

Abstract
Measurement of Aβ toxicity of cells in culture exposes a subpopulation of cells with resistance to Aβ, even at high concentrations and after long periods of treatment. The cell-selective toxicity of Aβ resembles the selective damage observed in cells of specific regions of the Alzheimer's disease (AD) brain and suggests that there must be particular characteristics or stages of these cells that make them exceptionally sensitive or resistant to the effect of Aβ. Using flow cytometry and cell sorting, we efficiently separated and analyzed the Aβ-sensitive and the Aβ-resistant subpopulations within a variety of neuronal cell lines (PC12, GT1–7) and primary cultured neurons (hippocampal, cortex). We found that this distinctive sensitivity to Aβ was essentially associated with cell membrane Aβ binding. This selective Aβ binding was correlated to distinctive cell characteristics, such as cell membrane exposure of the apoptotic signal molecule phosphatidyl serine, larger cell size, the G1 cell cycle stage, and a lower than normal cytosolic ATP level. The response to Aβ by the cells with high Aβ binding affinity was characterized by a larger calcium response and increased mortality, lactate dehydrogenase release, caspase activation, and DNA fragmentation. The distinctive sensitivity or resistance to Aβ of the different subpopulations was maintained even after multiple cell divisions. We believe that these distinctive cell characteristics are the determining factors for the selective attack of Aβ on cells in culture and in the AD brain.
Keywords: Aβ peptide, Alzheimer's disease, neurotoxicity, β-amyloid, Aβ binding, Aβ selectivity
Introduction
Alzheimer's disease (AD) symptoms are associated with selective damage in the neuronal circuits of the neocortex, hippocampus, and basal forebrain cholinergic system. Accumulations of β-amyloid (Aβ)-containing plaques and neurofibrillary tangles are the main pathological features of the neurons in the damaged AD brain.
Aβ is made normally by all cells throughout life (Wisniewski et al., 1997), and its concentration in human CSF is physiologically altered in normal aging (Shoji et al., 2001). Aβ is also released from cultured cells during normal metabolism (Haass et al., 1992; Seubert et al., 1992; Shoji et al., 1992). However, despite Aβ normal existence, abundant evidence has associated the cytotoxicity of Aβ peptides with the neurodegeneration observed in AD. External application of Aβ on cells, either in cultures or in animal brains, shows cell-selective toxic effects. It is generally observed that not all cells are equally affected by Aβ, and, interestingly, some cells are even found to be resistant. The resistance of some cells within the same culture to Aβ is observed even at high Aβ concentration and after long periods of exposure. Therefore, the selective attack of Aβ on only some of the cells in the cultures may embody the selective attack observed in cells of specific regions of the AD brain and suggest that there must be particular characteristics or stages of the cells that make them exceptionally sensitive or resistant to the effect of Aβ.
How and why specific sets of neurons in selected brain structures become dysfunctional, why some neurons and not others die, and what initiates these deleterious processes are yet to be explained. It is conceivable that when cells are externally exposed to Aβ, specific association between the Aβ and the cell membrane is the initial step of the chain of events that leads to toxicity. Therefore, the cell-selective effects of Aβ likely reside in this initial Aβ–membrane association and in characteristic factors that may endorse this initial association. To search for the characteristic cellular factors responsible for the Aβ–membrane association, we used flow cytometry and cell sorting to separate cells on the basis of the preferential binding to Aβ. Sorted cells were then evaluated on their extent of Aβ binding and intrinsic characteristics and responses to external exposure to Aβ.
Here we present data demonstrating that within populations of the neuronal cells lines PC12, GT1–7, and the primary cultured hippocampal and cortex neurons, cells can be efficiently separated into Aβ-sensitive and Aβ-resistant subpopulations on the basis of their capacity to bind Aβ. These subpopulations have distinct properties and maintain their differential properties and responses to Aβ, even after successive cell divisions. The most prominent of these properties was related to the level of phosphatidyl serine (PS) on the outer face of the surface membrane and the basal levels of cytosolic ATP. Based on the specific responses to Aβ, we believe that these distinct cell indicators are the basis for the selective attack of Aβ on some cells in culture and on cells of specific regions of the AD brain.
Materials and Methods
Cell cultures.
The following cell cultures were used: immortalized cell line PC12 differentiated and undifferentiated, derived from a transplantable rat pheochromocytoma (American Type Culture Collection, Manassas, VA); immortalized hypothalamic neurons GT1–7 (provided by Dr. M. Kawahara, National Institutes of Health, Bethesda, MD); and primary cultures of hippocampal and cortical neurons from prenatal day 18–19 rat brains. Cell lines and primary cultured cells were grown in the previously described media (Simakova and Arispe, 2006).
Flow cytometry analysis and cell sorting.
For flow cytometric analysis of Aβ binding, cells were incubated in PBS with 0.3% BSA containing 0.2–40 μm Aβ1–42 conjugated to the fluorescent probe fluorescein isothiocyanate (FITC; AnaSpec) for 10–120 min, washed three times to remove unbound Aβ-FITC, and analyzed (EPICs XL-MCL; Beckman Coulter, Villepinte, France). Forward and side scatter gating was used to exclude debris and dead cells. Cells with different Aβ-FITC affinity were separated by a fluorescence-activated cell sorter (FacsAria; BD Biosciences, San Jose, CA) after 80 min of 5 μm Aβ-FITC exposure and three washing steps. All single cells were included. We used low-pressure (20 psi) and low-speed (1000 events/s) cell sorting. The purity of sorted populations routinely exceeded 80–90%.
Intracellular free calcium measurements.
Sorted cells after 8 d in culture and plated on glass coverslips coated with collagen or poly-lysine were loaded with 2 μm fura-2 AM (Invitrogen, Carlsbad, CA) calcium-sensitive probe in the incubation buffer (in mm: 135 NaCl, 5 KCl, 2.5 CaCl2, 1.2 MgCl2, 10 glucose, 10 HEPES, pH 7.4). After a loading period (∼30 min), cells were treated with 5 μm Aβ (Bachem, Bubendorf, Switzerland) or 1 μm cation ionophore 4-BrA23187 (TEF Labs, Austin, TX). The time course of changes in the emission from fura-2 AM was observed using an inverted epifluorescence/phase contrast microscope equipped with CCD camera + microphotometer assembly (InCyt I/P-2 Imaging and Photometry System; Intracellular Imaging, Cincinnati, OH). Fifty to sixty cells were analyzed for each condition.
Cell viability assays.
Cell viability was measured using a colorimetric XTT assay (Cell Proliferation Kit II; Roche, Indianapolis, IN). The release of lactate dehydrogenase (LDH) from the cytosol into the media was used to report on the integrity of the cell membrane (Cytotoxicity Detection Kit-LDH; Roche).
Apoptosis assay.
Changes in the mitochondrial membrane potential of cells exposed to 15 μm Aβ (Bachem) for 3 d were determined with the J-aggregate-forming fluorochrome 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine iodide (JC-1; Invitrogen). This lipophilic dye forms red fluorescent aggregates inside healthy mitochondria. When the mitochondria membrane potential collapses, the dye cannot accumulate within the mitochondria and remains in the cytoplasm in a green fluorescent monomeric form. Analysis was done by flow cytometry (EPICs XL-MCL).
To determine surface membrane PS, the Annexin V-PE Apoptosis Detection Kit I (BD Biosciences) was used. Cells were exposed to 5 μm Aβ-FITC for 80 min, washed three times, stained with annexin V-phycoerythrin (PE), and analyzed by flow cytometry (FacsAria).
After sorting, cells were cultured for 7 d and then exposed for 24 h to 5 μm Aβ. The caspase 3/7 activation was quantified in Aβ-exposed and -unexposed cells, adjusted to have similar cell numbers, by the CellProbe HT caspase 3/7 whole cell assay kit (Beckman Coulter). To measure DNA fragmentation, we used terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) APO-BrdU kit (BD Biosciences). Cells were incubated with Br-dUTP and detected with FITC or PE-labeled anti-Br-dU monoclonal antibody and by flow cytometry (EPICs XL-MCL).
Cell cycle analysis.
Propidium iodide (PI) was used for cell cycle analysis immediately after cell sorting. Cells were fixed in 70% ethanol and washed twice, double-stranded RNA was removed by DNase-free RNase (Sigma), and the cells were stained with PI (Sigma) and analyzed by flow cytometry (EPICs XL-MCL) immediately. ModFit LT (Verity Software House, Topsham, ME) was used for the analysis.
Analysis of neurite outgrowth.
Sorted PC12 cells were differentiated by NGF for 1 week, then treated with 5 μm Aβ40 (Bachem) for an additional 3 d, and neurite characterization by Neurite Outgrowth Assay Kit (Millipore, Billerica, MA) was performed as described in the manufacturer's manual.
Determination of ATP.
ATP Bioluminescence Assay Kit CLS II (Roche) was used to evaluate the level of cytosolic ATP. PC12 cells and hippocampal neurons were sorted on the basis of Aβ binding and cultured. After 8 d, the different sorted groups of cells were detached, adjusted to have similar cell numbers, and boiled in 100 mm Tris, 4 mm EDTA, pH 7.75. Luciferase reagent was added to the samples, and luminescence was analyzed (Fluostar Optima).
Statistics.
All data are presented as means of three to five experiments ± SD. Origin scientific analysis software (OriginLab, Northampton, MA) was used to determine significance levels using Student's t test. The level of significance is denoted in each figure as follows: ns, no statistically significant difference; single and double asterisk, significant and highly significant p values, respectively.
Results
Not all cells within the same culture are equally affected by external addition of Aβ, and some cells are even found to be resistant
It is generally observed that not all cells within the same culture bind and are equally affected by Aβ. To illustrate these observations, we have chosen, from a pool of flow cytometry data, a series of dot plots and histograms of the distribution of cells after exposure to Aβ for different experimental conditions. These figures will describe some of the cellular processes known to be affected by Aβ and the levels of Aβ binding. Figure 1 illustrates the cell-specific toxicity of Aβ. The highly Aβ-sensitive neuronal cell line PC12 and rat hippocampal neurons were treated with an unequivocal toxic concentration of Aβ, and the DNA fragmentation and the mitochondrial membrane potential were measured. To enhance the interaction of Aβ with the surface membrane of the cells, an abnormally high concentration of freshly prepared Aβ40 (15 μm) was added to the culture medium, and apoptosis was evaluated 3 d later by flow cytometry TUNEL assay and mitochondrial membrane potential assay. The data in Figure 1 show that exposure of cells to Aβ clearly induced a remarkably high level of DNA fragmentation, (Fig. 1A) and a significant mitochondrial membrane potential disruption (Fig. 1B). However, despite the long exposure to a highly toxic concentration of Aβ, approximately one-half of the cells remained relatively unaffected.
Figure 1.
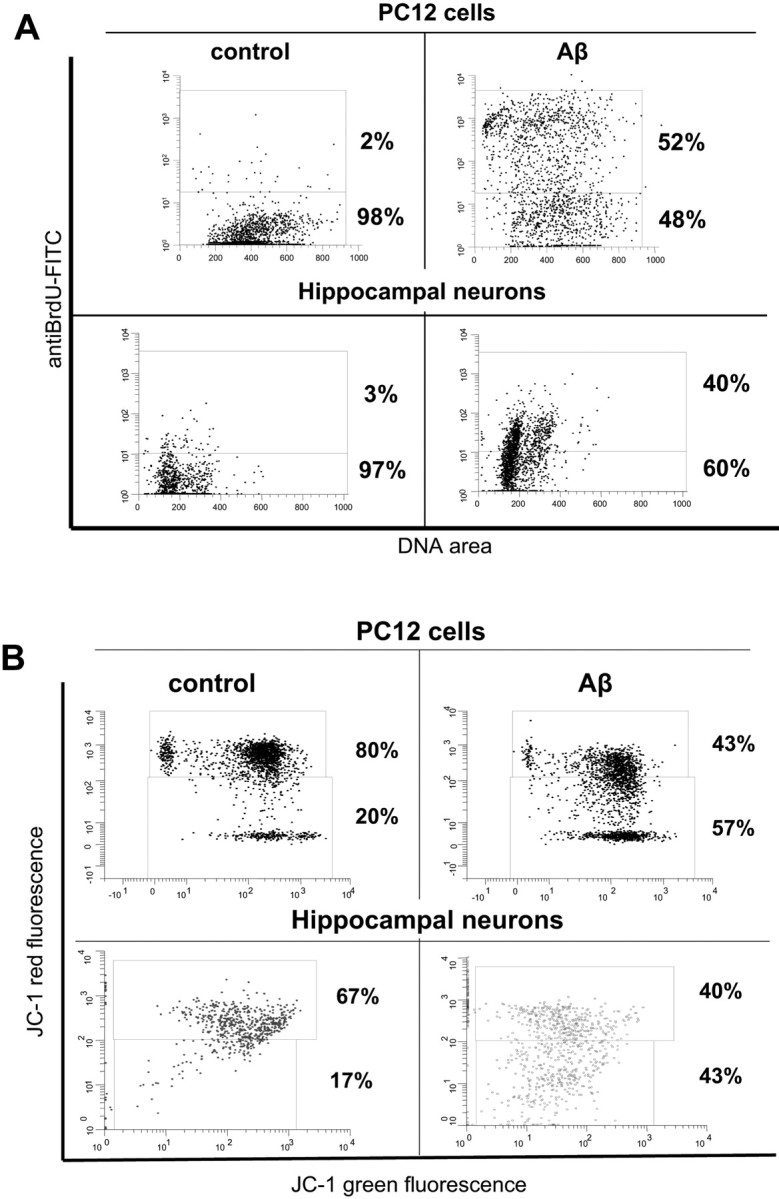
Apoptosis assays disclose cells sensitive and resistant to Aβ. PC12 cells and hippocampal neurons were grown for 3 d in a medium containing an abnormally high concentration (15 μm) of freshly prepared Aβ. The disruption of DNA fragmentation and the mitochondrial membrane were evaluated by TUNEL and mitochondrial membrane potential flow cytometry assays. After the third day of exposure to Aβ, significant cell damage is observed. However, despite the long exposure to a highly toxic concentration of Aβ, in terms of DNA fragmentation and mitochondrial function, mitochondrial function, 48 and 43% of PC12 cells and 60 and 40% of hippocampal neurons, respectively, remained unaffected.
Cell-selective Aβ binding: concentration and time dependence
The results in Figure 1 show that although all cells were exposed to the same conditions, a subpopulation of cells did not respond to the toxic effects of Aβ. The results from Aβ binding experiments in Figure 2 suggest that the resistance of these cells to respond to Aβ may reside, initially, in the failure of the cell surface membrane to bind Aβ. PC12 cells were incubated for different periods of time in media containing different concentrations of Aβ conjugated with the fluorescent probe FITC (Aβ-FITC). Cells were then washed three times with PBS/BSA buffer and analyzed for Aβ-FITC binding using flow cytometry. Figure 2A shows a progressive rightward shift of the fluorescence histograms with increasing Aβ concentration, in contrast to the saturation in the number of fluorescent cells. Figure 2B, constructed from the raw data in Figure 2A, shows that as the Aβ-FITC concentration is increased, and more cells bind Aβ, the percentage of cells in the M2 region, Aβ-FITC-positive, increases. At Aβ-FITC concentrations of >10 μm, a maximum percentage of fluorescent-positive cells were reached. The effect of increasing the time of exposure to Aβ-FITC on the level of Aβ binding to the cells was analyzed in the experiment shown in Figure 2C. PC12 cells were exposed to 2 μm Aβ-FITC for various periods of time before the cells were washed and analyzed by flow cytometry. The histograms show that increase of exposure to Aβ-FITC up to 2 h basically increases the number of positive-fluorescent cells, rather than the cell fluorescence intensity, as indicated by the very modest shift of the distribution curves toward higher intensity levels. Figure 2D shows that the percentage of Aβ-FITC-positive cells in the M2 region increased very rapidly during the first 20 min of exposure to Aβ-FITC. Thereafter, the rate of increase was very moderate and constant.
Figure 2.

Effect of concentration and time of exposure on Aβ binding to cells. PC12 cells were exposed for different periods of time to different concentrations of Aβ-FITC, and Aβ-FITC binding was analyzed by EPICs XL-MCL. A, Histograms of the number of cells versus Aβ-FITC fluorescence after 60 min exposure to different concentrations of Aβ-FITC. B, Percentages of cells (filled squares) and fluorescence intensity [geometric mean of fluorescence (GeoMean of Fluor), filled circles] of bound Aβ-FITC (M2 gate from A) as a function of the concentration of Aβ-FITC. C, Histograms of the number of cells versus Aβ-FITC fluorescence at different times of exposure to 2 μm Aβ-FITC. D, Percentages of cells with bound Aβ-FITC (M2 gate from C) as a function of time of exposure to 2 μm Aβ-FITC. Binding and the percentage of cells that bind Aβ-FITC increase as a function of time of exposure and concentration of Aβ to reach a saturation level.
Binding affinity for Aβ distinguishes subpopulations of cells within different neuronal cell lines and primary cultured neurons
The results described above clearly show that PC12 cells and hippocampal neurons can be grouped into subpopulations based in their surface membrane binding affinity toward Aβ. A similar approach was also applied to four other cell types, namely differentiated PC12, GT1–7, and primary cultured rat cortex and hippocampal neurons. We exposed the cells to Aβ-FITC and sorted them on the basis of their bound Aβ-FITC using a cell sorter FacsAria (BD Biosciences), which also enabled us to analyze the cells with high fluorescence sensitivity. Figure 3 summarizes the flow cytometric data from experiments in which the different types of cells were exposed for binding to 5 μm Aβ-FITC for a period of 80 min. Histograms illustrating the distribution for PC12 cells and for hippocampal neurons are shown in Figure 3a. The histograms show that cells are clearly distributed into three distinct subpopulations. One subpopulation (labeled A) of cells was completely Aβ-FITC negative (no Aβ binding affinity). The rest of the cells, Aβ-FITC positive, clearly separated into two subpopulations that exhibit high and extra-high affinity for Aβ-FITC (labeled B and C, respectively). The division of cells into subpopulations was observed in all cell types studied. The bar plot in Figure 3b summarizes the percentage of cells in each subpopulation according to the cell type. Most cells from the PC12 cell sample showed the highest affinity for Aβ-FITC compared with the other cell lines.
Figure 3.

Subpopulations of cells in different cell lines with different Aβ binding affinity. Different cell lines and primary neurons were exposed to 5 μm Aβ-FITC for 80 min. a, Histograms of Aβ-FITC binding to PC12 cells (left) and hippocampal neurons (right) show three distinct subpopulation of cells: subpopulation A (cells with no Aβ-FITC binding affinity), subpopulation B (cells with high Aβ-FITC binding affinity), and subpopulation C (cells with extra-high Aβ-FITC binding affinity). b, The bar plot summarizes the percentages of cells in the subpopulations A (left white bar), B (black bar), and C (right white bar) observed in five different cell types.
The clear separation of the cells into subpopulations observed in the Aβ-FITC fluorescence histograms made it possible to physically separate the cells based on their different Aβ-FITC binding affinity. The histograms in Figure 4 show the results of the application of the separation protocol on PC12 cells and hippocampal neurons. Before the sorting procedure was applied, the cells were exposed for 80 min for binding to Aβ-FITC (5 μm). The ranges of the sorting gates of the cell sorter, Figure 4, a and c, are indicated by the bars labeled A (cells with no Aβ-FITC affinity), B (cells with high Aβ-FITC affinity), and C (cells with extra-high Aβ-FITC affinity). The subpopulations of cells were collected in three separate tubes, and the efficiency of the separation process was confirmed by reexamining the cell-bound Aβ-FITC under the sorter. The histograms in Figure 4, b and d, show the distribution of the Aβ-FITC fluorescence from each one of the cell subpopulations and verify the purity of the sorted subpopulations of cells. The degree of subpopulation contamination was routinely <6% for PC12 cells and <20% for hippocampal neurons.
Figure 4.

Fluorescence-activated cell sorting of cells with different Aβ binding affinity. PC12 cells and hippocampal neurons were exposed to 5 μm Aβ-FITC for 80 min and sorted by FacsAria on the basis of their Aβ-FITC binding affinity. a, c, Histograms of Aβ-FITC binding to PC12 cells (a) and hippocampal neurons (c) show three distinct subpopulation of cells: subpopulation A (cells with no Aβ-FITC binding affinity), subpopulation B (cells with high Aβ-FITC binding affinity), and subpopulation C (cells with extra-high Aβ-FITC binding affinity). b, d, Histograms of Aβ-FITC binding to PC12 cells (b) and hippocampal neurons (d) to illustrate the efficiency of the sorting procedure. Each cell type was sorted and collected in separate subpopulations and immediately reanalyzed for the binding of Aβ-FITC. The purity of the sorted subpopulations in all experiments (n = 8) was routinely ∼90% for PC12 cells and ∼80% for hippocampal neurons.
Characteristics of cell subpopulations with different binding affinity for Aβ
Detailed examination of the cell subpopulations, with different binding affinity for Aβ-FITC, shows that these populations additionally possess distinct morphological and compositional characteristics that correlate with their level of affinity for Aβ-FITC.
Cell size
The forward scattergrams (proportional to cell size; FSCs), resulting from flow cytometry analysis of cells exposed to Aβ-FITC (5 μm, 80 min), point out that Aβ-FITC binding to cells appears to be correlated to cell size. The FSC histograms in Figure 5(left) show that the control population of cells, Aβ-FITC-untreated cells, for both PC12 and hippocampal neurons, exhibit relatively normal size distribution typical for viable cells. Most of the cells, ∼70%, were grouped in the medium sizes, limited by the bar II. When the subpopulations of cells, sorted by their affinity to bind Aβ-FITC as described above, are analyzed in terms of cell size distribution (Fig. 5, right), the highest percentage of PC12 cells and hippocampal neurons with no Aβ affinity (continuous curves) are found in the bar II of the medium-sized cells, 65 and 75%, respectively. PC12 cells and hippocampal neurons with high Aβ affinity (dashed curves) distributed preferentially within the large cell sizes limited by the bar III, 42 and 71%, and the medium cell sizes limited by the bar II, 54 and 26%, respectively. However, those cells with the extra-high Aβ-FITC affinity (dotted lines) were, for both types of cells, found preferentially within the smallest cell sizes limited by bar I, 61 and 38%, and the medium in sizes, 25 and 50%, respectively. This distribution probably corresponds to subpopulations of nonviable cells and cell bodies.
Figure 5.

Aβ binding correlates with cell size. Forward scattergrams (FSC) from flow cytometry analysis of subpopulation of PC12 cells (top) and hippocampal neurons (bottom) sorted on the basis of their affinity to bind Aβ-FITC. For sorting, cells were exposed to 5 μm Aβ-FITC for 80 min. Bars I, II, and III define the cells on the basis of their sizes, as small, medium, and large sizes, respectively. The continuous, dashed, and dotted curves correspond to cells with no Aβ affinity, high Aβ affinity, and extra-high Aβ affinity, respectively. Cells and neurons with high Aβ affinity distributed preferentially within the medium and large cell sizes. Cells and neurons with the extra-high Aβ-FITC affinity distributed preferentially within the smallest and the medium cell sizes.
Surface phosphatidyl serine
The capacity of cells to bind Aβ-FITC appears to be also associated with cells with expressed measurable PS in the surface membrane. This association has been previously suggested by the results of competitive experiments performed in PC12 cells (Lee et al., 2002) and can be directly construed from the flow cytometry analysis shown in Figure 6A. PC12 cells, which typically show a certain level of the apoptotic signal molecule PS on their cell surface (Lee et al., 2002), were grown in two separate groups of cells to be labeled with the specific PS-binder annexin V-PE. One group of cells, the Aβ treated, was preincubated for 80 min in a medium containing 5 μm Aβ-FITC and washed three times before being labeled with annexin V-PE. The other group, the control cells, remained untreated with Aβ. Flow cytometric analysis showed 37% of the control cells positive for annexin V-PE (Fig. 6A, left histogram). Within the groups of cells treated with Aβ-FITC, the group with no Aβ-FITC affinity (Fig. 6A, middle histogram) showed only 7% of cells positive for annexin V-PE. In contrast, cells with Aβ-FITC affinity (Fig. 6A, right histogram) showed 58% of cells positive for annexin V-PE. The flow cytometric analysis was made immediately after a very short period of time of exposure to Aβ and to a relatively low concentration of Aβ. Therefore, these conditions are sufficient for Aβ binding but are not sufficient to induce a measurable externalization of PS and apoptosis. Hence, we consider that most detected PS is prior PS on the surface of the cell membrane. This strong correlation between cells binding Aβ and cells exposing PS was confirmed in three additional experiments on PC12 and on hippocampal neurons (data not shown). In these neurons, which do not normally expose the apoptotic signal molecule PS, a mild externalization of PS was experimentally induced. Overall, the results clearly show that there seems to be a close correlation between the presence of PS on the surface of the cells and the affinity of the cell to bind Aβ.
Figure 6.

Aβ binding correlates with the level of phosphatidylserine on the cell surface membrane, with the cytosolic ATP level, and with the cell cycle phase. A, Surface PS in PC12 cell subpopulations, identified on the basis of their affinity to bind Aβ-FITC, was analyzed by flow cytometry. Cells were exposed to annexin V-PE, and the cell bound fluorescence was used as a sign of the level of PS in the surface of the cell membrane. Cells with affinity to Aβ-FITC showed the highest percentage of cells positive for annexin V-PE (58%), compared with cells with no Aβ-FITC affinity (7%). B, Subpopulations of PC12 cells and hippocampal neurons sorted based on their Aβ-FITC affinity were cultured for 8 d before their cytosolic ATP levels were evaluated. For both types of cells, cells with Aβ-FITC affinity showed ∼50% lower cytosolic ATP levels than cells with no Aβ-FITC affinity and control cells. Data shown are mean ± SD. ns, Nonsignificant; *p < 0.01 (significant); **p < 0.0001 (highly significant). C, Subpopulations of PC12 cells were sorted based on their Aβ-FITC affinity and their cell cycle stage was immediately analyzed. Cells with Aβ-FITC affinity were preferentially in G1 stage (57%) compared with cells with no Aβ affinity (41%) and control cells (42%). The percentage of Aβ-FITC affinity cells in the G2 stage was reduced (13%) compared with the percentage of cells with no Aβ-FITC affinity (33%) and the percentage of control cells (30%).
The experiments that follow were performed with a similar procedure. PC12 cells and hippocampal neurons were exposed for 80 min to 5 μm Aβ-FITC and separated in subpopulations based on their different Aβ binding affinity, as described in Figures 3 and 4. Cells were grouped into two subpopulations: non-Aβ-FITC binding (no Aβ affinity) and Aβ-FITC binding (Aβ affinity).
ATP content
After sorting, the groups of cells were allowed to grow, and in the case of PC12 cells allow to divide, for 8 d before cytosolic levels of ATP were evaluated. As shown in Figure 6B, after several divisions, PC12 cells with Aβ affinity showed ∼50% lower cytosolic levels of ATP compared with cells with no Aβ affinity. In addition, hippocampal neurons that exhibit Aβ affinity permanently possessed significantly lower cytosolic ATP content than that of cells with no Aβ affinity. In addition to the lower cytosolic levels of ATP, the other distinctive characteristic of cells with preferred binding to Aβ, the higher surface PS level observed immediately after cell sorting, was also found to be maintained after several cell divisions (our unpublished results).
Cell cycle
Evaluation of the cell cycle stage of cells binding Aβ suggests that there is a preferential cell cycle stage for Aβ binding. We analyzed the cell cycle stage of PC12 immediately after sorting based on their affinity to bind Aβ-FITC. The histograms in Figure 6C show that control cells, never exposed to Aβ, and cells exposed to Aβ-FITC but with no Aβ affinity, displayed similar cell cycle stage distribution. On the other hand, the subpopulation of cells with bound Aβ, Aβ affinity, displayed a different cell cycle stage distribution. Most of the cells with Aβ affinity are found in the G1 stage (57% compared with 41% in control), and there was a significant reduced percentage of cells in the G2 stage (13% compared with 33% in the no-Aβ-affinity subpopulation).
Characteristic responses of subpopulation of cells with Aβ binding affinity
The effective separation of subpopulations of cells with different affinity to Aβ-FITC led us to study comparatively the subpopulations of cells on the basis of their most common responses to Aβ, particularly the induction of intracellular calcium increase and the activation of apoptotic mechanisms.
Calcium response
The increase in the intracellular calcium is the first event observed after Aβ binds to the surface membrane of cells. Hence it should be expected that cells that show affinity to bind Aβ would more likely respond with calcium spikes after exposure to Aβ pulses, as opposed to cells that do not bind Aβ. Sorted PC12 cells and hippocampal neurons cells were allowed to grow for 8 d before the calcium response to Aβ was tested. Figure 7 shows the records and analysis of the intracellular calcium changes observed after the addition of either freshly prepared Aβ or the calcium ionophore 4-BrA23187. For each condition, 50–60 cells were analyzed, but for illustration purposes, the panels display only representative simultaneous records from five responding cells located at different positions of the optical field. Control records from PC12 cells in Figure 7A show that most Aβ-untreated cells (78%) displayed an intracellular calcium response (266 ± 25 nm) soon after exposure to fresh Aβ. The cells sorted for their resistance to binding Aβ (subpopulation with no Aβ affinity) either did not respond or a few (14%) showed only a mild calcium response (<25 nm). This percentage of cell responding is not much different from the percentage (11%) observed 1 d after sorting. In contrast, almost all cells (99%) sorted for their capacity to bind Aβ (Aβ affinity) showed a robust calcium response (1312 ± 76 nm). The calcium responses to Aβ from subpopulations of hippocampal neurons are also shown in Figure 7A. Neurons sorted for their affinity to bind Aβ also showed a significant calcium response to Aβ. Eight days in culture after sorting, the subpopulations of neurons displayed their original sensitivity to Aβ. Ninety-six percent of the neurons in the subpopulation with Aβ affinity responded to the addition of a pulse of Aβ with a long-duration strong calcium response (285 ± 10 nm), compared with a much milder and shorter calcium response (188 ± 14 nm) observed in 74% of the control neurons and compared with almost no calcium response (5 ± 2 nm) observed in only 8% of the no-Aβ-affinity subpopulation of neurons. For comparison, Figure 7B shows the calcium response of PC12 cells to the calcium ionophore 4-BrA23187. In contrast to the differential response to Aβ, sorted subpopulations with different Aβ-FITC affinity and Aβ-FITC-untreated control, 100% of cells responded with a similar calcium response to 4-BrA23187 (1 μm). Comparison between the averaged calcium responses from each subpopulation showed no statistically significant difference (ns; p > 0.5). Figure 7C summarizes the median of the percentage of PC12 cells responding to Aβ (left) and the median of the calcium changes (right) from three different experiments.
Figure 7.

Calcium response to Aβ and cell Aβ binding affinity are correlated. A, The calcium response to fresh Aβ of subpopulations of PC12 cells and hippocampal neurons, 8 d after being sorted on the basis of their Aβ-FITC affinity. Of 50–60 calcium responses analyzed, the figure shows five records from each subpopulation of cells. After application of Aβ (arrow), 99% of PC12 cells with high Aβ-FITC affinity showed marked calcium peak (1212 ± 76 nm), five times higher than the response of 78% of control cells (266 ± 25 nm). Only 14% of cells with no Aβ-FITC affinity showed a very mild calcium response (0 ± 10 nm). This number is not much different than the percentage of responding cells observed right after sorting (11%). Ninety-six percent of hippocampal neurons with Aβ-FITC affinity responded to Aβ with a significantly higher (p < 0.001) calcium peak (285 ± 10 nm) compared with 74% of control cells (188 ± 14 nm) and 8% of cells with no Aβ-FITC affinity (5 ± 2 nm). B, After 8 d of sorting, all cells from the sorted subpopulations of PC12 cells responded with calcium increases to the application of the calcium ionophore 4-BrA23187. The magnitude of the calcium response (data shown as median ± SE) from the cells in the different subpopulations was not significantly different (ns; p > 0.05). C, Summary of the calcium response induced by Aβ from three different experiments on PC12 cells. The plots show the median of the percentage of responding cells (left) and the median of the magnitude of the calcium change (right).
The differential cell response to Aβ correlated with the cell affinity to bind Aβ is also observed in the magnitude of the surface membrane and mitochondrial disruption, caspase activation, DNA fragmentation, and neurite degeneration induced by Aβ.
Cell membrane and mitochondrial disruption
The subpopulations of cells sorted on the basis of their Aβ-FITC binding not only display correlation with the prompt calcium response to Aβ, but they also show differential responses to Aβ in terms of the magnitude of the cell surface and mitochondrial membrane disruption. Subpopulations of PC12 cells (Fig. 8A) and hippocampal neurons (Fig. 8B) were allowed to grow for 7 d after sorting. Cells were then exposed for 1 d to fresh Aβ, and cell viability and LDH release were evaluated. The subpopulations of both PC12 cells and hippocampal neurons with affinity for Aβ showed a significantly (p < 0.03) higher percentage of mortality (left) and LDH release (right) than did the subpopulations of Aβ-FITC-untreated control and cells with no Aβ-FITC affinity.
Figure 8.

Characteristic responses of subpopulation of cells with high Aβ binding affinity. A, Subpopulations of PC12 cells sorted on the basis of their Aβ-FITC affinity were allowed to grow and divide for 8 d. Cells were then exposed for 1 d to fresh Aβ, and 48 h later, cell viability (left) and LDH release (right) were evaluated. B, Subpopulations of hippocampal neurons sorted on the basis of their Aβ-FITC affinity were allowed to grow for 7 d. Neurons were then exposed for 1 d to fresh Aβ, and 48 h later, cell viability (left) and LDH release (right) were evaluated. C, Left, The level of caspase 3/7 activity [in relative fluorescence light units (RFLUs)] in subpopulations of PC12 cells 8 d after cells were sorted on the basis of their Aβ-FITC affinity. Right, Aβ activation of caspases 3/7 of subpopulations of PC12 cells 8 d after being sorted on the basis of their Aβ-FITC affinity. At day 7 after sorting, cells were exposed to Aβ, and the activation of caspases 3/7 was evaluated the following day. Error bars are mean ± SD. *p < 0.04; **p < 0.0001.
Caspase activation
The activity of caspases 3/7 from PC12 cells collected in subpopulations on the basis of their affinity to bind Aβ-FITC was measured after allowing the cells to divide for 8 d in Aβ-free media. The results in Figure 8C (left) show that the level of caspase activity was significantly higher in the subpopulation of cells with Aβ-FITC affinity than in control cells never treated with Aβ and cells that exhibited no affinity to bind Aβ-FITC. At day 7, sorted cells were exposed to Aβ for 1 d, and the activation of caspases 3/7 was evaluated. The results in Figure 8C (right) show that the subpopulation of cells with Aβ-FITC affinity exhibits significantly higher (p < 0.03) caspase activation than the other two subpopulations.
DNA fragmentation
The results displayed in Figure 9A show that the cell subpopulation with Aβ-FITC binding affinity was also more sensitive to undergo DNA fragmentation when exposed to fresh Aβ. PC12 cells were collected in groups on the basis of their affinity to bind Aβ-FITC and allowed to divide for 8 d in separate cultures. DNA fragmentation was measured in a group of cells after 8 d in culture (left) and in another group of cells that at day 7 were exposed to Aβ for 1 d (right). After several cell divisions, high DNA fragmentation was observed only in the group containing cells with Aβ binding affinity (left). Among the subpopulations of cells exposed to Aβ for 1 d, DNA fragmentation was significantly higher in cells with Aβ-FITC affinity than in control cells. Almost no increase in DNA fragmentation was observed in the subpopulation with no Aβ affinity.
Figure 9.

Characteristic responses of subpopulation of cells with high Aβ binding affinity. A, PC12 cells were sorted in subpopulations on the basis of their affinity to bind Aβ-FITC and allowed to divide for 8 d in separate cultures. DNA fragmentation was measured after 8 d in culture (left). At day 7, a group of cells from each subpopulation was exposed to Aβ for 1 d before DNA fragmentation measurement (right). DNA fragmentation induced by Aβ was only observed in the subpopulation of cells with affinity to Aβ-FITC. B, Subpopulations of PC12 cells, sorted on the basis of their affinity to bind Aβ-FITC, were differentiated by NGF and exposed to fresh Aβ for 2 d, and the neurite outgrowth was analyzed. Aβ produced a 55% reduction of neurite outgrowth in sorted control cells (untreated). The reduction of neurite outgrowth in the subpopulation of cells with high affinity to Aβ-FITC was 82%, significantly higher (p < 0.0001) than the subpopulation of cells with no affinity to Aβ-FITC (20%) and control untreated cells (p < 0.04).
Neurite outgrowth
The effect of Aβ on the synaptic plasticity, as evaluated by degeneration of neurites, was also studied in the different subpopulations of cells sorted for their different affinity to bind Aβ-FITC. Sorted subpopulations of PC12 cells were treated with NGF to induce cell differentiation and neurite outgrowth. After cells were fully differentiated, the subpopulations of cells were exposed for 2 d to Aβ, and the neurite outgrowth was evaluated. The results in Figure 9 show that Aβ produced a noticeable (55 ± 9%) decrease in neurite outgrowth in the control Aβ-FITC-untreated group. When Aβ is added after cells are sorted on the basis of Aβ-FITC binding, the decrease (82 ± 12%) in neurite outgrowth is highly significant (p < 0.0001) in the subpopulation of cells that exhibited affinity to bind Aβ-FITC, compared with the decrease (20 ± 5%) observed in the subpopulation with no Aβ affinity.
Discussion
The results from this investigation demonstrate that the selective sensitivity of cells to neurotoxic effects of the Alzheimer's Aβ peptide is established by distinctive characteristics of the cell subpopulations that favor the specific binding of Aβ to the cell membrane. These distinctive characteristics are maintained in the cultured cell subpopulations even after multiple cell divisions, and may be the basis for the selective attack of Aβ on Aβ-sensitive cells. The specific membrane binding represents the first step in the series of events that leads to cell degeneration by Aβ. The principal finding of our study is the efficient separation of those cells that exhibit significant sensitivity to Aβ from those that show resistance, thereby permitting the study of their distinctive characteristics that correlate with their Aβ sensitivity. Our approach was based on the binding of Aβ to the membrane of the cell as the basic conditions to initiate the potentially cytotoxic cellular events. We found that most cells that exhibit high Aβ binding affinity preferentially display the apoptotic signal molecule PS, are the largest in size, are in the G1 stage of their cell cycle, and have significantly much lower than normal cytosolic ATP levels. Consequently the sensitivity of these cells to Aβ was accompanied by a higher increase in intracellular Ca2+ levels, release of LDH, activation of caspases, fragmentation of DNA, decrease in neurite outgrowth, and increased mortality (Table 1). We believe that this investigation provides the basis to assert that the selective attack of normal existing Aβ on some cells in culture and on limited regions of the AD brain is defined mainly by specific distinctive characteristics of the target cells rather than by the production and level of Aβ. The requirements for Aβ binding to membranes seem to be independent of the structural configuration that makes Aβ a toxic molecule, as suggested by our experiments (our unpublished observations). The survival status of cells after Aβ binding corroborates that Aβ specific binding to cell membrane is required and is the prelude to Aβ cytotoxicity. Additional investigation would be required to determine the implications that the stage of Aβ aggregation may have on Aβ binding to membranes.
Table 1.
Cell characteristics and response to Aβ of subpopulations of cells resistant or sensitive to the toxic action of Aβ
| Cell characteristics and response to Aβ | Subpopulations of cells |
|
|---|---|---|
| Cells resistant to Aβ | Cells sensitive to Aβ | |
| Aβ binding affinity | No | High |
| Surface phosphatidyl serine | Low or absent | High |
| Cytosolic ATP level | Normal | Low |
| Cell cycle | Normal distribution | G1 |
| Cell size | Normal distribution | Large + very small |
| Calcium response | None or very low | High |
| Caspase 3/7 activation | Low | High |
| Cell sensitivity | None or very low | High |
| Decrease of neurite outgrowth | None or very low | High |
Cells were sorted on the basis of their affinity to bind Aβ. The Aβ binding affinity of cells correlates with their sensitivity to Aβ.
Properties of the cells that preferentially bind Aβ
One of the most noteworthy features we found characterizing cells that preferentially bind Aβ is the presence of measurable surface PS. PS is normally found on the inner face of the surface membrane of healthy cells (Rothman and Lenard, 1977; Daleke and Lyles, 2000). In the early stage of apoptosis, or under specific stimulation, PS is translocated to the outer leaflets of the plasma membrane and exposed to the extracellular environment (Martin et al., 1995; Zwaal et al., 2005). PS exposure functions both as a recognition signal for cell removal in apoptosis (Fadok et al., 1992, 2000; Martin et al., 1995) and as a docking site for some proteins (Zwaal and Schroit, 1997). The correlation between Aβ binding and the detection of PS on the selected cells suggest that Aβ preferentially goes after cells that are distressed or in early-stage apoptosis.
The histograms originated from the flow cytometric analysis of cells exposed to Aβ show that Aβ associates with the largest-sized cells and to a lesser degree the smallest-sized cells. Swelling and shrinkage are recognized parameters of cells that are injured by either necrosis or apoptosis (Lang et al., 2004). The failure to find Aβ bound to average-sized healthy cells suggests that in this condition, the cell surface membrane does not have the specific surface receptors to facilitate the interaction with Aβ. In contrast, the correlation observed in our experiments between the levels of Aβ binding and the level of membrane-surfaced PS strongly suggests that PS, the early apoptotic signal molecule associated with injured cells (Zwaal et al., 2005), may be one of the surface receptors that establish the cell selectivity observed by Aβ.
PS asymmetry in healthy cells is maintained by the activity of the aminophospholipid translocases, or flippase, which shuttles PS (and phosphatidylethanolamine) from the outer to the inner monolayer using Mg2+-ATP (Devaux and Zachowski, 1994; Daleke and Lyles, 2000). PS exposure requires the concomitant activation of a PLS (phospholipid scramblase) and inhibition of an ATP-dependent aminophospholipid translocase (Verhoven et al., 1995; Bratton et al., 1997; Wolfs et al., 2005). Consequently, suppression of intracellular ATP levels potentiates PS exposure (Martin and Pagano, 1987; Gleiss et al., 2002). We found that cells with high affinity for Aβ chronically have a low cytosolic ATP level, which is maintained even after several cell divisions. Therefore, it could be speculated that the low cytosolic ATP levels, which may indicate metabolic distress, in these cells could maintain a chronically PS exposure, thereby facilitating the cell recognition by Aβ.
It is known that Aβ-induced cell death is related to cell cycle changes. Aβ activates an unscheduled cell cycle in neurons by inducing the expression of molecules necessary for the G1/S transition (Copani et al., 1999, 2006; Giovanni et al., 1999, 2000). Neurons enter the S phase before they die by apoptosis (Copani et al., 2001; Herrup et al., 2004). Flow cytometry testing is useful for quantifying the distribution of a population of cells into the different nuclear phases of the cell cycle. Our results on PC12 cells exposed to Aβ showed that most of the cells selected by Aβ for binding were in the G1 stage. Hence this finding may suggest that Aβ preferentially binds to cells in G1 rather than inducing G1. There are no reports indicating changes in the phospholipid composition of the membrane during the different phases of cell division. However, it has been shown that in PC12 cells the highest expression of the CTX (cholera toxin) receptor GM1 takes place during interphase and G1 (Majoul et al., 2002). Aβ selectively recognizes a GM1 cluster in membranes and binds to and accumulates on GM1-rich domains in membrane in a time- and concentration-dependent manner (Kakio et al., 2001; Wakabayashi et al., 2005). The GM1 enrichment of the cell surface membrane during the G1 stage would explain the preferential Aβ binding to cells during this stage observed after our flow cytometry analysis.
Some of the distinctive cells characteristics that we found associated to Aβ binding and to Aβ sensitivity are also characteristics that are enhanced after processes activated by cerebral ischemia or other means of trauma-induced apoptosis (Forrester and Williams, 1977; Petrov et al., 2000; Krijnen et al., 2002; Dutta et al., 2004; Cao et al., 2007). Cerebral ischemia has been suggested as an important factor facilitating the symptoms of Alzheimer's disease. It has been reported that cerebral ischemia and Aβ administration to rats significantly impaired spatial memory, probably because of induction of apoptosis in pyramidal neurons of the hippocampus (Iwasaki et al., 2006). The experimental conditions that we used to unveil the existence of subpopulations of cells with different binding affinity and sensitivity to Aβ are unlikely to be considered hypoxic. However, the similarities in the cellular processes activated by trauma and Aβ and the cell characteristics that associate with responses to Aβ make it worthwhile to pursue investigations into those experimental conditions that may exacerbate characteristics that endow cells with enhanced sensitivity to Aβ.
Response to Aβ by the cells that preferentially bind Aβ
There have been numerous reports describing the different intracellular disturbances produced after cells in culture are exposed to Aβ (Mattson et al., 1992; Loo et al., 1993; Shearman, 1996; Allen et al., 2001; Green and Peers, 2001; Morishima et al., 2001; Simakova and Arispe, 2006). Our experiments show that these disturbances manifest only in those cells that firmly bind Aβ. Therefore, when these cells are selected and analyzed separately, the magnitudes of the disturbances induced by Aβ show values that are larger than the values obtained from analysis of the whole population of cells. We observed that the average intracellular Ca2+ response of cells that possess affinity for Aβ is particularly higher than the average response from the whole population of cells. The uniform response of all cells to a Ca2+ ionophore suggests that the higher affinity to bind Aβ by some cells is accompanied by unique characteristics in the cellular mechanisms that control the Ca2+ homeostasis. This has been previously suggested and analyzed in many reports (LaFerla, 2002; Smith et al., 2005).
In summary, we have followed a sorting procedure to isolate cells sensitive and resistant to Aβ in various types of cells. We found distinctive cell characteristics that facilitate the binding of Aβ to the cell surface and bestow cells with differential responses to Aβ. These distinct cell characteristic can be normally conserved after cell division and can also be induced by agents promoting apoptosis. Based on our results, we can speculate that the cell-selective Aβ neurotoxicity in cultured cells and AD brains is determined by preexisting conditions or distress of target cells rather than by the production and levels of Aβ. We conclude that membrane binding by Aβ is essential for Aβ to express cytotoxicity and that the cell-selective neurotoxicity of Aβ peptide resides in distinctive cell characteristics.
Footnotes
This work was supported by The Alzheimer's Association of America and by Uniformed Services University of the Health Sciences Grant CO70TS. We thank Dr. Richard Siarey for invaluable comments and critically reading this manuscript.
References
- Allen JW, Eldabah BA, Huang X, Knoblach SM, Faden AI. Multiple caspases are involved in beta-amyloid-induced neuronal apoptosis. J Neurosci Res. 2001;65:45–53. doi: 10.1002/jnr.1126. [DOI] [PubMed] [Google Scholar]
- Bratton DL, Fadok VA, Richter DA, Kailey JM, Guthrie LA, Henson PM. Appearance of phosphatidylserine on apoptotic cells requires calcium-mediated nonspecific flip-flop and is enhanced by loss of the aminophospholipid translocase. J Biol Chem. 1997;272:26159. doi: 10.1074/jbc.272.42.26159. [DOI] [PubMed] [Google Scholar]
- Cao G, Xing J, Xiao X, Liou AK, Gao Y, Yin XM, Clark RS, Graham SH, Chen J. Critical role of calpain I in mitochondrial release of apoptosis-inducing factor in ischemic neuronal injury. J Neurosci. 2007;27:9278–9293. doi: 10.1523/JNEUROSCI.2826-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copani A, Condorelli F, Caruso A, Vancheri C, Sala A, Giuffrida Stella AM, Canonico PL, Nicoletti F, Sortino MA. Mitotic signaling by beta-amyloid causes neuronal death. FASEB J. 1999;13:2225–2234. [PubMed] [Google Scholar]
- Copani A, Uberti D, Sortino MA, Bruno V, Nicoletti F, Memo M. Activation of cell-cycle-associated proteins in neuronal death: a mandatory or dispensable path? Trends Neurosci. 2001;24:25–31. doi: 10.1016/s0166-2236(00)01663-5. [DOI] [PubMed] [Google Scholar]
- Copani A, Hoozemans JJ, Caraci F, Calafiore M, Van Haastert ES, Veerhuis R, Rozemuller AJ, Aronica E, Sortino MA, Nicoletti F. DNA polymerase-beta is expressed early in neurons of Alzheimer's disease brain and is loaded into DNA replication forks in neurons challenged with beta-amyloid. J Neurosci. 2006;26:10949–10957. doi: 10.1523/JNEUROSCI.2793-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daleke DL, Lyles JV. Identification and purification of aminophospholipid flippases. Biochim Biophys Acta. 2000;1486:108–127. doi: 10.1016/s1388-1981(00)00052-4. [DOI] [PubMed] [Google Scholar]
- Devaux PF, Zachowski A. Maintenance and consequences of membrane phospholipid asymmetry. Chem Phys Lipids. 1994;73:107–120. [Google Scholar]
- Dutta AK, Sabirov RZ, Uramoto H, Okada Y. Role of ATP-conductive anion channel in ATP release from neonatal rat cardiomyocytes in ischaemic or hypoxic conditions. J Physiol (Lond) 2004;559:799–812. doi: 10.1113/jphysiol.2004.069245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol. 1992;148:2207–2216. [PubMed] [Google Scholar]
- Fadok VA, Bratton DL, Rose DM, Pearson A, Ezekewitz RAB, Henson PM. A receptor for phosphatidylserine-specific clearance of apoptotic cells. Nature. 2000;405:85–90. doi: 10.1038/35011084. [DOI] [PubMed] [Google Scholar]
- Forrester T, Williams CA. Release of adenosine triphosphate from isolated adult heart cells in response to hypoxia. J Physiol (Lond) 1977;268:371–390. doi: 10.1113/jphysiol.1977.sp011862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovanni A, Wirtz-Brugger F, Keramaris E, Slack R, Park DS. Involvement of cell cycle elements, cyclin-dependent kinases, pRb, and E2F x DP, in B-amyloid-induced neuronal death. J Biol Chem. 1999;274:19011–19016. doi: 10.1074/jbc.274.27.19011. [DOI] [PubMed] [Google Scholar]
- Giovanni A, Keramaris E, Morris EJ, Hou ST, O'Hare M, Dyson N, Robertson GS, Slack RS, Park DS. E2F1 mediates death of B-amyloid-treated cortical neurons in a manner independent of p53 and dependent on Bax and caspase 3. J Biol Chem. 2000;275:11553–11560. doi: 10.1074/jbc.275.16.11553. [DOI] [PubMed] [Google Scholar]
- Gleiss B, Gogvadze V, Orrenius S, Fadeel B. Fas-triggered phosphatidylserine exposure is modulated by intracellular ATP. FEBS Lett. 2002;519:153–158. doi: 10.1016/s0014-5793(02)02743-6. [DOI] [PubMed] [Google Scholar]
- Green KN, Peers C. Amyloid beta peptides mediate hypoxic augmentation of Ca(2+) channels. J Neurochem. 2001;77:953–956. doi: 10.1046/j.1471-4159.2001.00338.x. [DOI] [PubMed] [Google Scholar]
- Haass C, Schlossmacher M, Hung AY, Vigo-Pelfrey C, Mellon A, Ostaszewski BL, Lieberburg I, Koo EH, Schenk D, Teplow DB, Selkoe DJ. Amyloid β-peptide is produced by cultured cells during normal metabolism. Nature. 1992;359:322–325. doi: 10.1038/359322a0. [DOI] [PubMed] [Google Scholar]
- Herrup K, Neve R, Ackerman SL, Copani A. Divide and die: cell cycle events as triggers of nerve cell death. J Neurosci. 2004;24:9232–9239. doi: 10.1523/JNEUROSCI.3347-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki K, Egashira N, Hatip-Al-Khatib I, Akiyoshi Y, Arai T, Takagaki Y, Watanabe T, Mishima K, Fujiwara M. Cerebral ischemia combined with β-amyloid impairs spatial memory in the eight-arm radial maze task in rats. Brain Res. 2006;1097:216–223. doi: 10.1016/j.brainres.2006.04.073. [DOI] [PubMed] [Google Scholar]
- Kakio A, Nishimoto S, Yanagisawa K, Kozutsumi Y, Matsuzaki K. Cholesterol-dependent formation of GM1 ganglioside-bound amyloid β-protein, an endogenous seed for Alzheimer amyloid. J Biol Chem. 2001;276:24985–24990. doi: 10.1074/jbc.M100252200. [DOI] [PubMed] [Google Scholar]
- Krijnen PAJ, Nijmeijer R, Meijer CJLM, Visser CA, Hack CE, Niessen HWM. Apoptosis in myocardial ischaemia and infarction. J Clin Pathol. 2002;55:801–811. doi: 10.1136/jcp.55.11.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFerla FM. Calcium dyshomeostasis and intracellular signaling in Alzheimer's disease. Nat Rev Neurosci. 2002;3:862–872. doi: 10.1038/nrn960. [DOI] [PubMed] [Google Scholar]
- Lang F, Gulbins E, Szabo I, Lepple-Wienhues A, Huber SM, Duranton C, Lang KS, Lang PA, Wieder T. Cell volume and the regulation of apoptotic cell death. J Mol Recognit. 2004;17:473–480. doi: 10.1002/jmr.705. [DOI] [PubMed] [Google Scholar]
- Lee G, Pollard HB, Arispe N. Annexin 5 and apolipoprotein E2 protect against Alzheimer's Amyloid β Peptide cytotoxicity by competitive inhibition at a common phosphatidylserine interaction site. Peptides. 2002;23:1249–1263. doi: 10.1016/s0196-9781(02)00060-8. [DOI] [PubMed] [Google Scholar]
- Loo DT, Copani A, Pike CJ, Whittemore RE, Walencewicz AJ, Cotman CW. Apoptosis is induced by β-amyloid in cultured central nervous system neurons. Proc Natl Acad Sci USA. 1993;90:7951–7955. doi: 10.1073/pnas.90.17.7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majoul I, Schmidt T, Pomasanova M, Boutkevich E, Kozlov Y, Soling HD. Differential expression of receptors for Shiga and Cholera toxin is regulated by the cell cycle. J Cell Sci. 2002;115:817–826. doi: 10.1242/jcs.115.4.817. [DOI] [PubMed] [Google Scholar]
- Martin OC, Pagano RE. Transbilayer movement of fluorescent analogs of phosphatidylserine and phosphatidylethanolamine at the plasma membrane of cultured cells. Evidence for a protein mediated and ATP dependent process(es) J Biol Chem. 1987;262:5890–5898. [PubMed] [Google Scholar]
- Martin SJ, Reutelingsperger CPM, McGahon AJ, Rader JA, Van Schie R, LaFace DM, Green DR. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. J Exp Med. 1995;182:1545–1556. doi: 10.1084/jem.182.5.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Cheng B, Davis D, Bryant K, Liberberg I, Rydel RE. β-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci. 1992;12:376–389. doi: 10.1523/JNEUROSCI.12-02-00376.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishima Y, Gotoh Y, Zieg J, Barrett T, Takano H, Flavell R, Davis RJ, Shirasaki Y, Greenberg ME. Beta-amyloid induces neuronal apoptosis via a mechanism that involves the c-Jun N-terminal kinase pathway and the induction of Fas ligand. J Neurosci. 2001;21:7551–7560. doi: 10.1523/JNEUROSCI.21-19-07551.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrov A, Acio ER, Narula N, Koldgie FD, Tait JF, Strauss HW, Narula J. Sarcolemmal phosphatidyl serine expression in ischemic myocardial syndromes can be detected by 99mTc-annexin V imaging. Circulation. 2000;102(Suppl 2):544. [Google Scholar]
- Rothman JE, Lenard J. Membrane asymmetry. Science. 1977;195:743–753. doi: 10.1126/science.402030. [DOI] [PubMed] [Google Scholar]
- Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, Sinha S, Schiossmacher M, Whaley J, Swindlehurst C, McCormack R, Wolfert R, Selkoe D, Lieberburg I, Schenk D. Isolation and quantification of soluble Alzheimer's β-peptide from biological fluids. Nature. 1992;359:325–327. doi: 10.1038/359325a0. [DOI] [PubMed] [Google Scholar]
- Shearman MS. Cellular MTT reduction distinguishes the mechanism of action of beta-amyloid from that of tachykinin receptor peptides. Neuropeptides. 1996;30:125–132. doi: 10.1016/s0143-4179(96)90079-7. [DOI] [PubMed] [Google Scholar]
- Shoji M, Golde TE, Ghiso J, Cheung TT, Estus S, Shaffer LM, Cai XD, McKay DM, Tintner R, Frangione B. Production of the Alzheimer amyloid β-protein by normal proteolytic processing. Science. 1992;258:126–129. doi: 10.1126/science.1439760. [DOI] [PubMed] [Google Scholar]
- Shoji M, Kanai M, Matsubara E, Tomidokoro Y, Shizuka M, Ikeda Y, Ykeda M, Harigaya Y, Okamoto K, Hirai S. The levels of cerebrospinal fluid Aβ40 and Aβ42(43) are regulated age-dependently. Neurobiol Aging. 2001;22:209–215. doi: 10.1016/s0197-4580(00)00229-3. [DOI] [PubMed] [Google Scholar]
- Simakova O, Arispe N. Early and late cytotoxic effects of external application of the Alzheimer's Aβ result from the initial formation and function of ion channels. Biochemistry. 2006;45:5907–5915. doi: 10.1021/bi060148g. [DOI] [PubMed] [Google Scholar]
- Smith IF, Green KN, LaFerla FM. Calcium dysregulation in Alzheimer's disease: recent advances gained from genetically modified animals. Cell Calcium. 2005;38:427–437. doi: 10.1016/j.ceca.2005.06.021. [DOI] [PubMed] [Google Scholar]
- Verhoven B, Schlegel R, Williamson P. Mechanisms of phosphatidylserine exposure, a phagocyte recognition signal, on apoptotic T lymphocytes. J Exp Med. 1995;182:1597–1601. doi: 10.1084/jem.182.5.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi M, Okada T, Kozutsumi Y, Matsuzaki K. GM1 ganglioside-mediated accumulation of amyloid β-protein on cell membranes. Biochem Biophys Res Comm. 2005;328:1019–1023. doi: 10.1016/j.bbrc.2005.01.060. [DOI] [PubMed] [Google Scholar]
- Wisniewski T, Ghiso J, Frangione B. Biology of Aβ amyloid in Alzheimer's disease. Neurobiol Dis. 1997;4:311–328. doi: 10.1006/nbdi.1997.0147. [DOI] [PubMed] [Google Scholar]
- Wolfs JLN, Comfurius P, Rasmussen JT, Keuren JFW, Lindhout T, Zwaal RFA, Bevers EM. Activated scramblase and inhibited aminophospholipid translocase cause phosphatidylserine exposure in a district platelet fraction. Cell Mol Life Sci. 2005;62:1514–1525. doi: 10.1007/s00018-005-5099-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwaal RFA, Schroit AJ. Pathophysiologic implications of membrane phospholipid asymmetry in blood cells. Blood. 1997;89:1121–1132. [PubMed] [Google Scholar]
- Zwaal RF, Comfurius P, Bevers EM. Surface exposure of phosphatidylserine in pathological cells. Cell Mol Life Sci. 2005;62:971–988. doi: 10.1007/s00018-005-4527-3. [DOI] [PubMed] [Google Scholar]