. IAHP was evoked by a 50 ms depolarizing voltage step to 0 mV from a holding potential of −50 mV. ImAHP and IsAHP amplitudes were measured at the peak of the current and 1 s after the end of the depolarizing pulse, respectively. Because unclamped APs during the depolarizing voltage step could enhance Ca2+ currents and consequently IAHP, we confirmed that there was not difference in the number of unclamped AP between different groups. Only a small proportion of neurons in both VP16–CREB strains and wild-type mice groups showed more than one unclamped AP (usually a second small spike) but these neurons did not exhibit bigger IsAHP or ImAHP, suggesting that most of the Ca2+ current is evoked by the command step and the contribution of the unclamped APs is not significant. For extracellular recordings transverse hippocampal slices (400 μm) were prepared, incubated in an interface chamber at 32° with oxygenated ACSF and allowed to equilibrate for at least 2 h. For recording of fEPSP in CA1 region both stimulating and recording electrodes were placed in the stratum radiatum of CA1 area. Extracellular activity was recorded simultaneously from stratum pyramidale of the CA1 and CA3 subregions with 1 MΩ pipettes filled with ACSF, filtered at 3 KHz and digitalized at 10 KHz. Power spectrum was calculated from a 250 s period and its area was measured between 0 and 1 KHz as an index of neuronal activity. LTP experiments were performed as previously described (Barco et al., 2002). In all electrophysiological experiments, ‘n‘ indicates the number of cells or slices tested. The number of mice is also indicated in figure legends and Table 1. Two-way ANOVA and Student's t test were used for data analysis. Experimenters were blind to mice genotype.
Table 1.
Electrophysiological properties of CA1 pyramidal neurons in VP16–CREBhigh mice
| Group of mice (n = neurons, mice) | Em (mV) | Rin (MΩ) | τ (ms) | AP amplitude (mV) | AP50 (ms) | dV/dtmax (V/s) | Threshold (mV) | Medium IAHP (pA) | Slow IAHP (pA) |
|---|---|---|---|---|---|---|---|---|---|
| VP16–CREBhigh On (n = 22, 7) | −56 ± 1.1 | 210 ± 20.7 | 21 ± 1.6 | 112 ± 1.6 | 0.99 ± 0.05 | 251 ± 10.4 | −33 ± 1.2 | 120 ± 13.1 | 15 ± 2.5 |
| Wild-type On (n = 16, 5) | −58 ± 2.1 | 181 ± 17.3 | 25 ± 2.7 | 119 ± 1.3 | 1.01 ± 0.03 | 309 ± 9.8 | −39 ± 0.7 | 251 ± 19.8 | 41 ± 6.7 |
| p | 0.40 | 0.29 | 0.13 | 0.004 | 0.71 | <0.001 | <0.001 | <0.001 | <0.001 |
| VP16–CREBhigh On/Off (n = 22, 5) | −60 ± 1.6 | 168 ± 12.5 | 23 ± 1.9 | 120 ± 1.3 | 0.99 ± 0.04 | 316 ± 9.9 | −39 ± 0.5 | 224 ± 15.9 | 36 ± 3.3 |
| Wild-type On/Off (n = 19, 4) | −61 ± 1.7 | 157 ± 10.4 | 23 ± 1.0 | 120 ± 1.2 | 1.01 ± 0.03 | 306 ± 6.0 | −39 ± 0.8 | 231 ± 15.6 | 44 ± 3.5 |
| p | 0.77 | 0.51 | 0.91 | 0.93 | 0.74 | 0.40 | 0.84 | 0.27 | 0.11 |
| VP16–CREBhigh Off (n = 13, 2) | −61 ± 2.1 | 137 ± 10.0 | 18 ± 1.5 | 116 ± 2.2 | 0.89 ± 0.04 | 310 ± 14.7 | −40 ± 1.1 | 211 ± 16.6 | 45 ± 8.6 |
| Wild-type Off (n = 19, 3) | −57 ± 1.4 | 134 ± 10.3 | 19 ± 1.2 | 119 ± 1.2 | 0.86 ± 0.03 | 321 ± 8.4 | −39 ± 1.0 | 243 ± 13.2 | 47 ± 7.6 |
| p | 0.13 | 0.95 | 0.61 | 0.14 | 0.43 | 0.40 | 0.45 | 0.12 | 0.88 |
τ, Membrane time constant; AP50, AP duration at 50% amplitude; dV/dtmax, maximum rate of depolarization. In VP16–CREBhigh On mice, the recordings were performed 10 d after transgene expression. In VP16–CREBhigh On/Off mice, the transgene was expressed for 10 d and then silenced for 10 additional days. In both cases, transgene expression was turned on when the mice were 4 weeks old. VP16–CREBhigh Off mice were 10 weeks old when the experiments were performed. In all these experiments, wild-type mice were housed and maintained in the same conditions than their mutant littermates.
Histological techniques.
Nissl staining was realized as previously described (Mayford et al., 1996). For Timm's staining, mice were anesthetized with ketamine/xylazine, transcardially perfused with buffered 0.37% Na2S solution, pH 7.2, for 5 min followed by 10% neutral buffered formalin (NBF) for 5 min. The brains were removed and postfixed overnight in 10% NBF. 50 μm thick sections were cut with a Leica vibration microtome, collected in 0.1M PB and mounted on the slides. Developer solutions were freshly prepared and mixed in darkness as described (Sloviter, 1982). The slides were incubated in developing solutions in total darkness for 60 min and then rinsed in dH2O and cleared with ethanol/xylene. For immunohistochemistry, mice were anesthetized, perfused with 4% paraformaldehyde, postfixed overnight, and 50 μm sections were obtained. The following primary antibodies were used: α-VP16 (Santa Cruz), α-Synaptophysin, α-MAP-2, α-Calbindin and α-GAP-43 (Sigma). Secondary biotinylated antibodies, streptavidin-peroxidase conjugate and DAB substrate were obtained from Sigma.
Quantitative RT-PCR.
qPCR was performed in an Applied Biosystems 7300 real-time PCR unit using the SYBR mix (Invitrogen) and primers specific for VP16 (forward: 5′ cctacggcgctctggatatg 3′, reverse: 5′cggtaaacatctgctcaaactcg 3′) and GAPDH (forward: 5′ cttcaccaccatggagaaggc 3′, reverse: 5′catggactgtggtcatgagcc 3′) sequences. Each independent sample was assayed in duplicate and VP16 levels were normalized using GAPDH.
Microarray analysis.
U74Av2 genechips were used to analyze changes in gene expression in the hippocampus of VP16–CREBhigh at different times postinduction, data were processed, normalized and statistically analyzed using GCOS 1.2 software as previously described (Barco et al., 2005). This dataset is accessible at the GEO database accession no. (GSE3965). I (increase), D (decrease), MI (mild increase), MD (mild decrease) and NC (no change) calls were obtained according to the statistical thresholds defined by the software. We filtered and sorted the list of genes using the change p value, change call, and Log Ratio Signal. For early time, we requested that the change call for wild-type samples or for those samples in which VP16–CREB expression was turned-off were NC, whereas the change call for samples in which VP16–CREB expression was turned-on for a week (2 samples) indicated a statistically significant change in expression. For late time, we used the same criteria applied to arrays corresponding to 3 or 5 weeks after induction (3 arrays). The two lists of genes so obtained were then analyzed for pathway building and GO group classification using Pathway Studio 5.0 software (Ariadne Genomic Inc.). Similar results were obtained when microarray data were normalized, modeled and filtered using DNA-Chip Analyzer (dChip) software instead of GCOS. Although there were significant differences in the composition of the gene lists generated with each one of these approaches, the results of Pathway Studio analysis were equivalent.
Results
Membrane properties and excitability of VP16–CREB expressing neurons
Our previous research demonstrated that enhanced CREB-mediated gene expression in the bitransgenic strain VP16–CREB reduced the threshold for obtaining the late phase of LTP in the Schaffer collateral pathway (Barco et al., 2002), whereas recent studies indicate that CREB may regulate neuronal excitability in striatal and brainstem neurons (Dong et al., 2006; Han et al., 2006). To investigate whether similar changes in excitability may also occur in the hippocampus, we examined the firing pattern and membrane properties of CA1 pyramidal neurons in VP16–CREB mice. From here on, we will refer to this bitransgenic strain as VP16–CREBhigh to facilitate its comparison with VP16–CREBlow, a new bitransgenic strain which we first describe in a later phase of this study.
The tTA/tetO system of double transgenic mice enabled us to obtain precise temporal regulation of transgene expression through the ability of doxycycline (dox) to block tTA binding to DNA. To avoid possible developmental problems attributable to early expression of CREB-activity, we raised VP16–CREBhigh animals in the presence of dox and induced the expression of the transgene at specific times by removing the drug from the mouse diet. Animals bred under these circumstances were indistinguishable from their wild-type littermates. Intracellular recordings in CA1 pyramidal neurons 10 d after dox removal revealed no differences in passive membrane properties between transgenic mice and their littermates (Table 1). However, the expression of VP16–CREB profoundly affected the firing properties of neurons, increasing the number of action potentials (APs) elicited by depolarizing current injections (Fig. 1A). Thus, CA1 pyramidal neurons from VP16–CREBhigh mice (VP16–CREB On) showed a reduced spike frequency adaptation during a depolarizing pulse and triggered on average more APs than their wild-type littermates (p < 0.001) (Fig. 1B). We also observed a significant increase in the threshold to elicit an AP and decreased AP amplitude and maximum rate of depolarization in CA1 pyramidal neurons from VP16–CREBhigh mice (Table 1). These later changes suggest reduced Na+ currents that could balance the increase in excitability observed in these neurons (Davis, 2006).
Figure 1.

Neuronal excitability is increased in VP16–CREB-expressing neurons. A, Representative CA1 pyramidal neuron response to 200 and 300 pA depolarizing pulse in a wild-type mouse (left), in a VP16–CREBhigh mouse (middle), and in a VP16–CREBhigh mouse that expressed the transgene for 10 d before turning off transgene expression for 10 additional days (right). In both experiments, transgene expression was turned on when the mice were 4 weeks old. Note the lack of spike frequency adaptation in CA1 VP16–CREB-expressing neurons. B, Average number of AP triggered in response to increasing depolarizing currents in neurons from VP16–CREBhigh (n = 22) and from wild-type littermate (n = 16) mice 10 d after inducing transgene expression (VP16-CREB On). This effect was completely reversed 10 d after turning off again transgene expression with dox (VP16–CREB On/Off; p = 0.22; n = 22).
These changes in firing pattern and membrane properties were reversed when VP16–CREB expression was repressed for 10 d with dox (Table 1, VP16–CREBhigh On/Off; Fig. 1A,B). We also recorded from CA1 pyramidal neurons of VP16–CREBhigh mice that were kept always in the presence of dox and never expressed the transgene. In these mice, we did not detect any difference between those transgenics and their littermates (Table 1, VP16–CREB Off). Therefore, the reduced spike frequency adaptation observed in neurons of VP16–CREBhigh mice results from the acute expression of this chimeric protein.
The expression of VP16–CREB reduces AHP in CA1 pyramidal neurons
What alterations in membrane properties underlie the reduction in spike frequency adaptation? Because firing frequency is importantly modulated by the amplitude and duration of the AHP (Madison and Nicoll, 1982; Peters et al., 2005), we investigated the AHP in CA1 pyramidal neurons of VP16–CREBhigh mice. Slow and medium AHP has been proposed to reduce temporal summation and prevent afterdepolarization and multiple action potential firing during intense synaptic events (Wu et al., 2004). We performed voltage-clamp recordings in CA1 pyramidal neurons of VP16–CREBhigh mice and found that the sAHP current were severely reduced (Table 1; Fig. 2A,B). IsAHP in neurons of VP16–CREBhigh mice was 36% of that observed in neurons of control littermates. This change was accompanied by a 52% reduction also in the ImAHP (Table 1; Fig. 2A,C). Both components of the AHP recovered when the expression of the transgene was turned off with dox (Fig. 2B,C, On/Off; Table 1, On/Off), suggesting that modulatory mechanisms for mAHP and sAHP can be downstream of CREB activation.
Figure 2.

Inhibition of AHP in VP16–CREB-expressing neurons. A, Currents underlying AHP in a CA1 pyramidal neuron of a VP16–CREBhigh mouse (black) and a wild-type littermate (gray). Medium (inset) and slow component of the IAHP were reduced in VP16–CREBhigh mice. Neurons were held at −50 mV. Averaged amplitude of slow (B) and medium (C) component of the IAHP in the same neurons presented in Figure 1B (VP16–CREBhigh, black; WT, white). Inhibition of the IsAHP and ImAHP were reversed by silencing again transgene expression for 10 d with dox (VP16-CREB On/Off). *p < 0.001.
The effect of VP16–CREB in AHP current is dose-dependent
To investigate whether the alterations in neuronal excitability and synaptic plasticity were dependent of the level of enhancement of CREB-activity, we compared our results in VP16–CREBhigh mice with those obtained in a lower-expressing strain, referred as VP16–CREBlow (for additional detail, see Materials and Methods). As with VP16–CREBhigh mice, VP16–CREBlow mice expressed the transgene in the hippocampus selectively in granular cells in the dentate gyrus (DG) and CA1 pyramidal neurons, excluding CA3 neurons (Fig. 3A). The quantification of VP16–CREB mRNA levels by real-time RT-PCR revealed that the hippocampal expression of this construct was fivefold higher in VP16–CREBhigh than in VP16–CREBlow mice (Fig. 3B) (p = 0.04). We found that, despite the reduced level of expression, these mice also exhibited a significant increase in neuronal excitability (Fig. 3C) (p < 0.001), although smaller than that observed in VP16–CREBhigh mice. Moreover, VP16–CREBlow mice also presented reduced IsAHP (Fig. 3D) (IsAHP in VP16–CREBlow = 21 ± 4.1 pA, n = 38, 5 mice; IsAHP in WT = 50 ± 6.2 pA, n = 42, 5 mice; p < 0.001). Interestingly, mAHP was, however, not affected in these mutants (Fig. 3D) (ImAHP in VP16–CREBlow = 265 ± 18.9 pA, n = 38, 5 mice; ImAHP in WT = 260 ± 11.7 pA, n = 42, 5 mice; p = 0.81).
Figure 3.
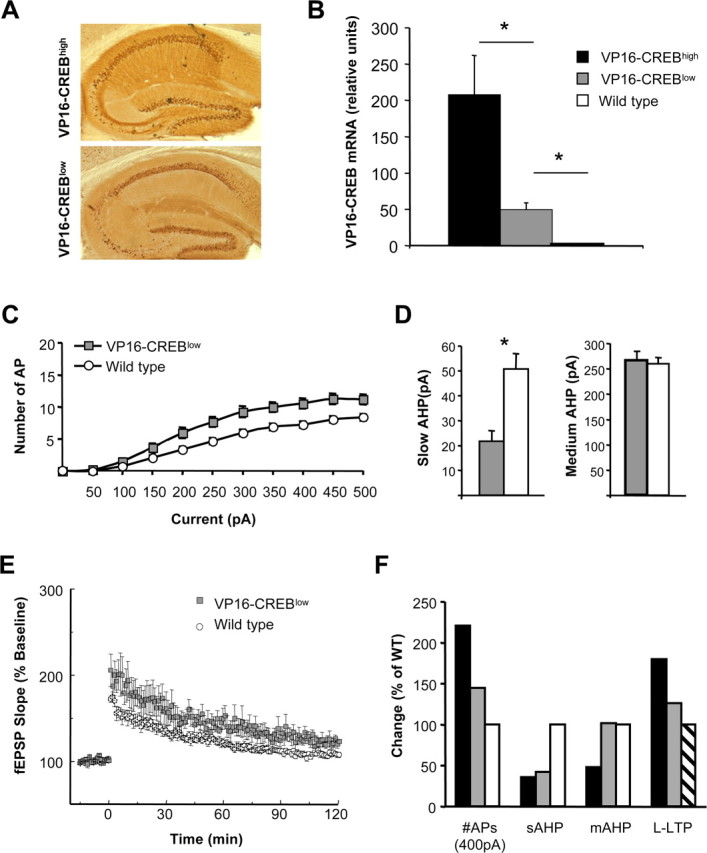
The increase in neuronal excitability and inhibition of IAHP is dose dependent. A, Comparison of the pattern of transgene expression in brain sagittal sections from VP16–CREBhigh and VP16–CREBlow mice using an antibody against the VP16 domain. DAB reaction was longer in the case of VP16–CREBlow to facilitate the identification of positive neurons; therefore, expression levels are not comparable between these two panels. B, Real-time quantitative PCR was used to compare the expression of VP16–CREB transcripts in the hippocampus of VP16–CREBhigh (3 weeks after induction) and VP16–CREBlow mice (VP16–CREBhigh vs pulled WT, p = 0.01; VP16–CREBlow vs pulled WT, p = 0.006; 3 mice per group). C, Average number of AP triggered in response to increasing depolarizing currents in VP16–CREBlow (n = 42, 5 mice) and wild-type littermate (n = 38, 5 mice) mice. D, Averaged amplitude of the IsAHP and ImAHP of the same neurons represented in C. Only IsAHP was significantly affected. *p < 0.001. E, A single 100 Hz train (1 s) evoked E-LTP in hippocampal slices from wild-type mice but a longer-lasting form of LTP in VP16–CREBlow mice. F, Comparison of electrophysiological phenotypes in the low and high expressing strains. Data are taken from Figures 1B and 3C (#APs 400 mA), from Figures 2B and 3D (IsAHP), from Figures 2C and 3D (ImAHP), and from Figure 3C by Barco et al. (2002) and 3E in this article (L-LTP, corresponding to the average amplitude response 90-120 min after LTP induction). Values are normalized to those obtained for wild-type littermates, except for L-LTP in which values are expressed as percentage of baseline response (dashed bar).
The reduction in IsAHP correlated with a reduced threshold for the induction of L-LTP in both VP16–CREBhigh and VP16–CREBlow mice. Like VP16–CREBhigh mice (Barco et al., 2002), VP16–CREBlow animals showed an enhanced response to one standard 100 Hz tetanus train of 1 s duration. This stimulation evoked a LTP with an enhanced and sustained long-lasting phase in mutant mice, but only produced a nonsaturating shorter-lasting LTP (E-LTP) in wild-type littermates (Fig. 3E) (90-120 min: VP16–CREBlow: 126 ± 11%, n = 4; WT: 109 ± 6%, n = 4; p = 0.034).
The comparison of the results obtained in both bitransgenic strains (Fig. 3F) shows that the magnitude of the alterations in neuronal excitability and synaptic plasticity correlates well with the level of expression of VP16–CREB.
Long-term consequences of enhanced CREB-mediated gene expression and excitability
We next turned to examine the long-term consequences of enhanced CREB-mediated gene expression and found that the continuous expression of this chimeric transcription factor for several weeks could cause the premature death of the mouse. Indeed, mice expressing VP16–CREB for more than 3-4 weeks occasionally showed spontaneous seizures during handling. These seizures frequently preceded the death of the mouse by 24-48 h (Fig. 4), although ∼50% of the animals survived for more than 8 weeks. In contrast, the low-expressing strain VP16–CREBlow showed a normal life span even when the transgene was expressed through embryonic development and the whole life of the animal (results not shown).
Figure 4.

Survival of VP16–CREBhigh mice. The survival of VP16–CREBhigh mice (black circles) is compromised after 3-4 weeks of transgene expression. The reduction in the lifespan and the occasional seizures were not observed in wild-type littermates (crosses), in mice maintained in the presence of dox (data not shown), or in mice with periodic induction of VP16–CREB expression (white circles). The upper schemes indicate the transgene induction protocol used for VP16–CREBhigh On (dox removal after 2 months of age) and for mice with periodic induction of VP16–CREB expression (VP16-CREBhigh On/Off/On… ), whose diet alternated every 2 weeks between regular food and food supplemented with dox.
The premature death of VP16–CREBhigh mice could be completely prevented by turning off transgene expression with dox during the first month of its induction (data not shown). Even when transgene expression was repeatedly induced several times during the life of the animal by switching between regular food and food supplemented with dox, the deleterious effects were only manifested when the expression was sustained for more than 3-4 weeks (Fig. 4). Conversely, when VP16–CREBhigh mice were kept off dox during their whole life, including embryonic development, most mice died before reaching the third moth of age. As described for mice in which we induced VP16–CREB expression during adulthood, we occasionally observed severe seizures that preceded the premature death of the animal.
The sustained activation of CRE-driven gene expression causes the loss of CA1 pyramidal neurons
At the time in which we performed the electrophysiological characterization of these mice (one-two weeks after transgene induction) no gross anatomical difference was observed in the brain of transgenic and control littermates (see time course in Fig. 5). In contrast, VP16–CREBhigh transgenic mice expressing this chimeric transcription factor for several weeks showed progressive cell loss in the CA1 and DG regions. This neurodegenerative process could be easily visualized by Nissl staining (Fig. 5A) and by immunostaining with a number of neuronal markers (Fig. 5B). In accordance with these results, the neurodegenerative process in the DG severely reduced the mossy fiber projection after 6 weeks of transgene expression as revealed by Timm's staining (Fig. 5C).
Figure 5.
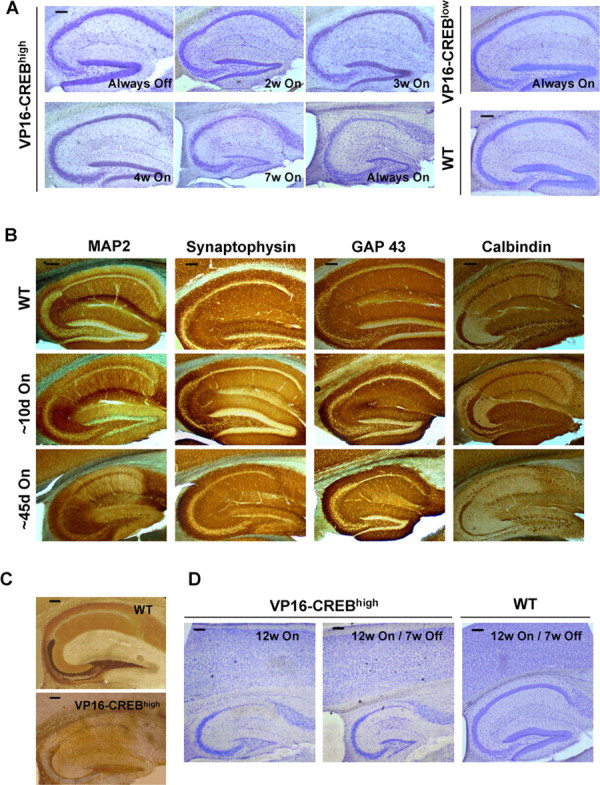
Sustained transgene expression causes cell loss in the hippocampus of VP16–CREBhigh mice. A, Nissl staining of the hippocampus of VP16–CREBhigh mice at different times after transgene induction expressed in weeks. Always On indicates that the animal was kept off dox through its whole life, including embryonic development. Panel VP16–CREBlow shows the hippocampus of a 1-year-old transgenic mouse. B, Immunohistochemical analysis of the hippocampus of transgenics (VC) and control mice. Floating vibratome sections (50 μm) were stained with antibodies against calbindin, GAP-43, synaptophysin, and MAP2. No significant differences were found in either hippocampus or other brain regions at early times (∼10d On), but significant neuronal loss was observed after more than 6 weeks of transgene expression (∼45d On). C, Timm's staining of brain vibratome sagittal sections of a wild-type and a VP16–CREBhigh mouse 6 weeks after dox removal. D, Nissl staining of the hippocampus of VP16–CREBhigh mice at different times after transgene induction and repression. All the experiments were realized in adult mice. Transgene expression was turned on when the animals were at least 2 month old. Scale bars, 140 μm.
Although we observed significant variability in the onset of neurodegeneration (from 3 to 5 weeks), all VP16–CREBhigh mice investigated exhibited severe cell loss 6 weeks after dox removal. The onset of cell loss in the CA1 pyramidal layer seemed to coincide with the occasional observation of epileptic seizures, suggesting that these two events might be related. Mice in which we alternated 2 weeks of regular mouse diet with 2 weeks of food supplemented with dox never showed epileptic attacks (Fig. 4) and exhibited normal hippocampal anatomy (data not shown) despite accumulating several months of transgene expression. Indeed, the degenerative process could be stopped at any time by turning transgene expression off, although this did not cause a reversal of the lesion produced during the expression of the transgene (Fig. 5D). Interestingly, no cell loss was observed in VP16–CREBlow mice even after one year of transgene expression, indicating that a milder increase in neuronal excitability was compatible with neuronal survival (Fig. 5A, VP16–CREBlow).
Physiological and pathological regulation of neuronal excitability and gene expression by VP16–CREB
What are the consequences of the early increase in excitability and the late cellular loss in the activity of hippocampal circuits? To investigate this issue we performed field recordings in acute hippocampal slices from VP16–CREBhigh mice 1 week after dox removal. We found that basal synaptic transmission in the Schaffer collateral pathway was normal (Fig. 6A), but there was a clear increase in spontaneous activity at the CA1 subfield (Fig. 6B) (p = 0.01), as estimated by the power spectrum analysis, that correlated with the increased neuronal excitability observed in CA1 pyramidal neurons. In contrast, activity in the CA3 subfield, in which the transgene is not expressed, was unaltered (Fig. 6B).
Figure 6.

Increased spontaneous activity in the hippocampal circuit. A, Input/output curve of fEPSP slope (millivolts per milliseconds) versus stimulus at the Schaffer collateral pathway of hippocampal slices from VP16–CREBhigh (n = 31) and wild-type (n = 36) littermates 1 week after induction (p = 0.27). B, Cumulative probability versus area of the power spectra calculated from 250 s recordings at the CA1 (left) and CA3 (right) subfields in the same slices used in A. C, Input/output curve of hippocampal slices from VP16–CREBhigh (n = 45) and wild-type (n = 37) littermates 3 weeks after induction (p < 0.001). D, Cumulative probability versus area of the power spectra calculated from 250 s recordings in the CA1 (left) and CA3 (right) subfields in the same slices used in C. In all cases, transgene expression was turned on when the mice were 4 weeks old.
When the expression of VP16–CREB was sustained for more than 3 weeks, we observed a significant reduction in the response of CA1 pyramidal neurons to stimulation of afferent CA3 axons (Fig. 6C), probably reflecting the incipient loss of synapses in the CA1 subfield. This reduction may compensate the previously observed shifting toward an increase in spontaneous activity of CA1 pyramidal neurons and, consequently, no increase in spontaneous activity in the CA1 subfield was observed after 3 weeks (Fig. 6D). Strikingly, we observed, however, an increase in spontaneous activity in the CA3 subfield (Fig. 6D) (p = 0.002), suggesting the existence of readjustments in the hippocampal circuit to compensate the ongoing neuronal loss.
Because sAHP can be inhibited by glutamate through kainate receptors activation (Melyan et al., 2002), we investigated whether an increase in extracellular glutamate concentration resulting from the neurodegenerative process initiated in VP16–CREBhigh mice could underlie the reduction of the sAHP. We measured IsAHP of neurons from slices of VP16–CREBhigh mice that were either incubated for 2 h in the presence of 5 μm NBQX, an AMPA/kainate antagonist, or maintained in normal ACSF. No differences were found between the two groups of neurons (NBQX: IsAHP = 26 ± 6.2pA, n = 15; veh: IsAHP = 26 ± 3.6 pA, n = 17; p = 0.99).Together, these results indicate that the increase in excitability precedes the onset and perhaps contributes to the neurodegenerative process observed in the hippocampus of VP16–CREBhigh mice.
The gene profiling analysis performed in VP16–CREBhigh mice (Barco et al., 2005) also supports a dynamic scenario in which a physiological CREB-mediated response out of context eventually triggers a neurodegenerative process. The re-analysis of our microarray data using Pathway Studio software comparing early and late times (1 week vs >3 weeks) after transgene induction revealed clear differences between these two stages. At early times, the groups more represented in the list of significantly altered genes were signal transduction, transport, transcriptional regulation and synaptic transmission. In contrast, at late times, there was a overrepresentation of gene groups related to pathological responses (immune and defense response, proteolysis, apoptosis, inflammation) (Fig. 7A). Moreover, we also used Pathway Studio software for pathway building and visualization and found that the overall connectivity of altered genes to CREB decreased significantly at later times (Fig. 7B), suggesting that these late changes result from indirect effects that could be mediated by the altered expression of some early target genes.
Figure 7.
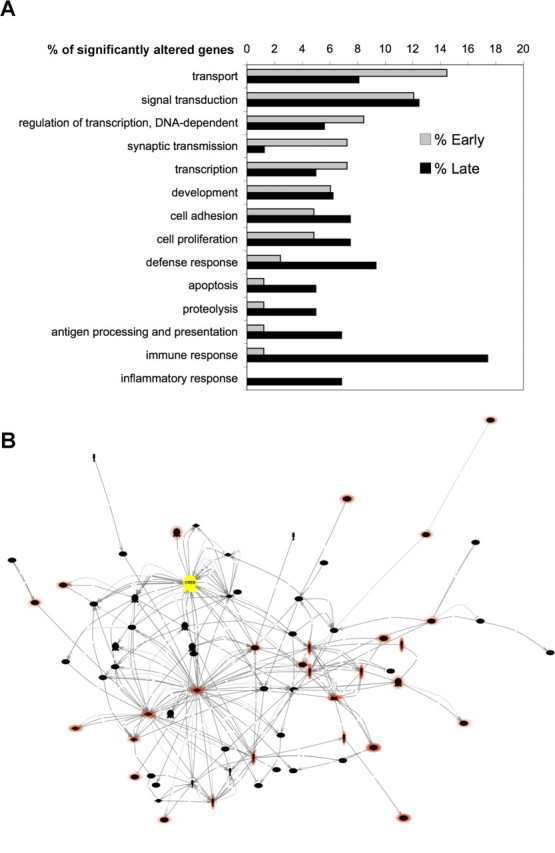
Altered gene expression in VP16–CREBhigh mice. A, Main cellular processes, as defined in gene ontology (GO), affected early (1 week) or late (>3 weeks) after transgene induction. We compared those groups that represent at least a 5% of significantly altered genes either early or late after expression. The main cellular process groups among the 84 probe sets significantly altered during the first week after dox removal included transport, transcription regulation, signal transduction, and synaptic transmission. However, the main GO groups represented in the 161 probe sets significantly altered at late times were related to defense and inflammatory responses, apoptosis, and proteolysis. B, Pathway created with the 161 probe sets significantly altered at late times using Pathways Studio 5.0 software; only direct interactions were considered and unlinked entities were excluded. CREB is labeled in yellow, whereas genes related to pathological response are highlighted in red (red intensities reflect the overlapping of pathology-related groups). Note that most red nodes are not directly connected to CREB. The comparison of this pathway with the pathway obtained for early changes (data not shown) revealed a reduced connectivity with CREB (CREB participates in 48% of early interactions but only in 17% of late interactions) and a much larger presence of genes related with pathological situations (for additional information, see supplemental Fig. S1, available at www.jneurosci.org as supplemental material).
Discussion
Modulation of intrinsic excitability of CA1 pyramidal neurons by CREB-mediated gene expression: Implications for learning and memory
Our results indicate that the enhanced expression of CRE-driven genes increases the neuronal excitability of CA1 pyramidal neurons and reduces the threshold for LTP in the Schaffer collateral pathway. These two effects might be at least partially mediated by a reduction of AHP currents. However, when a high level of CREB activity is maintained for a long period of time and a given threshold is reached, epileptic activity develops in the hippocampal circuit and causes neuronal loss. These deleterious effects are dose-dependent and were not observed in a transgenic strain that express VP16–CREB at lower levels, although these mice also showed increased excitability.
Two recent studies have found that a similar CREB variant, when expressed using viral vectors, increased the excitability of neurons in the locus ceruleus (Han et al., 2006) and the nucleus accumbens (Dong et al., 2006), suggesting that the modulation of intrinsic neuronal properties is a well conserved CREB function through different neuronal types in the CNS. However, the specific changes in gene expression underlying these increments in excitability remain unknown. We describe here that depression of AHP might underlie the changes in neuronal excitability triggered by enhanced CREB function in CA1 pyramidal neurons. In particular, sAHP was affected both in the low and in the high expression bitransgenic strains, whereas mAHP was reduced only in mice expressing high levels of CREB activity. AHP controls the responsiveness of neurons (Sah, 1996) and its modulation can be a powerful mechanism for regulating NMDA-R-mediated plasticity related to learning (Wu et al., 2004; Faber et al., 2005). Blockade of the sAHP or mAHP converts short-lasting forms of potentiation into L-LTP (Sah and Bekkers, 1996; Cohen et al., 1999; Haug and Storm, 2000; Faber et al., 2005), whereas the activation of the AHP increases the threshold for induction of LTP (Sah and Bekkers, 1996). Interestingly, learning hippocampus-dependent tasks, such as eye blinking conditioning or navigation in the Morris water maze, causes a reduction of sAHP in CA1 pyramidal neurons (Moyer et al., 2000; Oh et al., 2003), a phenomenon that may favor learning by reducing the threshold for late forms of LTP.
Although our knowledge of the molecular underpinnings of the plasticity of membrane properties is still limited, the prime candidates to regulate these changes primarily overlap with those thought to regulate LTP. These include adenylyl cyclase and a number of kinases known to phosphorylate CREB (Zhang and Linden, 2003). In particular, variations in cAMP levels and PKA activity, a signaling cascade directly linked to CREB activation in neurons, play a major role in the regulation of the sAHP current. Thus, forskolin inhibits the current underlying sAHP by 90%, whereas the PKA inhibitor Rp-cAMPS increased the current, suggesting that PKA maintains sAHP channels in the closed state (Vogalis et al., 2003). Indeed, suppression of sAHP seems to be mainly mediated by PKA in CA1 pyramidal neurons (Pedarzani and Storm, 1993, 1995; Haug and Storm, 2000). In the case of mAHP, the inhibition observed in VP16–CREBhigh mice might result from modulation of the SK current (Sah and Faber, 2002) or M current (Gu et al., 2005), whose suppression in transgenic animals also increased the excitability of CA1 pyramidal neurons (Peters et al., 2005). Although neither CREB nor de novo gene expression has been so far directly involved in the modulation of AHP, the persistence of the changes in neuronal excitability and sAHP observed during behavioral training suggests the participation of molecular mechanisms that can support long-lasting changes, such as de novo gene expression and protein synthesis. We therefore propose that CREB-mediated changes in excitability of different neuronal subtypes may play a relevant role regulating physiological neuronal responses during addiction and learning and memory. Indeed, a reemerging view in the learning and memory field proposes that although synaptic changes, such as LTP or LTD, are likely to represent the most important cellular mechanism for memory storage given its computational properties, other forms of plasticity, such as changes in neuronal intrinsic excitability, may also importantly contribute to memory storage (Kandel, 1967; Zhang and Linden, 2003). Although the encoding capacity of those mechanisms is significantly lower than synaptic alterations, intrinsic changes might function as a trigger for consolidation or adaptative generalization of memories. Thus, AHP, in addition to its established role in spike frequency adaptation, may work as an adjustable gain control (Sah and Bekkers, 1996). Ascending monoaminergic fibers may facilitate hippocampal LTP induction and LTP-dependent learning processes in a global manner during arousal and attention by suppressing this current (Deng et al., 2007; Reymann and Frey, 2007). This process might be mediated by CREB activation, which is known to be downstream of such inputs (Berke and Hyman, 2000).
We have proposed that the sustained activation of CREB-mediated gene expression causes a cell-wide facilitation that prime the synapses for subsequent induction of L-LTP by a single tetanus (Barco et al., 2002; Alarcon et al., 2006). Now, we found that in the same mice shortly after transgene induction there is a significant reduction in the value of the IsAHP, a current that has been related to learning and memory as much as has L-LTP. Are these two phenomena two manifestation of the same molecular process? The inhibition of sAHP may well represent the cellular mechanism underlying cell-wide facilitation in VP16–CREB mice, although it cannot explain the resistance to protein and RNA synthesis inhibitors of L-LTP in these mutants, suggesting that other processes also contribute to this phenotype. The specific CREB target genes controlling these changes are still unknown, but it has been suggested that BDNF, a well known CREB downstream gene and the main effector molecule that emerged from our gene profiling analysis of VP16–CREBhigh mice (Barco et al., 2005), contributes to control the coordinated regulation of synaptic and intrinsic properties aimed at allowing neuronal networks to adapt to long-lasting changes in activity (Desai et al., 1999).
Deleterious effects of the prolonged activation of CREB-mediated gene expression
During normal activity of the brain, the neurons receive transient signals and initiate transcriptional responses that are important for learning and memory. However, more intense or sustained stimulation (for example those used in the kindling model) may initiate inappropriate gene expression response and lead to the formation of epileptic neuronal circuits and disorders of neuronal excitability (McEachern and Shaw, 1999). A physiological process can thereby become pathological. The deleterious effects observed at late times in VP16–CREBhigh mice could be the undesired consequence of a physiological function of CREB missing regulatory negative feedback mechanisms. VP16–CREBhigh mice may, therefore, represent a useful model to investigate the cellular and molecular alterations associated with epileptogenic activity in the temporal lobe, as well as to assay possible therapeutic approaches. The reversibility and possibilities for regulation provided by the tTA/tetO system of double transgenics would be very useful in such studies.
The genechip analysis performed in VP16–CREBhigh mice provides some additional clues for understanding this neurodegenerative process. The list of genes upregulated by VP16–CREBhigh included BDNF, a neurotrophin that, as CREB itself, is known to play an important role in neuroprotection. Genetic and pharmacological studies have shown that increased expression of BDNF leads to hyperexcitability and epileptiform activities, whereas suppression of BDNF function inhibits the development of seizures (Kokaia et al., 1995; Elmer et al., 1997; Binder et al., 1999; Croll et al., 1999; Tandon et al., 1999; Lahteinen et al., 2002). These findings suggest that although BDNF may have a neuroprotective role, too much BDNF may also have adverse effects. VP16–CREBhigh mice also overexpress diverse MHC I molecules, which have also been involved in adult synaptic plasticity (Huh et al., 2000; Boulanger and Shatz, 2004; Goddard et al., 2007). Although under normal circumstances, the blood brain barrier prevents entry of leukocytes, antibodies, complement factors and cytokines into the brain parenchyma, a significant increase in MHCI expression, such as in chronic epilepsy or after prolonged VP16–CREB expression, might make the expressing neurons more susceptible to destruction by invading cells of the immune system. The prolonged overexpression of both BDNF and MHC I molecules, individually or in combination with other molecules, could trigger the neurodegenerative process and complex molecular changes observed in mice that expressed VP16–CREB for several weeks or months.
Therapeutic enhancement of the CREB pathway
CREB-dependent gene expression is necessary to maintain the survival of different neuronal subtypes both in vitro and in vivo (Bonni et al., 1999; Riccio et al., 1999; Lonze et al., 2002; Mantamadiotis et al., 2002; Papadia et al., 2005; Parlato et al., 2006). Moreover, the transient expression of the constitutively active CREB variant VP16–CREB has been shown to promote axon regeneration (Gao et al., 2004), neurogenesis (Zhu et al., 2004) and neuronal survival in vitro (Andreatta et al., 2004; Lee et al., 2005; Deng et al., 2006). Furthermore, transcriptional dysregulation in the CREB pathway has been proposed to play a central role in the pathogenesis of various neurodegenerative disorders, including Alzheimer's and Huntington's diseases. Based in these results, CREB activation has been proposed as a possible target of therapeutic approaches for neurodegenerative disorders (Barco et al., 2003; Tully et al., 2003).
Our results in CREB mutant mice support the critical role of CREB promoting neuronal survival and controlling learning-related plasticity, but also highlight the dangers that may be associated to the manipulation of a high level of activity in this pathway. This knowledge, however, does not reduce the promise of drugs targeted to this pathway for treating memory or neurodegenerative disorders. A better understanding of the molecular mechanisms underlying CREB activation and function should make possible the design and development of pharmaceuticals that enhance CREB-mediated gene induction at more physiological levels without excessively elevating basal activity. This type of enhancement should have a beneficial impact in situations in which this signaling cascade is impaired.
Footnotes
A.B., M.L.d.A., D.J., and R.O. are supported by European Commission Grants MEXT-CT-2003-509550 and MIRG-CT-2005-016343, Spanish Ministerio de Educación y Ciencia Grants BFU2005-00286 and SAF2005-24584-E, and Generalitat Valenciana Grant ACOMP06/061. E.R.K. and J.M.A. are supported by Howard Hughes Medical Institute and the Kavli Institute for Brain Sciences. We thank Eloisa Herrera, Juan Lerma, Pankaj Sah, Luis M. Valor, and Jose Viosca for critical reading of this manuscript and helpful comments.
Disclosure of financial interest: E.R.K. is one of four founders of Memory Pharmaceuticals and Chairman of its Scientific Advisory Board. Memory Pharmaceuticals is concerned with developing drugs for age-related memory loss. Some of these drugs are also potentially useful in depression and schizophrenia.
References
- Alarcon JM, Barco A, Kandel ER. Capture of the late phase of long-term potentiation within and across the apical and basilar dendritic compartments of CA1 pyramidal neurons: synaptic tagging is compartment restricted. J Neurosci. 2006;26:256–264. doi: 10.1523/JNEUROSCI.3196-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreatta CP, Nahreini P, Hanson AJ, Prasad KN. Regulated expression of VP16CREB in neuroblastoma cells: analysis of differentiation and apoptosis. J Neurosci Res. 2004;78:570–579. doi: 10.1002/jnr.20304. [DOI] [PubMed] [Google Scholar]
- Balschun D, Wolfer DP, Gass P, Mantamadiotis T, Welzl H, Schutz G, Frey JU, Lipp HP. Does cAMP response element-binding protein have a pivotal role in hippocampal synaptic plasticity and hippocampus-dependent memory? J Neurosci. 2003;23:6304–6314. doi: 10.1523/JNEUROSCI.23-15-06304.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barco A, Alarcon JM, Kandel ER. Expression of constitutively active CREB protein facilitates the late phase of long-term potentiation by enhancing synaptic capture. Cell. 2002;108:689–703. doi: 10.1016/s0092-8674(02)00657-8. [DOI] [PubMed] [Google Scholar]
- Barco A, Pittenger C, Kandel ER. CREB, memory enhancement and the treatment of memory disorders: promises, pitfalls and prospects. Expert Opin Ther Targets. 2003;7:101–114. doi: 10.1517/14728222.7.1.101. [DOI] [PubMed] [Google Scholar]
- Barco A, Patterson S, Alarcon JM, Gromova P, Mata-Roig M, Morozov A, Kandel ER. Gene expression profiling of facilitated L-LTP in VP16-CREB mice reveals that BDNF is critical for the maintenance of LTP and its synaptic capture. Neuron. 2005;48:123–137. doi: 10.1016/j.neuron.2005.09.005. [DOI] [PubMed] [Google Scholar]
- Berke JD, Hyman SE. Addiction, dopamine, and the molecular mechanisms of memory. Neuron. 2000;25:515–532. doi: 10.1016/s0896-6273(00)81056-9. [DOI] [PubMed] [Google Scholar]
- Binder DK, Routbort MJ, McNamara JO. Immunohistochemical evidence of seizure-induced activation of trk receptors in the mossy fiber pathway of adult rat hippocampus. J Neurosci. 1999;19:4616–4626. doi: 10.1523/JNEUROSCI.19-11-04616.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bito H, Deisseroth K, Tsien RW. CREB phosphorylation and dephosphorylation: a Ca2+- and stimulus duration-dependent switch for hippocampal gene expression. Cell. 1996;87:1203–1214. doi: 10.1016/s0092-8674(00)81816-4. [DOI] [PubMed] [Google Scholar]
- Blendy JA, Kaestner KH, Schmid W, Gass P, Schutz G. Targeting of the CREB gene leads to up-regulation of a novel CREB mRNA isoform. EMBO J. 1996;15:1098–1106. [PMC free article] [PubMed] [Google Scholar]
- Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science. 1999;286:1358–1362. doi: 10.1126/science.286.5443.1358. [DOI] [PubMed] [Google Scholar]
- Boulanger LM, Shatz CJ. Immune signalling in neural development, synaptic plasticity and disease. Nat Rev Neurosci. 2004;5:521–531. doi: 10.1038/nrn1428. [DOI] [PubMed] [Google Scholar]
- Bourtchuladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva AJ. Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell. 1994;79:59–68. doi: 10.1016/0092-8674(94)90400-6. [DOI] [PubMed] [Google Scholar]
- Cohen AS, Coussens CM, Raymond CR, Abraham WC. Long-lasting increase in cellular excitability associated with the priming of LTP induction in rat hippocampus. J Neurophysiol. 1999;82:3139–3148. doi: 10.1152/jn.1999.82.6.3139. [DOI] [PubMed] [Google Scholar]
- Croll SD, Suri C, Compton DL, Simmons MV, Yancopoulos GD, Lindsay RM, Wiegand SJ, Rudge JS, Scharfman HE. Brain-derived neurotrophic factor transgenic mice exhibit passive avoidance deficits, increased seizure severity and in vitro hyperexcitability in the hippocampus and entorhinal cortex. Neuroscience. 1999;93:1491–1506. doi: 10.1016/s0306-4522(99)00296-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GW. Homeostatic control of neural activity: from phenomenology to molecular design. Annu Rev Neurosci. 2006;29:307–323. doi: 10.1146/annurev.neuro.28.061604.135751. [DOI] [PubMed] [Google Scholar]
- Deisseroth K, Bito H, Tsien RW. Signaling from synapse to nucleus: postsynaptic CREB phosphorylation during multiple forms of hippocampal synaptic plasticity. Neuron. 1996;16:89–101. doi: 10.1016/s0896-6273(00)80026-4. [DOI] [PubMed] [Google Scholar]
- Deng P, Zhang Y, Xu ZC. Involvement of Ih in dopamine modulation of tonic firing in striatal cholinergic interneurons. J Neurosci. 2007;27:3148–3156. doi: 10.1523/JNEUROSCI.5535-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Neve RL, Rosenberg PA, Volpe JJ, Jensen FE. AMPA receptor subunit composition and CREB regulate oligodendrocyte excitotoxicity. J Biol Chem. 2006;281:36004–36011. doi: 10.1074/jbc.M606459200. [DOI] [PubMed] [Google Scholar]
- Desai NS, Rutherford LC, Turrigiano GG. BDNF regulates the intrinsic excitability of cortical neurons. Learn Mem. 1999;6:284–291. [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Green T, Saal D, Marie H, Neve R, Nestler EJ, Malenka RC. CREB modulates excitability of nucleus accumbens neurons. Nat Neurosci. 2006;9:475–477. doi: 10.1038/nn1661. [DOI] [PubMed] [Google Scholar]
- Elmer E, Kokaia M, Ernfors P, Ferencz I, Kokaia Z, Lindvall O. Suppressed kindling epileptogenesis and perturbed BDNF and TrkB gene regulation in NT-3 mutant mice. Exp Neurol. 1997;145:93–103. doi: 10.1006/exnr.1997.6478. [DOI] [PubMed] [Google Scholar]
- Faber ES, Delaney AJ, Sah P. SK channels regulate excitatory synaptic transmission and plasticity in the lateral amygdala. Nat Neurosci. 2005;8:635–641. doi: 10.1038/nn1450. [DOI] [PubMed] [Google Scholar]
- Gao Y, Deng K, Hou J, Bryson JB, Barco A, Nikulina E, Spencer T, Mellado W, Kandel ER, Filbin MT. Activated CREB is sufficient to overcome inhibitors in myelin and promote spinal axon regeneration in vivo. Neuron. 2004;44:609–621. doi: 10.1016/j.neuron.2004.10.030. [DOI] [PubMed] [Google Scholar]
- Gass P, Wolfer DP, Balschun D, Rudolph D, Frey U, Lipp HP, Schutz G. Deficits in memory tasks of mice with CREB mutations depend on gene dosage. Learn Mem. 1998;5:274–288. [PMC free article] [PubMed] [Google Scholar]
- Goddard CA, Butts DA, Shatz CJ. Regulation of CNS synapses by neuronal MHC class I. Proc Natl Acad Sci USA. 2007;104:6828–6833. doi: 10.1073/pnas.0702023104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu N, Vervaeke K, Hu H, Storm JF. Kv7/KCNQ/M and HCN/h, but not KCa2/SK channels, contribute to the somatic medium after-hyperpolarization and excitability control in CA1 hippocampal pyramidal cells. J Physiol (Lond) 2005;566:689–715. doi: 10.1113/jphysiol.2005.086835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han MH, Bolanos CA, Green TA, Olson VG, Neve RL, Liu RJ, Aghajanian GK, Nestler EJ. Role of cAMP response element-binding protein in the rat locus ceruleus: regulation of neuronal activity and opiate withdrawal behaviors. J Neurosci. 2006;26:4624–4629. doi: 10.1523/JNEUROSCI.4701-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haug T, Storm JF. Protein kinase A mediates the modulation of the slow Ca2+-dependent K+ current, I(sAHP), by the neuropeptides CRF, VIP, and CGRP in hippocampal pyramidal neurons. J Neurophysiol. 2000;83:2071–2079. doi: 10.1152/jn.2000.83.4.2071. [DOI] [PubMed] [Google Scholar]
- Huang YY, Pittenger C, Kandel ER. A form of long-lasting, learning-related synaptic plasticity in the hippocampus induced by heterosynaptic low-frequency pairing. Proc Natl Acad Sci USA. 2004;101:859–864. doi: 10.1073/pnas.2237201100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh GS, Boulanger LM, Du H, Riquelme PA, Brotz TM, Shatz CJ. Functional requirement for class I MHC in CNS development and plasticity. Science. 2000;290:2155–2159. doi: 10.1126/science.290.5499.2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummler E, Cole TJ, Blendy JA, Ganss R, Aguzzi A, Schmid W, Beermann F, Schutz G. Targeted mutation of the CREB gene: compensation within the CREB/ATF family of transcription factors. Proc Natl Acad Sci USA. 1994;91:5647–5651. doi: 10.1073/pnas.91.12.5647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Impey S, Mark M, Villacres EC, Poser S, Chavkin C, Storm DR. Induction of CRE-mediated gene expression by stimuli that generate long-lasting LTP in area CA1 of the hippocampus. Neuron. 1996;16:973–982. doi: 10.1016/s0896-6273(00)80120-8. [DOI] [PubMed] [Google Scholar]
- Josselyn SA, Nguyen PV. CREB, synapses and memory disorders: past progress and future challenges. Curr Drug Targets CNS Neurol Disord. 2005;4:481–497. doi: 10.2174/156800705774322058. [DOI] [PubMed] [Google Scholar]
- Kandel ER. Cellular studies of learning. In: Quarton GC, Melnechuk T, Schmitt FO, editors. The neurosciences: a study program. New York: Rockefeller UP; 1967. pp. xiii–962. [Google Scholar]
- Kokaia M, Ernfors P, Kokaia Z, Elmer E, Jaenisch R, Lindvall O. Suppressed epileptogenesis in BDNF mutant mice. Exp Neurol. 1995;133:215–224. doi: 10.1006/exnr.1995.1024. [DOI] [PubMed] [Google Scholar]
- Lahteinen S, Pitkanen A, Saarelainen T, Nissinen J, Koponen E, Castren E. Decreased BDNF signalling in transgenic mice reduces epileptogenesis. Eur J Neurosci. 2002;15:721–734. doi: 10.1046/j.1460-9568.2002.01897.x. [DOI] [PubMed] [Google Scholar]
- Lee B, Butcher GQ, Hoyt KR, Impey S, Obrietan K. Activity-dependent neuroprotection and cAMP response element-binding protein (CREB): kinase coupling, stimulus intensity, and temporal regulation of CREB phosphorylation at serine 133. J Neurosci. 2005;25:1137–1148. doi: 10.1523/JNEUROSCI.4288-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35:605–623. doi: 10.1016/s0896-6273(02)00828-0. [DOI] [PubMed] [Google Scholar]
- Lonze BE, Riccio A, Cohen S, Ginty DD. Apoptosis, axonal growth defects, and degeneration of peripheral neurons in mice lacking CREB. Neuron. 2002;34:371–385. doi: 10.1016/s0896-6273(02)00686-4. [DOI] [PubMed] [Google Scholar]
- Lu YF, Kandel ER, Hawkins RD. Nitric oxide signaling contributes to late-phase LTP and CREB phosphorylation in the hippocampus. J Neurosci. 1999;19:10250–10261. doi: 10.1523/JNEUROSCI.19-23-10250.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison DV, Nicoll RA. Noradrenaline blocks accommodation of pyramidal cell discharge in the hippocampus. Nature. 1982;299:636–638. doi: 10.1038/299636a0. [DOI] [PubMed] [Google Scholar]
- Mantamadiotis T, Lemberger T, Bleckmann SC, Kern H, Kretz O, Martin Villalba A, Tronche F, Kellendonk C, Gau D, Kapfhammer J, Otto C, Schmid W, Schutz G. Disruption of CREB function in brain leads to neurodegeneration. Nat Genet. 2002;31:47–54. doi: 10.1038/ng882. [DOI] [PubMed] [Google Scholar]
- Marie H, Morishita W, Yu X, Calakos N, Malenka RC. Generation of silent synapses by acute in vivo expression of CaMKIV and CREB. Neuron. 2005;45:741–752. doi: 10.1016/j.neuron.2005.01.039. [DOI] [PubMed] [Google Scholar]
- Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER. Control of memory formation through regulated expression of a CaMKII transgene. Science. 1996;274:1678–1683. doi: 10.1126/science.274.5293.1678. [DOI] [PubMed] [Google Scholar]
- McEachern JC, Shaw CA. The plasticity-pathology continuum: defining a role for the LTP phenomenon. J Neurosci Res. 1999;58:42–61. [PubMed] [Google Scholar]
- Melyan Z, Wheal HV, Lancaster B. Metabotropic-mediated kainate receptor regulation of IsAHP and excitability in pyramidal cells. Neuron. 2002;34:107–114. doi: 10.1016/s0896-6273(02)00624-4. [DOI] [PubMed] [Google Scholar]
- Moyer JR, Jr, Power JM, Thompson LT, Disterhoft JF. Increased excitability of aged rabbit CA1 neurons after trace eyeblink conditioning. J Neurosci. 2000;20:5476–5482. doi: 10.1523/JNEUROSCI.20-14-05476.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh MM, Kuo AG, Wu WW, Sametsky EA, Disterhoft JF. Watermaze learning enhances excitability of CA1 pyramidal neurons. J Neurophysiol. 2003;90:2171–2179. doi: 10.1152/jn.01177.2002. [DOI] [PubMed] [Google Scholar]
- Papadia S, Stevenson P, Hardingham NR, Bading H, Hardingham GE. Nuclear Ca2+ and the cAMP response element-binding protein family mediate a late phase of activity-dependent neuroprotection. J Neurosci. 2005;25:4279–4287. doi: 10.1523/JNEUROSCI.5019-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parlato R, Rieker C, Turiault M, Tronche F, Schutz G. Survival of DA neurons is independent of CREM upregulation in absence of CREB. Genesis. 2006;44:454–464. doi: 10.1002/dvg.20236. [DOI] [PubMed] [Google Scholar]
- Pedarzani P, Storm JF. PKA mediates the effects of monoamine transmitters on the K+ current underlying the slow spike frequency adaptation in hippocampal neurons. Neuron. 1993;11:1023–1035. doi: 10.1016/0896-6273(93)90216-e. [DOI] [PubMed] [Google Scholar]
- Pedarzani P, Storm JF. Dopamine modulates the slow Ca2+-activated K+ current IAHP via cyclic AMP-dependent protein kinase in hippocampal neurons. J Neurophysiol. 1995;74:2749–2753. doi: 10.1152/jn.1995.74.6.2749. [DOI] [PubMed] [Google Scholar]
- Peters HC, Hu H, Pongs O, Storm JF, Isbrandt D. Conditional transgenic suppression of M channels in mouse brain reveals functions in neuronal excitability, resonance and behavior. Nat Neurosci. 2005;8:51–60. doi: 10.1038/nn1375. [DOI] [PubMed] [Google Scholar]
- Pittenger C, Huang YY, Paletzki RF, Bourtchouladze R, Scanlin H, Vronskaya S, Kandel ER. Reversible inhibition of CREB/ATF transcription factors in region CA1 of the dorsal hippocampus disrupts hippocampus-dependent spatial memory. Neuron. 2002;34:447–462. doi: 10.1016/s0896-6273(02)00684-0. [DOI] [PubMed] [Google Scholar]
- Reymann KG, Frey JU. The late maintenance of hippocampal LTP: requirements, phases, “synaptic tagging,” “late-associativity” and implications. Neuropharmacology. 2007;52:24–40. doi: 10.1016/j.neuropharm.2006.07.026. [DOI] [PubMed] [Google Scholar]
- Riccio A, Ahn S, Davenport CM, Blendy JA, Ginty DD. Mediation by a CREB family transcription factor of NGF-dependent survival of sympathetic neurons. Science. 1999;286:2358–2361. doi: 10.1126/science.286.5448.2358. [DOI] [PubMed] [Google Scholar]
- Sah P. Ca2+-activated K+ currents in neurones: types, physiological roles and modulation. Trends Neurosci. 1996;19:150–154. doi: 10.1016/s0166-2236(96)80026-9. [DOI] [PubMed] [Google Scholar]
- Sah P, Bekkers JM. Apical dendritic location of slow afterhyperpolarization current in hippocampal pyramidal neurons: implications for the integration of long-term potentiation. J Neurosci. 1996;16:4537–4542. doi: 10.1523/JNEUROSCI.16-15-04537.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah P, Faber ES. Channels underlying neuronal calcium-activated potassium currents. Prog Neurobiol. 2002;66:345–353. doi: 10.1016/s0301-0082(02)00004-7. [DOI] [PubMed] [Google Scholar]
- Sloviter RS. A simplified Timm stain procedure compatible with formaldehyde fixation and routine paraffin embedding of rat brain. Brain Res Bull. 1982;8:771–774. doi: 10.1016/0361-9230(82)90104-6. [DOI] [PubMed] [Google Scholar]
- Tandon P, Yang Y, Das K, Holmes GL, Stafstrom CE. Neuroprotective effects of brain-derived neurotrophic factor in seizures during development. Neuroscience. 1999;91:293–303. doi: 10.1016/s0306-4522(98)00609-5. [DOI] [PubMed] [Google Scholar]
- Tully T, Bourtchouladze R, Scott R, Tallman J. Targeting the CREB pathway for memory enhancers. Nat Rev Drug Discov. 2003;2:267–277. doi: 10.1038/nrd1061. [DOI] [PubMed] [Google Scholar]
- Vogalis F, Harvey JR, Furness JB. PKA-mediated inhibition of a novel K+ channel underlies the slow after-hyperpolarization in enteric AH neurons. J Physiol (Lond) 2003;548:801–814. doi: 10.1113/jphysiol.2002.037325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu WW, Chan CS, Disterhoft JF. Slow afterhyperpolarization governs the development of NMDA receptor-dependent afterdepolarization in CA1 pyramidal neurons during synaptic stimulation. J Neurophysiol. 2004;92:2346–2356. doi: 10.1152/jn.00977.2003. [DOI] [PubMed] [Google Scholar]
- Zhang W, Linden DJ. The other side of the engram: experience-driven changes in neuronal intrinsic excitability. Nat Rev Neurosci. 2003;4:885–900. doi: 10.1038/nrn1248. [DOI] [PubMed] [Google Scholar]
- Zhu DY, Lau L, Liu SH, Wei JS, Lu YM. Activation of cAMP-response-element-binding protein (CREB) after focal cerebral ischemia stimulates neurogenesis in the adult dentate gyrus. Proc Natl Acad Sci USA. 2004;101:9453–9457. doi: 10.1073/pnas.0401063101. [DOI] [PMC free article] [PubMed] [Google Scholar]