

Abstract
Overexpression of the major myelin protein of the CNS, proteolipid protein (PLP), leads to late-onset degeneration of myelin and pathological changes in axons. Based on the observation that in white matter tracts of these mutants both CD8+ T-lymphocytes and CD11b+ macrophage-like cells are numerically elevated, we tested the hypothesis that these cells are pathologically involved in the primarily genetically caused neuropathy. Using flow cytometry of mutant brains, CD8+ cells could be identified as activated effector cells, and confocal microscopy revealed a close association of the T-cells with MHC-I+ (major histocompatibility complex class I positive) oligodendrocytes. Crossbreeding the myelin mutants with mice deficient in the recombination activating gene-1 (RAG-1) lacking mature T- and B-lymphocytes led to a reduction of the number of CD11b+ cells and to a substantial alleviation of pathological changes. In accordance with these findings, magnetic resonance imaging revealed less ventricular enlargement in the double mutants, partially because of more preserved corpora callosa. To investigate the role of CD8+ versus CD4+ T-lymphocytes, we reconstituted the myelin-RAG-1 double mutants with bone marrow from either CD8-negative (CD4+) or CD4-negative (CD8+) mice. The severe ventricular enlargement was only found when the double mutants were reconstituted with bone marrow from CD8+ mice, suggesting that the CD8+ lymphocytes play a critical role in the immune-related component of myelin degeneration in the mutants. These findings provide strong evidence that a primary glial damage can cause secondary immune reactions of pathological significance as it has been suggested for some forms of multiple sclerosis and other leukodystrophies.
Keywords: microglia, macrophages, T-lymphocytes, leukodystrophy, multiple sclerosis, animal model, inflammation
Introduction
Formed by Schwann cells in the peripheral nervous system (PNS) and by oligodendrocytes in the CNS, the myelin sheath is an important cellular component that mediates the correct function of axons and thus of the nervous system in general. It is well established that one function of the myelin sheath is to accelerate conductance by allowing saltatory propagation of action potentials. Furthermore, the myelin sheath plays important roles in regulating axon diameters and influencing axonal transport (de Waegh et al., 1992; Kirkpatrick and Brady, 1994; Martini, 2001; Lappe-Siefke et al., 2003; Edgar et al., 2004; Gould and Brady, 2004). Consequently, degeneration or malformation of myelin causes or at least facilitates axonal dysfunction or even axonal loss (Griffiths et al., 1998; Bjartmar et al., 1999; Wrabetz et al., 2000; Yin et al., 2000; Martini, 2001; Lappe-Siefke et al., 2003; Samsam et al., 2003; Edgar and Garbern, 2004).
The possible causes for myelin destruction are manyfold and imply metabolic disorders, intoxication, bacterial and viral infections, autoimmunity against myelin components, and mutations in myelin-related genes. A primary inflammatory attack to the oligodendrocytic myelin sheath is one of the most frequent causes for myelin loss in the CNS, leading to multiple sclerosis (MS) and related inflammatory disorders of the CNS (Lucchinetti et al., 2000; Bruck and Stadelmann, 2003; Lassmann, 2004, 2005). However, recent studies in biopsies and autopsies suggest the occurrence of subforms of MS that are primarily related to glial damage that secondarily rises an inflammatory reaction of putative pathogenetic relevance (Lucchinetti et al., 2000; Barnett and Prineas, 2004; Warshawsky et al., 2005).
In the present study, we tested the hypothesis that an intrinsic oligodendroglial abnormality could cause a pathologically relevant inflammation in the CNS. As a model for an intrinsic glial damage, we investigated a mouse mutant that mildly overexpresses the myelin component proteolipid protein (PLP) in oligodendrocytes causing a late-onset and slowly progressing demyelination that is associated with axonopathic changes (Anderson et al., 1998). The concept that inherited abnormalities in glial cells cause secondary inflammation with pathogenetic relevance has recently also been tested in the peripheral nervous system. Both in mice heterozygously deficient in P0 or homo/hemizygously deficient in Cx32 (GJB1), we could demonstrate that immune cells foster the degeneration of myelin (Schmid et al., 2000; Mäurer et al., 2001; Kobsar et al., 2003; Ip et al., 2006). Here, we show that immune cells contribute to the primarily genetically caused demyelination in PLP-overexpressing mice, confirming the concept that primary glial damage can lead to pathogenetically relevant inflammation in the CNS.
Materials and Methods
Animals and determination of genotypes.
PLP transgenic (PLP-tg) mice of the line 66 (Readhead et al., 1994) have been backcrossed to a C57BL/6 genetic background and mated with C57BL/6 wild-type (wt) mice to obtain heterozygous offsprings. These were investigated at 2, 4, 8, and 12 months of age. Additionally, transgenic PLP mutants were crossbred with recombination activating gene-1 (RAG-1)-deficient null mutants (also on a C57BL/6 genetic background) in analogy to previous experiments (Schmid et al., 2000; Kobsar et al., 2003).
CD4−/− (Rahemtulla et al., 1991) and CD8−/− (Fung-Leung et al., 1991) knock-out mice (on a C57BL/6 background) were purchased from The Jackson Laboratory (Bar Harbor, ME) and kept in our animal facility under specific pathogen-free conditions.
Presence of the autosomal plp transgene was displayed by a PCR amplification product obtained with a primer at the 3′ end of the PLP gene (5′-CAGGTGTTGAGTCTGATCTACACAAG-3′) and a primer T7cos (5′-GCATAATACGACTCACTATAGGGATC-3′) directed against the T7 promoter of the PLP cosmid cos901. The PCR was performed in a volume of 25 μl following a standard protocol with an annealing temperature of 55°C and an elongation time of 45 s at 72°C.
Determination of the RAG-1 status and of the presence of CD4+ and CD8+ cells in bone marrow-reconstituted RAG-1−/− mice has been performed by flow cytometry analysis on tail vein blood using CD4- and CD8-specific antibodies (BD Biosciences, Heidelberg, Germany) as described previously (Schmid et al., 2000; Kobsar et al., 2003).
Tissue preparation and immunohistochemistry.
For the identification of macrophage-like cells, mice were transcardially perfused with 0.1 m cacodylate buffer containing 4% paraformaldehyde. The brain, spinal cord, and the optic nerves were dissected and postfixed in the same fixative for 2 h and cryoprotected in 30% sucrose overnight. For T- and B-lymphocyte staining (CD45R/B220, see below) the animals were perfused with 0.1 m PBS only. After freezing in liquid nitrogen-cooled isopentane, 10-μm-thick coronal sections of the corpus callosum, transverse sections of the spinal cord, sagittal sections of the cerebellum, and longitudinal sections of the optic nerve were cut. Additionally, when the antibody PAX-5/L26 was applied for another B-cell staining (see below), 7-μm-thick paraffin sections of fixed tissue (see above) were used.
Sections were preincubated for 30 min in 5% normal bovine serum in 0.1 m PBS and then incubated in one of the following primary antibodies diluted in 1% normal bovine serum (overnight at 4°C). Rat anti-mouse CD11b (Serotec, Oxford, UK) was used for detection of activated macrophages; rat anti-mouse CD4 (Serotec) and rat anti-mouse CD8 (Chemicon, Temecula, CA) antibodies were used for the identification of T-lymphocytes. For detection of B-lymphocytes, rat anti-mouse CD45R/B220 (BD Biosciences, San Jose, CA) and mouse anti-mouse PAX-5/L26 (Biocare Medical, Walnut Creek, CA) were used. Monoclonal anti-mouse 2′,3′-cyclic nucleotide 3′ phosphodiesterase (CNPase; Sigma, St. Louis, MO) was used for oligodendrocytic myelin detection. To optimize immunolabeling for CD11b, sections were permeabilized with 0.3% Triton X-100. To visualize primary antibodies, a biotinylated secondary antibody to rat Igs (macrophage, T-cell antibodies, CD45R/B220) or to mouse Igs (PAX-5/L26, CNPase) was applied for 1 h, followed by avidin/biotin reagent (Dako, High Wycombe, UK) before incubation and staining with diaminobenzidine-HCl (DAB) and H2O2.
CD11b/myelin basic protein (MBP) double staining to identify phagocytosing macrophages was performed on 10 μm optic nerve cross sections at the region of 1300 μm caudal to the pigment epithelium of the eye bulb. Activated macrophages were stained with CD11b (Serotec) as described above by DAB reaction followed by fluorescence staining with rabbit anti-mouse MBP antibody (Massachusetts Biologic Laboratory, Woburn, MA) and cyanine 3 (Cy3)-labeled secondary goat anti-rabbit antibody (Dianova, Hamburg, Germany).
For CD8/CD3 double-immunofluorescence staining, rat anti-mouse CD8 (Chemicon) and rabbit anti-mouse CD3 (AB16044; Abcam, Cambridge, UK) was applied on 10-μm-thick acetone-treated fresh-frozen sections overnight at 4°C, followed by incubation with fluorescence-labeled secondary antibodies [goat anti-rat Alexa Fluor 488 (Invitrogen, Karlsruhe, Germany) and goat anti-rabbit Cy3 (Dianova)] for 1 h at room temperature.
For CD3/major histocompatibility complex class I (MHC-I)/pan-neurofilament and CD3/MHC-I/myelin protein triple-immunofluorescence staining rabbit anti-mouse CD3 (AB16044; Abcam), rat anti-mouse MHC-I (ER-HR 52; Dianova), and either mouse anti-mouse pan-neurofilament (SMI-311; Abcam) or a mouse antibody against myelin protein [CNPase (Sigma), myelin-associated glycoprotein (MAG; Chemicon), or PLP (MCA 839G; Serotec)] was applied on 10-μm-thick acetone-treated fresh-frozen sections overnight at 4°C, followed by incubation with fluorescence-labeled secondary antibodies [goat anti-rat Alexa Fluor 350 (Invitrogen), goat anti-mouse Cy2 (Dianova), and goat anti-rabbit Cy3 (Dianova)] for 1 h at room temperature.
The specificity of the immunoreaction was assessed by omission of the primary or secondary antibodies.
Tissue preservation for light and electron microscopy.
Optic nerves were processed for light and electron microscopy as reported recently for the PNS (Carenini et al., 2001). The mice were transcardially perfused using 4% paraformaldehyde and 2% glutaraldehyde in 0.1 m cacodylate buffer. The nerves stayed in the same fixative overnight, followed by osmification and embedding in Spurr’s medium. For light microscopic analysis, 0.5-μm-thick semithin sections were stained with alkaline methylene blue. Investigation of the pathological features was performed by blinded investigators (R.M. and I.K.) who were not aware of the RAG-1 immune status.
For electron microscopy, ultrathin sections (70 nm thick) were counterstained with lead citrate and investigated using a Proscan (Lagerlechfeld, Germany) slow-scan camera attached to the Zeiss (Oberkochen, Germany) EM 10CR with the corresponding software analySIS 3.0 Doku (Soft Imaging System, Muenster, Germany). For quantitative analysis, two wild-type mice, four PLP-tg RAG-1+/? and four PLP-tg RAG-1−/− mice were examined.
For each optic nerve cross section, a series of 70–90 electron micrographs were taken (at 2500×) extending from one border of the nerve through its center and from there to the opposite confinements of the optic nerve. On the resulting composed micrograph covering 6–8% (the optic nerve contains 80,000 axons in average, thus 4800–6400 axons per nerve were quantified) of the whole optic nerve cross-section area, myelinated and degenerated axons were marked and counted manually by using standard unbiased counting rules.
Quantification of immune cells in the CNS.
The corpus callosum, the white matter fiber tracts of the cerebellum, white and gray matter of the spinal cord (at level L1–L2), and the optic nerve of heterozygous PLP-tg and wild-type mice of 2, 4, 8, and 12 months of age were examined. Quantification of the number of macrophage-like cells was performed with a light microscope (BH2; Olympus, Hamburg, Germany) using an ocular grid covering a defined area (0.0361 mm2). For evaluation of the corpus callosum, white matter fiber tracts of the cerebellum, spinal cord white and gray matter a final magnification of 525× were chosen. For each animal, four to six sections in the optic nerve and 6–10 sections in the other regions were investigated. Within each section, measurement ranged between 5 and 12 ocular grids per region. To measure the cell density of macrophage-like cells in the optic nerve, we selected a final magnification of 600× using the ocular grid (0.0256 mm2). Two defined regions were chosen for quantification, namely the rostral region, covering the rostral 800–1500 μm of the optic nerve, and the remaining caudal area. The cell number of macrophage-like cells was then calculated per square millimeter.
Because of the low number of T-cells, the quantification was performed on a Zeiss Axiophot2 microscope using digital images acquired via a CCD camera (Visitron Systems, Tuchheim, Germany) and ImagePro 4.0 software (Media Cybernetics, Silver Spring, MD) at a final magnification of 300× measuring 8–10 consecutive sections. The entire region of the cerebellum, the spinal cord and the optic nerve, and a defined region of the corpus callosum were investigated for T-cell number, and the cell density was then calculated per square millimeter.
Confocal microscopy was performed using the laser-scanning unit TCS SP2 mounted to a DM RE-7 SDK microscope (both from Leica Microsystems, Wetzlar, Germany). The Leica Confocal Software 2.61 was applied.
Flow cytometry.
Extraction of CNS-invading lymphocytes was performed as described previously (Magnus et al., 2005). Briefly, 12-month-old wt and PLP-tg mice were killed with CO2, spinal cords were flushed out of the spinal column with sterile PBS, and the brain was prepared and used in toto. CNS tissue was homogenized and strained through a 70 μm nylon filter (BD Biosciences). After centrifugation, the cell pellet was resuspended in 9 ml of Percoll and mixed with 20 ml of PBS. The gradient was centrifuged at 600 × g for 25 min at room temperature (21–23°C). The interphase cells were collected and washed before staining. For flow cytometry, the cells were stained with the following directly labeled antibodies for 30 min at 4°C: CD4-PerCP (peridiniumchlorophyll-protein), CD8-PE (R-phycoerythrin), CD62L-APC (allophycocy), CD44-FITC, and CD69-FITC (BD Biosciences). Cells were washed and analyzed using a FACSCalibur with CellQuest software (both from BD Biosciences). Specific staining was assessed by applying accordant isotype control antibodies.
Transplantation of bone marrow cells into PLP/RAG-1 double mutants.
Bone marrow transplantation from wild-type, CD4−/−, or CD8−/− donor mice into PLP/RAG-1−/− mutants (hosts) has been performed in analogy to previous experiments in P0+/−/RAG-1−/− mutants (Mäurer et al., 2001). Control experiments were performed by transplanting wild-type bone marrow into PLP-wt mice. In brief, bone marrow from ∼8-week-old donors was isolated according to established protocols (Mueller et al., 2001), and ∼2 × 107 cells were injected intravenously in a volume of 500 μl into 8-week-old PLP/RAG-1−/− mutants (or into PLP-wt mice as controls). Hosts were allowed to survive for 4 months. To control the degree of bone marrow chimerism, blood samples were analyzed by fluorescence-assisted cell sorting using CD4 and CD8 antibodies. Hosts reconstituted with wild-type bone marrow expressed both antigens, and hosts reconstituted with CD4−/− bone marrow were CD4-negative and CD8-positive and vice versa.
Magnetic resonance imaging.
Magnetic resonance (MR) measurements were performed on a clinical 1.5 T unit (Magnetom Vision; Siemens, Erlangen, Germany). For all MR imaging (MRI) scans, animals were lying in a supine position with their heads fixed in a custom-made dual-channel surface coil specially designed for investigations of small rodent brains (A063HACG; Rapid Biomedical, Würzburg, Germany). Animals were anesthetized by inhalation narcosis with enflurane in a 2:1 N2O:O2 atmosphere via a face mask. The MR protocol included a T2-w turbo spin echo sequence [repetition time (TR), 2500 ms; echo time (TE), 80 ms; slice thickness, 2 mm] and a high-resolution three-dimensional constructed interference in steady state (CISS) sequence (TR, 16.4 ms; TE, 8.2 ms; slice thickness, 1 mm) in the coronal plane.
Image analysis was performed by an investigator (M.B.) blinded to the age of animals and genotype. On a four-step scale, ventricular size was assessed on CISS images (see Fig. 8A–D) from one (normal ventricular size) (see Fig. 8A) to four (massive ventricular enlargement) (see Fig. 8D).
Figure 8.
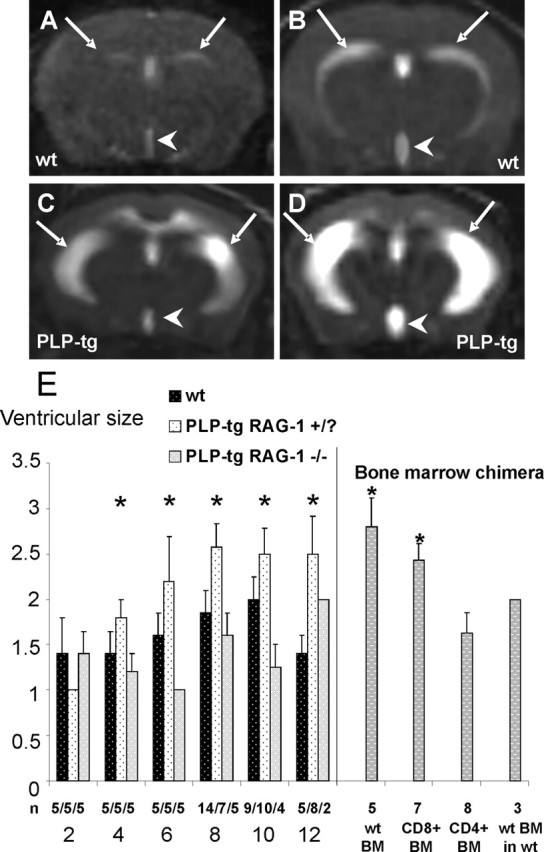
Ventricular sizes in wt mice and PLP mutants. A–D, MRI scans of brains from wt mice (A, B) and from PLP mutants (C, D). Note the abnormally extended ventricular sizes of the mutants, with sizes 3 (C) and 4 (D). Arrows, Lateral ventricles; arrowheads, third ventricle. E, Ventricular sizes of wt mice (black bars), of immune-competent PLP mutants (PLP-tg/RAG-1+/?; stippled white bars), and of immune-deficient PLP mutants (PLP-tg RAG-1−/−; gray bars) at 2–12 months of age. Ventricular sizes of immune-deficient PLP mutants reconstituted with bone marrow (BM) from wt, CD8+ (CD4−/−), and CD4+ (CD8−/−) mice are also indicated at 6 months of age. Note the enlarged ventricular sizes exceeding the normal size 2 of PLP mutants with intact immune systems and of immune-deficient PLP mutants reconstituted with bone marrow from wild-type mice or CD8+ (CD4−/−) mutants but not when reconstituted with bone marrow from CD4+ (CD8−/−) mutants. As a control experiment, reconstitution with wt BM (comprising CD8+/CD4+ stem cells) into wt mice also fails to increase ventricular sizes (score of 2 in 3 of 3 animals). Error bars indicate mean values + SEMs. The asterisks on the left side indicate a significant enlargement of ventricles in the PLP mutants with intact immune system (RAG-1+/?) with increasing age (p < 0.05, Kruskal–Wallis test), whereas asterisks on the right side (bone marrow chimera) indicate significant enlargement of ventricles [p < 0.05 compared with CD4+ (CD8−/−)] in bone marrow reconstituted immune-deficient mutants.
All animal experiments were approved by the local authorities at the Regierung von Unterfranken, Würzburg, Germany.
Statistical analysis.
Statistical analysis was performed by using the unpaired two-tailed Student’s t test for comparison of quantified profiles (immune cells, vacuoles). MRI scores were analyzed by using the nonparametric Mann–Whitney U test and the Kruskal–Wallis test.
Results
To investigate the putative impact of immune-related cells in CNS tissue of PLP-tg mice, we investigated the number of CD11b+ macrophages, B-lymphocytes, CD4+, and CD8+ T-lymphocytes in corpus callosum, cerebellum, in white and gray matter of the spinal cord, and in the optic nerve.
T-lymphocytes in the CNS of PLP transgenic mice express CD8, increase numerically with age, are directly attached to MHC-I+ oligodendrocytes, and carry markers of lymphocytic effector cells
In all parts of the CNS and at any age investigated, the number of B- and CD4+ T-lymphocytes was very low, both in wild-type mice and PLP transgenic mice. The very low numbers of CD4+ T-lymphocytes and B-cells in neural tissues of the PLP mutants was considered relevant because positive control stainings in sections from spleens revealed a strong reactivity of the respective antibodies (see Materials and Methods). In contrast, the number of CD8+ cells was low in wt mice but markedly upregulated in adult PLP mutants (Fig. 1A). At 2 months of age, none of the investigated CNS regions showed a significant difference comparing wild-type with mutant mice. After 4 months, a statistically significant increase of CD8+ cells was observed in the spinal cord gray matter and the optic nerve of the PLP mutants and a nonsignificant trend toward higher cell numbers was seen in the corpus callosum (Fig. 1A). At 8 and 12 months of age, the other examined regions (corpus callosum, cerebellum, and white matter of the spinal cord) of the PLP mutants also showed a significant increase in the number of CD8+ cells (Fig. 1A).
Figure 1.
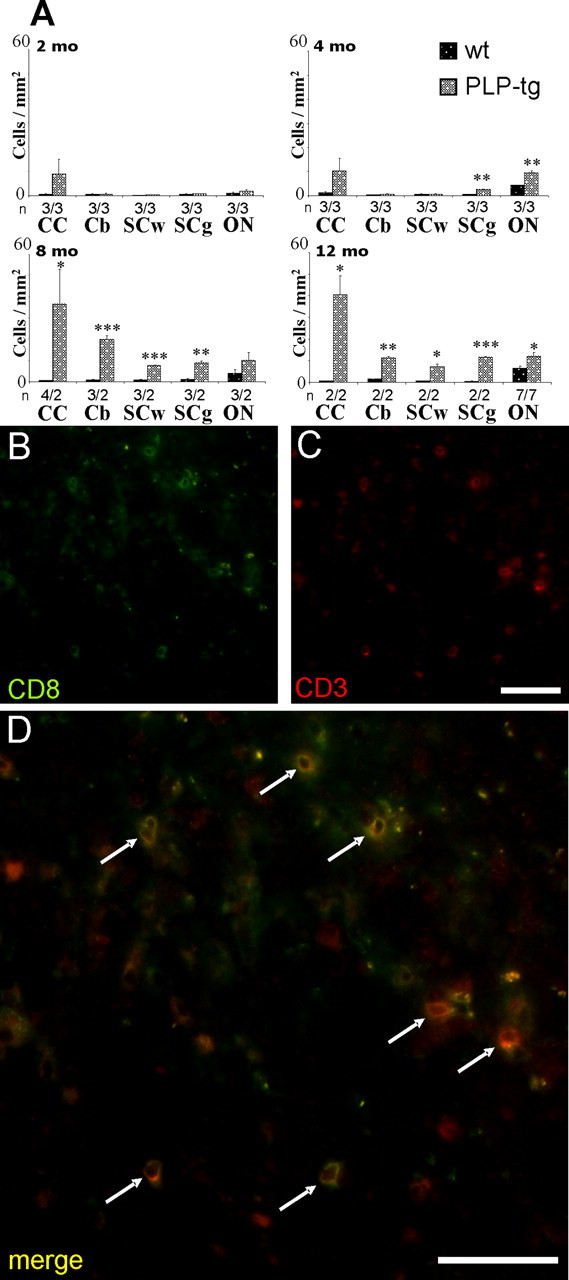
Elevated numbers of CD8+ T-lymphocytes in the CNS of PLP mutants. A, Quantification of CD8+ T-lymphocytes in corpus callosum (CC), cerebellum (Cb), white matter of the spinal cord (SCw), and gray matter of the spinal cord (SCg) and in the optic nerve (ON) of wt and PLP mutants (PLP-tg) at 2, 4, 8, and 12 months (mo) of age. Note age-dependent increase of the T-lymphocytes in the different regions of the CNS of the mutants. Mean values + SEM are indicated as well as the number of individuals investigated (n). Asterisks indicate statistical significance between values from the mutants compared with the values from wild-type mice: ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. B–D, Double-immunofluorescence staining for CD8+ cells (B, green color) and CD3+ T-lymphocytes (C, red color) in the corpus callosum of a PLP-tg mouse at 12 months of age. Note that all CD8+ cells are also CD3+, proving that the infiltrating CD8+ cells are T-lymphocytes (D, arrows). Scale bar, 50 μm.
To prove that the identified CD8+ cells were T-lymphocytes rather than other CD8+ cells (i.e., antigen-presenting cells or natural killer cells), we performed double-immunofluorescence stainings using CD8 antibodies and antibodies against the pan-T-lymphocytic marker CD3 (Fig. 1B,C). Nearly all CD8+ cells were CD3+, indicating that the CD8+ cells are indeed T-lymphocytes (Fig. 1D).
Having seen that nearly all CD8+ profiles are CD3+ T-lymphocytes, we investigated MHC-I-expressing structures in the mutant CNS that could be the corresponding targets for the identified CD8+ T cells. For this purpose, we performed immunofluorescence triple stainings with antibodies against CD3 (Fig. 2A), MHC-I (Fig. 2B), antibodies recognizing all neurofilament isoforms (Fig. 2C), or established myelin markers (anti-CNPase, anti-PLP, anti-MAG) (data not shown). A marked increase of MHC-I expression in the white matter of PLP mutants was observed (Fig. 2B), whereas substantial MHC-I expression was not seen in wt animals (data not shown). Strikingly, by confocal microscopy, ∼25–30% of the CD3+ T-lymphocytes were closely attached to the MHC-I-positive profiles (Fig. 2C), suggesting that CD3/CD8+ T-cells attack MHC-I+ structures. Confocal microscopic analysis clearly identified the MHC-I+ structures as being negative for all neurofilament isoforms (Fig. 2C) but expressing the above mentioned myelin markers. Thus, CD8+ T-lymphocytes may target mutant MHC-I+ oligodendrocytes rather than axons.
Figure 2.

CD3+ T-lymphocytes directly contact MHC-I+ profiles that resemble oligodendrocytic cell somata. Confocal microscopic images of triple-immunohistochemical stainings for CD3 (A), MHC-I (B), and pan-neurofilament (C, green in merged image). Note the direct contact of MHC-I+ profiles with CD3+ T-lymphocytes (C, arrowheads). The MHC-I+ profiles are not identical with neurofilament+ structures (C, arrows) but are closely associated with them being highly reminiscent of oligodendrocytic cell somata. Scale bar, 10 μm.
To substantiate our histological characterization of the CD8+ T-lymphocytes by an independent technique, we performed repeated flow cytometric analysis of cerebral lymphocytes in 12-month-old PLP mutants and assessed phenotypic markers of T-cell subset attribution and effector function in direct comparison with splenic lymphocytes (Fig. 3). In the brain of PLP mutants, ∼86% of the T-lymphocytes were CD8+/CD44+/CD62L− effector T-lymphocytes in contrast to only 32% in the spleen (Fig. 3A). In contrast, in the spleen, 23% of CD8+ cells expressed an immature phenotype as revealed by simultaneous CD62L positivity and CD44 negativity, and a substantial population (40%) were identified as memory T-cells (CD44+/CD62L+). In contrast, basically no CD62L+ cells were detectable in the brains of mutant mice.
Figure 3.
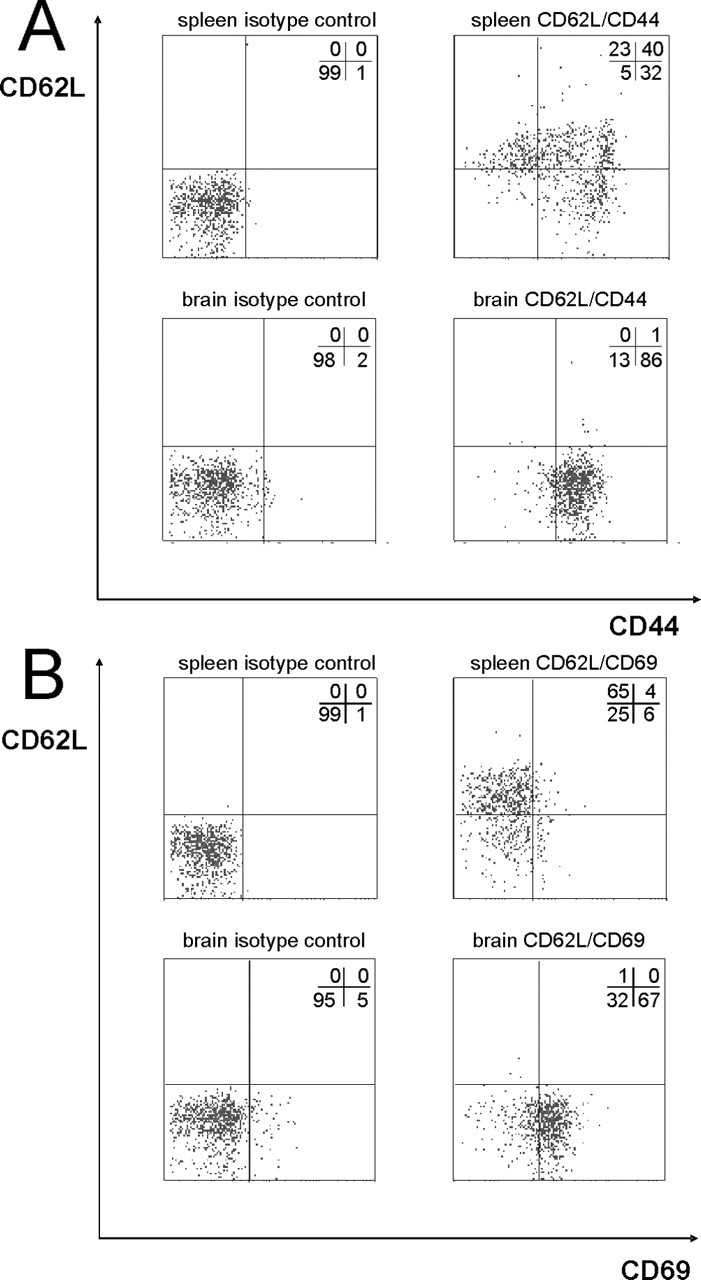
Flow cytometry of spleen and brain lymphocytes from PLP-tg mice. Spleen and brain lymphocytes were stained for CD8, CD62L and either CD44 (A) or CD69 (B). Only CD8-positive cells were gated and analyzed for CD44 (effector cell marker), CD69 (effector cell marker), and CD62L (marker for immature lymphocytes in this context) expression. Left blots, Isotype control. Right blots, CD44, CD69, and CD62L staining. Percentages of cells per quadrant are provided in each blot.
By using CD69 as another activation marker, we could confirm these results (Fig. 3B), showing that the cerebral CD8+ T-lymphocytes within the PLP mutant brain exhibit an effector T-cell phenotype. Thus, because of the different markers applied, the vast majority of CD8+ T-lymphocytes in the mutant brain are mature effector cells carrying markers of activation.
Macrophage-like cells express the activation marker CD11b, increase with age, and are spatially associated with demyelinated profiles
CD11b+ cells comprising macrophages and microglial cells (in the following designated as macrophages or macrophage-like cells) were always detectable both in wild-type and transgenic PLP mutants (Fig. 4). In 2-month-old transgenic mice, there were no differences with regard to the number of macrophage-like cells in all CNS structures investigated, whereas at 4 months, there was a statistically significant increase of macrophage numbers in the rostral part of the optic nerve (see below) and a nonsignificant tendency of elevation of macrophage numbers in the other regions of interest. The first statistically significant difference in addition to the optic nerve was seen in gray matter of spinal cord of 8–month-old PLP mutants compared with wild-type littermates. At 12 months of age, macrophage-like cells showed a statistically significant increase in all CNS tissues investigated, with the highest number of cells in the optic nerve. In contrast, in wild-type mice the number of macrophage-like cells remained at a similar basis level in all CNS regions examined, except in the optic nerve, in which we observed a steady increase of CD11b+ macrophage-like cells during aging, but at a degree lower than that in the mutants, with a maximum elevation at 12 months.
Figure 4.

Elevated numbers of macrophage-like cells in the CNS of PLP mutants. Quantification of CD11b+ macrophages in corpus callosum (CC), cerebellum (Cb), white matter of the spinal cord (SCw), and gray matter of the spinal cord (SCg) and in the optic nerve (ON) of wt and PLP mutants (PLP-tg) at 2, 4, 8, and 12 months (mo) of age. Note the age-dependent increase of the macrophages in the different regions of the CNS of the mutants. Mean values + SEM are indicated as well as the number of individuals investigated (n). Asterisks indicate statistical significance between values from the mutants in comparison to the values from wild-type mice: ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
To investigate the pathological changes in the mutants, we selected the optic nerve. This is a particularly interesting structure because of the clear subdivision into its unmyelinated part next to the retina, called lamina cribrosa or optic nerve head, and its myelinated region extending to the optic chiasm (Fig. 5A). Furthermore, in the optic nerve head, vascularization is unusual in that the vessels are surrounded by an extended perivascular space and the blood–brain barrier is leaky (Wolburg and Bäuerle, 1993). Macrophage staining using CD11b antibodies showed a homogeneous distribution of labeled cells along the optic nerve of the wild-type mice, as revealed by longitudinal frozen sections (Fig. 5B). In the mutants, in which the unmyelinated part of the optic nerve was diffusely confined (Fig. 5C), macrophages were more intensely stained than in the wild type and were more frequent, with the exception of the optic nerve head, in which only few CD11b+ cells were detectable at all ages investigated. Interestingly, there was a nonuniform distribution of macrophages in that CD11b+ cells were more frequent within the first 800–1500 μm of the rostral part of the myelinated compartment of the optic nerve (Fig. 5D). Based on the investigation of serial cross sections, this region corresponds to a particularly highly vascularized compartment of the optic nerve (B. Kohl, C. W. Ip, and R. Martini, unpublished observations), which has already been described in previous anatomical reports (Hayreh, 2001). In the remaining, more caudal part of the myelinated region of the optic nerve, there was an elevated number of macrophage-like cells compared with wild-type mice, yet at a lower level than in the rostral part (Fig. 5D).
Figure 5.
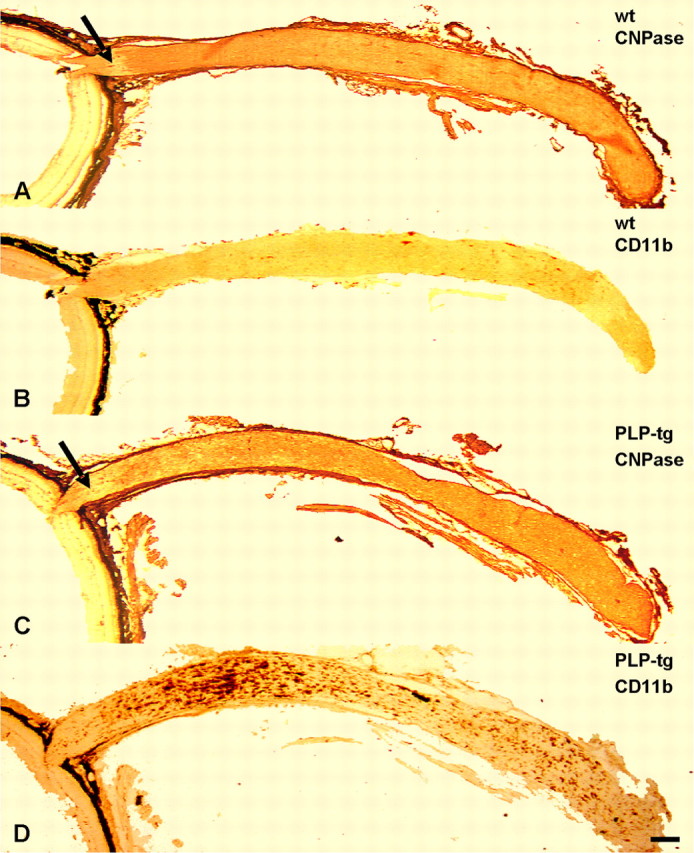
Macrophage-like cells in the optic nerves of wild-type and PLP mutants. A–D, Longitudinal, consecutive frozen sections of retinas (left) and optic nerves from wt mice (A, B) and PLP mutants (PLP-tg; C, D) immunolabeled with antibodies to CNPase (A, C) and with antibodies to the macrophage-specific marker CD11b (B, D). Note that particularly in the rostral part of the mutant optic nerve, there is a substantially increased number of CD11b+ cells compared with the optic nerves of the wild-type mice. In this region, myelin damage is particularly prominent, as reflected by a more patchy CNPase labeling. Arrows in A and C mark the unmyelinated part of the optic nerve, which is clearly confined by myelin labeling in the wild-type mice but diffusely in the mutants. Labeling of meninges and connective tissue of the eye cup in A and C is attributable to unspecific binding of the corresponding secondary antibody. The black appearance associated with the eye cup in all micrographs corresponds to the pigment epithelium. Scale bar, 200 μm.
In the rostral regions of the optic nerve with high numbers of macrophage-like cells, there was significantly reduced CNPase labeling (Fig. 5C). Light microscopic studies on semithin sections of this region revealed a demyelinating phenotype and, additionally, the occurrence of more or less large “holes” in the tissue (Fig. 6A, left panel). Electron microscopy in the same regions revealed myelinopathic changes, such as redundant myelin loops and demyelinated axons (Fig. 7A). The “holes” recognized in semithin sections could be identified as periaxonal vacuoles, reflecting axonopathic changes (Fig. 7A,C). Macrophage-like cells laden with myelin debris were frequently in close vicinity to these periaxonal vacuoles (Fig. 7C). These features were significantly more frequent in the rostral versus the caudal part of the optic nerve (Fig. 7A,B), thus showing a correlation of the number of activated macrophage-like cells with histopathological features.
Figure 6.
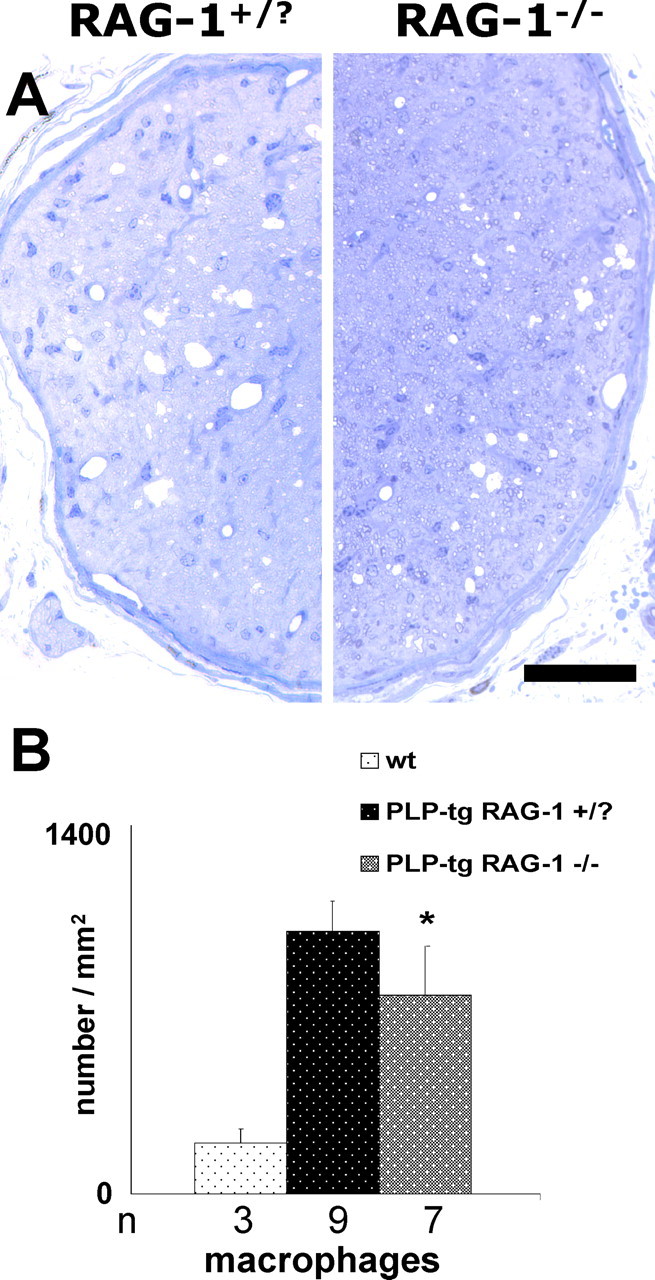
Histopathological changes in optic nerves of PLP mutants. Semithin sections of optic nerves (A) of immune-competent (RAG-1+/?) and of immune-deficient PLP mutants (RAG-1−/−) and quantification of macrophages (B) in optic nerves of wt, PLP-tg RAG-1+/?, and PLP-tg RAG-1−/− mutants at 12 months of age. Note the reduced number of periaxonal vacuoles (A) and macrophages (B) in RAG-1-deficient PLP mutants. ∗ p < 0.05. n, Number of individuals investigated. Scale bar, 50 μm.
Figure 7.
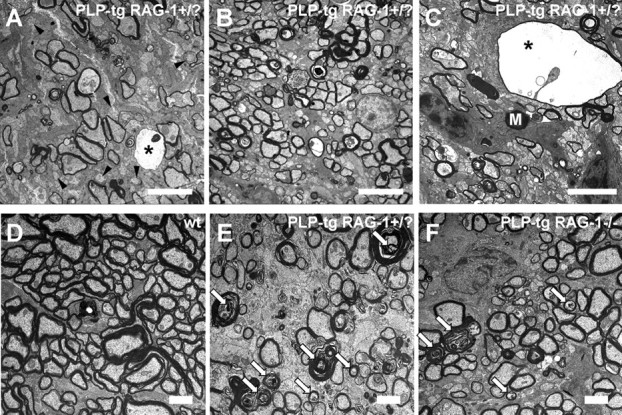
Pathological changes in optic nerves of PLP mutants. A–C, Electron microscopy of mutant optic nerves in the rostral (A, C) and in the caudal region (B). Note that demyelination and axonopathic changes are more pronounced in the rostral region, as reflected by many demyelinated axons (A, arrowheads), thin myelin, and periaxonal vacuoles (asterisks). C, Periaxonal vacuoles often contain the remnants of degenerating axons and are often in close vicinity of macrophage-like cells containing myelin debris (M). D–F, Electron microscopy of representative wt (D), PLP-tg RAG-1+/? (E), and PLP-tg RAG-1−/− (F) optic nerves from the rostral region. PLP-tg RAG-1−/− mice (F) show fewer demyelinating/axonopathic profiles (white arrows) than PLP-tg RAG-1+/? mutants (E). Wild-type optic nerves (D) do not show pathological changes. Scale bars: A–C, 5 μm; D–F, 2 μm.
MRI identifies enlargement of the lateral ventricles in PLP transgenic mice
To independently measure pathological changes in PLP transgenic mice, we performed MRI scans in both wild-type and mutant mice brain. As a first step, we scored the enlargement of the lateral ventricles at different ages on a 1–4 scale (Fig. 8A–D). In wild-type mice, ventricles constantly increased over time and reached a mean level of 2 of scoring at postnatal month 10 without additional increase at later times (Fig. 8E). PLP transgenic mice also increased their ventricular sizes during maturation but constantly breached level 2 at 6 months of age. Based on corresponding histological sections (data not shown) the abnormally increased ventricular sizes of the mutants were partially attributable to reduced sizes of white matter tracts, e.g., the corpora callosa. Similar observations have been made in CNPase-deficient mutants using conventional histological techniques (Lappe-Siefke et al., 2003). Statistical analysis using the nonparametric Kruskal–Wallis test showed that the enlargement of ventricles in the PLP mutants was significant, demonstrating that enlargement of ventricular sizes faithfully reflects the pathological changes in PLP transgenic mice (Fig. 8E).
RAG-1 deficiency ameliorates the pathological changes in PLP transgenic mice
To investigate the putative pathogenetic role of immune cells in the PLP mutants, we crossbred the PLP-tg mice with mice deficient in the RAG-1 that lack mature T- and B-lymphocytes (Mombaerts et al., 1992). As a first step, we quantified the T-lymphocytes and macrophage-like cells in optic nerves of 12-month-old PLP/RAG-1−/− double mutants. As expected, T-lymphocytes were completely absent. Additionally, there was a clear reduction in the number of macrophage-like cells in the optic nerves at this age (Fig. 6B). Moreover, the remaining macrophages in the PLP-tg RAG-1−/− mutants appeared to have a lower phagocytotic activity, as revealed by quantification of intracellular MBP+ myelin debris. Whereas 26% of the PLP-tg RAG-1 +/? macrophages in the optic nerve showed intracellular myelin debris, only 17% of PLP-tg RAG-1−/− macrophages had phagocytosed myelin. Most importantly, in semithin cross sections of rostral optic nerves, we identified significantly less periaxonal vacuoles in the RAG-deficient PLP mutants (5.25 ± 1.31; n = 5) than in the PLP mutants with an intact immune system (12.71 ± 1.56; n = 7, p < 0.050) (Fig. 6A), reflecting an amelioration in the demyelinating/axonopathic phenotype.
We additionally performed quantitative studies of myelin degeneration and axonopathic changes in optic nerves of four PLP/RAG-1+/? and four PLP/RAG-1−/− double mutants at the electron microscopic level. These studies revealed that in the PLP/RAG-1−/− double mutants, the number of demyelinated intact axons was decreased by 16%. Interestingly, axons displaying features indicative of degeneration (in the absence of periaxonal vacuoles) were even more reduced (36%, p < 0.05, Mann–Whitney U test) when compared with immune-competent PLP mutants. Representative micrographs reflecting these pathological features of the corresponding genotypes are shown in Figure 7D–F.
As a next step, we investigated the ventricular sizes in the PLP/RAG-1−/− double mutants by MRI. They increased over time similarly to PLP transgenic mice, but the ventricular score remained ≤2 over time, thus resembling the ventricular sizes seen in wt mice (Fig. 8E). Therefore, both at the histological and MRI level, RAG-1 deficiency ameliorated the pathological changes in a CNS-specific myelin mutant.
Bone marrow transfer from wt and CD4−/− but not from CD8−/− mice reverts the RAG-1-related amelioration of the pathological changes in PLP mutants
To prove that the RAG-1-related amelioration of the pathological changes in the PLP mutants is attributable to the absence of mature T-lymphocytes rather than to the RAG-1-mutation itself, we performed bone marrow transplantation from wild-type donor mice into the double mutants at 8 weeks of age. At 6 months, the MRI scan showed a clearly aggravated phenotype of the wild-type-transplanted RAG-1-deficient PLP mutants, with ventricular sizes larger than score level 2 (Fig. 8E).
Next, we investigated the pathological impact of CD4+ versus CD8+ T-lymphocytes. This was based on our finding that in the PLP mutants, CD8+ but not CD4+ cells were upregulated in the diseased CNS tissue (Fig. 1).
Bone marrow reconstitution from CD4+/CD8−/− mice resulted in ventricular sizes comparable with PLP/RAG-1−/− double mutants. In contrast, bone marrow reconstitution from CD8+/CD4−/− mice resulted in ventricular sizes comparable with genuine PLP mutants with an intact immune system or PLP/RAG-1−/− double mutants transplanted with bone marrow from wild-type mice (Fig. 8E). Double-immunofluorescence stainings in these transplanted mutants confirmed the presence of CD8+/CD3+ T-lymphocytes in the CNS (data not shown). This strongly suggests that CD8+ but not CD4+ T-lymphocytes play a pathogenic role in PLP transgenic mutants. Control experiments with PLP wild-type mice that received wild-type bone marrow comprising CD8+/CD4+ stem cells resulted in a normal ventricular size at 6 months of age (Fig. 8E).
Discussion
In our present study, we identified components of the immune system as being pathologically relevant for a primarily genetically mediated myelinopathy in the CNS of mice. We found CD8+ T-lymphocytes and CD11b+ macrophage-like cells elevated in white matter tracts of the mutants. The macrophage-like cells thereby outnumber the T-lymphocytes by a factor of ∼20, which is similar to our previous observations in demyelinating mutants of the peripheral nervous system (Schmid et al., 2000; Kobsar et al., 2003; Martini and Toyka, 2004; Ip et al., 2006). The CD8+ T-lymphocytes could often be found in close contact to MHC-I+ oligodendrocytes, which is highly reminiscent of the interaction of cytotoxic T-lymphocytes with their target structure. This view fits very well with the finding that the vast majority of isolated CD8+ cells from PLP mutant brains express markers for cytotoxic effector cells. Interestingly, abrogating the CD8+ cells by crossbreeding the myelin mutants with RAG-1-deficient mice leads to a substantial amelioration of the demyelinating phenotype. Transplantation of these double mutants with CD8+/CD4− but not with CD8−/CD4+ bone marrow reverts the ameliorated demyelinating phenotype, supporting the view that CD8+ cells are pathogenic effector cells.
It is of note that CD8+ cells have been identified as the most prominent cell type in multiple sclerosis lesions (Neumann et al., 2002; Lassmann, 2004), and experimental studies suggest their putative impact on axonal damage (Medana et al., 2001; Neumann et al., 2002). In the present model, it is highly probable that the PLP-overexpressing oligodendrocytes are the primary target of the CD8+ cells and that the axons may suffer secondarily from the myelin loss. This could be attributable to transcellular alterations resulting in, e.g., altered axonal transport, as has been shown in myelin mutants of the peripheral nervous system (de Waegh and Brady, 1990, 1991; de Waegh et al., 1992) and the CNS (Edgar and Garbern, 2004; Edgar et al., 2004). Additionally, it is possible that the loss of the “neuroprotective barrier” of the axons normally preserving them from adverse influences such as NO (nitric oxide), toxic radicals, and proinflammatory cytokines released by immune-related cells, supports the degeneration of the preinjured axons (Redford et al., 1997; Smith et al., 2001; Neumann, 2003). In addition, it is probable that the CD8+ cells operate as activators of resident macrophage-like cells (Brookes et al., 2003; Gurlo and von Grafenstein, 2003), which then mediate degenerative changes in the myelinated tracts (Hendriks et al., 2005). The hypothesis that CD8+ cells may be important activators for executing macrophage-like cells gains support from the observation that in our RAG-1-deficient PLP double mutants, not only are all T-lymphocytes absent but the number of macrophage-like cells is reduced in the ameliorated white matter tracts. Moreover, the remaining macrophages contain less myelin debris than in PLP mutants with an intact immune system, reflecting a lower activation status than in the presence of T-lymphocytes. Similar observations have been made in RAG-1-deficient myelin mutants of the peripheral nervous system (Schmid et al., 2000; Kobsar et al., 2003; Martini and Toyka, 2004; Ip et al., 2006). A direct proof that macrophage-like cells are pathogenetically relevant in our CNS mutants is provided by recent investigations focusing on PLP mutants that have been crossbred to mice deficient in the gene for the cell adhesion molecule sialoadhesin (Kobsar et al., 2006; Oetke et al., 2006). This molecule is strictly confined in its expression to macrophage-like cells and mediates the activation and effector functions of macrophages (Crocker, 2005). In the absence of this macrophage-restricted cell recognition molecule, fewer macrophages, but also fewer CD8+ T-lymphocytes, are found in the CNS of PLP mutants, reflecting the mutual interaction of the two immune-related cell types in the process of primarily genetically related demyelination. Most interestingly, the demyelinating/axonopathic phenotype is strongly alleviated in the PLP mutants deficient in the macrophage-restricted molecule, indicating that macrophages are important players of probably multiple functions (e.g., phagocytosis, activation of lymphocytes) in the pathogenesis of the PLP transgenic mice (Ip, A. Kroner, P. R. Crocker, K. A. Nave, and R. Martini, unpublished observations).
Pathogenetically relevant immune reactions after primary glial mutations appear to be a widespread mechanism in different types of leukodystrophic disorders. In the present model, the overexpression of a major myelin component leads to an activation of cellular components of the immune system. Although the exact mechanisms are still obscure, it is tempting to speculate that the overexpression of PLP induces intracellular stress because of an accumulation of the protein and cholesterol in intracellular compartments of the oligodendrocytes (Simons et al., 2002). This intracellular stress could cause an activation and attraction of immune cells via proinflammatory cytokines secreted by the mutant glial cells.
To our knowledge, one of the first experimental studies proving a pathogenetic role of microglial cells in a leukodystrophic disorder was the investigation of a mouse mutant deficient in the galactocerebrosidase mimicking the globoid cell leukodystrophy in humans. Crossbreeding these mice with mice deficient in MHC class II molecules led to a reduced frequency of microglial cells in the mutants and amelioration of the pathological features. In contrast to the PLP mutants, infiltrating lymphocytes have been suggested not to play a role in the pathogenesis (Matsushima et al., 1994), although the corresponding experiments have not been performed.
X-linked adrenoleukodystrophy, a serious neurological disorder in childhood, is primarily caused by mutations in a peroxisomal membrane protein [ALDP (adrenoleukodystrophy protein gene)] related to the not yet fully understood occurrence of very long-chain fatty acids in organs with high fatty acid turnover, such as the white matter tracts in the CNS (Moser, 2004). Although the severity of the disease can vary, the most detrimental and rapidly progressing forms are usually associated with inflammation (Berger et al., 2001). Interestingly, CD8+ cytotoxic T-lymphocytes displaying CD44 are often tightly attached to oligodendrocytes, as we have observed in the PLP mutants, suggesting cellular toxicity (Moser, 2004). The relevance of the immune cells cannot be investigated at present because animal models displaying the inflammatory component of adrenoleukodystrophy have not yet been established (Moser, 2004).
Another leukodystrophic disorder of primarily genetic origin and regular involvement of immune-related cells is Leber’s hereditary optic neuropathy, in which mitochondrial DNA mutations are the primary culprits and some mutations lead to inflammatory reactions not only in the optic tracts but also in other white matter regions (Kovacs et al., 2005). Based on clinical, histopathological, and MRI criteria, these forms appear to be similar to typical multiple sclerosis.
The primarily immune-independent damage or dystrophy of oligodendroglial cells has presently been viewed as one possible cause of MS. In a study focusing on biopsies and autopsies from a large cohort of MS patients, four different patterns of pathological features emerged. Whereas two patterns were categorized as typical primarily immune-mediated forms involving T-cell-mediated or T-cell- and antibody-mediated mechanisms, the other patterns were considered as primary oligodendrocyte damage, with a secondary involvement of the immune system (Lucchinetti et al., 2000). In another study focusing on two MS patients who died during or shortly after a relapse, the corresponding autopsies suggested a primary oligodendrocytic damage with some activated microglial cells and no lymphocytes within the area of the active lesion (Barnett and Prineas, 2004). In a recent case report, Warshawsky et al. (2005) described a 49-year-old female patient who met the criteria of a primary progressive MS of adult onset that was caused by a heterozygous Leu30Arg mutation in the PLP gene. Interestingly, the hemizygous mutation caused Pelizaeus-Merzbacher disease in the patient’s son (Warshawsky et al., 2005). These examples support the view that primary oligodendrocyte damage can cause secondary immune reactions of significant pathogenetic relevance, possibly originating from mutation- and/or damage-induced intracellular stress leading to the expression of proinflammatory molecules. In contrast, it is of note that a protective function of immune cells has been proposed for inflammatory diseases of the CNS (Kerschensteiner et al., 1999, 2003; Hohlfeld et al., 2000) and for CNS injury (Hauben et al., 2000; Moalem et al., 2000; Schwartz, 2001). It is well established that, after facial nerve injury, lymphocytes invade the facial nucleus (Raivich et al., 1998; Moran and Graeber, 2004), and it appears that CD4-positive T-cells protect facial motoneurons against lesion-induced apoptosis (Serpe et al., 1999, 2003; Byram et al., 2004; Jones et al., 2005). Corresponding functions have been suggested for T-lymphocytes invading dorsal root ganglia and spinal nerves after sciatic nerve injury (Hu and McLachlan, 2002, 2003). Additionally, both lymphocytes and microglial cells have been shown to foster remyelination in the CNS after lysolecithin damage (Bieber et al., 2003; Kotter et al., 2005), and microglial cells may be neuroprotective to excitotoxicity (Mitrasinovic et al., 2005). As an explanation for the dichotomal roles of inflammatory cells, it has been proposed that the balance between destructive or protective effects of neuroinflammation may vary in different disorders or even within different stages of a distinct disease (Kerschensteiner et al., 2003).
Our genetic model of a slowly progressing demyelinating disorder of the CNS shows a cryptic, yet pathogenetically highly relevant inflammatory reaction in white matter tracts. At least with regard to the established clinical and pathological criteria, these mice cannot be viewed as a model of MS. It is possible that the PLP overexpression is not the appropriate trigger to generate an MS-like response. It is also possible that a single glial damage is not sufficient to cause an MS-like disorder. A “second hit” might be necessary to modify the observed immune reactions in a way resembling typical MS. In this context, it is interesting to mention that in mouse EAE, the pathological features can be aggravated by an additional lack of CNTF that serves as a survival factor for oligodendrocytes (Linker et al., 2002). Indeed, MS patients devoid of CNTF preferentially develop early-onset forms of MS (Giess et al., 2002). It is well known that distinct polymorphisms or mutations in MHC or other immune-related genes can substantially modify the disease course and progression of MS (Hemmer et al., 2002; Ibrahim and Gold, 2005). An interesting concept has recently emerged implicating the role of inhibitory costimulatory molecules of the B7/CD28 family. Mutations in PD-1 (programmed death 1 gene) have been identified as being highly linked to progressive forms of MS (Kroner et al., 2005). It is, therefore, likely that additional genetic variations could modify the phenotypic outcome of a particular glial damage. Future studies are necessary to investigate the consequences of the combination of glial damage and mutations in immune-related genes.
Footnotes
This work was supported by the Gemeinnützige Hertie-Stiftung (Grants 1.319.110/03/04 and 1.01.1/05/010 to R.M.), MUCK e.V. (to R.M.), the German Research Foundation (Grant SFB581 to R.M.; Grant DFG1722/3-1 to H.W.), and the State of Bavaria. M.B. holds a professorship donated by Schering (Berlin, Germany), to the University of Wuerzburg (Wuerzburg, Germany). We are grateful to Heinrich Blazyca, Carolin Kiesel, Nadine Weckesser, and Tanja Horn for expert technical assistance and to Helga Brünner for animal care. We thank Katharina Kreymborg and Burkhard Becher for helpful advice concerning the flow cytometric analysis of brain lymphocytes and Florian Bender for his initial help with confocal microscopy. The constant support and advice by Klaus V. Toyka and contributions to initial experiments by Dr. Mathias Maurer are gratefully acknowledged.
References
- Anderson TJ, Schneider A, Barrie BA, Klugmann M, McCulloch MC, Kirkham D, Kyriakides E, Nave K-A, Griffiths IR (1998). Late-onset neurodegeneration in mice with increased dosage of the proteolipid protein gene. J Comp Neurol 394:506–519. [DOI] [PubMed] [Google Scholar]
- Barnett MH, Prineas JW (2004). Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann Neurol 55:458–468. [DOI] [PubMed] [Google Scholar]
- Berger J, Moser HW, Forss-Petter S (2001). Leukodystrophies: recent developments in genetics, molecular biology, pathogenesis and treatment. Curr Opin Neurol 14:305–312. [DOI] [PubMed] [Google Scholar]
- Bieber AJ, Kerr S, Rodriguez M (2003). Efficient central nervous system remyelination requires T cells. Ann Neurol 53:680–684. [DOI] [PubMed] [Google Scholar]
- Bjartmar C, Yin X, Trapp BD (1999). Axonal pathology in myelin disorders. J Neurocytol 28:383–395. [DOI] [PubMed] [Google Scholar]
- Brookes RH, Pathan AA, McShane H, Hensmann M, Price DA, Hill AV (2003). CD8+ T cell-mediated suppression of intracellular Mycobacterium tuberculosis growth in activated human macrophages. Eur J Immunol 33:3293–3302. [DOI] [PubMed] [Google Scholar]
- Bruck W, Stadelmann C (2003). Inflammation and degeneration in multiple sclerosis. Neurol Sci 24:[Suppl 5], S265–S267. [DOI] [PubMed] [Google Scholar]
- Byram SC, Carson MJ, DeBoy CA, Serpe CJ, Sanders VM, Jones KJ (2004). CD4-positive T cell-mediated neuroprotection requires dual compartment antigen presentation. J Neurosci 24:4333–4339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carenini S, Maürer M, Werner A, Blazyca H, Toyka KV, Schmid CD, Ravich G, Martini R (2001). The role of macrophages in demyleinating peripheral nervous system of mice heterozygously deficient in PO. J Cell Biol 152:301–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crocker PR (2005). Siglecs in innate immunity. Curr Opin Pharmacol 5:1–7. [DOI] [PubMed] [Google Scholar]
- de Waegh S, Brady ST (1990). Altered slow axonal transport and regeneration in a myelin-deficient mutant mouse: the trembler as an in vivo model for Schwann cell-axon interactions. J Neurosci 10:1855–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Waegh SM, Brady ST (1991). Local control of axonal properties by Schwann cells: neurofilaments and axonal transport in homologous and heterologous nerve grafts. J Neurosci Res 30:201–212. [DOI] [PubMed] [Google Scholar]
- de Waegh SM, Lee VMY, Brady ST (1992). Local modulation of neurofilament phosphorylation, axonal caliber, and slow axonal transport by myelinating Schwann cells. Cell 68:451–463. [DOI] [PubMed] [Google Scholar]
- Edgar JM, Garbern J (2004). The myelinated axon is dependent on the myelinating cell for support and maintenance: molecules involved. J Neurosci Res 76:593–598. [DOI] [PubMed] [Google Scholar]
- Edgar JM, McLaughlin M, Yool D, Zhang SC, Fowler JH, Montague P, Barrie JA, McCulloch MC, Duncan ID, Garbern J, Nave KA, Griffiths IR (2004). Oligodendroglial modulation of fast axonal transport in a mouse model of hereditary spastic paraplegia. J Cell Biol 166:121–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung-Leung WP, Schilham MW, Rahemtulla A, Kundig TM, Vollenweider M, Potter J, van Ewijk W, Mak TW (1991). CD8 is needed for development of cytotoxic T cells but not helper T cells. Cell 65:443–449. [DOI] [PubMed] [Google Scholar]
- Giess R, Mäurer M, Linker RA, Gold R, Warmuth-Metz M, Toyka KV, Sendtner M, Rieckmann P (2002). Association of a null mutation in the CNTF gene with early onset of multiple sclerosis. Arch Neurol 59:407–409. [DOI] [PubMed] [Google Scholar]
- Gould RM, Brady ST (2004). Neuropathology: many paths lead to hereditary spastic paraplegia. Curr Biol 14:R903–R904. [DOI] [PubMed] [Google Scholar]
- Griffiths I, Klugmann M, Anderson T, Yool D, Thomson C, Schwab MH, Schneider A, Zimmermann F, McCulloch M, Nadon N, Nave KA (1998). Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science 280:1610–1613. [DOI] [PubMed] [Google Scholar]
- Gurlo T, von Grafenstein H (2003). Antigen-independent cross-talk between macrophages and CD8+ T cells facilitates their cooperation during target destruction. Int Immunol 15:1063–1071. [DOI] [PubMed] [Google Scholar]
- Hauben E, Butovsky O, Nevo U, Yoles E, Moalem G, Agranov E, Mor F, Leibowitz-Amit R, Pevsner E, Akselrod S, Neeman M, Cohen IR, Schwartz M (2000). Passive or active immunization with myelin basic protein promotes recovery from spinal cord contusion. J Neurosci 20:6421–6430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayreh SS (2001). The blood supply of the optic nerve head and the evaluation of it–myth and reality. Prog Retin Eye Res 20:563–593. [DOI] [PubMed] [Google Scholar]
- Hemmer B, Archelos JJ, Hartung HP (2002). New concepts in the immunopathogenesis of multiple sclerosis. Nat Rev Neurosci 3:291–301. [DOI] [PubMed] [Google Scholar]
- Hendriks JJ, Teunissen CE, de Vries HE, Dijkstra CD (2005). Macrophages and neurodegeneration. Brain Res Brain Res Rev 48:185–195. [DOI] [PubMed] [Google Scholar]
- Hohlfeld R, Kerschensteiner M, Stadelmann C, Lassmann H, Wekerle H (2000). The neuroprotective effect of inflammation: implications for the therapy of multiple sclerosis. J Neuroimmunol 107:161–166. [DOI] [PubMed] [Google Scholar]
- Hu P, McLachlan EM (2002). Macrophage and lymphocyte invasion of dorsal root ganglia after peripheral nerve lesions in the rat. Neuroscience 112:23–38. [DOI] [PubMed] [Google Scholar]
- Hu P, McLachlan EM (2003). Distinct functional types of macrophage in dorsal root ganglia and spinal nerves proximal to sciatic and spinal nerve transections in the rat. Exp Neurol 184:590–605. [DOI] [PubMed] [Google Scholar]
- Ibrahim SM, Gold R (2005). Genomics, proteomics, metabolomics: what is in a word for multiple sclerosis? Curr Opin Neurol 18:231–235. [DOI] [PubMed] [Google Scholar]
- Ip CW, Kroner A, Fischer S, Berghoff M, Kobsar I, Mäurer M, Martini R (2006). Role of immune cells in animal models for inherited peripheral neuropathies. Neuromol Med 8:175–190. [DOI] [PubMed] [Google Scholar]
- Jones KJ, Serpe CJ, Byram SC, Deboy CA, Sanders VM (2005). Role of the immune system in the maintenance of mouse facial motoneuron viability after nerve injury. Brain Behav Immun 19:12–19. [DOI] [PubMed] [Google Scholar]
- Kerschensteiner M, Gallmeier E, Behrens L, Leal VV, Misgeld T, Klinkert WE, Kolbeck R, Hoppe E, Oropeza-Wekerle RL, Bartke I, Stadelmann C, Lassmann H, Wekerle H, Hohlfeld R (1999). Activated human T cells, B cells, and monocytes produce brain-derived neurotrophic factor in vitro and in inflammatory brain lesions: a neuroprotective role of inflammation? J Exp Med 189:865–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerschensteiner M, Stadelmann C, Dechant G, Wekerle H, Hohlfeld R (2003). Neurotrophic cross-talk between the nervous and immune systems: implications for neurological diseases. Ann Neurol 53:292–304. [DOI] [PubMed] [Google Scholar]
- Kirkpatrick LL, Brady ST (1994). Modulation of the axonal microtubule cytoskeleton by myelinating Schwann cells. J Neurosci 1412:7440–7450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobsar I, Berghoff M, Samsam M, Wessig C, Maurer M, Toyka KV, Martini R (2003). Preserved myelin integrity and reduced axonopathy in connexin32-deficient mice lacking the recombination activating gene-1. Brain 126:804–813. [DOI] [PubMed] [Google Scholar]
- Kobsar I, Oetke C, Kroner A, Wessig C, Crocker P, Martini R (2006). Attenuated demyelination in the absence of the macrophage-restricted adhesion molecule sialoadhesin (Siglec-1) in mice heterozygously deficient in P0. Mol Cell Neurosci 31:685–691. [DOI] [PubMed] [Google Scholar]
- Kotter MR, Zhao C, van Rooijen N, Franklin RJ (2005). Macrophage-depletion induced impairment of experimental CNS remyelination is associated with a reduced oligodendrocyte progenitor cell response and altered growth factor expression. Neurobiol Dis 18:166–175. [DOI] [PubMed] [Google Scholar]
- Kovacs GG, Hoftberger R, Majtenyi K, Horvath R, Barsi P, Komoly S, Lassmann H, Budka H, Jakab G (2005). Neuropathology of white matter disease in Leber’s hereditary optic neuropathy. Brain 128:35–41. [DOI] [PubMed] [Google Scholar]
- Kroner A, Mehling M, Hemmer B, Rieckmann P, Toyka KV, Maurer M, Wiendl H (2005). A PD-1 polymorphism is associated with disease progression in multiple sclerosis. Ann Neurol 58:50–57. [DOI] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, Nave KA (2003). Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet 33:366–374. [DOI] [PubMed] [Google Scholar]
- Lassmann H (2004). Cellular damage and repair in multiple sclerosis. In: Myelin biology and disorders (Lazzarini RA, Griffin JW, Lassmann H, Nave KA, Miller RH, Trapp BD, eds) pp. 733–762. Amsterdam: Elsevier Academic.
- Lassmann H (2005). Multiple sclerosis pathology: evolution of pathogenetic concepts. Brain Pathol 15:217–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linker RA, Mäurer M, Gaupp S, Martini R, Holtmann B, Giess R, Rieckmann P, Lassmann H, Toyka KV, Sendtner M, Gold R (2002). CNTF is a major protective factor in demyelinating CNS disease: a neurotrophic cytokine as modulator in neuroinflammation. Nat Med 8:620–624. [DOI] [PubMed] [Google Scholar]
- Lucchinetti C, Brück W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H (2000). Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol 47:707–714. [DOI] [PubMed] [Google Scholar]
- Magnus T, Schreiner B, Korn T, Jack C, Hong G, Antel J, Ifergan I, Chen L, Bischof F, Bar-Or A, Wiendl H (2005). Microglial expression of the B7-family member B7–H1 confers strong immune inhibition: implications for immune responses and autoimmunity in the CNS. J Neurosci 25:2537–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini R (2001). The effect of myelinating Schwann cells on axons. Muscle Nerve 24:456–466. [DOI] [PubMed] [Google Scholar]
- Martini R, Toyka KV (2004). Immune-mediated components of hereditary demyelinating neuropathies: lessons from animal models and patients. Lancet Neurol 3:457–465. [DOI] [PubMed] [Google Scholar]
- Matsushima GK, Taniike M, Glimcher LH, Grusby MJ, Frelinger JA, Suzuki K, Ting JPY (1994). Absence of MHC class II molecules reduces CNS demyelination, microglial/macrophage infiltration, and twitching in murine globoid cell leukodystrophy. Cell 78:645–656. [DOI] [PubMed] [Google Scholar]
- Mäurer M, Schmid CD, Bootz F, Zielasek J, Toyka KV, Oehen S, Martini R (2001). Bone marrow transfer from wild type mice reverts the beneficial effect of genetically-mediated immune deficiency in myelin mutants. Mol Cell Neurosci 17:1094–1101. [DOI] [PubMed] [Google Scholar]
- Medana I, Martinic MA, Wekerle H, Neumann H (2001). Transection of major histocompatibility complex class I-induced neurites by cytotoxic T lymphocytes. Am J Pathol 159:809–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitrasinovic OM, Grattan A, Robinson CC, Lapustea NB, Poon C, Ryan H, Phong C, Murphy GM Jr (2005). Microglia overexpressing the macrophage colony-stimulating factor receptor are neuroprotective in a microglial-hippocampal organotypic coculture system. J Neurosci 25:4442–4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moalem G, Gdalyahu A, Shani Y, Otten U, Lazarovici P, Cohen IR, Schwartz M (2000). Production of neurotrophins by activated T cells: implications for neuroprotective autoimmunity. J Autoimmun 15:331–345. [DOI] [PubMed] [Google Scholar]
- Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE (1992). RAG-1-deficient mice have no mature B and T lymphocytes. Cell 68:869–877. [DOI] [PubMed] [Google Scholar]
- Moran LB, Graeber MB (2004). The facial nerve axotomy model. Brain Res Brain Res Rev 44:154–178. [DOI] [PubMed] [Google Scholar]
- Moser HW (2004). Adrenoleukodystrophies. In: Myelin biology and disorders (Lazzarini RA, Griffin JW, Lassmann H, Nave KA, Miller RH, Trapp BD, eds) pp. 807–839. Amsterdam: Elsevier Academic.
- Mueller M, Wacker K, Ringelstein EB, Hickey WF, Imai Y, Kiefer R (2001). Rapid response of identified resident endoneurial macrophages to nerve injury. Am J Pathol 159:2187–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann H (2003). Molecular mechanisms of axonal damage in inflammatory central nervous system diseases. Curr Opin Neurol 16:267–273. [DOI] [PubMed] [Google Scholar]
- Neumann H, Medana IM, Bauer J, Lassmann H (2002). Cytotoxic T lymphocytes in autoimmune and degenerative CNS diseases. Trends Neurosci 25:313–319. [DOI] [PubMed] [Google Scholar]
- Oetke C, Vinson MC, Jones C, Crocker PR (2006). Sialoadhesin-deficient mice exhibit subtle changes in B- and T-cell populations and reduced immunoglobulin M levels. Mol Cell Biol 26:1549–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahemtulla A, Fung-Leung WP, Schilham MW, Kundig TM, Sambhara SR, Narendran A, Arabian A, Wakeham A, Paige CJ, Zinkernagel RM, Miller RG, Mak TW (1991). Normal development and function of CD8+ cells but markedly decreased helper cell activity in mice lacking CD4. Nature 353:180–184. [DOI] [PubMed] [Google Scholar]
- Raivich G, Jones LL, Kloss CUA, Werner A, Neumann H, Kreutzberg GW (1998). Immune surveillance in the injured nervous system: T-lymphocytes invade the axotomized mouse facial motor nucleus and aggregate around sites of neuronal degeneration. J Neurosci 18:5804–5816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Readhead C, Schneider A, Griffiths I, Nave K-A (1994). Premature arrest of myelin formation in transgenic mice with increased proteolipid protein gene dosage. Neuron 12:583–595. [DOI] [PubMed] [Google Scholar]
- Redford EJ, Kapoor R, Smith KJ (1997). Nitric oxide donors reversibly block axonal conduction: demyelinated axons are especially susceptible. Brain 120:2149–2157. [DOI] [PubMed] [Google Scholar]
- Samsam M, Mi W, Wessig C, Zielasek J, Toyka KV, Coleman MP, Martini R (2003). The wlds mutation delays robust loss of motor and sensory axons in a genetic model for myelin-related axonopathy. J Neurosci 23:2833–2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid CD, Stienekemeier M, Oehen S, Bootz F, Zielasek J, Gold R, Toyka KV, Schachner M, Martini R (2000). Immune deficiency in mouse models for inherited peripheral neuropathies leads to improved myelin maintenance. J Neurosci 20:729–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz M (2001). Protective autoimmunity as a T-cell response to central nervous system trauma: prospects for therapeutic vaccines. Prog Neurobiol 65:489–496. [DOI] [PubMed] [Google Scholar]
- Serpe CJ, Kohm AP, Huppenbauer CB, Sanders VM, Jones KJ (1999). Exacerbation of facial motoneuron loss after facial nerve transection in severe combined immunodeficient (scid) mice. J Neurosci 19:RC7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serpe CJ, Coers S, Sanders VM, Jones KJ (2003). CD4+ T, but not CD8+ or B, lymphocytes mediate facial motoneuron survival after facial nerve transection. Brain Behav Immun 17:393–402. [DOI] [PubMed] [Google Scholar]
- Simons M, Kramer EM, Macchi P, Rathke-Hartlieb S, Trotter J, Nave KA, Schulz JB (2002). Overexpression of the myelin proteolipid protein leads to accumulation of cholesterol and proteolipid protein in endosomes/lysosomes: implications for Pelizaeus-Merzbacher disease. J Cell Biol 157:327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KJ, Kapoor R, Hall SM, Davies M (2001). Electrically active axons degenerate when exposed to nitric oxide. Ann Neurol 49:470–476. [PubMed] [Google Scholar]
- Warshawsky I, Rudick RA, Staugaitis SM, Natowicz MR (2005). Primary progressive multiple sclerosis as a phenotype of a PLP1 gene mutation. Ann Neurol 58:470–473. [DOI] [PubMed] [Google Scholar]
- Wolburg H, Bäuerle C (1993). Astrocytes in the lamina cribrosa of the rat optic nerve: are their morphological peculiarities involved in an altered blood-brain barrier? J Hirnforsch 343:445–459. [PubMed] [Google Scholar]
- Wrabetz L, Feltri ML, Quattrini A, Imperiale D, Previtali S, D’Antonio M, Martini R, Yin X, Trapp BD, Zhou L, Chiu SY, Messing A (2000). P0 glycoprotein overexpression causes congenital hypomyelination of peripheral nerves. J Cell Biol 148:1021–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X, Kidd GJ, Wrabetz L, Feltri ML, Messing A, Trapp BD (2000). Schwann cell myelination requires timely and precise targeting of P0 protein. J Cell Biol 148:1009–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]