

Abstract
Polymorphisms in genetic copy number can influence gene expression, coding sequence, and zygosity, making them powerful actors in the evolutionary process. Copy number variants (CNVs) are however understudied, being more difficult to detect than single-nucleotide polymorphisms. We take advantage of the intense selective pressures on the major malaria vector Anopheles gambiae, caused by the widespread use of insecticides for malaria control, to investigate the role of CNVs in the evolution of insecticide resistance. Using the whole-genome sequencing data from 1142 samples in the An. gambiae 1000 genomes project, we identified 250 gene-containing CNVs, encompassing a total of 267 genes of which 28 were in gene families linked to metabolic insecticide resistance, representing significant enrichment of these families. The five major gene clusters for metabolic resistance all contained CNVs, with 44 different CNVs being found across these clusters and multiple CNVs frequently covering the same genes. These 44 CNVs are widespread (45% of individuals carry at least one of them) and have been spreading through positive selection, indicated by their high local frequencies and extended haplotype homozygosity. Our results demonstrate the importance of CNVs in the response to selection, highlighting the urgent need to identify the contribution of each CNV to insecticide resistance and to track their spread as the use of insecticides in malaria endemic countries intensifies and as the operational deployment of next-generation bed nets targeting metabolic resistance gathers pace. Our detailed descriptions of CNVs found across the species range provide the tools to do so.
Copy number variants (CNVs) are a form of genetic variation that occur when a genomic sequence is deleted or duplicated, potentially affecting both the structure and expression levels of coding sequences and playing a crucial role in evolution and adaptation (Redon et al. 2006; Chen et al. 2008; Emerson et al. 2008). Part of the importance of CNVs lies in the wide range of effects that they can have on the transcriptome. Increases in copy number (amplifications) encompassing the entire sequence of a gene can lead to elevated expression levels if new gene copies are associated with cis-regulatory sequences required for transcription (Handsaker et al. 2015). Alternatively, duplication or deletion of only part of a gene's sequence can lead to major changes in protein sequence. For example, in humans, a CNV spanning parts of two glycophorin genes creates a novel hybrid glycophorin associated with resistance to malaria (Leffler et al. 2017). CNVs can also allow alternative variants of a gene to appear in tandem on the same chromosome through heterogeneous gene duplication, creating constitutive heterozygotes. This can be seen in the mosquitoes Anopheles gambiae and Culex pipiens, where mutations in Acetylcholinesterase 1 (Ace1) cause resistance to carbamate and organophosphate insecticides, but carry a fitness cost in the absence of insecticide. This cost is mitigated in heterozygotes, leading to the spread of heterogeneous Ace1 duplications in which the mutant and wild-type alleles co-occur (Labbé et al. 2007; Assogba et al. 2015).
Although the importance of CNVs is widely recognized, they typically receive less attention than single-nucleotide polymorphisms (SNPs) in investigations of genetic variation, likely because they are harder to identify. Population-level genome-wide analyses of CNVs are thus rare, and the extent of their impact on evolution is poorly understood (although a worldwide study has been conducted in humans [Sudmant et al. 2015]).
The malaria mosquito An. gambiae and its close sister species An. coluzzii are excellent organisms in which to study the evolution of CNVs at a population level for three reasons. First, these species are the major vectors of malaria in Sub-Saharan Africa (SSA) and are highly anthropophilic (Kiszewski et al. 2004). Consequently, they are heavily targeted by insecticides used in malaria control programs, creating intense selection pressure that drives rapid contemporary evolution. CNVs thus have an opportunity to contribute to the selective response to these pressures, providing a context in which their importance can be assessed. Second, CNVs can play a key role in the evolution of insecticide resistance through their ability to affect gene expression and allow coexpression of wild-type and mutant alleles (Bass and Field 2011; Weetman et al. 2018). Despite nearly two decades of genetic research into insecticide resistance, known resistance-associated SNPs are still unable to explain much of the variance in insecticide resistance (Donnelly et al. 2016). CNVs potentially represent a crucial source of missing variation that can potentially fill this gap. Third, the Anopheles gambiae 1000 Genomes Project (Ag1000G) has produced whole-genome sequencing data from 1142 individual An. gambiae and An. coluzzii from multiple locations in SSA, providing a unique opportunity to conduct genome-wide searches for CNVs from across the species’ distributions (https://www.malariagen.net/data/ag1000g-phase-2-ar1).
The two major mechanisms of insecticide resistance are target site resistance and metabolic resistance (Liu 2015). CNVs have been found to affect all three major insecticide target site genes in insects: Ace1 (Assogba et al. 2015; Weetman et al. 2015), the para voltage-gated sodium channel (Vgsc) (Martins et al. 2013, 2017), and the gamma-aminobutyric acid (GABA) receptor Rdl (Remnant et al. 2013), usually combining resistant and wild-type alleles to provide resistance while mitigating its cost. However, duplications in Ace1 are the only CNVs so far shown to play a role in Anopheles insecticide resistance, being associated with a resistance-conferring mutation and either increasing the resistance which it confers (Assogba et al. 2016) or diminishing the fitness cost of the mutation (Assogba et al. 2015).
Metabolic genes whose expression levels are associated with insecticide resistance have been reported in a wide range of species (Li et al. 2007) and are particularly concerning because up-regulation of a single gene can provide cross-resistance to a range of insecticides with different targets. For example, in An. gambiae, up-regulation of the cytochrome P450s Cyp6m2 and Cyp6p3 is associated with resistance to pyrethroids and bendiocarb (Edi et al. 2014). Despite the importance of metabolic insecticide resistance, the causative genetic variants often remain unidentified, although focused studies have identified cases in which CNVs play a critical role in metabolic resistance in a range of species. In Drosophila, duplication of the detoxification gene Cyp6g1 has been implicated in resistance to DDT (Schmidt et al. 2010), whereas in Cx. quinquefasciatus resistance to permethrin is associated with increased expression of Cyp9m10, due in part to a duplication (Itokawa et al. 2011). Similarly, amplification of esterase genes leading to elevated expression provides increased resistance to organophosphates in the mosquitoes Cx. pipiens (Raymond et al. 2001) and Aedes albopictus (Grigoraki et al. 2015), and to several insecticides in the peach-potato aphid Myzus persicae (Devonshire et al. 1998; Field et al. 1999). In brown planthoppers, neofunctionalization of a duplicated copy of Cyp6er1 has even created a novel gene variant providing resistance to the neonicotinoid imidacloprid (Zimmer et al. 2018). In An. gambiae and An. coluzzii, the most important metabolic genes that have been identified as major insecticide resistance candidates to date are Gste2 (Mitchell et al. 2014), Cyp6p3 (Müller et al. 2008; Edi et al. 2014), Cyp6m2 (Stevenson et al. 2011; Mitchell et al. 2012; Edi et al. 2014), Cyp6z1 (Chiu et al. 2008), and Cyp9k1 (Main et al. 2015; Vontas et al. 2018). If CNVs play an important role in the evolution of insecticide resistance in An. gambiae and An. coluzzii, we would expect to find them among such genes.
Here, we perform an agnostic genome-wide scan of CNVs in An. gambiae and An. coluzzii using the whole-genome sequencing from Phase 2 of Ag1000G, which consists of 1142 wild-caught mosquitoes collected from 16 populations between 2000 and 2012 and sequenced with a mean coverage of 32×. We then focus on five regions of particular interest for metabolic insecticide resistance to investigate whether CNVs in these regions are under positive selection.
Results
Genome-wide CNV discovery
We first used short-read whole-genome sequencing data from the Ag1000G Phase 2 data set to find genomic regions with increased read coverage relative to the genomic average, controlling for GC richness and excluding 27 individual mosquitoes with unusually high coverage variance. Briefly, we filtered 300-bp genomic windows for mapping quality and rare levels of GC content (which prevented normalization) and identified regions covering at least five contiguous 300-bp windows of at least 1.5-fold increased coverage determined by a Hidden Markov Model (HMM). We then filtered this initial CNV set based on the relative likelihood of the observed read coverage in the absence of a CNV. After grouping identical CNVs between individuals according to their genomic locations and retaining only CNVs present in at least 5% of individuals in a population (or at least three individuals in small populations), we identified 1557 candidate CNVs (Fig. 1; Supplemental Data S1). The largest of these (203,100 bp) was the well-documented duplication in the insecticide target site Ace1. No CNVs were found in the other common insecticide target sites Vgsc or Rdl.
Figure 1.

Distribution of CNVs in different size ranges across the genome. Blue bars represent regions of the genome in which a CNV was found. Numbers on the left indicate the CNV size range. Numbers on the right show the number of CNVs in that size range. Simulations showed a significant enrichment of CNVs in heterochromatic regions (0 out of 10,000 simulations had as many CNVs in the heterochromatin).
To estimate the accuracy of the CNV detection method, we performed simulations in which the coverage values obtained at each 300-bp window were randomly shuffled. These simulations indicated a low rate of CNVs being the result of random chance (false discovery rate of 0.003 at the sample level; 0.013 at the population level) and good rates of recovery for CNVs spanning at least 10 windows (≥85% at the sample level; ≥83% after population-level filtering), with a lower rate of recovery for CNVs spanning only five windows (31.4% at an individual level; 42.2% after population-level filtering).
To determine the genomic factors associated with the presence of CNVs, we investigated how CNVs are distributed relative to chromatin type and gene content. We found that CNVs were particularly abundant in heterochromatic regions. Of the 1557 CNVs, 534 (34.3%) were found in the heterochromatin, which covers only 9.3% of the genome (P < 0.0002; from 10,000 simulations, the number of heterochromatic CNVs ranged from 44 to 112, with a mean of 77). CNVs in heterochromatin were also significantly larger than in euchromatin (median for heterochromatin: 7200 bp, median for euchromatin: 3300 bp; Wilcoxon test, n1 = 534, n2 = 1023, W = 377,190, P < 0.0001) (Supplemental Fig. S1).
CNVs were also more abundant in gene-containing regions. Of the 1557 CNVs, 250 contained at least one gene, significantly more than expected by chance (P < 0.0002; across 10,000 simulations, the number of CNVs that contained genes ranged from 107 to 187, with a mean of 145). Because euchromatin typically contains more genes than heterochromatin, we repeated the simulation, focusing on euchromatic CNVs and randomizing their position within the euchromatin. Of the 1023 euchromatic CNVs, 226 contained at least one gene, again representing a significant enrichment for genic regions (P < 0.0002; across 10,000 simulations, the number of CNVs that contained genes ranged from 59 to 127, with a mean of 92). This enrichment was consistent for all sizes of euchromatic CNVs (Supplemental Table S1).
CNVs are enriched in metabolic resistance gene families
We next explored whether CNVs were enriched for genes potentially involved in metabolic insecticide resistance. Overall, out of 10,939 genes included in the analysis, 267 (2.4%) were within at least one CNV (Supplemental Data S2). Of these 267 genes, 28 were candidate metabolic resistance genes (defined as a cytochrome P450, glutathione S-transferase or carboxylesterase, and referred to as “metabolic detox genes” from here on). Because many related genes occur in clusters, and are therefore not independently included in CNV events, we counted the number of CNVs that included at least one metabolic detox gene. Of the 250 CNVs that contained any genes, 27 contained at least one metabolic detox gene, significantly more than expected by chance (P < 0.0002; from 10,000 simulations, the number of gene-containing CNVs that contained detox genes ranged from 0 to 14, with a mean of 4). The same result was found when only euchromatic CNVs were considered, for all CNV sizes (Supplemental Table S2). Although there was some variation between populations in the number of metabolic detox genes found in CNVs (Table 1), this was not significant (Fisher's exact test: P = 0.08).
Table 1.
Number of CNVs detected either containing or not containing at least one cytochrome P450, glutathione S-transferase, or carboxylesterase (detox genes)

Genes found in CNVs were enriched for 13 molecular function Gene Ontology (GO) terms after multiple correction to a Q-value threshold of 0.05 (Supplemental Data S3), primarily reflecting an enrichment for two classes of genes: cytochrome P450s (significant GO terms included monooxygenase activity, heme binding, iron ion binding, oxidoreductase activity) and proteases (significant GO terms included several forms of peptidase activity). This result also held when heterochromatic CNVs were excluded, with 12 of the 13 GO terms remaining significant (Supplemental Data S3). No GO terms from biological process or cellular compartment ontologies were significantly enriched.
The 28 metabolic detox genes found inside CNVs were predominantly from gene clusters that have previously been implicated in insecticide resistance (Supplemental Data S2), with 16 of the 28 genes being members of the glutathione S-transferase epsilon cluster (Gste on Chromosome 3R) or one of four cytochrome P450 clusters (Cyp6p on Chromosome 2R, Cyp6m on Chromosome 3R, Cyp6z on Chromosome 3R, and Cyp9k1 on Chromosome X). This again indicates that genes involved in metabolic insecticide resistance have been the focus of amplification events. We therefore performed a detailed analysis of the CNVs around these five gene clusters (Fig. 2A). Because the Cyp6aa1/Cyp6aa2 genes, which are adjacent to the Cyp6p cluster, were also highly represented in the list of amplified genes (Supplemental Data S2), we extended the study region around Cyp6p3 to include these genes.
Figure 2.

(A) CNVs in gene clusters known to be associated with metabolic insecticide resistance were found on all three chromosomes. (B) Of the 15 CNVs in Cyp6aa1–Cyp6p2, 13 include Cyp6aa1 and five include Cyp6p3. Inset maps show countries in which at least 5% of individuals carried a CNV in Cyp6aa1 (blue) and Cyp6p3 (red), and the countries absent from the data set are shown in gray. (C) Of the 11 duplications in Gstu4–Gste3, 10 include Gste2. Inset map shows countries in which at least 5% of individuals carried a CNV in Gste2 (red). Black rectangles and vertical gray bars show the positions of the genes in the cluster, with Cyp6aa1, Cyp6p3, and Gste2 highlighted in color. Purple horizontal bars show the extent of each CNV, with the gap in Gstue_Dup5 showing the deletion within this amplification. CNV names are abbreviated to Dup# and refer to Cyp6aap_Dup# and Gstue_Dup# in panels B and C, respectively. Further details on each of these CNVs, and of those from the other gene clusters, are presented in Supplemental Data S5–S8.
To more accurately determine the number of CNVs in each of these five gene clusters, we used discordant read pairs and reads aligning to CNV breakpoints to distinguish different CNVs. Three of the five gene clusters showed high numbers of different CNVs. We identified 16 CNV alleles in Cyp9k1 (named Cyp9k1_Dup1–16) (Supplemental Data S7; Supplemental Fig. S4), 15 in the Cyp6aa1–Cyp6p2 cluster (Cyp6aap_Dup1–15) (Fig. 2B; Supplemental Data S4; Supplemental Fig. S1), 11 in the Gstu4–Gste3 cluster (Gstue_Dup1–11) (Fig. 2C; Supplemental Data S5; Supplemental Fig. S2), one in Cyp6m2 (Cyp6m_Dup1) (Supplemental Data S6; Supplemental Fig. S3), and one in Cyp6z3–Cyp6z1 (Cyp6z_Dup1) (Supplemental Data S6; Supplemental Fig. S3).
Several CNV alleles were found across different populations (for example, Cyp6aap_Dup7 was found in An. coluzzii from Burkina Faso, Côte d'Ivoire, Ghana, and Guinea) (Supplemental Data S4), although none were found in all populations of either species (Supplemental Data S8). Furthermore, multiple CNV alleles covering the same genes could be found in the same population (for example, Cyp9k1_Dup4, Dup11, and Dup15 in An. gambiae from Burkina Faso) (Supplemental Data S7). For the Cyp6aa/p cluster, CNVs were primarily found in An. coluzzii from Burkina Faso, Côte d'Ivoire, and Ghana (Table 2). In Cyp9k1, CNVs were primarily found in An. gambiae from Burkina Faso, Ghana, and Guinea. Some genes were amplified at very high frequency in certain populations (Table 2). For example, >92% of An. gambiae from Burkina Faso had a CNV in Cyp9k1, and >90% of An. coluzzii from Côte d'Ivoire had a CNV covering genes in the Cyp6aa/p cluster. Overall, 511 of the 1142 samples in this study (45%) carried at least one of the CNVs described in these five gene clusters.
Table 2.
Number (and proportion) of individuals with a CNV covering Cyp6aa1, Cyp6p3, Gste2, Cyp6m2, Cyp6z1, or Cyp9k1

We used the patterns of discordant reads supporting each of the CNVs to putatively determine the type of duplication event that caused them (Fig. 3). Of the 44 CNVs, 33 are supported by read pairs mapping facing away from each other in the genome and spanning the region of increased coverage, indicating a tandem duplication (Supplemental Data S4–S7), and two CNVs were supported by read pairs mapping in the same orientation, indicating a tandem inversion (Supplemental Data S4, S7). The remaining CNVs were supported by reads whose mate or soft-clipped bases mapped elsewhere on the genome, often in multiple genomic locations, suggesting a role for transposable elements (TEs) in the duplication event (Supplemental Data S4–S7). Two of these CNVs had flanking sequences that matched known transposable elements in An. gambiae. The soft-clipped bases of reads mapping to the breakpoints of Gstue_Dup7 and Gstue_Dup8, respectively, returned significant BLAST hits to a hAT DNA transposon (HATN1_AG; E-value = 2 × 10−26) and a transposon bearing similarity to mariner/Tc1-like elements (IKIRARA1; E-value = 10−34) (Leung and Romans 1998), in the Repbase database (Bao et al. 2015).
Figure 3.
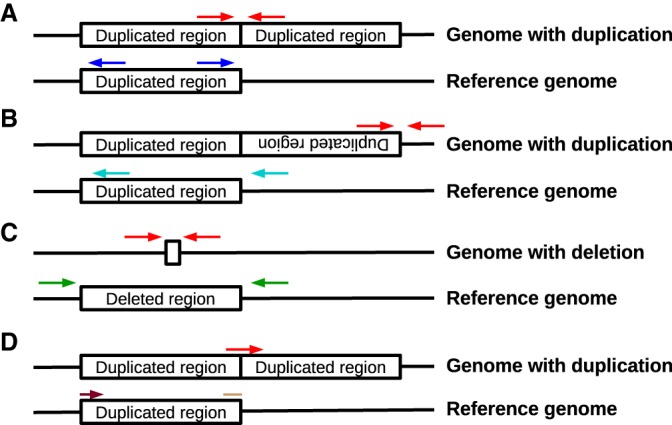
Three types of discordant read pairs (A–C) and breakpoint reads (D) were used to identify different CNV alleles. (A) In tandem duplications, read pairs derived from segments spanning the CNV breakpoint (red arrows) align facing away from each other around the breakpoint on the reference genome (dark blue arrows). (B) In tandem inversions, read pairs derived from segments spanning the end of the inverted segment (red arrows) align facing in the same direction as each other around the breakpoint on the reference genome (cyan arrows). (C) In deletions, read pairs derived from segments spanning the deleted sequence (red arrows) align in the correct orientation around the breakpoint, but farther apart than expected given the insert size of the sequencing library (green arrows). (D) In any of the aforementioned types of CNV (tandem duplication shown here as an example), reads crossing the breakpoint (red arrow) will only partially align on either side of the breakpoint. For the tandem duplication shown here, the start of the read (light brown start of an arrow) aligns at the end of the duplicated region, whereas the end of the read (dark brown end of an arrow) aligns at the start of the duplication.
The CNVs were differentiated based on their patterns of discordant read pairs, which are influenced by the position of the CNV breakpoints and the orientation of the duplication (Fig. 3). It is possible that independent duplication events with very similar breakpoints could appear to be the same CNV (for example, Cyp6aap_Dup1 may in fact represent two different CNVs) (Supplemental Data S4), making the real number of CNV events even greater than reported. Equally, a mutation that disrupts the breakpoint of a CNV could affect its pattern of discordant reads and lead to a single CNV event appearing as two different CNV alleles. Although we cannot exclude this possibility, analysis of the haplotype backgrounds of the different CNVs suggested that this is not the case for at least most of the CNV alleles (Supplemental Data S8).
CNVs in metabolic resistance genes are under positive selection
Several CNV alleles were found at high local frequencies (Supplemental Data S9), suggesting that they are likely to be under positive selection. To investigate this possibility, we phased the CNV genotype calls onto the Ag1000G Phase 2 haplotype scaffold and calculated extended haplotype homozygosity (EHH) for the CNV alleles present in at least 5% of individuals in a population.
Rates of EHH decay around CNV alleles were consistently lower than for wild-type (WT) haplotypes (Fig. 4; Supplemental Figs. S6, S8, S10), supporting our contention that these alleles are reaching high frequency through positive selection. Furthermore, the median length of shared haplotypes was significantly higher between pairs of haplotypes carrying the same CNV allele than between wild-type haplotypes from the same population (bootstrapped 95% confidence intervals for the medians did not overlap) (Fig. 5; Supplemental Figs. S7, S9, S11).
Figure 4.

Evidence for prolonged linkage disequilibrium around CNVs in the Cyp6aa1–Cyp6p2 gene cluster. Extended haplotype homozygosity (EHH) decay was calculated around CNV and non-CNV (WT) haplotypes using SNPs from outside the region containing CNVs (break in the x-axis): (BF) Burkina Faso; (CI) Côte d'Ivoire; (UG) Uganda; (col) An. coluzzii; (gam) An. gambiae.
Figure 5.

Lengths of pairwise shared haplotypes are greater between samples sharing a CNV allele than between wild-type samples. Shared haplotype lengths were calculated on either side of the CNV-containing region of the Cyp6aa/p gene cluster. Non-CNV (WT) samples were taken from the same populations as the focal CNV alleles. Bars show the distribution of shared haplotype lengths between all haplotype pairs with the same core haplotype. Bar limits show the interquartile range, fliers show the 5th and 95th percentiles, horizontal black lines show the median, and notches in the bars show the bootstrapped 95% confidence interval for the median. The names of the CNVs (Cyp6aap_Dup#) are abbreviated as Dup#: (BF) Burkina Faso; (CI) Côte d'Ivoire; (UG) Uganda; (col) An. coluzzii; (gam) An. gambiae.
Phasing of CNV genotype calls was only possible for simple duplications, where the zygosity of the CNV alleles could be determined from the copy number estimates. For CNV alleles with higher copy numbers (triplications and above), this was not possible; thus the EHH decay could not be calculated. In the case of Cyp9k1, the CNV with the highest frequency (Cyp9k1_Dup11, found in An. gambiae from Burkina Faso, Ghana, and Guinea) could not be phased. We therefore investigated whether this CNV was associated, at the sample level, with haplotypes under selection. Hierarchical clustering of the haplotypes in these three populations revealed two large cross-population haplotype clusters around Cyp9k1, indicating selective sweeps (Supplemental Fig. S12). Cluster 1 was very strongly associated with Cyp9k1_Dup11 in both males (Fisher's exact test, P < 0.0001) (Supplemental Table S3) and females (Spearman's rank correlation: ρ = 0.9, P < 0.0001) (Fig. 6A). Cluster 2 was associated with the presence of Cyp9k1_Dup15, but the correlation was not as strong as between Cluster 1 and Cyp9k1_Dup11 (Spearman's rank correlation: ρ = 0.65, P < 0.0001) (Fig. 6B; Supplemental Table S4).
Figure 6.

The two main haplotype clusters around Cyp9k1 in Burkina Faso, Ghana, and Guinea are associated with respective CNV alleles. Points are jittered to show overlapping data. Lines show least-squares regression through the data. (A) Strong correlation between Cyp9k1_Dup11 and haplotype Cluster 1. Most of the points lie on a line of slope 0.5, indicating that Cyp9k1_Dup11 is found most frequently as a triplication (two extra copies per chromosome), although both lower and higher copy number versions of this CNV exist. (B) Weaker correlation between Cyp9k1_Dup15 and haplotype Cluster 2.
A Gste2 duplication in Burkina Faso is associated with the resistance-conferring I114T mutation
Ten of the 11 CNV alleles found in the Gstu4–Gste3 cluster included Gste2 (Fig. 2), perhaps reflecting the known importance of this gene in insecticide resistance. The well-characterized I114T mutation in Gste2 is known to confer DDT resistance (Mitchell et al. 2014) and could be associated with gene duplications in a similar fashion to other mutations such as Ace1 G119S. We therefore investigated whether any of the CNV alleles in Gste2 were associated with this mutation. Gste2-114T is present across Africa and in both An. gambiae and An. coluzzii (The Anopheles gambiae 1000 Genomes Consortium 2017) but was only associated with Gstue_Dup1 in our data. Gstue_Dup1 was found in 16 An. coluzzii samples from Burkina Faso, all of which were at least heterozygote for 114T (Supplemental Table S5). The presence of 114T homozygotes, together with the ratio of reads supporting the I114 and 114T alleles in heterozygotes (roughly 1:2), indicate that both copies of Gste2 in the Gstue_Dup1 CNV carry the 114T mutation.
Cyp6aa1 is more strongly associated with CNVs than Cyp6p3
Of the 15 CNV alleles found in the Cyp6aa1–Cyp6p2 cluster, five included Cyp6p3 but 13 included Cyp6aa1 (Fig. 2B). Cyp6p3 CNVs were found at high (>50%) frequency in one population (Côte d'Ivoire An. coluzzii: 90%), whereas Cyp6aa1 CNVs were found at high frequency in An. coluzzii from Burkina Faso (91%), Côte d'Ivoire (89%) and Guinea (75%), and in An. gambiae from Uganda (64%).
Discussion
Our study detected 1557 CNVs in 16 populations of An. gambiae and An. coluzzii, with CNVs being more abundant and larger in heterochromatin than in euchromatin. This parallels results in humans and rats, in which CNVs have been found to be particularly abundant in centromeres and telomeres (Nguyen et al. 2006; Guryev et al. 2008), typically composed of heterochromatin. The size distribution of CNVs was strongly right-skewed, with small CNVs being the most abundant in both euchromatin and heterochromatin, although we did not look for CNVs smaller than 1500 bp to avoid false discoveries. CNVs were biased toward gene-containing regions both overall and after excluding heterochromatic regions. This bias for genic regions has similarly been found in duplications in humans (Nguyen et al. 2006) and rats (Guryev et al. 2008), but not Drosophila (Schrider et al. 2016). Our results therefore indicate that abundance of CNVs around genes is not restricted to mammals.
The genes found in CNVs were themselves enriched for families involved in metabolic insecticide resistance. These results mirror findings in Drosophila, where cytochrome P450s were disproportionately represented in CNVs (Schrider et al. 2016). Similarly, in Ae. aegypti, cytochrome P450s were enriched among genes showing evidence of higher copy number in populations resistant to deltamethrin compared to susceptible populations (Faucon et al. 2015). In our study, the five metabolic genes most strongly associated with insecticide resistance in the literature for An. gambiae and An. coluzzii, and which have been shown to metabolize insecticides in vitro (Gste2, Cyp6p3, Cyp6m2, Cyp6z1, and Cyp9k1), were all found to be amplified in at least one population. Furthermore, three of these genes showed evidence of repeated CNV events within and between populations, with a total of 44 different CNVs across the five clusters, and as many as 16 in Cyp9k1. Of 44 CNVs, the majority (33) were tandem duplications, two were tandem inversions, and two (both in the Gstu4–Gste3 cluster) showed evidence of association with known DNA transposons. The remaining seven could not be classified, but the presence of unmappable sequences around the breakpoints suggests that they may also be associated with repetitive elements.
Evidence for the importance of Gste2 in insecticide resistance comes from studies showing its up-regulation in DDT resistant An. gambiae (Ding et al. 2003; David et al. 2005) and An. funestus (Riveron et al. 2014a) compared to susceptible mosquitoes, and from transgenic expression of An. gambiae/An. funestus Gste2 in Drosophila providing resistance to DDT (Mitchell et al. 2014; Riveron et al. 2014a). Nonsynonymous SNPs in Gste2 have also been shown to be associated with resistance to DDT in An. funestus (Riveron et al. 2014a) and to both DDT and deltamethrin in An. gambiae (Mitchell et al. 2014; Opondo et al. 2016). In our study, Gste2 was amplified in Kenya, in An. coluzzii from Angola, Burkina Faso, and Ghana, and in An. gambiae from Gabon and Uganda.
Cyp6p3 is up-regulated in mosquitoes resistant to pyrethroids, DDT, and bendiocarb (Djouaka et al. 2008; Müller et al. 2008; Fossog Tene et al. 2013; Kwiatkowska et al. 2013; Edi et al. 2014; Ngufor et al. 2015), metabolizes permethrin and deltamethrin (Müller et al. 2008), and provides resistance to pyrethroids when expressed in Drosophila (Edi et al. 2014). Cyp6m2 is also up-regulated in mosquitoes with resistance to permethrin, DDT, and bendiocarb (Müller et al. 2007; Djouaka et al. 2008; Mitchell et al. 2012; Edi et al. 2014), metabolizes pyrethroids and DDT (Stevenson et al. 2011; Mitchell et al. 2012), and provides resistance to pyrethroids, DDT, and bendiocarb when expressed in Drosophila (Edi et al. 2014). In our study, Cyp6p3 and Cyp6m2 were found amplified primarily in An. coluzzii from Côte d'Ivoire, a population with known up-regulation of both Cyp6p3 and Cyp6m2 compared to susceptible populations (Edi et al. 2014). Particularly in the case of Cyp6m2, this up-regulation may not be driven solely by CNVs because the CNV frequency and copy number are not sufficient to explain the expression levels, but the selective pressure to up-regulate these genes may have played a part in maintaining these CNVs in the population.
Cyp6z1 was amplified in An gambiae from Burkina Faso and Guinea. Cyp6z1 is up-regulated in mosquitoes with resistance to pyrethroids and DDT (Nikou et al. 2003; David et al. 2005) and metabolizes DDT and carbaryl (Chiu et al. 2008). Finally, Cyp9k1 was the most widely amplified gene of the five clusters that we studied, with CNVs found in more than half the populations in our data set. Cyp9k1 is up-regulated in mosquitoes resistant to pyrethroids and DDT (Fossog Tene et al. 2013; Thomsen et al. 2014; Ngufor et al. 2015) and metabolizes deltamethrin (Vontas et al. 2018). Furthermore, a selective sweep in the Cyp9k1 region has been associated with insecticide resistance in An. coluzzii (Main et al. 2015).
In-depth investigation of the CNVs around these five genes revealed strong evidence that they provide a selective advantage. First, some of the CNV alleles were found at high frequencies and across several populations. Second, the CNV alleles consistently showed evidence of being under positive selection as haplotype homozygosity was extended further for the CNVs than for wild-type haplotypes. Evidence for positive selection was also found in a CNV where the EHH score could not be calculated. Cyp9k1_Dup11, which exists as both duplications and triplications and thus could not be phased onto a haplotype scaffold for homozygosity calculation, was consistently found in the same samples as the haplotype of a large selective sweep around Cyp9k1 in An. gambiae from Burkina Faso, Guinea, and Ghana, raising the strong possibility that this CNV is the focus of the selective sweep. Although Cyp9k1_Dup11 may be in linkage disequilibrium with other mutations, the high frequency of the triplicated version of Cyp9k1_Dup11 compared to the duplicated version, both of which are associated with the swept haplotype cluster, suggests that higher-order amplifications of Cyp9k1 provide a selective advantage. The changes in allele frequencies in the different amplification levels in this CNV will need to be monitored to determine whether the triplication eventually replaces the duplication entirely.
A major current consideration for malaria control programs is the value of investing in next-generation bed nets that incorporate piperonyl butoxide (PBO), which interfere with cytochrome P450-mediated insecticide resistance, and which have been shown to be effective at reducing mosquito abundance and malaria incidence in at least some regions (Protopopoff et al. 2018). However, in most cases, it is not clear whether local insecticide resistance is cytochrome P450-mediated, partly due to the lack of molecular markers for identifying metabolic resistance. Our results highlight areas where mosquitoes are likely to exhibit cytochrome P450-based resistance. For example, CNVs in the Cyp6aa/p cluster were primarily found in An. coluzzii from Burkina Faso, Côte d'Ivoire, and Ghana, whereas CNVs in Cyp9k1 were primarily found in An. gambiae from Burkina Faso, Ghana, and Guinea. Although the presence of cytochrome P450 CNVs indicates the likely presence of cytochrome P450-mediated resistance, we emphasize that their absence does not necessarily indicate that such resistance does not exist. Other forms of genetic variation that can increase gene expression, such as mutations in the regulatory region (Schmidt et al. 2010), need to be investigated to obtain a complete set of molecular markers for detecting metabolic resistance.
All but one of the 11 CNV alleles in the Gstu/e cluster included Gste2, indicating that this is the major target of gene amplification in this cluster. Given the body of evidence linking Gste2 to DDT and pyrethroid resistance across multiple species—An. gambiae (Mitchell et al. 2014), An. funestus (Riveron et al. 2014a), Aedes aegypti (Lumjuan et al. 2011)—the focus of amplifications on this gene is likely to be linked to its importance in resistance.
The Gstue_Dup1 duplication in Burkina Faso occurs on the background of the Gste2_114T SNP, associated with DDT resistance in An. gambiae (Mitchell et al. 2014). The duplication may therefore serve to increase the dosage of Gste2, whose detoxifying activity has already been elevated by the 114T mutation. Alternatively, the role of Gstue_Dup1 may be to compensate for any negative fitness effects of 114T. Although impaired Gste2 activity may be compensated by increasing the expression of the gene, Gstue_Dup1 is homogeneous for 114T, excluding the possibility of compensation by pairing of mutant and wild-type alleles as found in heterogeneous Ace1 duplications (Assogba et al. 2015).
In the Cyp6aa/p cluster, only five of the 15 CNVs included Cyp6p3, and these were only found at appreciable frequency in An. coluzzii from Côte d'Ivoire. In contrast, 13 of the 15 CNVs included Cyp6aa1, with high CNV frequencies found in An. coluzzii from Burkina Faso, Côte d'Ivoire, and Guinea, and in An. gambiae from Uganda. Furthermore, the five high-frequency CNVs that include Cyp6aa1 (Cyp6aap_Dup1, Cyp6aap_Dup7, Cyp6aap_ Dup10, Cyp6aap_Dup14, Cyp6aap_Dup15) all show evidence of positive selection. Although Cyp6aa1 has received substantially less attention than Cyp6p3, it has previously been implicated in insecticide resistance. Expression of Cyp6aa1 is higher in populations of An. gambiae and An. coluzzii that are resistant to pyrethroids and DDT compared to susceptible laboratory colonies (Kwiatkowska et al. 2013; Thomsen et al. 2014). There is also strong evidence for a link between Cyp6aa1 and insecticide resistance in two congeneric species. In An. funestus, expression of Cyp6aa1 is higher in mosquitoes that have survived permethrin exposure compared to a susceptible strain (Riveron et al. 2014b; Ibrahim et al. 2018), and the protein has been shown to metabolize pyrethroids and drive resistance when expressed in Drosophila (Ibrahim et al. 2018). In An. minimus, the ortholog of Cyp6aa1 is up-regulated as a result of selection for resistance to deltamethrin (Rodpradit et al. 2005), and the protein has been shown to metabolize pyrethroids (Duangkaew et al. 2011). The ability of An. gambiae Cyp6aa1 to metabolize insecticides has not been tested empirically, although theoretical modeling suggests that it should effectively bind to permethrin and deltamethrin (Ibrahim et al. 2018). The high frequency of amplifications in Cyp6aa1 and the signals of selection associated with them suggest that the importance of this gene for insecticide resistance in An. gambiae and An. coluzzii has been underappreciated.
In conclusion, our results show a key role for CNVs in the adaptive response to strong and recent selective pressure. In populations of Anopheles mosquitoes across Africa, genes involved in metabolic resistance to insecticides have been duplicated, and these duplications have been driven to high frequencies by positive selection. These results highlight CNVs as a form of variation that can act as a rapid response to selective pressures requiring changes in expression levels. The widespread emergence of CNVs as a means of metabolic insecticide resistance reinforces the need to develop novel insecticidal compounds to tackle cross-resistance and highlights the potential value of PBO-treated bed nets. Our findings also highlight Cyp6aa1 as a gene that should be more closely investigated for its importance in An. gambiae, having been so far overlooked in preference to its genomic neighbor Cyp6p3. More broadly, the focus on SNPs in An. gambiae insecticide resistance research has allowed the emergence and selective spread of copy number mutations in key insecticide resistance genes to go unnoticed. Our findings demonstrate the importance of surveillance and investigation of CNVs in these genes. To this end, the breakpoint descriptions provided in our study will allow these CNVs to be screened and monitored in mosquito populations, allowing the spread of these mutations to be tracked and providing the groundwork for future studies investigating their resistance profile.
Methods
Population sampling and whole-genome sequencing
We analyzed data from 1142 individual wild-caught specimens of An. gambiae and An. coluzzii collected and sequenced in Phase 2 of Ag1000G (https://www.malariagen.net/data/ag1000g-phase-2-ar1) (metadata provided in Supplemental Data S10). The specimens were collected from sites in 13 African countries (Angola An. coluzzii n = 78, Burkina Faso An. coluzzii n = 75, Burkina Faso An. gambiae n = 92, Cameroon An. gambiae n = 297, Côte d'Ivoire An. coluzzii n = 71, Equatorial Guinea (Bioko) An. gambiae n = 9, Gabon An. gambiae n = 69, Ghana An. coluzzii n = 55, Ghana An. gambiae n = 12, Guinea An. coluzzii n = 4, Guinea An. gambiae n = 40, Guinea-Bissau (mixed ancestry) n = 91, Kenya (undetermined ancestry) n = 48, Mayotte An. gambiae n = 24, The Gambia (mixed ancestry) n = 65, Uganda An. gambiae n = 112). Individual specimens were sequenced using the Illumina HiSeq platform to obtain 100-bp paired-end reads with a target coverage of 30×. Further details of population sampling, sample preparation, sequencing, alignment, species identification, and data production are reported elsewhere (The Anopheles gambiae 1000 Genomes Consortium 2017). Metadata for all samples, as well as detailed collection methods for populations not included in the aforementioned publication, are provided in Supplemental Methods SM1 and Supplemental Data S10.
Calculation and normalization of coverage
For each individual, we used the pysam software package (https://github.com/pysam-developers/pysam) to count the number of aligned reads (coverage) in nonoverlapping 300-bp windows over the nuclear genome. The position of each read was considered to be its alignment start point; thus, each read was only counted once. Sequencing coverage can be biased by variation in local nucleotide composition. To account for this, we computed a normalized coverage from the read counts based on the expected coverage of each window given its GC content (Abyzov et al. 2011). For each 300-bp window we computed the percentage of (G + C) nucleotides to the nearest percentage point within the reference sequence and then divided the read counts in each window by the mean read count over all autosomal windows with the same (G + C) percentage. To minimize the impact of copy number variation when calculating these normalizing constants, we excluded windows from the calculation of mean read counts for which previous analyses of genome accessibility have found evidence for excessively high or low coverage or ambiguous alignment (windows with <90% accessible bases according to the Ag1000G Phase 2 genome accessibility map, referred to as “accessible windows”) (https://www.malariagen.net/data/ag1000g-phase-2-ar1). The normalized coverage values were then multiplied by a factor of 2, so that genome regions with a normal diploid copy number should have an expected normalized coverage of 2.
Before examining the normalized coverage data for evidence of copy number variation, we applied two filters to exclude windows for which coverage may be an unreliable indicator of copy number. The first filter removed windows in which >2% of reads were aligned with mapping quality 0 (Supplemental Fig. S13), which indicates that a read is mapped ambiguously and could be mapped equally well to a different genomic location. This filter removed 159,587 (20.8%) of 768,225 windows. The second filter removed windows for which the percentage (G + C) content was extreme and rarely represented within the accessible reference sequence, that is, fewer than 100 accessible windows with the same (G + C) percentage, because the small number of windows makes the calculation of a (G + C) normalizing constant unreliable. This filter removed 13,484 (2.2%) of the 608,638 remaining windows. Windows retained for analysis were referred to as “filtered windows.”
Genome-wide copy number variation discovery
To detect the most likely copy number state (CNS) at each window in each individual, we applied a Gaussian HMM to the individual's normalized windowed coverage data, following a similar approach to Miles et al. (2016) and Leffler et al. (2017) (for details, see Supplemental Methods SM2). Because we are primarily interested in amplifications rather than deletions, we obtained a raw set of CNV calls for each sample by locating contiguous runs of at least five windows with amplified CNS (CNS > 2, or CNS > 1 for Chromosome X in males).
CNV filtering
From the raw CNV call set, we created a quality-filtered list of CNV calls. We first removed samples with very high coverage variance, because high variance could lead to erratic CNV calls. We therefore removed 27 samples for which the variance in normalized coverage was greater than 0.2 (Supplemental Fig. S14), retaining 1115 samples for further analysis.
We then applied two filters to the raw CNV calls from these 1115 samples. For the first filter, we computed likelihoods for each raw CNV call for both the copy number state predicted by the HMM and for a null model of copy number = 2, and removed CNV calls for which the likelihood ratio was <1000 (Supplemental Methods SM3). For the second filter, we removed CNVs with low population frequencies. To do this, the raw CNV calls needed to be matched so that the same CNV in different individuals could be identified. We classed any two CNVs as identical if the breakpoints predicted by their copy number state transitions were within one window of each other. We then removed CNVs that were not found in at least 5% of individuals in at least one population (or at least three individuals for populations smaller than 40).
We determined the sensitivity and specificity of the CNV detection method using simulations in which coverage values were randomly shuffled across genomic windows. The details of these simulations are described in Supplemental Data S11.
CNVs in chromatin types
Regions of heterochromatin and euchromatin were taken from Sharakhova et al. (2010). A CNV was considered to be heterochromatic if any part of it overlapped the heterochromatin regions. We performed simulations to determine whether the CNVs that we detected were evenly distributed between heterochromatin and euchromatin. For each run of the simulation, we randomly reallocated the start positions of every detected CNV, keeping the number of filtered windows covered by the CNVs unchanged, and calculated the number of CNVs that overlapped the heterochromatin. This simulation was run 10,000 times to obtain the distribution of the null model. Two-tailed P-values were obtained by calculating the proportion of simulations that produced at least as many heterochromatic CNVs as observed in the real data and multiplying this by two.
Discovery of gene duplications and gene enrichment analysis
To determine the genes contained within each CNV, we compared the start and end points of the CNVs to the start and end points of all genes listed in the AgamP4.2 gene annotations (Anopheles-gambiae-PEST_BASEFEATURES_AgamP4.2.gff3). The start/end points of each CNV were calculated as the median of the start/end points of all the raw CNV calls that were matched to it. To keep only genes for which good coverage data were available, we retained only genes containing at least 50% filtered windows. We classed a retained gene as copied by a CNV if all the filtered windows within the gene were inside the CNV. To determine whether the CNVs that we detected contained more genes than expected by chance, we performed simulations as described for investigating enrichment for chromatin types, this time counting the number of CNVs that contained at least one gene. We also repeated these simulations after excluding the heterochromatic regions by only randomizing the euchromatic CNVs and preventing their randomized positions from being in the heterochromatin.
We identified genes that could potentially be involved in metabolic resistance through detoxification (“metabolic detox genes”) by finding genes whose annotations contained the terms “P450,” “glutathione S-transferase,” or “carboxylesterase” in the AgamP4 transcript annotations (Anopheles-gambiae-PEST_ TRANSCRIPTS_AgamP4.2.fa). We performed simulations to determine whether genes copied by CNVs were enriched for metabolic detox genes. For each run of the simulation, we randomized each genic CNV by reallocating the genes encompassed by the CNV, keeping the number of consecutive genes covered by each CNV unchanged, and calculated the number of CNVs that included at least one metabolic detox gene. This simulation was run 10,000 times to obtain the distribution of the null model. Two-tailed P-values were obtained by calculating the proportion of simulations that produced at least as many genic CNVs containing detox genes as observed in the real data and multiplying this by two.
GO term analysis of genes included in CNVs was performed using the topGO package in R (https://bioconductor.org/packages/release/bioc/html/topGO.html; R Core Team 2015). False discovery rates were calculated from the P-values using the R package fdrtool (https://cran.r-project.org/web/packages/fdrtool/index.html).
Identifying CNV alleles at candidate metabolic insecticide resistance loci
We characterized in detail the different duplication events (CNV alleles) at five gene clusters of particular interest (Cyp6aa1–Cyp6p2, Gstu4–Gste3, Cyp6m2–Cyp6m4, Cyp6z3–Cyp6z1, Cyp9k1) using their unique patterns of discordant read pairs and reads crossing the CNV breakpoint (breakpoint reads, see Fig. 3; Supplemental Methods SM4). We manually inspected the five regions of interest in all 1142 samples to identify patterns of discordant and breakpoint reads (“diagnostic reads”) consistently associated with changes in coverage (Supplemental Figs. S2–S5). The start and end point of each CNV allele could usually be precisely determined by the breakpoint reads and was otherwise determined by discordant read pairs or the point of change in coverage (Supplemental Data S4–S7). Once the diagnostic reads were identified for a CNV allele, we recorded the presence of that allele in all samples with at least two supporting diagnostic reads.
To identify TEs that may be implicated in CNVs that did not appear to be tandem inversions or tandem duplications, we took the soft-clipped bases from the reads mapping to the breakpoints of these CNVs and used discontiguous megablast to compare them against the Repbase database (version 24.10) of An. gambiae transposable and repeat elements (www.girinst.org/repbase/ [Bao et al. 2015]).
Detecting signals of selection on CNV alleles
We used the phased haplotypes to calculate the pairwise shared haplotype length and the EHH for each CNV allele (Sabeti et al. 2002), using only SNPs from outside the region in which CNVs were found. EHH calculations were performed using the scikit-allel Python package (https://zenodo.org/record/3238280).
Haplotype clusters in the Cyp9k1 region were obtained with scikit-allel, using the first 1000 SNPs on the centromeric side of Cyp9k1 (the telomeric side of this gene has low levels of accessibility). A distance matrix between haplotypes was computed using the proportion of accessible SNPs that differed between pairwise haplotype combinations. This was used to perform hierarchical clustering, with haplotype clusters defined using a cutoff of 0.001.
Statistics
Statistical analysis was performed in R (R Core Team 2015). Contingency tables were analyzed with Fisher's exact test. Where the sample size was too large for the Fisher's exact test, P-values were obtained using the “simulated.p.value” option with 106 replicates.
Estimating allele-specific copy numbers and phasing CNV genotypes onto the Ag1000G haplotype scaffolds
To determine the allele-specific copy number in a sample, we estimated the change in coverage associated with each CNV allele (Supplemental Fig. S15; Supplemental Methods SM5). Thus, even when overlapping CNV alleles were present in a single sample, we were usually able to determine the number of copies of each allele.
For single-copy CNVs, it is possible to determine the genotype of a sample from the copy numbers (copy numbers of 1 indicate a heterozygote, copy numbers of 2 indicate a homozygote for the CNV). For higher-order CNVs, this is not possible because a heterozygote triplication cannot be distinguished from a homozygote duplication. We therefore applied two filters to retain only single-copy CNV alleles. The first filter removed CNV alleles for which the allele-specific copy number was found to rise above 2 in the data (if only a single sample rose as high as 2.5, we assumed that this could be an error and classed it as 2). This filter removed five CNV alleles (Cyp6aap_Dup11, Gstue_Dup2, Gstue_Dup8, Cyp9k1_Dup11, Cyp9k1_Dup15). For the second filter, we classed each sample as homozygote wild-type, heterozygote or homozygote CNV based on their copy numbers, and then removed CNV alleles that were inconsistent with Hardy-Weinberg expectations within the populations in which they were found. This filter removed four CNV alleles (Cyp6aap_Dup4, Gstue_Dup5, Gstue_Dup7, Cyp9k1_Dup10). Three CNV alleles (Cyp9k1_Dup7, Cyp9k1_Dup13, and Cyp9k1_Dup14) were also excluded because of difficulties in calling allele-specific copy number (Supplemental Data S7). In one case (Cyp6m_Dup1), all individuals were found to have a copy number of 2, indicating that the CNV is a triplication, with no duplications present in the population. This CNV was therefore retained, with all samples carrying the CNV classed as heterozygotic.
CNV alleles that passed both filters were phased onto the Ag1000G Phase 2 haplotype scaffolds using the MVNcall software v1.0 (Menelaou and Marchini 2013), using default parameters apart from setting λ = 0.1 to ensure that none of the input CNV genotype calls were changed during phasing. For each of the five gene clusters, phasing was performed using the 200 nonsingleton SNPs either side of the region in which CNVs were found, thus avoiding the inclusion of SNPs found inside any of the CNVs. Haplotypes that contained more than one CNV allele were rare and therefore excluded from subsequent calculations of haplotype homozygosity.
Supplementary Material
Acknowledgments
We thank three anonymous reviewers for useful comments on this manuscript. This work was supported by the Wellcome Trust (090770/Z/09/Z; 090532/Z/09/Z; 098051), Medical Research Council United Kingdom (MR/P02520X/1; MR/M006212/1), and the National Institute of Allergy and Infectious Diseases ([NIAID] R01-AI116811). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIAID or National Institutes of Health (NIH).
Author contributions: E.R.L., A.M., M.K.N.L., D.P.K., D.W., and M.J.D. designed the study. E.R.L., A.M., N.J.H., and C.S.C. carried out the analysis. The Ag1000G Consortium undertook collection, preparation, sequencing, and primary analysis of the samples. E.R.L., A.M., D.W., and M.J.D. wrote the manuscript. All authors read and approved the final manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.245795.118.
Freely available online through the Genome Research Open Access option.
References
- Abyzov A, Urban AE, Snyder M, Gerstein M. 2011. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res 21: 974–984. 10.1101/gr.114876.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Anopheles gambiae 1000 Genomes Consortium. 2017. Genetic diversity of the African malaria vector Anopheles gambiae. Nature 552: 96–100. 10.1038/nature24995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assogba BS, Djogbénou LS, Milesi P, Berthomieu A, Perez J, Ayala D, Chandre F, Makoutodé M, Labbé P, Weill M. 2015. An ace-1 gene duplication resorbs the fitness cost associated with resistance in Anopheles gambiae, the main malaria mosquito. Sci Rep 5: 14529 10.1038/srep14529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assogba BS, Milesi P, Djogbénou LS, Berthomieu A, Makoundou P, Baba-Moussa LS, Fiston-Lavier A, Belkhir K, Labbé P, Weill M. 2016. The ace-1 locus is amplified in all resistant Anopheles gambiae mosquitoes: fitness consequences of homogeneous and heterogeneous duplications. PLoS Biol 14: e2000618 10.1371/journal.pbio.2000618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao W, Kojima KK, Kohany O. 2015. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob DNA 6: 11 10.1186/s13100-015-0041-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass C, Field LM. 2011. Gene amplification and insecticide resistance. Pest Manag Sci 67: 886–890. 10.1002/ps.2189 [DOI] [PubMed] [Google Scholar]
- Chen Z, Cheng CH, Zhang J, Cao L, Chen L, Zhou L, Jin Y, Ye H, Deng C, Dai Z, et al. 2008. Transcriptomic and genomic evolution under constant cold in Antarctic notothenioid fish. Proc Natl Acad Sci 105: 12944–12949. 10.1073/pnas.0802432105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu T, Wen Z, Rupasinghe SG, Schuler MA. 2008. Comparative molecular modeling of Anopheles gambiae CYP6Z1, a mosquito P450 capable of metabolizing DDT. Proc Natl Acad Sci 105: 8855–8860. 10.1073/pnas.0709249105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- David J, Strode C, Vontas J, Nikou D, Vaughan A, Pignatelli PM, Louis C, Hemingway J, Ranson H. 2005. The Anopheles gambiae detoxification chip: a highly specific microarray to study metabolic-based insecticide resistance in malaria vectors. Proc Natl Acad Sci 102: 4080–4084. 10.1073/pnas.0409348102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devonshire AL, Field LM, Foster SP, Moores GD, Williamson MS, Blackman RL. 1998. The evolution of insecticide resistance in the peach–potato aphid, Myzus persicae. Philos Trans R Soc Lond B Biol Sci 353: 1677–1684. 10.1098/rstb.1998.0318 [DOI] [Google Scholar]
- Ding Y, Ortelli F, Rossiter LC, Hemingway J, Ranson H. 2003. The Anopheles gambiae glutathione transferase supergene family: annotation, phylogeny and expression profiles. BMC Genomics 4: 35 10.1186/1471-2164-4-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djouaka RF, Bakare AA, Coulibaly ON, Akogbeto MC, Ranson H, Hemingway J, Strode C. 2008. Expression of the cytochrome P450s, CYP6P3 and CYP6M2 are significantly elevated in multiple pyrethroid resistant populations of Anopheles gambiae s.s. from Southern Benin and Nigeria. BMC Genomics 9: 538 10.1186/1471-2164-9-538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly MJ, Isaacs AT, Weetman D. 2016. Identification, validation, and application of molecular diagnostics for insecticide resistance in malaria vectors. Trends Parasitol 32: 197–206. 10.1016/j.pt.2015.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duangkaew P, Pethuan S, Kaewpa D, Boonsuepsakul S, Sarapusit S, Rongnoparut P. 2011. Characterization of mosquito CYP6P7 and CYP6AA3: differences in substrate preference and kinetic properties. Arch Insect Biochem Physiol 76: 236–248. 10.1002/arch.20413 [DOI] [PubMed] [Google Scholar]
- Edi CV, Djogbénou L, Jenkins AM, Regna K, Muskavitch MAT, Poupardin R, Jones CM, Essandoh J, Kétoh GK, Paine MJI, et al. 2014. CYP6 P450 enzymes and ACE-1 duplication produce extreme and multiple insecticide resistance in the malaria mosquito Anopheles gambiae. PLoS Genet 10: e1004236 10.1371/journal.pgen.1004236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerson JJ, Cardoso-Moreira M, Borevitz JO, Long M. 2008. Natural selection shapes genome-wide patterns of copy-number polymorphism in Drosophila melanogaster. Science 320: 1629–1631. 10.1126/science.1158078 [DOI] [PubMed] [Google Scholar]
- Faucon F, Dusfour I, Gaude T, Navratil V, Boyer F, Chandre F, Sirisopa P, Thanispong K, Juntarajumnong W, Poupardin R, et al. 2015. Identifying genomic changes associated with insecticide resistance in the dengue mosquito Aedes aegypti by deep targeted sequencing. Genome Res 25: 1347–1359. 10.1101/gr.189225.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field LM, Blackman RL, Tyler-Smith C, Devonshire AL. 1999. Relationship between amount of esterase and gene copy number in insecticide-resistant Myzus persicae (Sulzer). Biochem J 339: 737–742. 10.1042/bj3390737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fossog Tene B, Poupardin R, Costantini C, Awono-Ambene P, Wondji CS, Ranson H, Antonio-Nkondjio C. 2013. Resistance to DDT in an urban setting: common mechanisms implicated in both M and S forms of Anopheles gambiae in the city of Yaoundé Cameroon. PLoS One 8: e61408 10.1371/journal.pone.0061408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoraki L, Lagnel J, Kioulos I, Kampouraki A, Morou E, Labbé P, Weill M, Vontas J. 2015. Transcriptome profiling and genetic study reveal amplified carboxylesterase genes implicated in temephos resistance, in the Asian Tiger Mosquito Aedes albopictus. PLoS Negl Trop Dis 9: e0003771 10.1371/journal.pntd.0003771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guryev V, Saar K, Adamovic T, Verheul M, Van Heesch SA, Cook S, Pravenec M, Aitman T, Jacob H, Shull JD, et al. 2008. Distribution and functional impact of DNA copy number variation in the rat. Nat Genet 40: 538–545. 10.1038/ng.141 [DOI] [PubMed] [Google Scholar]
- Handsaker RE, Van Doren V, Berman JR, Genovese G, Kashin S, Boettger LM, McCarroll SA. 2015. Large multiallelic copy number variations in humans. Nat Genet 47: 296–303. 10.1038/ng.3200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim SS, Amvongo-Adjia N, Wondji MJ, Irving H, Riveron JM, Wondji CS. 2018. Pyrethroid resistance in the major malaria vector Anopheles funestus is exacerbated by overexpression and overactivity of the P450 CYP6AA1 across Africa. Genes (Basel) 9: 140 10.3390/genes9030140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itokawa K, Komagata O, Kasai S, Masada M, Tomita T. 2011. Cis-acting mutation and duplication: history of molecular evolution in a P450 haplotype responsible for insecticide resistance in Culex quinquefasciatus. Insect Biochem Mol Biol 41: 503–512. 10.1016/j.ibmb.2011.04.002 [DOI] [PubMed] [Google Scholar]
- Kiszewski A, Mellinger A, Spielman A, Malaney P, Sachs SE, Sachs J. 2004. A global index representing the stability of malaria transmission. Am J Trop Med Hyg 70: 486–498. 10.4269/ajtmh.2004.70.486 [DOI] [PubMed] [Google Scholar]
- Kwiatkowska RM, Platt N, Poupardin R, Irving H, Dabire RK, Mitchell S, Jones CM, Diabaté A, Ranson H, Wondji CS. 2013. Dissecting the mechanisms responsible for the multiple insecticide resistance phenotype in Anopheles gambiae s.s., M form, from Vallée du Kou, Burkina Faso. Gene 519: 98–106. 10.1016/j.gene.2013.01.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbé P, Berticat C, Berthomieu A, Unal S, Bernard C, Weill M, Lenormand T. 2007. Forty years of erratic insecticide resistance evolution in the mosquito Culex pipiens. PLoS Genet 3: e205 10.1371/journal.pgen.0030205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leffler EM, Band G, Busby GBJ, Kivinen K, Le QS, Clarke GM, Bojang KA, Conway DJ, Jallow M, Sisay-Joof F, et al. 2017. Resistance to malaria through structural variation of red blood cell invasion receptors. Science 356: eaam6393 10.1126/science.aam6393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung SS, Romans P. 1998. Excisions of the Ikirara1 transposon in an Anopheles gambiae cell line. Insect Mol Biol 7: 241–248. 10.1111/j.1365-2583.1998.00068.x [DOI] [PubMed] [Google Scholar]
- Li X, Schuler MA, Berenbaum MR. 2007. Molecular mechanisms of metabolic resistance to synthetic and natural xenobiotics. Annu Rev Entomol 52: 231–253. 10.1146/annurev.ento.51.110104.151104 [DOI] [PubMed] [Google Scholar]
- Liu N. 2015. Insecticide resistance in mosquitoes: impact, mechanisms, and research directions. Annu Rev Entomol 60: 537–559. 10.1146/annurev-ento-010814-020828 [DOI] [PubMed] [Google Scholar]
- Lumjuan N, Rajatileka S, Changsom D, Wicheer J, Leelapat P, Prapanthadara L, Somboon P, Lycett G, Ranson H. 2011. The role of the Aedes aegypti ε glutathione transferases in conferring resistance to DDT and pyrethroid insecticides. Insect Biochem Mol Biol 41: 203–209. 10.1016/j.ibmb.2010.12.005 [DOI] [PubMed] [Google Scholar]
- Main BJ, Lee Y, Collier TC, Norris LC, Brisco K, Fofana A, Cornel AJ, Lanzaro GC. 2015. Complex genome evolution in Anopheles coluzzii associated with increased insecticide usage in Mali. Mol Ecol 24: 5145–5157. 10.1111/mec.13382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins AJ, Brito LP, Linss JGB, Rivas GB, Machado R, Bruno RV, Lima JBP, Valle D, Peixoto AA. 2013. Evidence for gene duplication in the voltage-gated sodium channel gene of Aedes aegypti. Evol Med Public Health 2013: 148–160. 10.1093/emph/eot012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins WFS, Subramaniam K, Steen K, Mawejje H, Liloglou T, Donnelly MJ, Wilding CS. 2017. Detection and quantitation of copy number variation in the voltage-gated sodium channel gene of the mosquito Culex quinquefasciatus. Sci Rep 7: 5821 10.1038/s41598-017-06080-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menelaou A, Marchini J. 2013. Genotype calling and phasing using next-generation sequencing reads and a haplotype scaffold. Bioinformatics 29: 84–91. 10.1093/bioinformatics/bts632 [DOI] [PubMed] [Google Scholar]
- Miles A, Iqbal Z, Vauterin P, Pearson R, Campino S, Theron M, Gould K, Mead D, Drury E, O'Brien J, et al. 2016. Indels, structural variation, and recombination drive genomic diversity in Plasmodium falciparum. Genome Res 26: 1288–1299. 10.1101/gr.203711.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell SN, Stevenson BJ, Müller P, Wilding CS, Egyir-Yawson A, Field SG, Hemingway J, Paine MJI, Ranson H, Donnelly MJ. 2012. Identification and validation of a gene causing cross-resistance between insecticide classes in Anopheles gambiae from Ghana. Proc Natl Acad Sci 109: 6147–6152. 10.1073/pnas.1203452109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell SN, Rigden DJ, Dowd AJ, Lu F, Wilding CS, Weetman D, Dadzie S, Jenkins AM, Regna K, Boko P, et al. 2014. Metabolic and target-site mechanisms combine to confer strong DDT resistance in Anopheles gambiae. PLoS One 9: e92662 10.1371/journal.pone.0092662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller P, Donnelly MJ, Ranson H. 2007. Transcription profiling of a recently colonised pyrethroid resistant Anopheles gambiae strain from Ghana. BMC Genomics 8: 36 10.1186/1471-2164-8-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller P, Warr E, Stevenson BJ, Pignatelli PM, Morgan JC, Steven A, Yawson AE, Mitchell SN, Ranson H, Hemingway J, et al. 2008. Field-caught permethrin-resistant Anopheles gambiae overexpress CYP6P3, a P450 that metabolises pyrethroids. PLoS Genet 4: e1000286 10.1371/journal.pgen.1000286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngufor C, N'Guessan R, Fagbohoun J, Subramaniam K, Odjo A, Fongnikin A, Akogbeto M, Weetman D, Rowland M. 2015. Insecticide resistance profile of Anopheles gambiae from a phase II field station in Cové, southern Benin: implications for the evaluation of novel vector control products. Malar J 14: 464 10.1186/s12936-015-0981-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen D, Webber C, Ponting CP. 2006. Bias of selection on human copy-number variants. PLoS Genet 2: e20 10.1371/journal.pgen.0020020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikou D, Ranson H, Hemingway J. 2003. An adult-specific CYP6 P450 gene is overexpressed in a pyrethroid-resistant strain of the malaria vector, Anopheles gambiae. Gene 318: 91–102. 10.1016/S0378-1119(03)00763-7 [DOI] [PubMed] [Google Scholar]
- Opondo KO, Weetman D, Jawara M, Diatta M, Fofana A, Crombe F, Mwesigwa J, D'Alessandro U, Donnelly MJ. 2016. Does insecticide resistance contribute to heterogeneities in malaria transmission in The Gambia? Malar J 15: 166 10.1186/s12936-016-1203-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protopopoff N, Mosha JF, Lukole E, Charlwood JD, Wright A, Mwalimu CD, Manjurano A, Mosha FW, Kisinza W, Kleinschmidt I, et al. 2018. Effectiveness of a long-lasting piperonyl butoxide-treated insecticidal net and indoor residual spray interventions, separately and together, against malaria transmitted by pyrethroid-resistant mosquitoes: a cluster, randomised controlled, two-by-two factorial design trial. Lancet 391: 1577–1588. 10.1016/S0140-6736(18)30427-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2015. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna: http://www.R-project.org/. [Google Scholar]
- Raymond M, Berticat C, Weill M, Pasteur N, Chevillon C. 2001. Insecticide resistance in the mosquito Culex pipiens: What have we learned about adaptation? Genetica 112/113: 287–296. 10.1023/A:1013300108134 [DOI] [PubMed] [Google Scholar]
- Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, et al. 2006. Global variation in copy number in the human genome. Nature 444: 444–454. 10.1038/nature05329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remnant EJ, Good RT, Schmidt JM, Lumb C, Robin C, Daborn PJ, Batterham P. 2013. Gene duplication in the major insecticide target site, Rdl, in Drosophila melanogaster. Proc Natl Acad Sci 110: 14705–14710. 10.1073/pnas.1311341110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riveron JM, Yunta C, Ibrahim SS, Djouaka R, Irving H, Menze BD, Ismail HM, Hemingway J, Ranson H, Albert A, et al. 2014a. A single mutation in the GSTe2 gene allows tracking of metabolically based insecticide resistance in a major malaria vector. Genome Biol 15: R27 10.1186/gb-2014-15-2-r27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riveron JM, Ibrahim SS, Chanda E, Mzilahowa T, Cuamba N, Irving H, Barnes KG, Ndula M, Wondji CS. 2014b. The highly polymorphic CYP6M7 cytochrome P450 gene partners with the directionally selected CYP6P9a and CYP6P9b genes to expand the pyrethroid resistance front in the malaria vector Anopheles funestus in Africa. BMC Genomics 15: 817 10.1186/1471-2164-15-817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodpradit P, Boonsuepsakul S, Chareonviriyaphap T, Bangs MJ, Rongnoparut P. 2005. Cytochrome P450 genes: molecular cloning and overexpression in a pyrethroid-resistant strain of Anopheles minimus mosquito. J Am Mosq Control Assoc 21: 71–79. 10.2987/8756-971X(2005)21[71:CPGMCA]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Sabeti PC, Reich DE, Higgins JM, Levine HZ, Richter DJ, Schaffner SF, Gabriel SB, Platko JV, Patterson NJ, McDonald GJ, et al. 2002. Detecting recent positive selection in the human genome from haplotype structure. Nature 419: 832–837. 10.1038/nature01140 [DOI] [PubMed] [Google Scholar]
- Schmidt JM, Good RT, Appleton B, Sherrard J, Raymant GC, Bogwitz MR, Martin J, Daborn PJ, Goddard ME, Batterham P, et al. 2010. Copy number variation and transposable elements feature in recent, ongoing adaptation at the Cyp6g1 locus. PLoS Genet 6: e1000998 10.1371/journal.pgen.1000998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrider DR, Hahn MW, Begun DJ. 2016. Parallel evolution of copy-number variation across continents in Drosophila melanogaster. Mol Biol Evol 33: 1308–1316. 10.1093/molbev/msw014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharakhova MV, George P, Brusentsova IV, Leman SC, Bailey JA, Smith CD, Sharakhov IV. 2010. Genome mapping and characterization of the Anopheles gambiae heterochromatin. BMC Genomics 11: 459 10.1186/1471-2164-11-459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson BJ, Bibby J, Pignatelli P, Muangnoicharoen S, O'Neill PM, Lian L, Müller P, Nikou D, Steven A, Hemingway J, et al. 2011. Cytochrome P450 6M2 from the malaria vector Anopheles gambiae metabolizes pyrethroids: sequential metabolism of deltamethrin revealed. Insect Biochem Mol Biol 41: 492–502. 10.1016/j.ibmb.2011.02.003 [DOI] [PubMed] [Google Scholar]
- Sudmant PH, Mallick S, Nelson BJ, Hormozdiari F, Krumm N, Huddleston J, Coe BP, Baker C, Nordenfelt S, Bamshad M, et al. 2015. Global diversity, population stratification, and selection of human copy-number variation. Science 349: aab3761 10.1126/science.aab3761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen EK, Strode C, Hemmings K, Hughes AJ, Chanda E, Musapa M, Kamuliwo M, Phiri FN, Muzia L, Chanda J, et al. 2014. Underpinning sustainable vector control through informed insecticide resistance management. PLoS One 9: e99822 10.1371/journal.pone.0099822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vontas J, Grigoraki L, Morgan J, Tsakireli D, Fuseini G, Segura L, Niemczura de Carvalho J, Nguema R, Weetman D, Slotman MA, et al. 2018. Rapid selection of a pyrethroid metabolic enzyme CYP9K1 by operational malaria control activities. Proc Natl Acad Sci 115: 4619–4624. 10.1073/pnas.1719663115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weetman D, Mitchell SN, Wilding CS, Birks DP, Yawson AE, Essandoh J, Mawejje HD, Djogbenou LS, Steen K, Rippon EJ, et al. 2015. Contemporary evolution of resistance at the major insecticide target site gene Ace-1 by mutation and copy number variation in the malaria mosquito Anopheles gambiae. Mol Ecol 24: 2656–2672. 10.1111/mec.13197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weetman D, Djogbenou LS, Lucas E. 2018. Copy number variation (CNV) and insecticide resistance in mosquitoes: evolving knowledge or an evolving problem? Curr Opin Insect Sci 27: 82–88. 10.1016/j.cois.2018.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer CT, Garrood WT, Singh KS, Randall E, Lueke B, Gutbrod O, Matthiesen S, Kohler M, Nauen R, Davies TGE, et al. 2018. Neofunctionalization of duplicated P450 genes drives the evolution of insecticide resistance in the brown planthopper. Curr Biol 28: 268–274.e5. 10.1016/j.cub.2017.11.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.