

Abstract
Amyloid fibrils in Gerstmann–Sträussler–Scheinker (GSS) disease are composed of a fragment of the prion protein (PrP), the N and C termini of which correspond to ragged residues 81–90 and 144–153. A synthetic peptide spanning the sequence 82–146 (PrP 82–146) polymerizes into protease-resistant fibrils with the tinctorial properties of amyloid. We investigated the biological activity of PrP 82–146 and of two nonamyloidogenic variants of PrP 82–146 with scrambled amino acid sequence 106–126 or 127–146. Cortical neurons prepared from rat and mouse embryos were chronically exposed to the PrP 82–146 peptides (10–50 μm). PrP 82–146 and the partially scrambled peptides induced neuronal death with a similar dose–response pattern, indicating that neurotoxicity was independent of amyloid fibril formation. Neurotoxicity was significantly reduced by coadministration of an anti-oligomer antibody, suggesting that PrP 82–146 oligomers are primarily responsible for triggering cell death. Neurons from PrP knock-out (Prnp0/0) mice were significantly less sensitive to PrP 82–146 toxicity than neurons expressing PrP. The gliotrophic effect of PrP 82–146 was determined by [methyl-3H]-thymidine incorporation in cultured astrocytes. Treatment with PrP 82–146 stimulated [methyl-3H]-thymidine uptake 3.5-fold. This activity was significantly less when the 106–126 or 127–146 regions were disrupted, indicating that PrP 82–146 amyloid activates the gliotrophic response. Prnp0/0 astrocytes were insensitive to the proliferative stimulus of PrP 82–146. These results underline the role of cerebral accumulation of abnormally folded PrP fragments and indicate that cellular PrP governs the pathogenic process.
Keywords: prion, amyloid, apoptosis, neuron, astrocytes, neurotoxicity
Introduction
The posttranslational modification of the prion protein (PrP) from its cellular isoform (PrPC) to a disease-specific species (PrPSc) is the molecular signature of prion-related encephalopathies (PREs) (Prusiner, 1991). PrPC is an almost ubiquitous glycosylphosphatidylinositol-anchored membrane protein, highly expressed in neurons, glia, and lymphoid cells, the biological function of which is still uncertain. The transition from PrPC to PrPSc involves conformational changes with a decrease in α-helical secondary structure and a significant increase in β-sheet content (Caughey et al., 1991; Pan et al., 1993; Safar et al., 1993). PrP amyloidogenesis occurs consistently in genetic forms of prion disease, such as Gerstmann-Sträussler-Scheinker (GSS) syndrome and PrP cerebral amyloid angiopathy (Ghetti et al., 1996), as well as in the new variant of Creutzfeldt-Jakob disease (vCJD), which is causally linked to bovine spongiform encephalopathy (Will et al., 1996; Bruce et al., 1997). In all of these conditions, amyloid fibrils coexist with PrP aggregates that lack ultrastructural properties of amyloid (Giaccone et al., 1992), suggesting that different PrP peptides or protein conformers may form fibrillar and nonfibrillar aggregates, possibly with neurotoxic properties. In view of the common topological distribution of neuropathological changes and the PrPSc deposits and the temporal correlation in experimental scrapie, PrPsc is considered the primary cause of neurodegeneration in prion diseases.
Biochemical studies have shown that amyloid fibrils purified from the brains of GSS patients carrying the PrP A117V, F198S, and Q217R mutations contain a major PrP fragment of ∼7 kDa, spanning residues 81–82 through 144–153 of PrP (Tagliavini et al., 1991, 1994, 2001). Low molecular weight fragments have been detected in GSS patients with other mutations and are also seen in areas without amyloid deposits (Piccardo et al., 1996, 1998; Parchi et al., 1998). All of these fragments contain the PrP region spanning residues 89–140, which plays a central role in the conformational transition of PrPC into PrPSc (Muramoto et al., 1996; Supattapone et al., 1999).
We previously generated a synthetic peptide corresponding to the smallest amyloid subunit found in GSS brains (PrP 82–146wt) (Salmona et al., 2003). To investigate the contribution of different amino acid regions to the physicochemical properties and fibrillogenic capacity of this disease-specific protein, we generated partially scrambled peptides in which either the 106–126 sequence (PrP 82–146106-126scr) or 127–146 (PrP 82–146127-146scr) was disrupted. An entirely scrambled peptide (PrP 82–146scr) was used as control. Chemicophysical analyses showed that PrP 82–146wt has a high intrinsic ability to form amyloid fibrils indistinguishable from those extracted from GSS brains, and that the formation of ordered structures with a parallel β-sheet organization depends on the integrity of the C-terminal region (Salmona et al., 2003).
In the present study, we investigated the biological effects of the PrP 82–146 peptides. Their neurotoxic and gliotrophic activities were investigated in primary cultures from rat and mouse brains. The contribution of cellular PrP to the biological activity of peptides was studied by comparing the effects of the peptides on wild-type or PrP knock-out (Prnp0/0) cells.
Materials and Methods
Synthesis and purification of PrP peptides.
The following peptides were chemically synthesized: PrP 82–146 wild-type (PrP 82–146wt: GQPHGGGWGQGGGTHSQWNKPSKPKTNMKHMAGAAAAGAVVGGLGGYMLGSAMSRPIIHFGSDYE); PrP 82–146 with a scrambled sequence in the region spanning residues 106–126 (PrP 82–146106-126scr: GQPHGGGWGQGGGTHSQWNKPSKPNGAKALMGGHGATKVMVGAAAGYMLGSAMSRPIIHFGSDYE); PrP 82–146 with a scrambled sequence in the region spanning residues 127–146 (PrP 82–146127-146scr: GQPHGGGWGQGGGTHSQWNKPSKPKTNMKHMAGAAAAGAVVGGLGSMYPASHGLMEFYGIGSIR); PrP 82–146 scrambled (PrP 82–146scr: EADQFALGGSKHGNGMQQVAGHGGSMGAKAWGANGHPSGTGIPTAKMVPYKIYGGGWAGMGRPSS). In some experiments, peptides PrP 106–126wt (KTNMKHMAGAAAAGAVVGGLG) and PrP 127–146wt (GYMLGSAMSRTII-HFGSDYE) were also used.
Peptides were prepared by solid-phase synthesis on a 433A synthesizer (Applied Biosystems, Foster City, CA) at 0.1 mm scale with 4-hydromethylphenoxyacetic acid resin using N-(9-fluoroenyl)methoxycarbonyl protected l-amino acid derivatives. Amino acids were activated by reaction with 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluroniumhexafluorophosphate/1-hydroxybenzotriazole and N,N,-diiso propylethylamine. Peptides were cleaved from the resin with a mixture of phenol/thioanisole/ethandiol/trifluoroacetic acid, precipitated with cold diethyl ether, and washed several times with the same solvent.
The purification was performed by reverse-phase HPLC on a semipreparative C18 column (190 × 300 mm, 300 Å pore size, 15 μm particle size; Delta Pack, Nihon Waters, Tokyo, Japan) with a mobile phase of 0.1% TFA/water (eluent A) and 0.08% trifluoracetic acid (TFA)/acetonitrile (eluent B) using a linear gradient of 0–80% eluent B in 60 min with a flow rate of 3 ml/min. The fractions containing PrP peptides were separately collected, lyophilized, and kept at −80°C. Their purity and composition were determined by amino acid sequencing (46600 Prosequencer; Milligen, Bedford, MA) and electrospray mass spectrometry (model 5989 A; Hewlett-Packard, Palo Alto, CA).
Animals.
CD1 rats, C57BL/6J, and C57BL/6J × CBA/J F1 mice were purchased from Charles River Laboratories (Calco, Italy). The Prnp0/0 mice were derived from the Zürich I Prnp0/0 line (Bueler et al., 1992), which had been bred subsequently with C57BL/6J × CBA/J hybrid mice (Chiesa et al., 1998). Procedures involving animals and their care were conducted in conformity with the institutional guidelines that are in compliance with national and international laws and policies.
Cortical cultures.
Primary cultures of cortical neurons were prepared from rat and mouse embryos [embryonic day 17 (E17) and E14.5, respectively] or neonatal mouse pups [postnatal day 1 (P1) or P2] as described previously (Forloni et al., 1993; Chiesa et al., 2004). Cortical cells were dissociated and centrifuged twice at 1500 rpm for 2 min, resuspended by gentle trituration, counted, and plated at 150,000/cm2 in modified Eagle's medium (MEM)–FBS on poly-l-lysine-coated plates. After 3 h, the medium was replaced with Neurobasal medium containing B27 supplement (Invitrogen, Carlsbad, CA), 0.5 mm glutamine, 120 mg/L penicillin, and 260 mg/L streptomycin and incubated at 37°C in a humidified incubator with 5% CO2/95% air. Half the medium was replaced every 5–7 d. Aphidicolin (3 μg/ml) was added to the cultures 4 d after plating to inhibit glial growth. Under these culture conditions, the survival and growth of non-neuronal cells was minimized. Cells were maintained at 37°C in 95% air, 5% CO2 and treated with the peptides (10–50 μm) the day after plating (day 1); then, the medium was replaced and the treatment was repeated on day 4, and cell viability was assayed on day 7. The immunostaining with mouse anti-neuronal nuclei monoclonal antibody (NeuN) (Chemicon–Millipore, Billerica, MA) showed that in our preparation, a large part of the cells were neurons (see Fig. 3). The counts of NeuN-positive cells in three wells from two different preparations showed that 79 ± 2.5% of the cells are stained with the neuron-specific antibody.
Figure 3.
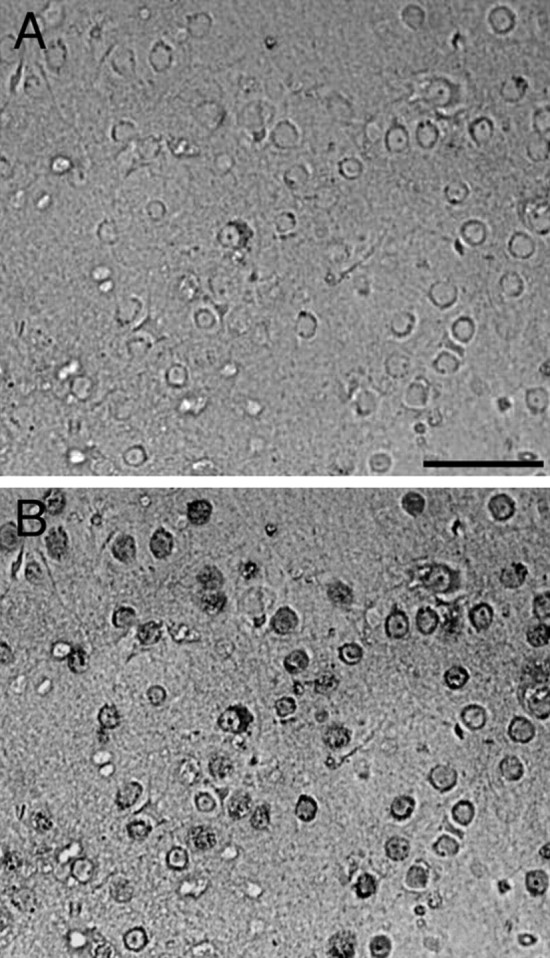
Neuronal preparation from neonatal rat cortical cells. A, Phase contrast photomicrograph of the neuronal preparation. B, Immunostaining of the same field with mouse anti-NeuN (dark gray) examined with fluorescent microscope. Scale bar, 30 μm
N2a neuroblastoma cells.
N2a mouse neuroblastoma cells were grown in DMEM and MEMα 1:1 supplemented with 10% FBS, nonessential amino acids and penicillin/streptomycin, and maintained in an atmosphere of 5% CO2, 95% air. The cells were plated at 20,000 cells/cm2 and treated the day after being plated, for the times indicated.
Astrocytes.
Astrocytes were prepared by dissociating cerebral cortices of newborn rats or mice (Forloni et al., 1994; Chiesa et al., 2004). Brains from P0–P5 pups were dissected, and the meninges were removed. Tissue was disrupted by pipetting, and resuspended cells were seeded on poly-l-lysine-coated 75 cm2 flasks. Cultures were maintained at 37°C with 5% CO2 for 2 weeks. When cultures were confluent, microglial cells were mechanically isolated by shaking the flasks at 260 rpm for 18 h. Astrocytes, which remained attached to the flask, were detached by trypsinization. After short incubation with 0.025% trypsin in PBS at 37°C, medium containing serum was added to stop the activity of the enzyme, and cells were dissociated by several passages through a 5 ml pipette, counted, and plated on appropriate plates. In this preparation, the presence of astrocytes GFAP positive is >90% of the cell population (Forloni et al., 1994)
Cell viability.
Cell viability was assessed by measuring the level of 3-(4,5 dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) to formazan. Cells were incubated for 3 h at 37°C with 0.4 mg/ml MTT, dissolved in 0.04N HCl in 2-propanol, and analyzed spectrophotometrically at 540 nm with an automatic microplate reader (Multiskan MS; Labsystems Italia, Helsinki, Finland). The data on cell viability were confirmed by crystal violet staining (Andreoni et al., 1997) and microscopic examination.
Apoptosis.
To investigate the apoptotic effect of PrP peptides, cells were seeded on poly-l-lysine-coated glass coverslips in 24-well plates (150,000 cells/well) and treated for the appropriate time. To visualize apoptotic nuclei, the bisbenzimide dye (Hoechst 33258; Sigma, St. Louis, MO) was used. Cells were washed with PBS and fixed in methanol/acetic acid (3:1) for 5 min at 4°C. They were then rinsed with PBS and incubated with 0.05 μg/ml Hoechst 33258 in PBS for 15 min. Slides were washed in distilled water, mounted with 30% glycerol in PBS, and viewed on an Olympus (Tokyo, Japan) FV500 laser confocal scanning system.
Incorporation of [methyl-3H]-thymidine.
Astrocytes were cultured with serum-containing medium until they reached ∼50% confluency. The medium was then replaced with serum-free medium, and the cells were cultured for 24 h before treating them with the different peptides. At the same time as treatment with the peptides, [methyl-3H]-thymidine (ICN Biomedicals, Irvine, CA) was added to the culture medium (0.025 mCi/ml). After 24 h, cells were washed twice with PBS and lysed in 100 μl of distilled water, and the lysate was transferred to a vial containing 4 ml of scintillating liquid (Ready SolvTM CP; Beckman Coulter, Fullerton, CA). The radioactive content of each vial was determined using a β-counter (LSC1; PerkinElmer, Wellesley, MA).
Results
The chemicophysical characteristics of the synthetic peptides used in this study are summarized in Figure 1. Circular dichroism analysis indicated that when dissolved in Tris buffer, pH 7.0, PrP 82–146wt is primarily composed of β-sheet (54%) with a significant content of α-helix (22%) and β-turn (31%). The self-aggregation capacity pf the peptides was established by sedimentation experiments. After 96 h, incubation at 37°C, and centrifugation, 90% of the peptide was found in the pellet. Staining with thioflavine and Congo red and EM analysis showed that PrP 82–146wt aggregates are structured in amyloid fibers. As described previously, PrP 82–146wt is seen as long straight, unbranched fibrils that form a dense meshwork (Fig. 2) (Salmona et al., 2003).
Figure 1.

A, Schematic representations of PrP with the regions considered for the PrP 82–146 peptide synthesis. B, Summary of the chemicophysical features of the PrP 82–146 peptides (percentage ± SE). The differences between fibrillar and amorphous aggregates were defined by EM examination. +, ++, and +++ indicate minimal, equal, and a large part of the preparation in amorphous or structured form, respectively. nd, Not detectable.
Figure 2.
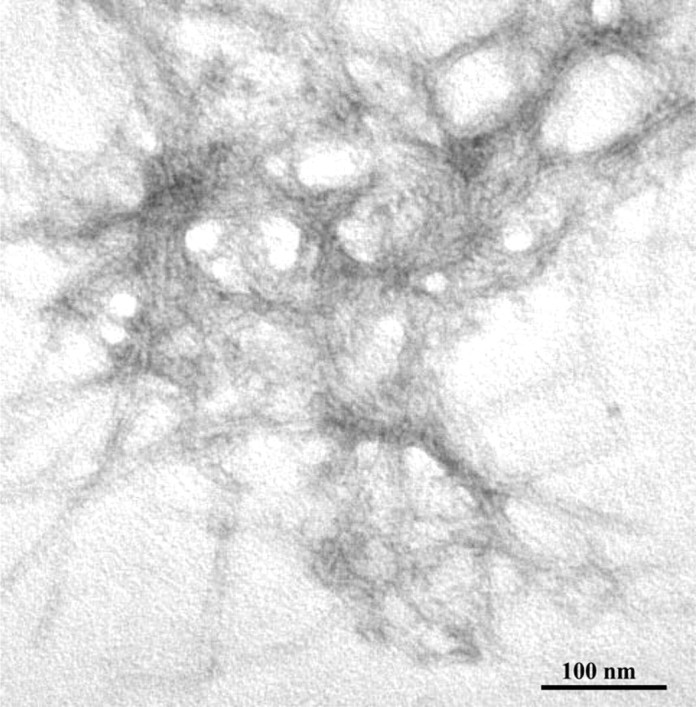
Electron micrograph of the aggregates generated by PrP 82–146, as by negative staining of peptide suspensions after 48 h of incubation indicated. Scale bar, 100 nm.
The self-aggregation capacity was associated with partial resistance to proteinase-K (PK) digestion. The entire scrambled peptide, PrP 82–146scr, did not exhibit any self-aggregation capacity and showed only minimal resistance to PK digestion. Both partial disruptions of the sequence markedly influenced the spontaneous fibrillogenesis. PrP 82–146106–126scr maintained its aggregation capacity after long incubation and even increased the resistance to PK digestion, but the pellet after centrifugation contained mainly electron-dense amorphous material and a few short filamentous structures. The aggregation capacity of PrP 82–146127–146scr was greatly reduced, and no structured filaments were evident in the pellet. Thus, chemicophysical analysis indicated that both sequences influence the structural properties of 82–146, although the 127–146 region had the most effect on the secondary structure and aggregation.
We then investigated the biological activity of all four peptides. Chronic treatment of cortical rat cultures (Fig. 3) with PrP 82–146wt induced dose-dependent neuronal death: the effect was already significant at 10 μm and increased progressively to ∼70% at 50 μm (Fig. 4B). The totally scrambled peptide was not toxic, and the neurotoxicity of both PrP 82–146106-126scr and PrP 82–146127-146scr was indistinguishable from that of the wild-type sequence. Thus, despite the profound changes in the peptide structures induced by disruption of the 106–126 and 127–146 sequences, the neurotoxicity was unaffected. This result was confirmed using mouse cortical cultures (Fig. 5).
Figure 4.

Amyloid and nonamyloid forms of PrP 82–146 are toxic to rat cortical neurons. Cortical cells from rat embryos (E17) were treated with PrP 82–146 peptides at the concentrations indicated, and cell viability was determined after 7 d by MTT assay. All peptides, except of PrP 82–146scr, significantly reduced viability. Data are the mean ± SEM of 20–30 replicates from three to four independent t tests. *p < 0.01 versus control group by Dunnett's test.
Figure 5.

PrP 82–146 peptides are toxic to murine cortical neurons. Cortical cells from mice (E14.5) were treated with PrP 82–126 peptides at the indicated concentrations for 7 d, and the effect on cell viability was determined by MTT assay. Except for PrP 82–146scr, all peptides dose-dependently reduced viability. Data are the mean ± SEM of 18–24 replicates from three to four independent experiments. *p < 0.01 versus control group by Dunnett's test.
Previous studies showed that the toxicity of the synthetic peptide PrP 106–126 depends on cellular PrP expression (Brown et al., 1994; Fioriti et al., 2005), cells from Prnp0/0 mice that are resistant to its toxicity. To investigate whether PrP 82–146 toxicity also depended on PrPC expression, murine cortical cells from control Prnp+/+ or Prnp0/0 mice were chronically treated with the different peptides. Similarly to rat cultures, all peptides except the totally scrambled one, caused a dose-dependent reduction of cell viability of PrP-expressing neurons, which resulted in ∼50% cell death at 10 μm (Fig. 5). In a parallel experiment, we tested the toxicity of the four peptides toward Prnp0/0 cortical neurons (Fig. 6). PrP 82–146 (25 μm) toxicity toward Prnp0/0 was reduced by ∼40% compared with Prnp+/+ cultures. Results were similar with the two partially scrambled peptides, whereas the total scrambled peptide did not affect the viability of either to Prnp+/+ or Prnp0//0 cells. It has been reported that PrP 106–126 kills neurons by apoptosis (Forloni et al., 1993). Because PrP 106–126 and PrP 82–146 shared some features, we verified whether this mechanism was also operative for the latter. After 5–7 d of treatment with PrP 82–146, a subset of cells showed the typical apoptotic morphology with condensation of the chromatin and fragmentation of the nucleus; cells treated with the scrambled peptide showed no such changes (Fig. 7). The effect was similar in N2a mouse neuroblastoma cells exposed to PrP 82–146 (Fig. 8). Because the toxicity of PrP 106–126 is antagonized by the anti-oligomers antibody A11 developed by Kayed et al. (2003), we cheeked whether this was also the case for PrP 82–146 neurotoxicity. At 1 μg/ml, the A11 antibody partially prevented the PrP 82–146wt toxicity, and the antagonism was almost complete at a higher antibody concentration (4 μg/ml) (Fig. 9).
Figure 6.

PrP 82–146 toxicity is partially dependent on PrPC expression. Murine cortical cells from either control or Prnp0/0 mice were treated with PrP 82–146 peptides (25 μm), and cell viability was determined after 7 d by MTT assay. All of the peptides reduced cell viability of PrP-expressing neurons (Prnp+/+) by ∼60%. PrP knock-out (Prnp0/0) neurons were significantly less vulnerable than PrP-expressing cells, showing 35–40% cell death at the end of the treatment. Data are the mean ± SEM of 12–20 replicates from three to four independent experiments. *p < 0.01 by Dunnett's test.
Figure 7.

PrP 82–146wt induces nuclear fragmentation of cortical neurons. Cortical neurons from C57BL/6J × CBA/J pups were exposed to the vehicle (A), PrP 82–146wt (B), or PrP 82–146scr (C) 25 μm for 7 d. Cells were analyzed by fluorescence microscopy after staining with the DNA-binding fluorochrome bisbenzimide (Hoechst 33258). The arrows indicate nuclei with typical apoptotic features. Scale bar, 20 μm.
Figure 8.

Time course of the PrP 82–146 induced nuclear fragmentation of N2a cells. N2a cells (A, control condition) were exposed to PrP 82–146wt for 24 h (B), 48 h (C), and 96 h (D). Cells were analyzed by fluorescence microscopy after staining with the DNA-binding fluorochrome (Hoechst 33258). The percentage of fragmented nuclei progressively increased with time. The arrows indicate nuclei with typical apoptotic features. Scale bar, 20 μm.
Figure 9.

Anti-oligomeric antibody antagonized the toxicity of PrP 82–146wt. Cortical cells from rat embryos (E17) were treated with PrP 82–146 (10 μm) and anti-oligomeric antibody (ab, A11) at two concentrations: 1 and 4 μg/ml. The cell viability was determined after 7 d by MTT assay. Data are the mean ± SEM of 8–10 determinations. Ab 4 μg/ml, ANOVA (2 × 2) analysis: Fint = p < 0.01; *p < 0.05; **p < 0.01 versus control group by Tukey's test.
Neuronal death in transmitted spongiform encephalopathies (TSE) is associated with glial proliferation. This neuropathological feature can be recapitulated in culture by PrP 106–126 (Forloni et al., 1994; Florio et al., 1996; Thellung et al., 2000). We investigated the effect of PrP 82–146 on astroglial proliferation in astrocytes prepared from cortices of 2-d-old rats. Cells were treated with the four peptides (25 μm) for 24 h in serum-free medium, and the rate of proliferation was determined by [methyl-3H]-thymidine uptake. As illustrated in Figure 10A, PrP 82–146wt induced glial proliferation. This effect was completely abolished by partial or total disruption of the wild-type sequence. Thus, the gliotrophic activity strongly depends on the integrity of the peptide sequence and its structural characteristics. Similarly, PrP 82–146wt induced a fourfold increase of [methyl-3H]-thymidine uptake by mouse cortical astrocytes; again, the entirely or partially scrambled peptides had less proliferative activity. Astrocytes from Prnp0/0 mice were insensitive to the proliferative stimulus of the peptides, indicating that PrPC plays a role in the gliotrophic activity of PrP 82–146.
Figure 10.
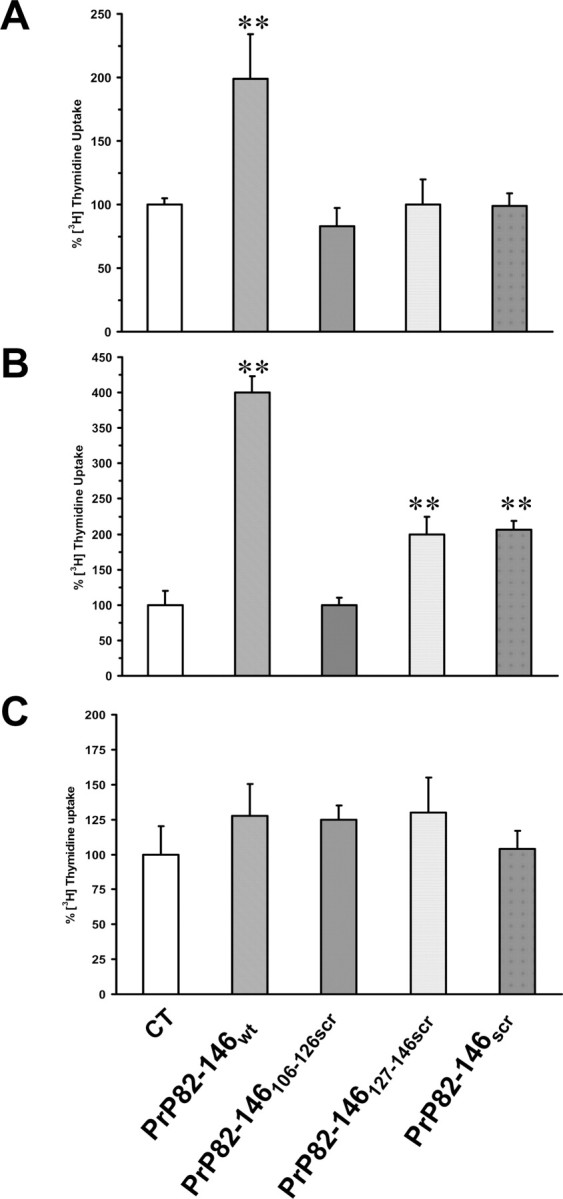
PrP 82–146 amyloid stimulates PrPC-dependent proliferation of astrocytes. A, Astrocytes prepared from cortices of 2-d-old rats were treated with the peptides (25 μm) in serum-free medium, and cell proliferation was assayed by [methyl-3H]-thymidine incorporation after 24 h. PrP 82–146wt stimulated [methyl-3H]-thymidine uptake by twofold, whereas entirely or partially scrambled peptides did not. B, C, Cell proliferation was assayed in mouse astrocytes from Prnp+/+ (B) or Prnp0/0 (C) pups as described above. PrP 82–146wt stimulated [methyl-3H]-thymidine incorporation by fourfold, whereas the partially or entirely scrambled peptides had little or no activity. Prnp0/0 astrocytes were insensitive to the proliferative stimulus of the peptides. Data are the mean ± SEM of 10–20 replicates from three to four independent experiments. *p < 0.01 versus control group by Dunnett's test.
Discussion
We explored the neurotoxic and gliotrophic potential of PrP 82–146, a synthetic peptide homologous to the major PrP fragment found in GSS amyloid. To investigate the relationship between the structure and the biological activity, we used nonamyloidogenic variants of PrP 82–146. The data presented indicate that both amyloid and nonamyloid forms are toxic to neurons, whereas only the amyloid form stimulates astroglial proliferation. The biological effects of PrP 82–146 were influenced by expression of cellular PrP. These results highlight the importance of amyloidogenic PrP fragments in GSS pathogenesis and argue for a role of cellular PrP in the pathogenic process.
The kinetics of aggregation of PrP 82–146 was investigated by Gobbi et al. (2006), who proposed a “dock-and-lock” model to explain the mechanism of polymerization. Structural analysis found profound differences between the wild-type and partially scrambled PrP 82–146 peptides in their propensity to aggregate (Salmona et al., 2003), amyloid formation being significantly reduced or prevented when either region 106–126 or 127–146 was disrupted. These structural differences, however, did not affect the neurotoxicity, because partially scrambled peptides caused neuronal death in a dose-dependent manner similar to wild-type PrP 82–146. This is consistent with a recent report that both amyloid and oligomeric forms of recombinant PrP are toxic to cultured neurons (Novitskaya et al., 2006) and that indicates that assembly of PrP into highly polymerized fibrils is not a prerequisite for toxicity. The fact that amyloid deposits of PrP are only seen in a subset of prion diseases and that in some forms protease-resistant PrP deposits are absent suggests that other PrP species, perhaps small PrP oligomers, are the primary neurotoxic culprit.
The neurotoxicity of protein oligomers was originally proposed by Hartley et al. (1999), who reported that protofibrillar forms of Alzheimer amyloid β (Aβ) were neurotoxic. We also found that Aβ 25–35 toxicity was not affected by amidation of its C terminus, although this chemical modification significantly reduced the propensity of the peptide to assemble into amyloid fibrils (Forloni et al., 1997). More recent studies have found that oligomeric forms of Aβ can directly alter synaptic structure and function, confirming the role of oligomers in Alzheimer's disease (AD) (Cleary et al., 2005; Huang et al., 2006; Townsend et al., 2006). It is possible that in AD, senile plaques serve as the reservoir of aggregated Aβ that can continuously release diffusible oligomers and protofibrils to injure not only neighboring cells but also at a certain distance from the senile plaques.
Studies with synthetic PrP peptides support the potential pathogenic role of oligomers in prion diseases too. With amidation at the C terminus of PrP 106–126, we noted strong inhibition of the PrP peptide fibrillogenic capacity with no changes in its cytotoxicity (Forloni et al., 1996a). This was confirmed by Corsaro et al. (2003) using PrP 106–126 with two GlyAla substitutions at the residues 114 and 119, generating a soluble nonamyloidogenic peptide that retained its toxic properties. Unlike our study, Bergström et al. (2005) found that amidation at the C terminal of PrP 106–126 influenced its neurotoxicity. However, as these authors mention, this might reflect some methodological differences. In their conditions, the neurotoxic effect in primary cultures was limited at the higher concentration of the peptide (80 μm). In contrast, Novitskaya et al. (2006) showed recently that recombinant PrP can be toxic in the fibrillar or the oligomeric state with similar mechanisms.
The neurotoxic role of soluble oligomers is particularly important in TSE, where the formation of amyloid deposits is evident only in GSS and in vCJD, whereas in CJD and familial fatal insomnia, amyloid deposits are rare or absent. PrP 106–126 and PrP 82–146 toxicity is significantly less when oligomers are depleted by preincubation with an oligomer-specific antibody (Kayed et al., 2003; our study). Moreover, the potential pathogenic role of PrP oligomers is supported by evidence in transgenic mice. For example, a conformationally altered form of PrP that is weakly protease-resistant and aggregated into small oligomers accumulates in the brains of transgenic mice that express a nine-octapeptide PrP insertion and develop a fatal neurological disorder (Chiesa et al., 1998). Similarly, a soluble PrP isoform with an immunoreactive profile similar to PrPSc is found in neurologically ill mice expressing a mouse PrP homolog of the P102L mutation associated with GSS (Nazor et al., 2005).
Our previous studies recognized the primary neurotoxic domain of PrP in region 106–126 (Forloni et al., 1993, 1996b, 1999). It was therefore surprising that disruption of the 106–126 sequence in PrP 82–146106-126scr did not affect neurotoxicity. This might be explained by the evidence that PrP regions encompassing residues 127–146 and 118–135 are also neurotoxic (Rymer and Good, 2000; Pillot et al., 2000). Thus, the 106–126 and 127–146 regions may contribute equally to the toxicity of PrP 82–146.
Our results point to the role played by cellular PrP in mediating the biological effects of the PrP 82–146. It is known that PrPC expression is required for propagation of infectivity (Büeler et al., 1993) and pathogenesis (Brandner et al., 1996; Mallucci et al., 2003) in mouse models of TSE. The expression of PrP is also essential for the toxicity of PrP 106–126 as shown originally by Brown et al. (1994) and confirmed by others (Jobling et al., 1999; Chabry et al., 2003; Fioriti et al., 2005). Novitskaya et al. (2006) found that silencing PrPC expression by the small RNA interference technique blocked recombinant PrP neurotoxicity. We found that in the absence of endogenous PrP, PrP 82–146 had significantly less neurotoxicity, although it was not completely abolished. This suggests that the toxicity results from multiple effects, only some of which are mediated by PrPC. For instance, amyloidogenic peptides share a common mechanism of toxicity based on membrane destabilization and the formation of ion-permeable pores (Demuro et al., 2005). We found that both wild-type and partially scrambled PrP 82–146 peptides form ion channels in artificial membranes (Bahadi et al., 2003) and alter the membrane fluidity (Salmona et al., 2003). In addition, the squalene synthase inhibitor squalestatin, which reduces plasma membrane cholesterol, antagonized the toxic effect of PrP 82–146 (Bate et al., 2004). The idea that certain PrP domains exert toxic effects independently of PrPC expression is supported by evidence that the synthetic peptide PrP 118–135 induces apoptotic cell death by destabilizing membrane properties, independently of PrPC expression (Chabry et al., 2003). Thus, PrPC-mediated neurotoxicity and PrPC-independent effects, possibly induced by different domains of PrP 82–146, might explain its pathogenic features.
In TSE, PrPSc deposition is associated with activation of astroglia, and this precedes neuronal dysfunction That abnormal PrP has a direct role in triggering astroglial proliferation is supported by the evidence that the amyloidogenic fragment PrP 106–126 induces astrogliosis in primary cultures (Forloni et al., 1994; Hafiz and Brown, 2000; Ning et al., 2005). Here, we also found that PrP 82–146 induced astroglial proliferation, and this effect was abolished when the 106–126 and 127–146 regions were disrupted. Interestingly, the gliotrophic effect of PrP 106–126 was abolished by C-terminal amidation, which strongly reduced its self-aggregation capacity, suggesting that the propensity to assume an amyloid structure is a prerequisite for the effect on astroglial proliferation, also in the case of PrP 82–146 (Rizzardini et al., 1997; Peyrin et al., 1999).
The gliotrophic effect of PrP 82–146 was completely abolished by PrPC deletion, arguing for a specific role of PrP in the activity of this peptide. Astroglial proliferation induced by PrP 106–126 was also dependent on the expression of PrPC, and it was proposed this was attributable to microglial cells (Brown et al., 1998). We and others, however, have shown that PrP 106–126 has direct effects on astrocytes (Forloni et al., 1994; Rizzardini et al., 1997; Peyrin et al., 1999; Ning et al., 2005). Because PrP 82–146 can activate microglial cells (Bate et al., 2006), it is possible that both direct and microglia-mediated effects contribute to the gliotrophic activity.
In conclusion, this study has shown that profound alterations of the PrP 82–146 sequence and amyloidogenic properties do not significantly affect the peptide neurotoxicity that appears to depend on the formation of oligomeric species. In contrast, the gliotrophic effect strictly depends on the sequence integrity and amyloidogenic properties of the peptide. Lack of endogenous PrP expression partially influences the neurotoxicity and completely abolishes the astrogliotic effect. Thus, the synthetic analog of the minimal amyloid subunit purified from GSS brains induced both neurotoxic and astrogliotic effects through a mechanism partly depending on PrPC. Our results shed light on the structural and chemicophysical features underlying PrP neurotoxicity and confirm the validity of this PrP peptide model as an effective tool for defining the cellular mechanisms of neuronal damage in prion diseases.
Footnotes
This work was supported in part by Telethon-Italy Grant S00083, European Community Grant QLG-CT-2001-2353 (R.C.), and NoE NeuroPrion FOOD-CT-2004-50679 (G.F., M.S., and F.T.). L.F. was supported by a fellowship from the Fondazione Monzino. R.C. is an Assistant Telethon Scientist (Dulbecco Telethon Institute, Fondazione Telethon).
References
- Andreoni G, Angeretti N, Lucca E, Forloni G. Densitometric quantification of neuronal viability by computerized image analysis. Exp Neurol. 1997;148:281–287. doi: 10.1006/exnr.1997.6647. [DOI] [PubMed] [Google Scholar]
- Bahadi R, Farrelly PV, Kenna BL, Kourie JI, Tagliavini F, Forloni G, Salmona M. Channels formed with a mutant prion protein PrP (82–146) homologous to a 7-kDa fragment in diseased brain of GSS patients. Am J Physiol Cell Physiol. 2003;285:C862–C872. doi: 10.1152/ajpcell.00077.2003. [DOI] [PubMed] [Google Scholar]
- Bate C, Salmona M, Diomede L, Williams A. Squalestatin cures prion-infected neurons and protects against prion neurotoxicity. J Biol Chem. 2004;279:14983–14990. doi: 10.1074/jbc.M313061200. [DOI] [PubMed] [Google Scholar]
- Bate C, Kempster S, Williams A. Prostaglandin D2 mediates neuronal damage by amyloid-beta or prions which activates microglial cells. Neuropharmacology. 2006;50:229–237. doi: 10.1016/j.neuropharm.2005.09.008. [DOI] [PubMed] [Google Scholar]
- Bergström AL, Cordes H, Zsurger N, Heegaard PM, Laursen H, Chabry J. Amidation and structure relaxation abolish the neurotoxicity of the prion peptide PrP106–126 in vivo and in vitro. J Biol Chem. 2005;280:23114–23121. doi: 10.1074/jbc.M500210200. [DOI] [PubMed] [Google Scholar]
- Brandner S, Isenmann S, Raeber A, Fischer M, Sailer A, Kobayashi Y, Marino S, Weissmann C, Aguzzi A. Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature. 1996;379:339–343. doi: 10.1038/379339a0. [DOI] [PubMed] [Google Scholar]
- Brown DR, Herms J, Kretzschmar HA. Mouse cortical cells lacking cellular PrP survive in culture with a neurotoxic PrP fragment. NeuroReport. 1994;5:2057–2060. doi: 10.1097/00001756-199410270-00017. [DOI] [PubMed] [Google Scholar]
- Brown DR, Schmidt B, Kretzschmar HA. A prion protein fragment primes type 1 astrocytes to proliferation signals from microglia. Neurobiol Dis. 1998;4:410–422. doi: 10.1006/nbdi.1998.0169. [DOI] [PubMed] [Google Scholar]
- Bruce ME, Will RG, Ironside JW, McConnell I, Drummond D, Suttie A, McCardle L, Chree A, Hope J, Birkett C, Cousens S, Fraser H, Bostock CJ. Transmissions to mice indicate that “new variant” CJD is caused by the BSE agent. Nature. 1997;389:498–501. doi: 10.1038/39057. [DOI] [PubMed] [Google Scholar]
- Büeler H, Fischer M, Lang Y, Bluethmann H, Lipp HP, DeArmond SJ, Prusiner SB, Aguet M, Weissmann C. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature. 1992;356:577–582. doi: 10.1038/356577a0. [DOI] [PubMed] [Google Scholar]
- Büeler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, Weissmann C. Mice devoid of PrP are resistant to scrapie. Cell. 1993;73:1339–1347. doi: 10.1016/0092-8674(93)90360-3. [DOI] [PubMed] [Google Scholar]
- Caughey BW, Dong A, Bhat KS, Ernst D, Hayes SF, Caughey WS. Secondary structure analysis of the scrapie-associated protein PrP 27-30 in water by infrared spectroscopy. Biochemistry. 1991;30:7672–7680. doi: 10.1021/bi00245a003. [DOI] [PubMed] [Google Scholar]
- Chabry J, Ratsimanohatra C, Sponne I, Elena PP, Vincent JP, Pillot T. In vivo and in vitro neurotoxicity of the human prion protein (PrP) fragment P118–135 independently of PrP expression. J Neurosci. 2003;23:462–469. doi: 10.1523/JNEUROSCI.23-02-00462.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiesa R, Piccardo P, Ghetti B, Harris DA. Neurological illness in transgenic mice expressing a prion protein with an insertional mutation. Neuron. 1998;21:1339–1351. doi: 10.1016/s0896-6273(00)80653-4. [DOI] [PubMed] [Google Scholar]
- Chiesa R, Fioriti L, Tagliavini F, Salmona M, Forloni G. Cytotoxicity of prion peptides. In: Lehmann S, Grassi P, editors. Methods and tools in biosciences and medicine: techniques in prion research. Basel: Birkhäuser Verlag; 2004. pp. 176–197. [Google Scholar]
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- Corsaro A, Thellung S, Villa V, Principe DR, Paludi D, Arena S, Millo E, Schettini D, Damonte G, Aceto A, Schettini G, Florio T. Prion protein fragment 106–126 induces a p38 MAP kinase-dependent apoptosis in SH-SY5Y neuroblastoma cells independently from the amyloid fibril formation. Ann NY Acad Sci. 2003;1010:610–622. doi: 10.1196/annals.1299.114. [DOI] [PubMed] [Google Scholar]
- Demuro A, Mina E, Kayed R, Milton SC, Parker I, Glabe CG. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J Biol Chem. 2005;280:17294–17300. doi: 10.1074/jbc.M500997200. [DOI] [PubMed] [Google Scholar]
- Fioriti L, Quaglio E, Massignan T, Colombo L, Stewart RS, Salmona M, Harris DA, Forloni G, Chiesa R. The neurotoxicity of prion protein (PrP) peptide 106–126 is independent of the expression level of PrP and is not mediated by abnormal PrP species. Mol Cell Neurosci. 2005;28:165–176. doi: 10.1016/j.mcn.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Florio T, Grimaldi M, Scorziello A, Salmona M, Bugiani O, Tagliavini F, Forloni G, Schettini G. Intracellular calcium rise through L-type calcium channels, as molecular mechanism for prion protein fragment 106–126-induced astroglial proliferation. Biochem Biophys Res Commun. 1996;228:397–405. doi: 10.1006/bbrc.1996.1673. [DOI] [PubMed] [Google Scholar]
- Forloni G, Angeretti N, Chiesa R, Monzani E, Salmona M, Bugiani O, Tagliavini F. Neurotoxicity of a prion protein fragment. Nature. 1993;362:543–546. doi: 10.1038/362543a0. [DOI] [PubMed] [Google Scholar]
- Forloni G, Del Bo R, Angeretti N, Chiesa R, Smiroldo S, Doni R, Ghibaudi E, Salmona M, Porro M, Verga L, Giaccone G, Bugiani O, Tagliavini F. A neurotoxic prion protein fragment induces rat astroglial proliferation and hypertrophy. Eur J Neurosci. 1994;6:1415–1422. doi: 10.1111/j.1460-9568.1994.tb01003.x. [DOI] [PubMed] [Google Scholar]
- Forloni G, Bugiani O, Tagliavini F, Salmona M. Apoptosis-mediated neurotoxicity induced by beta-amyloid and PrP fragments. Mol Chem Neuropathol. 1996a;28:163–171. doi: 10.1007/BF02815218. [DOI] [PubMed] [Google Scholar]
- Forloni G, Tagliavini F, Bugiani O, Salmona M. Amyloid in Alzheimer's disease and prion-related encephalopathies: studies with synthetic peptides. Prog Neurobiol. 1996b;49:287–315. doi: 10.1016/0301-0082(96)00013-5. [DOI] [PubMed] [Google Scholar]
- Forloni G, Lucca E, Angeretti N, Della Torre P, Salmona M. Amidation of beta-amyloid peptide strongly reduced the amyloidogenic activity without alteration of the neurotoxicity. J Neurochem. 1997;69:2048–2054. doi: 10.1046/j.1471-4159.1997.69052048.x. [DOI] [PubMed] [Google Scholar]
- Forloni G, Angeretti N, Malesani P, Peressini E, Rodriguez Martin T, Della Torre P, Salmona M. Influence of mutations associated with familial prion-related encephalopathies on biological effects of PrP peptides. Ann Neurol. 1999;45:489–494. doi: 10.1002/1531-8249(199904)45:4<489::aid-ana10>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Ghetti B, Piccardo P, Spillantini MG, Ichimiya Y, Porro M, Perini F, Kitamoto T, Tateishi J, Seiler C, Frangione B, Bugiani O, Giaccone G, Prelli F, Goedert M, Dlouhy SR, Tagliavini F. Vascular variant of prion protein cerebral amyloidosis with tau-positive neurofibrillary tangles: the phenotype of the stop codon 145 mutation in PRNP. Proc Natl Acad Sci USA. 1996;93:744–748. doi: 10.1073/pnas.93.2.744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaccone G, Verga L, Bugiani O, Frangione B, Serban D, Prusiner SB, Farlow MR, Ghetti B, Tagliavini F. Prion protein preamyloid and amyloid deposits in Gerstmann-Straussler-Scheinker disease, Indiana kindred. Proc Natl Acad Sci USA. 1992;89:9349–9353. doi: 10.1073/pnas.89.19.9349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobbi M, Colombo L, Morbin M, Mazzoleni G, Accardo E, Vanoni M, Del Favero E, Cantu L, Kirschner DA, Manzoni C, Beeg M, Ceci P, Ubezio P, Forloni G, Tagliavini F, Salmona M. Gerstmann-Straussler-Scheinker disease amyloid protein polymerizes according to the “dock-and-lock” model. J Biol Chem. 2006;281:843–849. doi: 10.1074/jbc.M506164200. [DOI] [PubMed] [Google Scholar]
- Hafiz FB, Brown DRA. Model for the mechanism of astrogliosis in prion disease. Mol Cell Neurosci. 2000;16:221–232. doi: 10.1006/mcne.2000.0868. [DOI] [PubMed] [Google Scholar]
- Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, Teplow DB, Selkoe DJ. Protofibrillar intermediates of amyloid β-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci. 1999;19:8876–8884. doi: 10.1523/JNEUROSCI.19-20-08876.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SM, Mouri A, Kokubo H, Nakajima R, Suemoto T, Higuchi M, Staufenbiel M, Noda Y, Yamaguchi H, Nabeshima T, Saido TC, Iwata N. Neprilysin-sensitive synapse-associated amyloid-beta peptide oligomers impair neuronal plasticity and cognitive function. J Biol Chem. 2006;281:17941–17951. doi: 10.1074/jbc.M601372200. [DOI] [PubMed] [Google Scholar]
- Jobling MF, Stewart LR, White AR, McLean C, Friedhuber A, Maher F, Beyreuther K, Masters CL, Barrow CJ, Collins SJ, Cappai R. The hydrophobic core sequence modulates the neurotoxic and secondary structure properties of the prion peptide 106–126. J Neurochem. 1999;73:1557–1565. doi: 10.1046/j.1471-4159.1999.0731557.x. [DOI] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- Mallucci G, Dickinson A, Linehan J, Klohn PC, Brandner S, Collinge J. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science. 2003;302:871–874. doi: 10.1126/science.1090187. [DOI] [PubMed] [Google Scholar]
- Muramoto T, Scott M, Cohen FE, Prusiner SB. Recombinant scrapie-like prion protein of 106 amino acids is soluble. Proc Natl Acad Sci USA. 1996;93:15457–15462. doi: 10.1073/pnas.93.26.15457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazor KE, Kuhn F, Seward T, Green M, Zwald D, Purro M, Schmid J, Biffiger K, Power AM, Oesch B, Raeber AJ, Telling GC. Immunodetection of disease-associated mutant PrP, which accelerates disease in GSS transgenic mice. EMBO J. 2005;24:2472–2480. doi: 10.1038/sj.emboj.7600717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning ZY, Zhao DM, Liu HX, Yang JM, Han CX, Cui YL, Meng LP, Wu CD, Liu ML, Zhang TX. Altered expression of the prion gene in rat astrocyte and neuron cultures treated with prion peptide 106–126. Cell Mol Neurobiol. 2005;25:1171–1183. doi: 10.1007/s10571-005-8357-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novitskaya V, Bocharova OV, Bronstein I, Baskakov IV. Amyloid fibrils of mammalian prion protein are highly toxic to cultured cells and primary neurons. J Biol Chem. 2006;281:13828–13836. doi: 10.1074/jbc.M511174200. [DOI] [PubMed] [Google Scholar]
- Pan KM, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, Mehlhorn I, Huang Z, Fletterick RJ, Cohen FE, Prusiner S. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci USA. 1993;90:10962–10966. doi: 10.1073/pnas.90.23.10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parchi P, Chen SG, Brown P, Zou W, Capellari S, Budka H, Hainfellner J, Reyes PF, Golden GT, Hauw JJ, Gajdusek DC, Gambetti P. Different patterns of truncated prion protein fragments correlate with distinct phenotypes in P102L Gerstmann-Straussler-Scheinker disease. Proc Natl Acad Sci USA. 1998;95:8322–8327. doi: 10.1073/pnas.95.14.8322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyrin JM, Lasmezas CI, Haik S, Tagliavini F, Salmona M, Williams A, Richie D, Deslys JP, Dormont D. Microglial cells respond to amyloidogenic PrP peptide by the production of inflammatory cytokines. NeuroReport. 1999;10:723–729. doi: 10.1097/00001756-199903170-00012. [DOI] [PubMed] [Google Scholar]
- Piccardo P, Seiler C, Dlouhy SR, Young K, Farlow MR, Prelli F, Frangione B, Bugiani O, Tagliavini F, Ghetti B. Proteinase-K-resistant prion protein isoforms in Gerstmann-Sträussler-Scheinker disease (Indiana kindred) J Neuropathol Exp Neurol. 1996;55:1157–1163. doi: 10.1097/00005072-199611000-00007. [DOI] [PubMed] [Google Scholar]
- Piccardo P, Dlouhy SR, Lievens PM, Young K, Bird TD, Nochlin D, Dickson DW, Vinters HV, Zimmerman TR, Mackenzie IR, Kish SJ, Ang LC, De Carli C, Pocchiari M, Brown P, Gibbs CJ, Jr, Gajdusek DC, Bugiani O, Ironside J, Tagliavini F, Ghetti B. Phenotypic variability of Gerstmann-Sträussler-Scheinker disease is associated with prion protein heterogeneity. J Neuropathol Exp Neurol. 1998;57:979–988. doi: 10.1097/00005072-199810000-00010. [DOI] [PubMed] [Google Scholar]
- Pillot T, Drouet B, Pincon-Raymond M, Vandekerckhove J, Rosseneu M, Chambaz JA. Nonfibrillar form of the fusogenic prion protein fragment [118–135] induces apoptotic cell death in rat cortical neurons. J Neurochem. 2000;75:2298–2308. doi: 10.1046/j.1471-4159.2000.0752298.x. [DOI] [PubMed] [Google Scholar]
- Prusiner SB. Molecular biology of prion diseases. Science. 1991;252:1515–1522. doi: 10.1126/science.1675487. [DOI] [PubMed] [Google Scholar]
- Rizzardini M, Chiesa R, Angeretti N, Lucca E, Salmona M, Forloni G, Cantoni L. Prion protein fragment 106–126 differentially induces heme oxygenase-1 mRNA in cultured neurons and astroglial cells. J Neurochem. 1997;68:715–720. doi: 10.1046/j.1471-4159.1997.68020715.x. [DOI] [PubMed] [Google Scholar]
- Rymer DL, Good TA. The role of prion peptide structure and aggregation in toxicity and membrane binding. J Neurochem. 2000;75:2536–2545. doi: 10.1046/j.1471-4159.2000.0752536.x. [DOI] [PubMed] [Google Scholar]
- Safar J, Roller PP, Gajdusek DC, Gibbs CJ., Jr Conformational transitions, dissociation, and unfolding of scrapie amyloid (prion) protein. J Biol Chem. 1993;268:20276–20284. [PubMed] [Google Scholar]
- Salmona M, Morbin M, Massignan T, Colombo L, Mazzoleni G, Capobianco R, Diomede L, Thaler F, Mollica L, Musco G, Kourie JJ, Bugiani O, Sharma D, Inouye H, Kirschner DA, Forloni G, Tagliavini F. Structural properties of Gerstmann-Straussler-Scheinker disease amyloid protein. J Biol Chem. 2003;278:48146–48153. doi: 10.1074/jbc.M307295200. [DOI] [PubMed] [Google Scholar]
- Supattapone S, Bosque P, Muramoto T, Wille H, Aagaard C, Peretz D, Nguyen HO, Heinrich C, Torchia M, Safar J, Cohen FE, DeArmond SJ, Prusiner SB, Scott M. Prion protein of 106 residues creates an artifical transmission barrier for prion replication in transgenic mice. Cell. 1999;96:869–878. doi: 10.1016/s0092-8674(00)80596-6. [DOI] [PubMed] [Google Scholar]
- Tagliavini F, Prelli F, Ghiso J, Bugiani O, Serban D, Prusiner SB, Farlow MR, Ghetti B, Frangione B. Amyloid protein of Gerstmann-Straussler-Scheinker disease (Indiana kindred) is an 11 kd fragment of prion protein with an N-terminal glycine at codon 58. EMBO J. 1991;10:513–519. doi: 10.1002/j.1460-2075.1991.tb07977.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliavini F, Prelli F, Porro M, Rossi G, Giaccone G, Farlow MR, Dlouhy SR, Ghetti B, Bugiani O, Frangione BA. Amyloid fibrils in Gerstmann-Straussler-Scheinker disease (Indiana and Swedish kindreds) express only PrP peptides encoded by the mutant allele. Cell. 1994;79:695–703. doi: 10.1016/0092-8674(94)90554-1. [DOI] [PubMed] [Google Scholar]
- Tagliavini F, Lievens PM, Tranchant C, Warter JM, Mohr M, Giaccone G, Perini F, Rossi G, Salmona M, Piccardo P, Ghetti B, Beavis RC, Bugiani O, Frangione B, Prelli F. A 7-kDa prion protein (PrP) fragment, an integral component of the PrP region required for infectivity, is the major amyloid protein in Gerstmann-Straussler-Scheinker disease A117V. J Biol Chem. 2001;276:6009–6015. doi: 10.1074/jbc.M007062200. [DOI] [PubMed] [Google Scholar]
- Thellung S, Florio T, Corsaro A, Arena S, Merlino M, Salmona M, Tagliavini F, Bugiani O, Forloni G, Schettini G. Intracellular mechanisms mediating the neuronal death and astrogliosis induced by the prion protein fragment 106–126. Int J Dev Neurosci. 2000;18:481–492. doi: 10.1016/s0736-5748(00)00005-8. [DOI] [PubMed] [Google Scholar]
- Townsend M, Shankar GM, Mehta T, Walsh DM, Selkoe DJ. Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: a potent role for trimers. J Physiol (Lond) 2006;572:477–492. doi: 10.1113/jphysiol.2005.103754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will RG, Ironside JW, Zeidler M, Cousens SN, Estibeiro K, Alperovitch A, Poser S, Pocchiari M, Hofman A, Smith PG. A new variant of Creutzfeldt-Jakob disease in the UK. Lancet. 1996;347:921–925. doi: 10.1016/s0140-6736(96)91412-9. [DOI] [PubMed] [Google Scholar]