

Abstract
Predicting biosynthetic gene clusters (BGCs) is critically important for discovery of antibiotics and other natural products. While BGC prediction from complete genomes is a well-studied problem, predicting BGCs in fragmented genomic assemblies remains challenging. The existing BGC prediction tools often assume that each BGC is encoded within a single contig in the genome assembly, a condition that is violated for most sequenced microbial genomes where BGCs are often scattered through several contigs, making it difficult to reconstruct them. The situation is even more severe in shotgun metagenomics, where the contigs are often short, and the existing tools fail to predict a large fraction of long BGCs. While it is difficult to assemble BGCs in a single contig, the structure of the genome assembly graph often provides clues on how to combine multiple contigs into segments encoding long BGCs. We describe biosyntheticSPAdes, a tool for predicting BGCs in assembly graphs and demonstrate that it greatly improves the reconstruction of BGCs from genomic and metagenomics data sets.
Although there exist many tools for assembling microbial genomes or metagenomes (Simpson et al. 2009; Li et al. 2015; Nurk et al. 2017), they all have limitations with respect to assembling contigs that contain long genes encoding proteins with repetitive domains. Since long genes are often scattered between multiple contigs in fragmented assemblies, the existing gene prediction tools (Besemer and Borodovsky 2005; Delcher et al. 2007; Hyatt et al. 2010; Pati et al. 2010) cannot predict them. The challenge of assembling long genes in a single contig is illustrated by genes encoding Nonribosomal Peptides Synthetases (NRPSs), Polyketide Synthases (PKSs), and other genes that are parts of biosynthetic gene clusters (BGCs) encoding the production of antibiotics and other natural products. BGCs usually include multiple consecutive genes that participate in a single metabolic pathway responsible for synthesizing a natural product. NRPS BGCs encode Nonribosomal Peptides (NRPs) built from amino acids, and PKS BGCs encode polyketides (PSs) built from keto groups. Mixed NRPS/PKS BGCs contain both NRPS-specific and PKS-specific domains, and their natural products represent fusions of peptides and polyketides (Cane and Walsh 1999). Klassen and Currie (2012) showed that long and repetitive NRPSs and PKSs are responsible for a large fraction of fragmentation in microbial assemblies.
This paper focuses on NRPSs because NRPs represent an important class of natural product drugs (Newman and Cragg 2016) that is most amenable to downstream peptidogenomics analysis as compared to other classes of natural products (Kersten et al. 2011; Medema et al. 2014a; Mohimani et al. 2014b). NRPS BGCs constitute 34% of all BGCs in publicly available genomes, as found in the antiSMASH database (https://antismash-db.secondarymetabolites.org/#!/stats). Since NRPSs are very common (albeit elusive) in diverse bacterial data sets (Mukherjee et al. 2017) and since the downstream peptidogenomics analysis of NRPs is greatly impaired by fragmented assemblies, most examples in this paper refer to NRPs. In addition to NRPS BGCs, biosynteticSPAdes is also applicable to PKS BGCs and mixed NRPS-PKS BGCs (NRPS, PKS, and mixed NRPS-PKS BGCs constitute the majority of BGCs in the MIBiG database). Klassen and Currie (2012) have shown that fragmented ORFs in genome assemblies are highly enriched in NRPSs and PKSs, which thus constitute a prominent source of breakpoints in (meta)genome assemblies. The fact that the vast majority of genomes contain either an NRPS or a PKS or a mixed NRPS-PKS BGC (for some species, over 30% of the genome is allocated to these BGCs) and direct interest to a large research community is a good reason to provide a specialized assembler for these BGCs.
NRPSs are large modular protein complexes containing multiple highly similar adenylation domains (A-domains) responsible for recruiting amino acids that form NRPs according to the substrate specificity of each A-domain (Stachelhaus et al. 1999). NRPSs are often accompanied by other adjacently located genes that together form NRP BGCs and contribute to NRP synthesis, transport, and regulation. NRP BGCs are typically long, with an average length of ∼60 kb and some exceeding 100 kb in length. Assembling NRP BGCs into single contigs is a crucial step in natural product discovery by genome mining (Weber et al. 2015) and peptidogenomics (Mohimani et al. 2014a, 2017; Mohimani and Pevzner 2016; Gurevich et al. 2018).
The recent Genomic Encyclopedia of Bacteria and Archaea (GEBA) study of over 1000 bacterial genomes revealed over 23,000 BGCs (Mukherjee et al. 2017). An average GEBA genome devotes nearly 10% of its genome to BGCs (some genomes devote >30%). However, the vast majority of predicted BGC products remain unknown, in part due to difficulties in predicting long BGCs (Hadjithomas et al. 2015).
The recently proposed genome mining and peptidogenomic approaches elucidate the amino acid sequences of NRPs by matching tandem mass spectra against predicted NRP synthetases in the assembled genomes (Medema et al. 2014b; Mohimani et al. 2014a, 2017). The success of these approaches depends on accurate prediction of genes encoding NRP synthetases followed by machine-learning algorithms to predict their substrate specificities and matching mass spectral data sets against the predicted NRP amino acid sequences. This is a challenging task requiring the recovery of the complete NRPS genes and the corresponding NRP BGCs in a single contig.
This challenge is further amplified in metagenomics assemblies, because NRP synthetases from different species within a microbial community often share similar domains. This makes it difficult to assemble them in a single contig in cases when multiple domains are collapsed into a single edge in the assembly graph (Coates et al. 2014). Therefore, while metagenomes represent a gold mine for antibiotics discovery, a limited number of antibiotics have been discovered from metagenomics data sets so far (Freeman et al. 2012; Donia et al. 2014; Donia and Fischbach 2015).
Despite the fact that it is difficult to reconstruct long NRPS BGCs from individual contigs, the structure of the assembly graph often provides clues on how to combine various contigs into intact BGCs. We describe the biosyntheticSPAdes tool for assembling NRPS BGCs in assembly graphs constructed by SPAdes (Bankevich et al. 2012) and metaSPAdes (Nurk et al. 2017) assemblers. Below, we show how biosyntheticSPAdes contributes to the discovery of NRPS BGCs in various genomes and metagenomes.
Results
The challenge of assembling BGCs
Contrary to the standard practice in existing gene prediction tools that attempt to reconstruct genes from individual contigs/scaffolds, biosyntheticSPAdes analyzes the assembly graph to join fragments of long BGCs (scattered over multiple contigs) into a single contig. Below, we describe the biosyntheticSPAdes algorithm and illustrate how it works using the genome of Streptomyces coelicolor A3(2) (referred to as S. coelicolor for brevity), a well-studied antibiotics-producing bacterium, which encodes four NRP BGCs (Bentley et al. 2002), including calcium-dependent antibiotic (CALC).
We illustrate the challenge of assembling long repetitive genes using a subgraph of the S. coelicolor assembly graph encoding the CALC BGC (Fig. 1). To generate this graph, we simulated error-free short paired-end reads (Huang et al. 2012) from the S. coelicolor genome using the ART read simulator (Huang et al. 2012). The reads from the resulting data set with coverage 180× (referred to as the STREP data set and containing paired reads of length 150 bp with a mean insert size of 300 bp) were assembled using the SPAdes assembler (Bankevich et al. 2012). The assembly graph constructed from these simulated reads contains 626 vertices and 697 edges (484 of them are longer than 1000 bp). The total edge length in the assembly graph is 8,598,860 with N50 = 41 kb. SPAdes uses paired reads to resolve repeats in the genome and combines some edges in the assembly graphs into contigs/scaffolds using exSPAnder (Prjibelski et al. 2014). exSPAnder constructed 145 scaffolds longer than 1000 bp with N50 = 135 kb after the repeat resolution step.
Figure 1.

Subgraph of the assembly graph of S. coelicolor corresponding to the CALC NRP BGC. (Top) Edges of the assembly graph traversed by the CALC BGC. Nodes of the assembly graph are shown as white circles. After applying exSPAnder, the CALC BGC remains scattered over 10 scaffolds. Three of them are shown as red, blue, and green paths through the assembly graph; the remaining seven consist of a single edge each (shown in black and marked with letters a through g). The positions of eleven A-domains (with their indices) along the CALC BGC are shown by violet boxes. Edges with low and high coverage by reads are shown as thin and thick edges, respectively. The edge harboring three A-domains 4, 5, and 7 has approximately triple coverage by reads as compared to other domain-harboring edges. The 11 A-domains in CALC are split over three NRP synthetases with 6, 3, and 2 A-domains, respectively. (Middle) A simplified representation of the graph with all short edges (shorter than 300 bp) contracted into single vertices. The two contracted subgraphs of the assembly graph (formed by short edges) are represented by yellow vertices. The brown dashed path illustrates how the CALC NRP synthetase traverses the contracted assembly graph. (Bottom) The bubble restoration procedure described below transforms the collapsed edge harboring three A-domains (A-domains 4, 5, and 7) into three edges, each of them harboring a single A-domain. Applying exSPAnder to the modified assembly graph results in seven scaffolds that differ from scaffolds before bubble restoration (shown as red, blue, green, and orange paths as well as three black edges). Gray squares show the starting and ending positions of the CALC BGC.
AntiSMASH (Weber et al. 2015) is a popular genome mining tool for detecting and annotating BGCs. AntiSMASH revealed 29 BGCs in the S. coelicolor genome, including four NRP BGCs. The CALC BGC with eleven A-domains traverses 25 edges in the assembly graph. exSPAnder (Prjibelski et al. 2014) combined some of these edges into single contigs, but even after applying exSPAnder, CALC was split into seven scaffolds (Fig. 1). This illustrates the challenge of reconstructing long genes even for isolated bacteria, let alone metagenomes. Note that 11 A-domains in CALC are represented by only nine A-domains in Figure 1 because three out of 11 A-domains got collapsed into a single edge in the assembly graph.
The CALC BGC illustrates just one example of the difficulties with assembling long and repetitive genes in genomic and metagenomic data sets. Supplemental Table S1 illustrates that 285 out of 7910 genes (≈ 3%) in the S. coelicolor genome are split over multiple edges in the assembly graph. The fraction of split genes further increases when we consider long genes: 11 out of the 100 longest genes (length > 3200 bp) traverse multiple edges and 17 out of these 100 longest genes correspond to BGCs (Supplemental Table S2). While the repeat resolution step in SPAdes (Prjibelski et al. 2014) captures some of the split genes in a single contig/scaffold, many long genes remain split even after repeat resolution, and three of them correspond to BGC genes (Supplemental Table S3). The fraction of such split genes further increases in metagenomics assemblies.
BiosyntheticSPAdes outline
The biosyntheticSPAdes pipeline includes six steps (Fig. 2) that are described in the Methods section:
assembling genomic/metagenomic reads with SPAdes/metaSPAdes;
identifying domain-edges in the assembly graph;
extracting BGC subgraphs from the assembly graph;
restoring collapsed domains in the assembly graph;
constructing the scaffolding graph; and
constructing putative BGCs by solving the Rural Postman Problem in the scaffolding graph.
Figure 2.
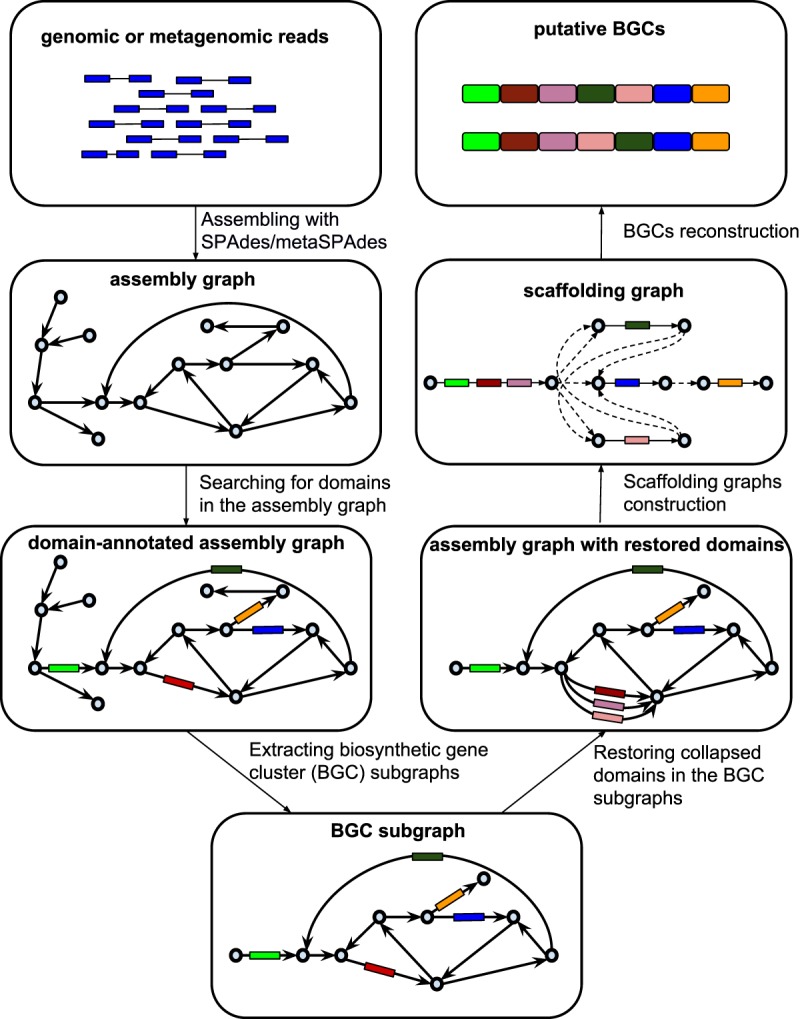
The biosyntheticSPAdes pipeline. Six steps of the biosyntheticSPAdes pipeline: (1) assembling genomic/metagenomic reads with SPAdes/metaSPAdes; (2) searching for edges harboring biosynthetic domains in the assembly graph; (3) extracting biosynthetic gene cluster subgraphs from the assembly graph; (4) restoring the collapsed domains in the BGC-subgraphs; (5) constructing the scaffolding graph; and (6) generating putative BGC by solving the Rural Postman Problem in the scaffolding graph.
Benchmarking design
To benchmark biosyntheticSPAdes, we compared its output (a single or multiple contigs) against the reference genome(s). Since the downstream applications, such as NRPquest (Mohimani et al. 2014a), do not require a single contig output and work equally well when a small set of output contigs contain a correct one, we classify the biosyntheticSPAdes output as correct if at least one of the reported contigs is contained in one of the reference genomes (with percent identity exceeding 95%).
In the case when the reference genomes are not available, we check whether a BGC subgraph contains a rural postman path. If it is the case, it is likely that one of the reported contigs is contained in an unknown reference genome.
Data sets
We analyzed the following data sets assembled using SPAdes or metaSPAdes with k-mer sizes varying from 21 to 55 nucleotides during the iterative assembly.
Pseudomonas data sets (PSEUDO). The PSEUDO data set (accession number ERR1890333) contains ≈4.5 million paired reads from the isolate of Pseudomonas protegens (fluorescens) Pf-5 (read length 100 bp, a mean insert size 440 bp, and a standard deviation of the insert size 140 bp). The genome sequence was finished using a combination of primer walking, generation and sequencing of transposon-tagged libraries, and multiplex PCR (Paulsen et al. 2005).
Cyanobacteria data set (CYANO). The CYANO data set contains genomic reads from cultured marine bacteria Moorea producens JHB (referred to as JHB below) described in Kleigrewe et al. (2015). The sample is contaminated with heterotrophic bacteria and thus represents a low-complexity metagenome. The JHB strain encodes various NRPs, PKs, and mixed NRP-PKs, including hectochlorin (Marquez et al. 2002) and jamaicamides (Edwards et al. 2004). The JHB data set contains ≈6 million paired reads (length 150 bp, a mean insert size 292 bp, and a standard deviation of the insert size 74 bp).
MIBiG data sets (MIBIG). The Minimum Information about a Biosynthetic Gene Cluster (MIBiG) database contains information about BGCs and their products (Medema et al. 2015). Each entry in the MIBiG database contains the nucleotide sequence of a BGC, the natural product type (NRPs, PKs, and other types), and its annotation. In order to benchmark biosyntheticSPAdes on a wide range of BGCs, we extracted all MIBiG entries describing NRPSs and PKSs with complete BGC sequences (665 entries) and used the ART read simulator (Huang et al. 2012) to simulate reads from BGC sequences with the default MiSeq parameters. Admittedly, generating reads from BGCs results in a simpler problem than simulating reads from the entire genome. However, since entire genomes are not available for many MIBiG entries, we simulated reads from BGCs only. We define the complexity of a BGC as the total number of A-domains and AT-domains in this BGC. Note that this is a very naive definition of complexity (e.g., trans-AT PKSs have few AT domains). Out of 665 BGCs in the MIBiG data set, 139 have complexity 10 and larger.
HMP data sets (HMP). The HMP data set consists of 20 metagenomic sub-data sets from seven parts of the human body that included keratinized gingiva, buccal mucosa, stool, gingival plaque, supragingival plaque, tongue dorsum, and throat (Supplemental Table S4). The description of these data sets is given in The Human Microbiome Project Consortium (2012).
Analyzing the PSEUDO data set
AntiSMASH (Weber et al. 2015) identified 12 BGCs in the Pseudomonas protegens Pf-5 genome, including seven NRP and PK BGCs. SPAdes assembled each of them into a single contig with the exception of the pyoverdine NRP BGC (with eight A-domains), which was assembled into four contigs that revealed only seven A-domains (Fig. 3, top left). In contrast, the domain restoration procedure in biosyntheticSPAdes succeeded in reconstructing two A-domains that were collapsed on a single edge by SPAdes (Fig. 3, top right). The resulting scaffolding graph contains a single rural postman route that revealed the correct arrangement of A-domains (Fig. 3, bottom). The reconstructed pyoverdine NRP BGC aligns to the Pseudomonas protegens Pf-5 genome with 99.9% identity.
Figure 3.

Subgraph of the assembly graph of Pseudomonas protegens Pf-5 corresponding to the pyoverdine NRP BGC. (Top left) The pyoverdine BGC is scattered over four scaffolds in the SPAdes assembly. Two scaffolds traversing single edges are shown by black color, and two scaffolds traversing multiple edges are shown by red and green colors. The repeat edges traversed by both red and green scaffolds are shown by brown color. Edges with low and high depth of coverage by reads are shown as thin and thick edges, respectively. Some A-domains span multiple edges (starting and ending positions of such domains are shown with dashed lines). (Top right) The domain restoration procedure restored two A-domains (5 and 6) in the assembly (SPAdes collapsed these domains into a single edge). Four scaffolds in the assembly graph are shown by red, green, blue, and black colors. (Bottom) The scaffolding graph of the pyoverdine BGC with a single rural postman route (dashed edges in this route are shown in blue).
Analyzing the CYANO data set
Kleigrewe et al. (2015) assembled the CYANO data set using SPAdes. metaSPAdes assembled the CYANO data set into the assembly graph with 217,826 vertices and 116,066 edges (8454 of them are longer than 1 kb). metaSPAdes assembled the jamaicamide BGC with complexity 9 into a single contig but failed to assemble the hectochlorin BGC with complexity 5 into a single contig.
biosyntheticSPAdes extracted 781 BGC subgraphs, including 12 nontrivial BGC subgraphs with complexities 21, 20, 11, 9, 6, 6, 5, 5, 5, 5, 4, and 4. The hectochlorin BGC contains 22 domains (four A-domains, one AT-domain, four C-domains, one KS-domain, three KR domains, and several others; one of them was also identified by HMMER as an A-domain). biosyntheticSPAdes assembled the hectochlorin BGCs into a single contig (Fig. 4) that aligns with the Moorea producens JHB genome with 99.9% identity. The jamaicamide BGC contains 42 domains (three A-domains, six AT-domains, four KR-domains, seven KS-domains, two C-domains, one TE-domain, and several others). The jamaicamide scaffolding graph contains a single solid edge (usually, this means that the entire BGC was recovered after the repeat resolution step with exSPAnder).
Figure 4.

biosyntheticSPAdes assembly of the hectochlorin BGCs (the CYANO data set). (Top) The subgraph of the assembly graph corresponding to the hectochlorin BGC. metaSPAdes assembly results in four scaffolds shown by a red path, a green path, and two black edges. The repeat edges traversed by both red and green scaffolds are shown by the brown color. The domain restoration procedure had no effect on this graph. (Bottom) The scaffolding graph of the hectochlorin BGC has only one rural postman route that revealed the correct domain order.
Besides reconstructing the hectochlorin and the jamaicamide BGCs, biosyntheticSPAdes recovered sequences for five more putative NRP BGCs that were missed in previous studies (see Supplemental Information, Appendix: “Putative NRP BGCs in the CYANO data set”; Supplemental Figs. S3, S4).
Analyzing the MIBiG data sets
For each of 665 MIBiG data sets corresponding to a single known NRP or PK, we launched biosyntheticSPAdes on the SPAdes assembly graph. We also compared them with the other popular assemblers: MEGAHIT v.1.1.3 (Li et al. 2015) and ABySS assembler v.2.1.0 (Simpson et al. 2009). For each assembler and each MIBiG data set, the assembly was classified as successful if it met the following criteria: (1) One of the contigs in the assembly covers more than 95% of the BGC and has at least 95% identity with the BGC being assembled; and (2) this contig has no misassemblies as identified by QUAST (Gurevich et al. 2013). biosyntheticSPAdes failed to successfully assemble only 11% of BGCs versus 22% for SPAdes, 35% for MEGAHIT, and 34% for ABySS (Table 1). For 139 out of 665 BGCs with complexity >10, biosyntheticSPAdes failed to successfully assemble 22% of BGCs versus 58% for SPAdes, 79% for MEGAHIT, and 83% for ABySS.
Table 1.
Results of SPAdes, biosyntheticSPAdes, MEGAHIT, and ABySS assemblies on 665 MIBiG data sets

Analyzing the HMP data sets
To reconstruct BGCs in the human microbiome, we assembled each HMP data set with biosyntheticSPAdes. We define the biosynthetic capacity of an assembly as the number of A and AT domains identified in this corresponding assembly. The biosynthetic capacity of the HMP data sets varies from 60 to over 400 across various human body sites (see Supplemental Information, Appendix: “Biosynthetic capacity of the HMP data sets”; Supplemental Table S4), suggesting that many HMP samples may encode over a dozen of NRP and PK BGCs. However, the amount of high-complexity BGC subgraphs suggests that sequencing depth in some data sets from the HMP project may be insufficient to capture the diversity of BGCs.
Below, we focus on analyzing the supragingival plaque metagenome (data set SRS013723) with large biosynthetic capacity. The assembly graph of this data set contains 1540 BGC subgraphs, including seven nontrivial BGC subgraphs. We analyzed one of the complex BGC subgraphs, with six predicted A-domains, five C-domains, and two TE-domains, that was assembled into six contigs. Figure 5 shows the BGC subgraph and two rural postman routes in the scaffolding graph generated by biosyntheticSPAdes. A nucleotide BLAST search of two predicted BGCs against the nt/rt database revealed only the short regions of similarity (<200 bp) with various Pseudomonas species, suggesting that Figure 5 represents a still unknown BGC. See Supplemental Information, Appendices: “Biosynthetic capacity of the HMP data sets,” “Putative NRP BGCs in supragingival plaque data sets,” Supplemental Figure S5, and Supplemental Table S5 for detailed analysis of the supragingival plaque data sets.
Figure 5.

The BGC subgraph and the scaffolding graph for the supragingival plaque metagenome (SRS013723) in the HMP data set. (1,2) The BGC subgraph and the scaffolding graph. (3,4) Two rural postman routes in the scaffolding graph. The duplicated C-domain is highlighted with red border and is traversed twice in the rural postman routes. The numbers labeling the dashed edges indicate their order in the resulting tour. (5) Since biosyntheticSPAdes and antiSMASH use different thresholds and filtering options, antiSMASH identified only five (rather than six) A-domains in the NRP BGC predicted by biosyntheticSPAdes. The three most likely amino acids for each A-domain are shown along with their NRPSpredictor2 (Röttig et al. 2011) scores for the first of two rural postman routes.
Discussion
While the human microbiome encodes natural products with great biomedical potential, little is known about these abundant small molecules, despite the fact that the human host is chronically exposed to them (Donia et al. 2014). One of the bottlenecks in discovering natural products from human and other metagenomes is deriving full-length BGCs from short metagenomics reads (Donia and Fischbach 2015). This bottleneck negatively affects various genome mining efforts. Indeed, although the discovery of coelichelin (Challis and Ravel 2000) was one of the first successes of genome mining that was followed by the characterization of many NRPs from sequenced genomes, genome mining in fragmented assemblies remains challenging.
The discovery of the bioactive peptides teixobactin (Wilson et al. 2014) and polytheonamides (Freeman et al. 2012) marks a new era of natural product discovery from uncultivated bacteria. However, while various metagenomes serve as a rich source of natural products (Cragg and Newman 2013; Katz et al. 2016), reconstructing complex BGCs from metagenomic assemblies is nearly impossible with short-read sequencing technologies. Since gene prediction of BGCs scattered between multiple contigs is challenging, the full-length BGC reconstruction is usually difficult without additional biological experiments and extensive manual analysis (Kleigrewe et al. 2015).
biosyntheticSPAdes is a step toward enabling high-throughput natural product discovery by coupling metagenomics and mass spectrometry projects using tools such as NRPquest (Mohimani et al. 2014a). It represents the first automated pipeline for BGC reconstruction from genomic and metagenomic sequencing data sets that takes advantage of the assembly graph rather than individual contigs. While we demonstrated that biosyntheticSPAdes is able to recover long BGCs, it can also be extended to other types of long and highly repetitive genes, such as 16S rRNA genes or insecticide toxins (Palma et al. 2014). Although biosyntheticSPAdes currently has the predefined options only for the most important classes of BGCs (NRPS, PKSs, and their fusions), we plan to create presets for other BGCs so that it can be extended for other BGCs with different domain compositions. A user can replace the default HMM-profiles with any profiles of interest, such as TPR-proteins, mucus-binding proteins, etc. However, we currently do not have plans to develop a version of SPAdes for generic operon prediction since it is not clear how to account for a wide diversity of genes within operons in general.
We emphasize that, similarly to all gene prediction tools, a putative BGC predicted by biosyntheticSPAdes may be incorrect and should be used with caution. In particular, the homology-based mode of biosyntheticSPAdes is most useful when one or more closely related reference genomes are available that have well-annotated BGCs. In the case when multiple feasible paths exist in the assembly graph, we recommend to experimentally verify biosyntheticSPAdes predictions, e.g., using targeted PCR amplification or matching against mass spectrometry data. Also, peptidogenomics tools (Mohimani et al. 2014a) can be applied to all feasible paths in the assembly graph rather than to a single highest-scoring path.
Third generation sequencing technologies have greatly improved isolate bacterial sequencing, thus turning BGC assembly into a relatively simple task. However, they have not yet had a large impact on metagenomic sequencing due to the relatively high cost of long-read technologies and difficulties in assembly (no specialized long-read metagenomic assembler has been released yet). Since most new natural products are analyzed through metagenomics (or mini-metagenomics) rather than isolate data sets, short reads remain the workhorse of genome mining for natural products.
Some researchers use hybrid approaches for metagenomics assemblies by combining short and long reads (Frank et al. 2016; Tsai et al. 2016). biosyntheticSPAdes is implemented in a manner that allows one to use new sequencing technologies as long as they are supported by the SPAdes pipeline. Since both SPAdes and metaSPAdes support hybrid data sets (Illumina + Pacific Bioscience/Oxford Nanopores), biosyntheticSPAdes can also assemble BGCs in hybrid data sets.
Methods
Below, we describe the six steps of the biosyntheticSPAdes pipeline (Fig. 2) and illustrate them using reconstruction of the CALC BGC (Fig. 1).
Step 1: assembling genomic/metagenomic reads with SPAdes/metaSPAdes
BiosyntheticSPAdes starts with launching SPAdes (Bankevich et al. 2012) or metaSPAdes (Nurk et al. 2017) assemblers. SPAdes and metaSPAdes first construct a de Bruijn graph (Compeau et al. 2011) of all reads and subsequently perform various graph simplification procedures (e.g., bubble collapsing and tip removal) to transform it into an assembly graph. Both SPAdes and metaSPAdes use exSPAnder (Prjibelski et al. 2014) to utilize the read-pair information for repeat resolution and scaffolding in the assembly graph.
Step 2: identifying domain-edges in the assembly graph
The first step toward reconstructing the nucleotide sequence of a BGC is reconstruction of the arrangement of its biosynthetic domains. In many cases, this arrangement alone provides sufficient information for predicting the structure of the core scaffold of a natural product encoded by the BGC.
To identify edges harboring biosynthetic domains in the assembly graph, contigs generated by SPAdes/metaSPAdes are searched for the domain motifs using HMMER (Zhang and Wood 2003; Eddy 2011). For illustration purposes, here, we analyze only A-domains. After mapping contigs back to the assembly graph, biosyntheticSPAdes identifies the positions of all detected domains in the assembly graph (Fig. 1, top). Mapping the A-domains from the CALC BGC back to the assembly graph reveals that three A-domains (4, 5, and 7) map to the same positions on a single edge of the assembly graph. The edge harboring these positions has approximately three times higher coverage than the average coverage of edges that contain only a single copy of an A-domain. Supplemental Figure S1 illustrates that these three domains are similar to each other and share identical repeats of length ≈100 bp and longer. Sequences of these domains are collapsed during assembly, because the assembly graph was constructed from k-mers that are shorter than 100 nucleotides.
Step 3: extracting BGC subgraphs from the assembly graph
BGCs contain various domains and multiple biosynthetic genes in close proximity to each other. Analysis of all complete NRP BGCs from the MIBiG repository of BGCs (Medema et al. 2015) revealed that the distances between consecutive NRPS- or PKS-related domains do not exceed 20 kb in 95% of the cases (Supplemental Fig. S2).
Hence, we consider all edges in the assembly graph within 10 kb from the positions of domains on the domain edges identified in the previous step to capture all consecutive domains separated by, at most, 20 kb. The subgraph of the assembly graph formed by these edges, referred to as the BGC assembly graph, usually consists of multiple connected components, where each component, referred to as a BGC subgraph, usually corresponds to a single BGC. For example, four NRP BGCs in the S. coelicolor genome are represented by four different connected components of the BGC assembly graph. However, in some cases a single component of the BGC assembly graph may combine multiple BGCs, particularly when these BGCs share very similar domains with identical sequences exceeding the maximum default k-mers size in SPAdes. The complexity of the BGC subgraph is defined as the total number of A-domains and AT-domains in this subgraph. We define nontrivial BGC subgraphs as BGC subgraphs of complexity at least 3.
The BGC assembly graph for S.coelicolor consists of 24 BGC subgraphs. Three of them are nontrivial BGC subgraphs with complexities 9 (for the CALC BGC), 4, and 3. The BGC subgraph corresponding to the CALC BGC with 11 A-domains revealed only nine A-domains, since three A-domains were collapsed into a single edge.
Step 4: restoring collapsed domains in the assembly graph
Figure 1 reveals a limitation of existing assemblers (repeat collapsing) that negatively affects gene prediction tools: Three A-domains sharing long identical segments are collapsed into a single edge in the assembly graph. As a result, valuable information about the differences between these A-domains is lost (Supplemental Fig. S1). This effect is amplified in metagenomics assemblies since they aggressively collapse bubbles to improve contiguity of the assembly (Nurk et al. 2017), particularly in the case of metagenomes containing similar strains. A side effect of the bubble collapsing procedure is collapsing similar domains, which leads to a high number of mismatches and indels in reconstructed BGC sequences (referred to as an “assembly deterioration”).
This limitation of the existing assemblers can be remedied by restoring subtle variations in the collapsed repeats to enable better repeat resolution. Since SPAdes and metaSPAdes map each read to the assembly graph, we consider all reads mapped to edges of all BGC subgraphs and compute the median depth of coverage of each edge. Given an edge with coverage cov in a BGC subgraph, we extract all k-mers from the reads mapped to this edge. A k-mer is defined as solid if it does not belong to the edge but appears in at least α*cov reads mapped to this edge (the default value α = 0.2). Solid k-mers reveal variations in repeats (rather than sequencing errors), as the expected frequency of erroneous k-mers is typically below α*cov. We define a path formed by solid k-mers as a solid bubble if it forms an alternative path in a BGC subgraph. We restore all such solid bubbles in a BGC subgraph and rerun the exSPAnder repeat resolution on the modified BGC subgraphs with restored solid bubbles. We emphasize that we applied the domain restoration step to the domain edges in the BGC subgraphs only, since applying it to the entire assembly graph leads to deterioration of the assembly and reduced N50 statistics.
Note that the consensus sequence of the edge harboring three similar but not identical A-domains in the CALC assembly (Fig. 6) differs from the sequences of each of these A-domains. Therefore, it provides slightly inaccurate sequences for each of these three domains. However, after the domain restoration procedure, these three A-domains correspond to three different and 100% accurate consensus sequences. In some cases, the domain restoration procedure even enables exSPAnder to utilize the restored variations between domains for further repeat resolution by utilizing variations between long imperfect repeats.
Figure 6.

Effect of bubble restoration on the reconstruction of the CALC BGC. Schematic representation of repeat collapsing and consensus deterioration in the case of the CALC BGC assembly. While SPAdes outputs a single (and incorrect) consensus sequence of all three collapsed A-domains, these three sequences are not identical. In contrast, biosyntheticSPAdes utilized restored domains and reconstructed their distinct sequences with 100% accuracy (as compared to 99.6% accuracy for SPAdes). Numbers near dashed vertical lines represent the column numbers in the multiple alignment of three A-domain.
We note that, although the described bubble restoration procedure has a potential to resolve close strains in metagenomics assemblies, it has not been implemented in metaSPAdes yet.
Running exSPAnder on the modified BGC subgraph with restored bubbles often results in a more accurate estimate of the total number of domains (Fig. 6). In contrast to the initial BGC subgraph with only nine identified A-domains for the CALC BGC, all 11 A-domains are now captured in five resulting contigs in the modified BGC subgraph.
Step 5: constructing the scaffolding graph
We represent each domain-containing contig as an isolated solid edge in the scaffolding graph (Fig. 7). Given solid edges e and e′, we connect the ending vertex of e with the starting vertex of e′ by a dashed edge if the last domain on e and the first domain of e′ are close in the BGC assembly graph, i.e., the distance between them is <10 kb. Given a directed graph with solid and dashed edges, the Rural Postman Problem is to find a rural postman route, i.e., a path visiting all solid edges of the graph (Orloff 1974).
Figure 7.

The scaffolding graph of the CALC BGC. (Left) Five solid edges in the scaffolding graph correspond to five contigs shown in Figure 4 (bottom) that contain A-domains. These contigs are shown as a red edge (A-domains 1, 2, 3, 4, and 5), a green edge (A-domain 6), a pink edge (A-domain 7), a blue edge (A-domains 8, 9, and 10), and a black edge (A-domain 11). Eight dashed edges in the scaffolding graph connect solid edges that contain closely located domains in the BGC subgraph. (Right) Two rural postman routes in the CALC scaffolding graph. The first tour contains all violet dashed edges and results in the (1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11) arrangement of A-domains, while the second tour contains all brown dashed edges and results in the (1, 2, 3, 4, 5, 7, 6, 8, 9, 10, 11) arrangement of A-domains.
Step 6: constructing putative BGCs by solving the Rural Postman Problem
Inferring the arrangement of domains in an NRP BGC is crucial for identifying the NRP encoded by this NRPS. Since each NRP synthetase corresponds to a rural postman route in the scaffolding graph, biosyntheticSPAdes searches for all rural postman routes in the scaffolding graph using a brute force algorithm (most scaffolding subgraphs have less than 20 vertices). Figure 7 shows two rural postman routes in the CALC scaffolding graph.
Some bacterial genomes contain 100% identical domains that are collapsed into a single edge even after domain restoration. As the result, a rural postman route may visit the collapsed solid edges in some scaffolding graphs multiple times. For each solid edge in the scaffolding graph, the approximate number of times it should be traversed is defined by the ratio of the coverage of the corresponding domain-edge in the BGC subgraph to the median coverage across all edges of the BGC subgraph.
As Figure 7 illustrates, biosyntheticSPAdes may output multiple arrangements of A-domains, each arrangement corresponding to a rural postman route. For each rural postman route, biosyntheticSPAdes reconstructs a path in the BGC assembly graph corresponding to this route and its nucleotide sequence. Dashed edges in a rural postman route may correspond to multiple paths in the BGC assembly graph, and we report the path with the length closest to any of distances from the set of 550, 1500, and 2400 bp, the values of the three pronounced peaks in the distribution of the distances between consecutive domains in known NRPSs (Supplemental Fig. S2).
biosyntheticSPAdes and NRPquest for PNP reconstruction
Even when biosyntheticSPAdes fails to assemble a BGC into a single contig, it typically reduces the number of contigs as compared to SPAdes, e.g., outputs two contigs A and B without providing one of two possible orders to concatenate these contigs (B after A or A after B). This feature is important for natural product researchers since they often perform additional experiments to reconstruct the correct order of contigs (Kleigrewe 2015). For example, in the case of NRP BGC, one can generate all possible concatenates, predict putative NRPs for each concatenate, and match a spectral data set against all putative NRPs to find a concatenate with the best match. Supplemental Information, Appendix: “Output format of biosyntheticSPAdes” specifies the details of the biosyntheticSPAdes output. Supplemental Information, Appendix: “Coupling biosyntheticSPAdes and NRPquest for PNP reconstruction” presents an example of combining genomic and mass spectrometry data to infer the correct arrangement of A-domains.
Extending biosyntheticSPAdes from NRP BGCs to other BGCs
In addition to the A-domains, biosyntheticSPAdes analyzes other domains in NRP BGCs such as C–condensation domains (C-domains) and thioesterase domains (TE-domains), among others. Moreover, biosyntheticSPAdes is not limited to NRP BGCs and also works with BGCs encoding PKS BGCs (Robinson 1991). PKSs are built from various domains including acyltransferase domains (AT-domains), keto-synthase domains (KS-domains), keto-reductase domains (KR-domains), and acyl carrier protein domains (ACP-domains).
Reference-based BGC ranking algorithm
When a database of reference genomes is available, it can help to predict the correct order of contigs by identifying a genome with a similar BGC. This is especially relevant when assembling genomes that are related to an already sequenced species or during studies of microbial communities from which individual strains have been isolated and sequenced. biosyntheticSPAdes includes a pipeline that matches all possible orders of multiple putative BGC sequences to gene clusters in antiSMASH-DB (Blin et al. 2017) and ranks them based on how well the order of the matching domains corresponds to the domain order in the most similar BGC.
Note that the reference-based BGC ranking algorithm is an optional module in biosyntheticSPAdes that should be called only in cases when there is more than one plausible path in the assembly graph. In most of our test cases, biosyntheticSPAdes leads to a single plausible path through the assembly graph and thus a single BGC architecture. In all such cases, reference genomes are not required to infer the correct assembly.
In the case when a BGC-subgraph is not resolved into a single BGC, biosyntheticSPAdes generates multiple putative BGCs (pBGCs) and ranks them based on their similarity to BGCs from reference genomes from antiSMASH-DB (Blin et al. 2017). Each pBGC is compared to each reference BGCs (rBGCs) and scored according to the similarity between the pBGC and the rBGCs with respect to sequence similarity, domain composition, and domain order. See Supplemental Information, Appendices: “Reference-based putative BGC ranking algorithm,” “Ranking putative BGCs from Streptomyces coelicolor A3(2) and Streptomyces avermitilis MA-4680,” Supplemental Figures S6, S7, and S8, and Supplemental Tables S6 and S7 for details.
Software availability
biosyntheticSPAdes will be included in the next version of the SPAdes toolkit available from http://cab.spbu.ru/software/spades starting from version 3.14. The prerelease version, that was used for benchmarking in this paper and the biosyntheticSPAdes ranking pipeline, is available in Supplemental Material. BiosyntheticSPAdes source code is alternatively available from http://dx.doi.org/10.6084/m9.figshare.6948260.v2, and the biosyntheticSPAdes ranking pipeline is alternatively available from https://git.wur.nl/medema-group/biosyntheticSpadesRankingPipeline.
Supplementary Material
Acknowledgments
We thank Alexey Gurevich, Sergey Nurk, Bahar Behsaz, and Jeremy Owen for useful discussions and assistance with data analysis. A.K. was supported by the Russian Science Foundation (grant 19-14-00172). D.M. was supported by Saint Petersburg State University (grant 15.61.951.2015), and the Tri-Institutional Training Program in Computational Biology and Medicine (National Institutes of Health grant 1T32GM083937). H.M. was supported by a start-up package from Carnegie Mellon University, and research fellowship from the Alfred P. Sloan foundation. V.T. was supported by the research program NWO-Groen, which is jointly funded by the Netherlands Organization for Scientific Research (NWO), BASF SE and Baseclear BV (project ALWGR.2015.1). M.H.M. was supported by VENI grant 863.15.002 from the Netherlands Organization for Scientific Research (NWO). The work of P.A.P. was supported by the U.S. National Institutes of Health grant 2-P41-GM103484.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.243477.118.
References
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19: 455–477. 10.1089/cmb.2012.0021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley SD, Chater KF, Cerdeño-Tárraga AM, Challis GL, Thomson NR, James KD, Harris DE, Quail MA, Kieser H, Harper D, et al. 2002. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 417: 141–147. 10.1038/417141a [DOI] [PubMed] [Google Scholar]
- Besemer J, Borodovsky M. 2005. GeneMark: web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res 33: W451–W454. 10.1093/nar/gki487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blin K, Medema MH, Kottmann R, Lee SY, Weber T. 2017. The antiSMASH database, a comprehensive database of microbial secondary metabolite biosynthetic gene clusters. Nucleic Acids Res 45: D555–D559. 10.1093/nar/gkw960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cane DE, Walsh CT. 1999. The parallel and convergent universes of polyketide synthases and nonribosomal peptide synthetases. Chem Biol 6: R319–R325. 10.1016/S1074-5521(00)80001-0 [DOI] [PubMed] [Google Scholar]
- Challis GL, Ravel J. 2000. Coelichelin, a new peptide siderophore encoded by the Streptomyces coelicolor genome: structure prediction from the sequence of its non-ribosomal peptide synthetase. FEMS Microbiol Lett 187: 111–114. 10.1111/j.1574-6968.2000.tb09145.x [DOI] [PubMed] [Google Scholar]
- Coates RC, Podell S, Korobeynikov A, Lapidus A, Pevzner P, Sherman DH, Allen EE, Gerwick L, Gerwick WH. 2014. Characterization of cyanobacterial hydrocarbon composition and distribution of biosynthetic pathways. PLoS One 9: e85140 10.1371/journal.pone.0085140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compeau PE, Pevzner PA, Tesler G. 2011. How to apply de Bruijn graphs to genome assembly. Nat Biotechnol 29: 987–991. 10.1038/nbt.2023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cragg GM, Newman DJ. 2013. Natural products: a continuing source of novel drug leads. Biochim Biophys Acta 1830: 3670–3695. 10.1016/j.bbagen.2013.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcher AL, Bratke KA, Powers EC, Salzberg SL. 2007. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23: 673–679. 10.1093/bioinformatics/btm009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donia MS, Fischbach MA. 2015. Small molecules from the human microbiota. Science 349: 1254766 10.1126/science.1254766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donia MS, Cimermancic P, Schulze CJ, Brown LCW, Martin J, Mitreva M, Clardy J, Linington RG, Fischbach MA. 2014. A systematic analysis of biosynthetic gene clusters in the human microbiome reveals a common family of antibiotics. Cell 158: 1402–1414. 10.1016/j.cell.2014.08.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy SR. 2011. Accelerated profile HMM searches. PLoS Comput Biol 7: e1002195 10.1371/journal.pcbi.1002195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards DJ, Marquez BL, Nogle LM, McPhail K, Goeger DE, Roberts MA, Gerwick WH. 2004. Structure and biosynthesis of the jamaicamides, new mixed polyketide-peptide neurotoxins from the marine cyanobacterium Lyngbya majuscula. Chem Biol 11: 817–833. 10.1016/j.chembiol.2004.03.030 [DOI] [PubMed] [Google Scholar]
- Frank JA, Pan Y, Tooming-Klunderud A, Eijsink VG, McHardy AC, Nederbragt AJ, Pope PB. 2016. Improved metagenome assemblies and taxonomic binning using long-read circular consensus sequence data. Sci Rep 6: 25373 10.1038/srep25373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman MF, Gurgui C, Helf MJ, Morinaka BI, Uria AR, Oldham NJ, Sahl HG, Matsunaga S, Piel J. 2012. Metagenome mining reveals polytheonamides as posttranslationally modified ribosomal peptides. Science 338: 387–390. 10.1126/science.1226121 [DOI] [PubMed] [Google Scholar]
- Gurevich A, Saveliev V, Vyahhi N, Tesler G. 2013. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29: 1072–1075. 10.1093/bioinformatics/btt086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich A, Mikheenko A, Shlemov A, Korobeynikov A, Mohimani H, Pevzner PA. 2018. Increased diversity of peptidic natural products revealed by modification-tolerant database search of mass spectra. Nat Microbiol 3: 319–327. 10.1038/s41564-017-0094-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjithomas M, Chen IMA, Chu K, Ratner A, Palaniappan K, Szeto E, Huang J, Reddy TBK, Cimermančič P, Fischbach MA, et al. 2015. IMG-ABC: a knowledge base to fuel discovery of biosynthetic gene clusters and novel secondary metabolites. MBio 6: e00932-15 10.1128/mBio.00932-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Li L, Myers JR, Marth GT. 2012. ART: a next-generation sequencing read simulator. Bioinformatics 28: 593–594. 10.1093/bioinformatics/btr708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Human Microbiome Project Consortium. 2012. A framework for human microbiome research. Nature 486: 215–221. 10.1038/nature11209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt D, Chen GL, LoCascio PF, Land ML, Larimer FW, Hauser LJ. 2010. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11: 119 10.1186/1471-2105-11-119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz M, Hover BM, Brady SF. 2016. Culture-independent discovery of natural products from soil metagenomes. J Ind Microbiol Biotechnol 43: 129–141. 10.1007/s10295-015-1706-6 [DOI] [PubMed] [Google Scholar]
- Kersten RD, Yang Y-L, Xu Y, Cimermancic P, Nam SJ, Fenical W, Fischbach MA, Moore BS, Dorrestein PC. 2011. A mass spectrometry-guided genome mining approach for natural product peptidogenomics. Nat Chem Biol 7: 794–802. 10.1038/nchembio.684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klassen JL, Currie CR. 2012. Gene fragmentation in bacterial draft genomes: extent, consequences and mitigation. BMC Genomics 13: 14 10.1186/1471-2164-13-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleigrewe K, Almaliti J, Tian IY, Kinnel RB, Korobeynikov A, Monroe EA, Duggan BM, Di Marzo V, Sherman DH, Dorrestein PC, et al. 2015. Combining mass spectrometric metabolic profiling with genomic analysis: a powerful approach for discovering natural products from cyanobacteria. J Nat Prod 78: 1671–1682. 10.1021/acs.jnatprod.5b00301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Liu CM, Luo R, Sadakane K, Lam TW. 2015. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31: 1674–1676. 10.1093/bioinformatics/btv033 [DOI] [PubMed] [Google Scholar]
- Marquez BL, Watts KS, Yokochi A, Roberts MA, Verdier-Pinard P, Jimenez JI, Hamel E, Scheuer PJ, Gerwick WH. 2002. Structure and absolute stereochemistry of hectochlorin, a potent stimulator of actin assembly. J Nat Prod 65: 866–871. 10.1021/np0106283 [DOI] [PubMed] [Google Scholar]
- Medema MH, Cimermancic P, Sali A, Takano E, Fischbach MA. 2014a. A systematic computational analysis of biosynthetic gene cluster evolution: lessons for engineering biosynthesis. PLoS Comput Biol 10: e1004016 10.1371/journal.pcbi.1004016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema MH, Paalvast Y, Nguyen DD, Melnik A, Dorrestein PC, Takano E, Breitling R. 2014b. Pep2Path: automated mass spectrometry-guided genome mining of peptidic natural products. PLoS Comput Biol 10: e1003822 10.1371/journal.pcbi.1003822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema MH, Kottmann R, Yilmaz P, Cummings M, Biggins JB, Blin K, de Bruijn I, Chooi YH, Claesen J, Coates RC, et al. 2015. Minimum information about a biosynthetic gene cluster. Nat Chem Biol 11: 625–631. 10.1038/nchembio.1890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohimani H, Pevzner PA. 2016. Dereplication, sequencing and identification of peptidic natural products: from genome mining to peptidogenomics to spectral networks. Nat Prod Rep 33: 73–86. 10.1039/c5np00050e [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohimani H, Liu WT, Kersten RD, Moore BS, Dorrestein PC, Pevzner PA. 2014a. NRPquest: coupling mass spectrometry and genome mining for nonribosomal peptide discovery. J Nat Prod 77: 1902–1909. 10.1021/np500370c [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohimani H, Kersten RD, Liu WT, Wang M, Purvine SO, Wu S, Brewer HM, Pasa-Tolic L, Bandeira N, Moore BS, et al. 2014b. Automated genome mining of ribosomal peptide natural products. ACS Chem Biol 9: 1545–1551. 10.1021/cb500199h [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohimani H, Gurevich A, Mikheenko A, Garg N, Nothias LF, Ninomiya A, Takada K, Dorrestein PC, Pevzner PA. 2017. Dereplication of peptidic natural products through database search of mass spectra. Nat Chem Biol 13: 30–37. 10.1038/nchembio.2219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S, Seshadri R, Varghese NJ, Eloe-Fadrosh EA, Meier-Kolthoff JP, Göker M, Coates RC, Hadjithomas M, Pavlopoulos GA, Paez-Espino D, et al. 2017. 1,003 reference genomes of bacterial and archaeal isolates expand coverage of the tree of life. Nat Biotechnol 35: 676–683. 10.1038/nbt.3886 [DOI] [PubMed] [Google Scholar]
- Newman DJ, Cragg GM. 2016. Natural products as sources of new drugs from 1981 to 2014. J Nat Prod 79: 629–661. 10.1021/acs.jnatprod.5b01055 [DOI] [PubMed] [Google Scholar]
- Nurk S, Meleshko D, Korobeynikov A, Pevzner PA. 2017. metaSPAdes: a new versatile metagenomic assembler. Genome Res 27: 824–834. 10.1101/gr.213959.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orloff CS. 1974. A fundamental problem in vehicle routing. Networks 4: 35–64. 10.1002/net.3230040105 [DOI] [Google Scholar]
- Palma L, Muñoz D, Berry C, Murillo J, Caballero P. 2014. Bacillus thuringiensis toxins: an overview of their biocidal activity. Toxins (Basel) 6: 3296–3325. 10.3390/toxins6123296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pati A, Ivanova NN, Mikhailova N, Ovchinnikova G, Hooper SD, Lykidis A, Kyrpides NC. 2010. GenePRIMP: a gene prediction improvement pipeline for prokaryotic genomes. Nat Methods 7: 455–457. 10.1038/nmeth.1457 [DOI] [PubMed] [Google Scholar]
- Paulsen IT, Press CM, Ravel J, Kobayashi DY, Myers GS, Mavrodi DV, DeBoy RT, Seshadri R, Ren Q, Madupu R, et al. 2005. Complete genome sequence of the plant commensal Pseudomonas fluorescens Pf-5. Nat Biotechnol 23: 873–878. 10.1038/nbt1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prjibelski AD, Vasilinetc I, Bankevich A, Gurevich A, Krivosheeva T, Nurk S, Pham S, Korobeynikov A, Lapidus A, Pevzner PA. 2014. ExSPAnder: a universal repeat resolver for DNA fragment assembly. Bioinformatics 30: i293–i301. 10.1093/bioinformatics/btu266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JA. 1991. Polyketide synthase complexes: their structure and function in antibiotic biosynthesis. Philos Trans R Soc Lond B Biol Sci 332: 107–114. 10.1098/rstb.1991.0038 [DOI] [PubMed] [Google Scholar]
- Röttig M, Medema MH, Blin K, Weber T, Rausch C, Kohlbacher O. 2011. NRPSpredictor2—a web server for predicting NRPS adenylation domain specificity. Nucleic Acids Res 39: W362–W367. 10.1093/nar/gkr323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, Birol I. 2009. ABySS: a parallel assembler for short read sequence data. Genome Res 19: 1117–1123. 10.1101/gr.089532.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stachelhaus T, Mootz HD, Marahiel MA. 1999. The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases. Chem Biol 6: 493–505. 10.1016/S1074-5521(99)80082-9 [DOI] [PubMed] [Google Scholar]
- Tsai YC, Conlan S, Deming C, Segre JA, Kong HH, Korlach J, Oh J, NISC Comparative Sequencing Program. 2016. Resolving the complexity of human skin metagenomes using single-molecule sequencing. MBio 7: e01948-15 10.1128/mBio.01948-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber T, Blin K, Duddela S, Krug D, Kim HU, Bruccoleri R, Lee SY, Fischbach MA, Müller R, Wohlleben W, et al. 2015. antiSMASH 3.0—a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res 43: W237–W243. 10.1093/nar/gkv437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MC, Mori T, Rückert C, Uria AR, Helf MJ, Takada K, Gernert C, Steffens UA, Heycke N, Schmitt S, et al. 2014. An environmental bacterial taxon with a large and distinct metabolic repertoire. Nature 506: 58–62. 10.1038/nature12959 [DOI] [PubMed] [Google Scholar]
- Zhang Z, Wood WI. 2003. A profile hidden Markov model for signal peptides generated by HMMER. Bioinformatics 19: 307–308. 10.1093/bioinformatics/19.2.307 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.