

Abstract
Lasting changes in neuronal connectivity require calcium-dependent gene expression. Here we report the identification of LIM domain-only 4 (LMO4) as a mediator of calcium-dependent transcription in cortical neurons. Calcium influx via voltage-sensitive calcium channels and NMDA receptors contributes to synaptically induced LMO4-mediated transactivation. LMO4-mediated transcription is dependent on signaling via calcium/calmodulin-dependent protein (CaM) kinase IV and microtubule-associated protein (MAP) kinase downstream of synaptic stimulation. Coimmunoprecipitation experiments indicate that LMO4 can form a complex with cAMP response element-binding protein (CREB) and can interact with cofactor of LIM homeodomain protein 1 (CLIM1) and CLIM2. To evaluate the role of LMO4 in vivo, we examined the consequences of conditional loss of lmo4 in the forebrain, using the Cre-Lox gene-targeting strategy. The organization of the barrel field in somatosensory cortex is disrupted in mice in which lmo4 is deleted conditionally in the cortex. Specifically, in contrast to controls, thalamocortical afferents in conditional lmo4 null mice fail to segregate into distinct barrel-specific domains. These observations identify LMO4 as a calcium-dependent transactivator that plays a key role in patterning thalamocortical connections during development.
Keywords: LMO4, calcium, transcription, barrel cortex, development, cortex
Introduction
Neuronal activity is necessary for the development and function of many aspects of the visual and somatosensory systems. The somatosensory system consists of trigeminal and dorsal column pathways that convey sensory experience from the periphery to somatosensory cortex (S1) via relay stations in the brainstem and ventrobasal thalamus. Thalamic afferents are distributed diffusely in S1 of newborn rats, but after the first few days they are refined into spatially discrete barrel-like clusters (Schlaggar and O’Leary, 1993; O’Leary et al., 1994). The barrel is a histologically defined structure that is formed by the cell bodies of layer IV cortical neurons. The barrel interior is composed of synapses made by thalamocortical axons onto the dendrites of cortical neurons. Peripheral lesions in the somatosensory system perturb the somatotopic organization of cortical barrels and their subcortical relay stations. For example, ablation of a whisker results in loss of the corresponding barrel in the cortex and expansion of the adjacent barrels (Van der Loos and Woolsey, 1973) (for review, see O’Leary et al., 1994).
Molecular interactions between thalamocortical axons and cortical neurons establish an initial somatotopic pattern in S1 that subsequently is refined by neural activity in the early postnatal period (Goodman and Shatz, 1993; Katz and Shatz, 1996). Electrophysiological and anatomical studies on rats that have subdural implantation of aminophosphonovalerate-impregnated (APV-impregnated) Elvax reveal that this developmental plasticity requires NMDA receptor (NMDAR) activation (Schlaggar et al., 1993). Other studies have shown that glutamate receptor blockade inhibits refinement of the somatotopic map in S1 (Schlaggar et al., 1993; Fox et al., 1996; Mitrovic et al., 1996). Furthermore, deletion of NMDARs and metabotropic glutamate receptors (mGluRs) suggest that glutamatergic synaptic transmission is necessary for patterning of thalamocortical connections (Iwasato et al., 1997, 2000; Hannan et al., 2001; Lu et al., 2003). For example, conditional deletion of the NR1 subunit of the NMDAR disrupts formation of barrels in the somatosensory cortex (Iwasato et al., 2000) (S. Konur and A. Ghosh, unpublished observations). These experiments suggest that neural activity is essential for the development and refinement of thalamocortical projections in the somatosensory system much like in the visual system (Constantine-Paton et al., 1990).
Because NMDARs are a major route for calcium entry into neurons, the involvement of NMDARs in refinement of the somatosensory map (Iwasato et al., 1997, 2000) suggests that calcium influx plays a critical role in barrel cortex development. Although the acute effects of calcium influx are mediated by post-translational modifications of proteins that are already present in the cell, the long-term effects of calcium signaling involve calcium-dependent transcription and expression of new genes (Ghosh and Greenberg, 1995; West et al., 2001). It is therefore likely that activity-dependent development of cortical connections involves calcium-dependent transcription.
Our laboratory has designed an expression cloning strategy to identify proteins involved in calcium-dependent transcription. This screen is called “Transactivator Trap” and was used to identify a calcium-responsive transactivator (CREST) that is necessary for normal neuronal dendrite development (Aizawa et al., 2004). Here we describe the identification and characterization of LIM domain-only 4 (LMO4), using the Transactivator Trap screen. We show that LMO4 can drive transcription in response to calcium influx in cortical neurons. Furthermore, using the Cre-Lox system, we have generated mice with conditional deletion of lmo4 in cortical neurons. We report that deletion of lmo4 within the cortex causes abnormal development of somatosensory barrels in the mouse cortex. These observations show that LMO4 mediates activity-dependent transcription in response to NMDAR activation and is necessary for proper segregation of thalamocortical afferents in layer IV of mouse somatosensory cortex.
Materials and Methods
Cell culture.
Cortical neurons from embryonic day 18 (E18) Long–Evans rat embryos were cultured as previously described (Shieh et al., 1998). Briefly, timed-pregnant E18 rat embryos were removed from the pregnant female and decapitated in ice-cold HBSS. Next the cortices were dissected out and digested in 10 U/ml papain (Worthington, Freehold, NJ) in dissociation medium and then triturated until few or no cell clumps were left. The dissociated neurons were plated on 60 mm plates precoated with poly-l-lysine and laminin (BD Biosciences, San Diego, CA) at a density of 3 × 106 cells per plate. Neurons were maintained in either glutamine-free basal modified Eagle's supplemented with fetal bovine serum (FBS; to 5%) and N2 supplement (to 1%) or Neurobasal medium (NBM) supplemented with B-27 (to 1%). Both media were supplemented with glutamine (to 1 mm). Unless otherwise indicated, all tissue culture reagents were obtained from Invitrogen (Carlsbad, CA). NBM medium was used for all cultures that were maintained >7 d. For these mature cultures the medium was supplemented with fluorodeoxyuridine (FDU; Sigma, St. Louis, MO) at 3 d in vitro (3 DIV) to prevent glial proliferation. Drugs were used at the indicated concentrations and diluted according to the manufacturer's directions. Drugs were obtained from the following sources: TTX (Sigma); ω-agatoxin IVA (ω-AgaIVA) conotoxin (Tocris, Ellisville, MO); KN62, KN92, and KN93 (Sigma); EGTA (Sigma); APV (Calbiochem, La Jolla, CA; and Tocris); nifedipine (Calbiochem); glutamate (Sigma); and bicuculline (Sigma).
Transfections.
Transfections were performed by using a calcium phosphate method as previously described (Threadgill et al., 1997). Briefly, the culture medium was removed and replaced with DMEM ∼1 h before transfection. The calcium phosphate/DNA precipitate was formed in HEPES-buffered saline, pH 7.07, and added drop-wise to the DMEM. After a 50–60 min transfection period a sandy precipitate covered the cells and was washed off with 4–5 vol changes of fresh DMEM. Then the cells were returned to the incubator in their original culture medium. Transfection efficiency was typically between 1 and 5%.
Chloramphenicol acetyltransferase reporter assay.
Cells were harvested 10–12 h after stimulation in isotonic TNE (10 mm Tris, pH 7.8, 150 mm NaCl, 1 mm EDTA). Cells were spun down gently and subjected to three cycles of freeze–thaw lysis. Lysis supernatant was incubated with 0.5 μCi 14C-labeled chloramphenicol (Amersham Biosciences, Arlington Heights, IL) and 0.8 mm acetyl-coenzyme A (acetyl-CoA; Boehringer Mannheim, Indianapolis, IN), pH 7.8, at 37°C for 1 h. Reaction mixtures were extracted with ethyl acetate, reduced in a speed vacuum, resuspended in chloroform, spotted on a thin-layer chromatography plate (TLC; J.T. Baker Chemical, Phillipsburg, NJ), and separated by ascending chromatography for 1–2 h (in 95% chloroform/5% methanol). For measurements of relative CAT activity the levels of 14C emission on TLC plates were quantified by phosphoimager scans. Histograms shown are mean values from experiments repeated three or more times, with error bars representing the SEM. All values were normalized to unstimulated or empty vector controls as appropriate. Each trial of experiments was performed on cells that simultaneously were cultured, transfected, and assayed to minimize variability caused by differences in experimental procedures. Asterisks indicate significance at the p < 0.05 level, as determined by paired Student's t test.
Library screening and cloning of lmo4.
The construction of the library and screening procedure has been described previously (Aizawa et al., 2004). Briefly, mRNA was isolated from postnatal day 1 (P1) cerebral cortices of Long–Evans rat pups. A cDNA library was generated from the isolated mRNA and subcloned into pcDNA3-galactosidase-4-DNA-binding domain (GAL4-DBD), using EcoR1 and NotI sites. The ligation products were transformed into DH10β electrocompetent cells and grown at a density of 1000 colonies per 15 cm plate. A total of 200,000 colonies were grown in 200 dishes. The bacterial colonies in each dish were harvested into Luria–Bertani (LB) solution and stored at −70°C as a 50% glycerol stock. lmo4 was isolated by screening and cloning of pool number 17 as described here. The glycerol stock from pool number 17 was plated on 36 agar dishes at a density of 200 colonies per dish. The bacterial colonies from each plate were harvested and grown in LB overnight. The DNA from this broth then was isolated (miniprep kit; Eppendorf, Westbury, NY) and transfected into dissociated cortical neurons at 3 DIV. Next the neurons were stimulated overnight at 5 DIV and subsequently were fixed and stained for chloramphenicol acetyltransferase (CAT) immunoreactivity. To facilitate the screening process, we combined and screened the DNA from three dishes collectively so that only 12 initial screens were performed. After a positive group was identified, the miniprep DNA of those three dishes comprising the positive group was screened individually. Once a positive dish of 200 colonies was identified, the colonies were transferred to four or five agar dishes in a matrix of 7 × 7, and the DNA from each row and column was harvested collectively and screened as described previously. A positive matrix cell, which was at the intersection of a positive row and column, was identified as the positive clone and rescreened for accuracy. The cDNA isolated in this manner from pool number 17 was sequenced and identified as full-length rat lmo4.
Plasmids.
The following plasmids were used in this study: GAL4-DBD, GAL4-cAMP response element-binding protein (CREB), and upstream-activating sequence-CAT (UAS-CAT) (Shieh et al., 1998; Hu et al., 1999). lmo4 was isolated and cloned as described above. Full-length lmo4 was PCR-amplified and subcloned into a plasmid containing a cDNA clone (RK5), the pRK5-GAL4-DBD vector, by using SalI and NotI sites. A mammalian expression vector for LIM homeobox 2 (LHX2) was obtained from H. Westphal (National Institutes of Health, Bethesda, MD). This construct was PCR-amplified and subcloned into pRK5-GAL4-DBD, using SalI and NotI sites. All plasmids were sequenced for accuracy.
Immunofluorescence and immunocytochemistry.
Dissociated cells were fixed for 20 min or more with 4% paraformaldehyde and 4% sucrose in PBS, blocked for 2 h in 3% bovine serum albumin and 0.3% Triton X-100 in PBS, and incubated overnight with rabbit anti-CAT (5′→3′, Boulder, CO) antibody at 1:5000 dilution. After 1 h of incubation with anti-rabbit IgG antibody (Vector Laboratories, Burlingame, CA) at 1:200 dilution the avidin–biotin complex reaction was performed by using the ABC kit (Vector Laboratories), and cells were stained with 0.5 mg/ml 3,3-diaminobenzadine. For immunofluorescence on cortical slices and dissociated cells the blocking was performed in the same manner as for immunohistochemistry. Cells and slices were incubated overnight with goat anti-LMO4 antibodies at 1:100 concentration (C-15; Santa Cruz Biotechnology, Santa Cruz, CA), washed several times with PBS, and incubated in donkey anti-goat Alexa Fluor-conjugated secondary antibody (Invitrogen) for 2 h. The cells were washed again with PBS several times and mounted for visualization. For immunofluorescence on tissue sections the tissue was obtained from animals that were perfused with 7–10 vol of ice-cold PBS, followed by 4% paraformaldehyde in PBS. Then each brain was removed and post-fixed overnight in 4% paraformaldehyde in PBS. The next day the fixing solution was replaced with 30% sucrose, and the tissue was left shaking at 4°C until the brains sank to the bottom of the container. Then the brains were embedded in OCT compound (Tissue-Tek–Miles, Elkhart, IN) and mounted on a cryostat (CM3050S; Leica, Nussloch, Germany) for sectioning. Tissue slices were 30–40 μm thick. Other antibodies were used at the following concentrations: mouse anti-microtubule-associated protein 2 (anti-MAP2) 1:1000 (Sigma), mouse anti-neuronal marker (anti-NeuN) 1:1000 (Chemicon, Temecula, CA), and rabbit anti-GABA 1:1000 (Sigma). Cell nuclei were stained for 1 h, using Hoechst 33258 (1:10,000; Invitrogen). Images were acquired by using an LSM 510 Zeiss (Oberkochen, Germany) confocal microscope equipped with helium–neon and argon lasers or a Nikon TE300 (Tokyo, Japan) equipped with mercury arc lamp and filter sets for green fluorescent protein, Texas Red, and 4′,6-diamidino-2-phenylindole (DAPI; Chroma Technology, Rockingham, VT).
Generation and genotyping of conditional lmo4 null mice.
Conditional lmo4 null animals were generated by crossing mice in which the lmo4 gene is flanked by loxP sites (Lee at al., 2005) with a mouse line expressing the Cre-recombinase as a “knock-in” under the control of the neuronal differentiation factor annexin (nex) promoter (Schwab et al., 1998; Wu et al., 2005). This promoter is expressed in the cortex and hippocampus from E14 through adulthood (Schwab et al., 1998, 2000) (see Fig. 6). Genotyping PCR was performed by using Platinum Taq polymerase (Invitrogen) and the following primers: for the nexCre mice, nex 148.s forward primer (GAGTCCTGGAATCAGTCTTT TC), nex as reverse primer (AGAATGTGGAGTAGGGTGAC), and Cre as reverse primer (CCGCATAACCAGTGAAACAG); for the floxed lmo4 mice, floxed lmo4 forward (GACAGTTTAAAGACCTAGGGC) and floxed lmo4 reverse (TGAGACTCTGGAACCAGGACCG). Both PCRs were performed with the following protocol on a PTC-100 programmable thermal controller (MJ Research, Waltham, MA): 95°C for 2 min, 95°C for 30 s, 55°C for 30 s, 68°C for 2 min, cycled 35 times, 55°C for 1 min, and 68°C for 5 min. PCR products were run on 2% agarose gel.
Figure 6.

Targeted deletion of LMO4 in cortex. A, Cre-recombinase activity in the whole brain of floxed LacZ reporter mice crossed with nexCre transgenics. Recombination is seen in the cortex (cx) and hippocampus (hip). la, Lateral amygdaloid nucleus; pir, piriform cortex. B, High magnification of LacZ expression in the cortex after nexCre-mediated recombination. C, D, LMO4 expression (green) and MAP2 expression (red) in P15 cortex in control mice (C; n = 3) and LMO4 conditional knock-out mice (D; n = 3) detected by immunofluorescence. Genotypes are indicated in each panel (nc, nexCre; f, floxed lmo4). E, F, Immunofluorescence for neuronal (green) and GABAergic (red) markers in cortices of control (E) and LMO4 conditional knock-outs (F). G, Western blot detection of LMO4 protein from isolated cortices of control, lmo4 conditional heterozygous, and lmo4 conditional knock-out animals. H, Genotyping PCR from sample litter of mice bearing various lmo4 alleles (top band, 265 nt floxed allele; bottom band, 180 nt wild-type allele) and nexCre alleles (top band, 770 nt wild-type allele; bottom band, 520 nt Cre-recombination allele). The left panel represents PCR results from two lmo4 conditional knock-outs (nc/nc; f/f). The right panel shows PCR results from heterozygous and wild-type animals.
Cytochrome oxidase staining.
Cytochrome oxidase staining was performed on tissue prepared as described above for immunofluorescence. After being sectioned, the tissue was stored in phosphate buffer (PB) at 4°C until use. The tissue was stained for cytochrome oxidase by incubation with solution containing 20 mg of 3,3-diaminobenzadine, 1.5 mg of cytochrome c (Sigma), and 2 g of sucrose in 50 ml total vol of PB (Wong-Riley, 1979). Incubation was performed while shaking at 37°C and monitored every 30–40 min for ∼3–4 h until the desired intensity was observed. The reaction was stopped with two successive washes of fresh PB, and the sections were dehydrated and mounted for visualization.
Western blot analysis.
Cortices from wild-type, conditional heterozygote, and conditional lmo4 null animals were dissected out in ice-cold HBSS and homogenized by using a 2 ml Dounce homogenizer in lysis buffer containing 1% Triton X-100, 0.2% SDS, 150 mm NaCl, 50 mm Tris-HCl, pH 7.5, Complete protease inhibitor mixture tablet (Roche Bioscience, Palo Alto, CA), and 1 μm PMSF. Lysis was allowed to go on for 1–2 h at 4°C. The lysate was centrifuged to eliminate cellular debris, and the supernatant was isolated for use. The supernatant then was purified for LMO4 by the addition of LMO4 antibodies (Sigma) for 1 h at 4°C and subsequently with Protein A/G plus agarose beads (Santa Cruz Biotechnology) for another 1 h at 4°C. Next the sample was spun down briefly, and the beads were isolated. The beads were treated with 2× loading buffer containing β-mercaptoethanol and DTT and boiled for 5 min to release proteins from the beads. The sample was loaded and run on 10% PAGE, transferred to nitrocellulose paper, blocked with 5% nonfat dried milk in TBS with Tween 20 (TBST), and stained for LMO4 by using guinea pig anti-LMO4 (kindly provided by Gordon Gill, University of California, San Diego, La Jolla, CA) at 1:100.
Northern blot analysis.
Developmental Northerns were performed with total RNA extracts from cortices isolated from E18, P0, P7, P14, and adult mice. The cortices were isolated in cold HBSS, and total RNA was extracted via Trizol reagent (Invitrogen) per the manufacturer's instructions. Total RNA was assayed by using spectrophotometric analysis at 260 and 280 nm wave lengths and ethidium bromide gel electrophoresis to resolve 18S and 28S RNA species. Total RNA (10 μg) was loaded per lane onto a 1% agarose/formaldehyde gel, transferred to a nylon membrane after electrophoresis, and hybridized with 32P-labeled cDNA probes. Probes were used as follows: LMO4 was detected by using a full-length probe generated from full-length cDNA with the use of Ready-to-Go-DNA labeling beads (Amersham Biosciences) per the manufacturer's instructions. Cofactor of LIM homeodomain protein (CLIM) was detected by using a 300 bp long N-terminal probe generated from full-length CLIM2 by restriction enzyme digest and subsequent cloning into pcDNA3.
Results
Identification of LMO4 as a mediator of calcium-dependent transcription
To identify transcription factors involved in activity-dependent gene expression, the members of our laboratory have developed a strategy called Transactivator Trap (Aizawa et al., 2004). Transactivator Trap uses a rat cortical cDNA library fused to the DNA-binding domain of the yeast transcription factor GAL4. Discrete pools of the GAL4-cDNA fusion library are screened by transfection into dissociated cortical neurons with a UAS-driven reporter (CAT). Under baseline conditions the GAL4-cDNA fusion protein will be translated and targeted to the UAS site of the reporter without driving reporter activity. However, if any one of the cDNAs in the library encodes an activity-dependent transcription factor, then it will drive reporter expression after depolarization and calcium influx. This fusion library was split into 200 pools, each of which was cotransfected with reporter plasmid into E18 dissociated rat cortical neurons at 3 DIV, using calcium phosphate transfection. After 24 h the E18 cultures were stimulated by increasing the extracellular potassium concentration to 50 mm by the addition of KCl to the culture medium and were incubated at 37°C for 10–12 h. This manipulation depolarizes cells and opens voltage-sensitive calcium channels (VSCCs), allowing calcium influx. Because the GAL4-cDNA fusion proteins already are targeted to the UAS-CAT reporter, the activation of a potential calcium-responsive transcription factor by depolarization should result in transcription of the CAT gene and production of the CAT protein. Breaking down one of the pools identified by this method led to the isolation of LMO4 as the cDNA responsible for calcium-dependent transcription of the reporter.
As shown in Figure 1A, under baseline conditions GAL4-LMO4 does not drive the expression of a UAS-CAT reporter, but depolarization with 50 mm KCl leads to robust expression of the reporter (Fig. 1B). LMO4 is a 165 aa nuclear LMO protein characterized by two tandem 55 aa LIM domains and is expressed predominantly in the cerebral cortex during the early postnatal period (Bulchand et al., 2003) (Fig. 1D–F). Consistent with a potential role in transcriptional regulation, immunofluorescence of E18 dissociated cortical neurons showed that LMO4 was localized to neuronal nuclei (Fig. 1G–I). Northern analysis of LMO4 and its interactor CLIM2 (also called nuclear LIM interactor, NLI) confirmed their expression in the early postnatal cortex, with peak expression at approximately P7 (Fig. 1C).
Figure 1.

Depolarization-induced activation of LMO4-mediated transcription and expression pattern of LMO4. A, B, E18 dissociated cortical neurons at 5 DIV immunostained for CAT after being transfected with GAL4-LMO4 and UAS-CAT and either left untreated or stimulated with 50 mm KCl. C, Northern blot of RNA isolated from cortices at different developmental ages hybridized with probes to LMO4 and CLIM2. Then, the same blot was stripped and hybridized with a glyceraldehyde-3-phosphate dehydrogenase (GAPDH) probe for loading control. D–F, Distribution of LMO4 in the developing cortex detected by immunofluorescence with the use of goat anti-LMO4 (green) and mouse anti-MAP2 (red) antibodies on coronal slices of P15 mouse cortex. G–I, Subcellular localization of LMO4 (green) in E18 dissociated cortical cultures counterstained with a nuclear Hoechst dye (red).
To characterize better the calcium inducibility of LMO4, we transfected dissociated cortical neurons with GAL4-LMO4 and UAS-CAT reporter plasmids and stimulated the cells under various conditions. Activation of VSCCs by the application of 50 mm KCl resulted in a robust and reliable increase in CAT reporter activity (Fig. 2A). The response was comparable to GAL4-CREB, a well characterized calcium-activated transcription factor (Fig. 2A). KCl stimulation did not cause reporter expression in neurons transfected with a GAL4 fusion of LHX2 (Fig. 2B). LHX2 is a LIM homeodomain protein that is localized to layers II–IV of the postnatal cortex (Bulchand et al., 2003). This suggests that the inducibility of LMO4 by calcium influx is not a generic response of LIM domains to depolarization or an artifact of the GAL4 fusion protein but, rather, a specific property of LMO4.
Figure 2.

Characterization of LMO4-mediated transcription. E18 dissociated cortical neurons were transfected with the indicated constructs at 3 DIV and treated as indicated at 5 DIV. A, Relative CAT activity in neurons transfected with GAL4-DBD, GAL4-CREB, or GAL4-LMO4 and UAS-CAT and stimulated with 50 mm KCl. B, Relative CAT activity of neurons transfected with GAL4-LHX2 and UAS-CAT and stimulated with 50 mm KCl. C, Relative CAT activity of neurons transfected with GAL4-LMO4 and UAS-CAT and pretreated with pharmacologic inhibitors as indicated, followed by 50 mm KCl stimulation. D, Relative CAT activity of neurons transfected with wild-type GAL4-LMO4 or constructs with cysteine-to-serine point mutations at either cysteine 23 or 87, as indicated, and stimulated with 50 mm KCl. Asterisks indicate significance at p < 0.05. Error bars represent ± SEM.
Stimulating cortical neurons with 50 mm KCl induces calcium influx via VSCCs. KCl induction of GAL4-LMO4-mediated transcription was eliminated when the extracellular calcium was buffered with EGTA or calcium influx was blocked with cadmium chloride (Fig. 2C) (data not shown), indicating that calcium influx is necessary for LMO4 transactivation. Blocking L-type VSCCs (L-VSCCs) by preincubation with nifedipine also strongly inhibited reporter activity in neurons transfected with GAL4-LMO4 (Fig. 2C). In contrast, the P/Q-type calcium channel blocker, ω-AgaIVA conotoxin, had no significant effect (Fig. 2C). Thus calcium influx via L-VSCCs is required for depolarization-induced activation of LMO4. Blocking action potentials with the voltage-sensitive sodium channel blocker TTX also attenuated KCl-induced LMO4-mediated transactivation, indicating that the effects of depolarization may require action potentials (Fig. 2C).
We next examined whether the LIM domains of LMO4 were required for calcium-dependent activation of LMO4. To investigate the role of these domains, we generated GAL4-LMO4 constructs with cysteine-to-serine mutations of the first conserved cysteine in each LIM domain. These constructs are termed GAL4-LMO4(C23S) and GAL4-LMO4(C87S) to denote the mutation in either amino acid position 23 or position 87 of LMO4. Mutation of any of the conserved cysteines within the LIM domain prevents zinc coordination and ultimately interferes with the tertiary structure of the protein (Feuerstein et al., 1994). Neither of these constructs was capable of significant transactivation in response to 50 mm KCl stimulation (Fig. 2D). These data suggest that the integrity of both LIM domains is necessary for LMO4-mediated transactivation.
LMO4-mediated transcription requires calcium/calmodulin-dependent protein kinase activity
Calcium influx activates multiple second messengers, including the calcium/calmodulin-dependent protein (CaM) kinase (CaMK) and MAP kinase (MAPK) pathways (Ghosh and Greenberg, 1995; Shaywitz and Greenberg, 1999; Ahn et al., 2000; Wu et al., 2001). To determine whether calcium influx induces LMO4-mediated transactivation by one of these pathways, we examined reporter activity in neurons that were transfected with GAL4-LMO4 as well as constitutively active forms of CaMKII, CaMKIV, or the MAPK activator mitogen-activated and extracellular-regulated protein kinase (MEK) (Sun et al., 1994). Constitutively active CaMKIV significantly increased LMO4 transactivation even in the absence of KCl stimulation, but constitutively active CaMKII or MEK had no noticeable effect on LMO4 transactivation (Fig. 3A). These results suggest that activation of the CaMK pathway, particularly CaMKIV, is sufficient for LMO4-mediated transcription.
Figure 3.

Signaling pathways involved in calcium activation of LMO4-mediated transcription. A, Relative CAT activity in E18 dissociated cortical neurons transfected with GAL4-LMO4 and either wild-type or constitutively active forms of CaMK II (CKII), CaMK IV (CKIV), or MEK at 3 DIV and assayed for CAT activity at 5 DIV. B, D, F, Relative CAT activity in E18 dissociated cortical neurons transfected with GAL4-LMO4 at 3 DIV and pretreated with pharmacologic inhibitors, followed by 50 mm KCl stimulation at 5 DIV. C, E, Relative CAT activity in E18 dissociated cortical neurons transfected with GAL4-LMO4 and UAS-CAT, together with indicated constructs, at 3 DIV and stimulated as indicated at 5 DIV. Asterisks indicate significance at p < 0.05. Error bars represent ± SEM.
To determine whether CaMK function was necessary for LMO4-mediated transactivation, we preincubated cortical neurons with KN62, a CaMK inhibitor. This treatment dramatically reduced KCl-induced transactivation of LMO4, indicating that CaMKs are necessary for LMO4 activation (Fig. 3B). To control for any nonspecific effects of KN62 on calcium influx, we repeated these experiments with another CaMK inhibitor, KN93, and its inactive analog KN92. KN93, but not KN92, also markedly reduced KCl-induced transactivation of LMO4 (data not shown). These results suggest that the CaMK pathway is necessary for LMO4-mediated transcription.
To determine the relative contributions of CaMKII and CaMKIV to calcium-dependent activation of LMO4, we examined the consequences of expressing constitutively active and dominant-negative forms of CaMKII and CaMKIV on KCl-induced activation of GAL4-LMO4. Whereas dominant-negative CaMKII did not affect calcium activation of GAL4-LMO4 significantly, constitutively active CaMKII blocked calcium-dependent activation (Fig. 3C). This unexpected result is similar to the effect of constitutively active CaMKII on CREB-mediated transcription (Sun et al., 1994) and suggests that LMO4 and CREB might function in the same complex. Indeed, as described later (see Fig. 5), we have found evidence for an interaction between LMO4 and CREB. In contrast to the effects of dominant-negative CaMKII, expression of dominant-negative CaMKIV completely abolished calcium activation of GAL4-LMO4 (Fig. 3C). These observations, together with the effects of constitutively active CaMKIV and pharmacological CaMK inhibitors, suggest that calcium signaling activates LMO4-mediated transcription via CaMKIV.
Figure 5.
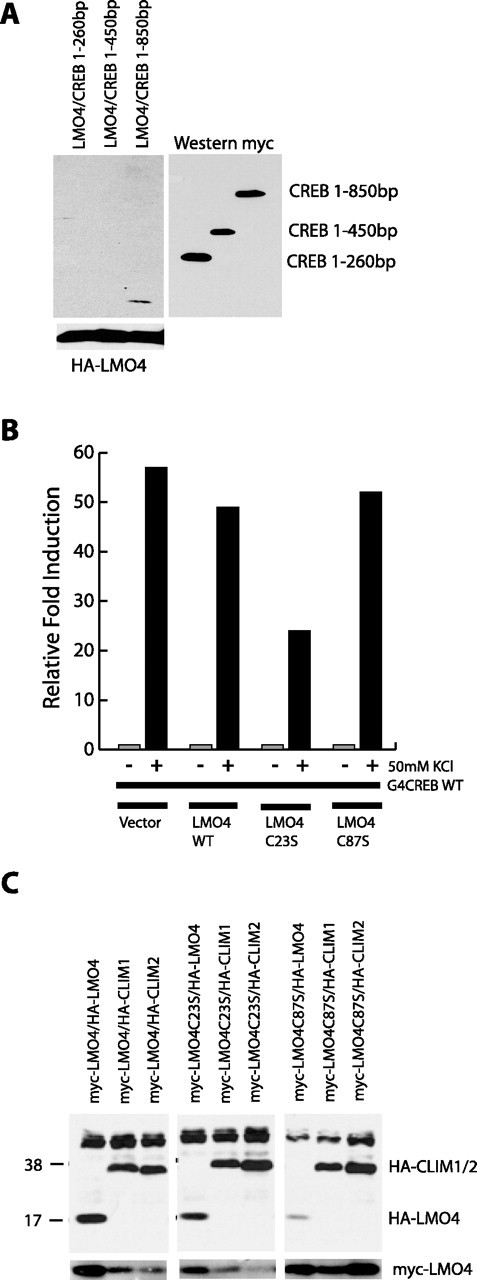
LMO4 interacts with CREB, CLIM1, and CLIM2. A, Coimmunoprecipitation of HA-tagged LMO4 with myc-tagged CREB deletion constructs in 293T-cells. Myc immunoprecipitations followed by anti-HA Western blots show the interaction of LMO4 with CREB base pairs 1–850, but not with CREB deletion constructs containing base pairs 1–260 and 1–450 (top left). Equivalent transfection of HA-LMO4 in all plates is indicated by the HA Western blot on whole-cell lysates (bottom left). Equivalent expression of CREB constructs is indicated by the myc-Western blot (bottom right). B, Relative CAT activity in E18 neurons transfected with GAL4-CREB and UAS-CAT, together with indicated constructs, at 3 DIV and stimulated at 5 DIV. C, Coimmunoprecipitation assays in 293T-cells transfected with myc-tagged LMO4, together with HA-LMO4, HA-CLIM1, or HA-CLIM2. LMO4 can interact with LMO4 as well as with both CLIM1 and CLIM2. Mutation of cysteine 87 to serine [LMO4(C87S)] in the LMO4 protein prevents LMO4 dimerization. LMO4(C23S) and LMO4(C87S) indicate constructs in which the conserved cysteine at position 23 or 87, respectively, was mutated to a serine.
To explore the potential contribution of other calcium-regulated second messenger systems, we performed transactivation assays in cortical neurons in the presence of pharmacological inhibitors of the protein kinase A (PKA) and MAPK pathways. Preincubation of neurons with 10 μm UO126, a specific blocker of MEK1 and MEK2 (Favata et al., 1998; Davies et al., 2000), substantially inhibited LMO4 transactivation (Fig. 3D), but preincubation with 1 μm KT 5720 (an inhibitor of PKA; Sigma) had only a modest effect on LMO4-mediated transactivation (Fig. 3D). Similarly, GAL4-LMO4-mediated transcription was not inhibited in neurons cotransfected with the PKA inhibitor (PKI) (Fig. 3E). These results suggest that MAPK activity, but not PKA activity, is necessary for calcium-dependent activation of LMO4.
To determine whether PKA activation was sufficient to induce LMO4-mediated transcription, we treated neurons with 10 μm forskolin (Fig. 3F). This led to robust expression of reporter activity, indicating that PKA can induce LMO4-mediated transactivation. Together, our results suggest that calcium influx induces LMO4-mediated transcription principally via activation of CaMKIV and MAPK. PKA also can induce LMO4-dependent transcription, but it is not required for calcium-dependent activation.
NMDAR function is necessary for synaptic activation of LMO4-mediated transcription
In addition to L-VSCCs, NMDARs regulate increases in intracellular calcium levels, and the activation of both of these channels has been linked to changes in gene expression (Bading et al., 1993; Deisseroth et al., 1996; Hardingham et al., 2001). Because of the central role of NMDARs in activity-dependent development and plasticity, we next investigated the potential role of NMDARs in LMO4-mediated transactivation.
Exogenous application of glutamate to cortical neurons transfected with GAL4-LMO4 and UAS-CAT resulted in a robust increase in reporter activity, indicating that glutamate receptor activation can drive LMO4-mediated transactivation (Fig. 4A). This stimulus paradigm, however, does not differentiate between the contribution of synaptic and nonsynaptic receptors. To determine whether excitatory synaptic stimulation can drive LMO4-mediated transcription, we examined the effects of disinhibiting mature cortical cultures with the GABAA receptor antagonist bicuculline. This is an effective method to increase the excitatory synaptic drive in 12–14 DIV cultures (Hardingham et al., 2001) (A. Kashani, Ghosh, and B. Hall, unpublished observations). Bicuculline treatment of 12–14 DIV cultures resulted in a significant increase in reporter activity when compared with controls (Fig. 4B,C), indicating that synaptic activity can drive LMO4-mediated transcription.
Figure 4.

Synaptic stimulation induces LMO4-mediated transcription via NMDAR and L-VSCC activation. A, Relative CAT activity in E18 dissociated cortical neurons transfected with GAL4-LMO4 and UAS-CAT at 3 DIV and stimulated with either 50 mm KCl or 10 μm glutamate at 5 DIV, as indicated. B, C, Relative CAT activity in E18 cortical cultures transfected with GAL4-LMO4 and UAS-CAT at 3 DIV and treated as indicated at 12–14 DIV. Asterisks indicate significance at p < 0.05. Error bars represent ± SEM.
Bicuculline treatment leads to a marked increase in cellular activity, which would lead to the opening of VSCCs along with NMDARs. To assess the contribution of L-VSCCs and NMDARs to synaptic activation of LMO4, we examined the effect of blocking these channels on bicuculline-induced LMO4-mediated transactivation. As shown in Figure 4C, preincubation with the L-VSCC antagonist nifedipine or the NMDAR antagonist APV substantially suppressed activity-induced reporter expression. This indicates that both NMDARs and L-VSCCs contribute to synaptically induced LMO4-dependent transcription. As in the case of KCl-induced transactivation, bicuculline-induced LMO4 transactivation also was suppressed by the CaMK inhibitor KN62 (Fig. 4C).
Interaction of LMO4 with CREB and CLIM1/CLIM2
The activation properties of LMO4 are strikingly similar to those of CREB. Both are activated by calcium influx via VSCCs and NMDARs, both require CaMK and MAPK activity for transactivation, and both are inhibited by a constitutively active form of CaMKII and are activated by a constitutively active form of CaMKIV. These similarities motivated us to examine the possibility that LMO4 and CREB might be part of the same transcription complex. This possibility also seemed plausible given the reported interaction between CREB and CREM and the LIM domain-only protein four-and-half LIM domain 2 (FHL2) (Fimia et al., 2000).
To determine whether LMO4 can interact with CREB, we coexpressed hemagglutinin-tagged (HA-tagged) LMO4 together with various myelocytomatosis-tagged (myc-tagged) deletions of CREB in human embryonic kidney 293 T-cells (293T-cells). As shown in Figure 5A, myc immunoprecipitation followed by HA Western blots indicated that HA-LMO4 could interact with full-length myc-CREB, but not with the fragments lacking the C terminus of CREB. These observations suggest that LMO4 can interact with CREB via a mechanism that requires the C terminus of CREB.
To determine whether LMO4 can influence depolarization-induced activation of CREB, we expressed GAL4-CREB together with wild-type LMO4 or LMO4 mutants with disrupted LIM domains. As shown in Figure 5B, KCl activation of GAL4-CREB was not affected by wild-type LMO4 or LMO4(C87S) but was attenuated significantly by coexpression of LMO4(C23S). This suggests that the first LIM domain of LMO4 contributes to calcium activation of CREB-mediated transcription.
To characterize the LMO4 complex additionally, we examined the ability of LMO4 to form homodimers and to interact with the previously identified coactivators CLIM1 and CLIM2. Cotransfection in 293T-cells followed by immunoprecipitation revealed that LMO4 interacts strongly with LMO4 as well as with CLIM1 and CLIM2 (Fig. 5C). Although mutation of the first LIM domain [LMO4(C23S)] does not affect these interactions, disruption of the second LIM domain [LMO4(C87S)] reduces the affinity of LMO4 for itself but does not affect binding to CLIM1 or CLIM2. These observations suggest the existence of a CREB-LMO4 and an LMO4-CLIM interaction. The first LIM domain of LMO4 contributes to CREB-mediated transcription, and the second LIM domain contributes to LMO4–LMO4 interactions (Fig. 5B,C). The fact that disruption of either LIM domain leads to a loss of calcium activation of LMO4 suggests the possibility that the active LMO4 complex might contain CREB, LMO4, and CLIM and that LMO4 might need to interact with both LMO4 and CREB for calcium-dependent activation.
Effect of conditional deletion of lmo4 on cortical development
The calcium activation properties of LMO4 and expression pattern suggest that it might contribute to activity-dependent cortical development (Kenny et al., 1998). To explore this possibility, we decided to examine the consequences of the loss of lmo4 on the development of barrel cortex, which is known to depend on NMDAR signaling (Iwasato et al., 1997, 2000). Because a null mutation of the lmo4 gene causes perinatal lethality caused by anencephaly or exencephaly (Hahm et al., 2004; Tse et al., 2004; Lee et al., 2005), we examined the effects of conditional loss of lmo4 in the cortex.
Conditional lmo4 null animals were generated by crossing floxed lmo4 mice with a mouse line expressing the Cre-recombinase as a knock-in under the control of the nex promoter [for full description, see Wu et al. (2005) and Schwab et al. (1998)]. This promoter is primarily active in the cortex and hippocampus from E14 through adulthood (Schwab et al., 1998, 2000). To characterize the spatial and temporal specificity of Cre-mediated recombination in these mice, we crossed nexCre/Cre mice with a floxed β-galactosidase (LacZ) reporter mouse. nexCre/LacZ heterozygotes displayed strong X-gal staining throughout all layers of the cortex and hippocampus from E14 through adulthood (Fig. 6A,B). During embryogenesis LacZ expression was restricted to the cortical plate, indicating that the promoter is active in postmitotic neurons.
nexCre/Cre and nexCre/+ mice were crossed with lmo4flox/flox or lmo4flox/+ mice to generate four types of mice: (1) nexCre/Cre;lmo4flox/flox, (2) nexCre/+;lmo4flox/flox, (3) nexCre/+;lmo4flox/+, and (4) nex+/+;lmo4flox/flox. Recombination at the floxed lmo4 locus in both of the first two lines eliminates the starting ATG and the first coding exon of both lmo4 alleles and prevents the production of any LMO4 protein (Fig. 6C,D,G,H). Conditional lmo4 null mice were viable through adulthood and were indistinguishable from wild-type littermates. Immunofluorescence analysis of cortical sections from control and conditional lmo4 null animals with antibodies to glial fibrillary acidic protein (GFAP), MAP2, neuronal antigen NeuN, and the inhibitory neuronal marker GABA showed no gross differences in cortical organization among the genotypes (Fig. 6E,F). Hoechst staining revealed no differences in apoptotic profiles between control and conditional lmo4 null mice (data not shown).
To determine whether LMO4 plays a role in barrel cortex development, we assessed the development of somatosensory barrels in conditional lmo4 null animals. The barrels are formed by discrete groups of layer IV cell bodies, which form the walls of the barrels, and invading thalamocortical afferents, which occupy the center of the barrels. Conditional lmo4 null mice had no abnormalities in the arrangement of whiskers on the snout or in the somatotopic map formed in the thalamus as assessed by cytochrome oxidase staining, indicating that the subcortical somatosensory system forms normally in these animals (data not shown). Examination of cortical barrels by using cytochrome oxidase staining in coronal and tangential sections revealed that the development of the barrel field was disrupted in conditional lmo4 null mice. In contrast to control animals in which discrete barrels in layer IV are clearly visible (Fig. 7A,B,E,F), in conditional lmo4 nulls the barrels were poorly differentiated, the barrel edges were not defined, and, when identifiable, the barrels were smaller than those in controls (Fig. 7C,D,G,H).
Figure 7.

Mice with a conditional deletion of LMO4 have abnormal development of somatosensory barrels. Shown is cytochrome oxidase staining of coronal and tangential sections from control (A, B, E, F; n = 15) and lmo4 conditional knock-out (cKO) cortices (C, D, G, H; n = 9) at P15. Barrels consist of denser patches of staining interspersed with lighter areas (arrows in A and B). Control animals have clearly defined barrel patterns in both coronal and tangential planes, whereas conditional lmo4 knock-outs have either no barrels or very small patches of staining. Genotypes of animals are indicated in the bottom left corner of each panel. Similar results were seen in nine conditional lmo4 knock-out mice and 15 control mice between P15 and P30, indicating that this is not attributable to a developmental delay.
Although cytochrome oxidase staining is a useful method to assess the overall organization of barrel cortex, it does not provide specific information about the distribution of thalamocortical innervation of barrel cortex. To determine whether the segregation of thalamocortical afferents in barrel cortex was disrupted in conditional lmo4 null mice, we used 5-hydroxytryptamine (5-HT; serotonin) immunohistochemistry, which can be used to assess the distribution of thalamic afferents in the cortex during the first 10 d of postnatal life. 5-HT immunocytochemistry labels thalamic afferents because of the presence of the 5-HT serotonin transporter (SerT) on these terminals, which concentrate 5-HT in thalamic terminals (Lebrand et al., 1996). As shown in Figure 8A–C, thalamocortical afferents are well segregated in control mice in both the coronal and tangential planes. In contrast, the segregation of thalamocortical afferents is affected in conditional lmo4 null mice. Thalamocortical afferents in conditional lmo4 null mice do not segregate into distinct terminal patches, and, when they are distinguishable, they tend to be significantly smaller than control barrels (Fig. 8D–F). Quantification of the cross-sectional area of barrels in rows B–D in control and conditional lmo4 null mice reveals this decrease in barrel cross section (Fig. 8G). These results show that conditional loss of lmo4 does not prevent thalamocortical projections from reaching the cortex, but it does affect their patterning in the target tissue.
Figure 8.
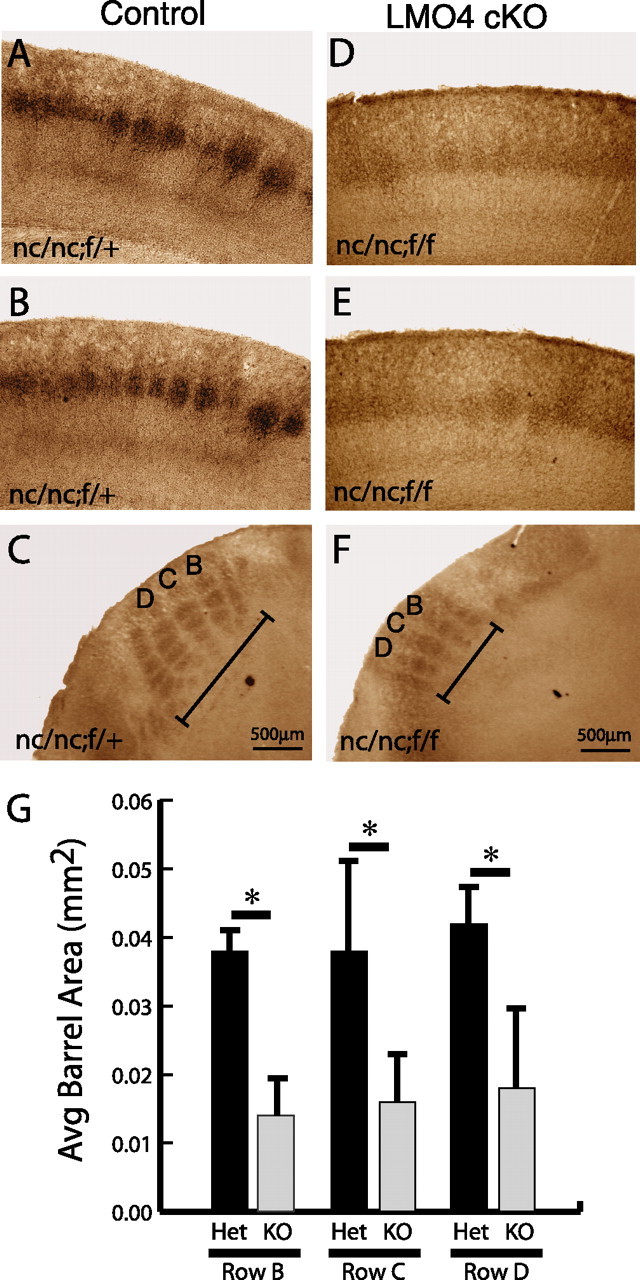
Segregation of thalamocortical afferents is disrupted in conditional LMO4 null mice. P7 mice cortices were fixed, sectioned, and stained for 5-HT immunohistochemistry. Coronal (A, B) and tangential (C) sections of control mouse (genotype nc/nc;f/+) reveal normal patterning of thalamocortical afferents as revealed by 5-HT immunohistochemistry. Coronal (D, E) and tangential (F) sections of conditional LMO4 null mice (genotype nc/nc;f/f) reveal poor segregation of thalamocortical afferents with small barrels, poorly defined boundaries, and overall decreased cross-sectional area (G; labeled in C and F as rows B, C, D) compared with controls. Asterisks indicate significance at p < 0.05 by paired Student's t test. Error bars represent ± SEM.
As an alternate method to examine the distribution of thalamocortical afferents in lmo4 mutant mice, we injected DiI into the ventrobasal nucleus in the thalamus to label thalamocortical projections to the barrel cortex in wild-type and conditional lmo4 null mice. As shown in supplemental Figure 1 (available at www.jneurosci.org as supplemental material), segregation of thalamocortical axon terminals into discrete cortical barrels is clearly visible in wild-type mice. In contrast, the labeling of thalamocortical axon terminals in conditional lmo4 null mice is diffuse. The defect in segregation in DiI-labeled brains (analyzed at P15) appears to be more severe than that seen in 5-HT-stained brain sections (analyzed at P7). This may reflect an increasing severity of the phenotype with age.
Because barrel boundaries are defined by the distribution of layer IV neurons, we performed Hoechst staining in coronal and tangential sections to determine whether layer IV neurons could form normal barrel walls in conditional lmo4 null mice. This staining revealed that conditional lmo4 null mice lack the distinct barrel-like organization of layer IV neuronal cell bodies normally seen in controls (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Thus both presynaptic and postsynaptic organization of the barrel field is disrupted in the absence of cortical LMO4.
To determine whether loss of lmo4 affects other aspects of neuronal development, we examined the effects of the loss of lmo4 on dendritic development, which we had shown previously to require calcium-dependent transcription (Redmond et al., 2002). Retrograde labeling of layer 2/3 neurons in wild-type and conditional lmo4 null mice at P11 revealed no major defects in neuronal morphology (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Although the number of clearly labeled neurons did not allow for detailed morphometric analysis, LMO4 appears not to have a major role in regulating dendritic growth in layer 2/3 neurons. This is in contrast to mice lacking CREST or neuronal determination factor 2 (Neuro D2), which have major defects in dendritic patterning (Aizawa et al., 2004) (G. Ince-Dunn and Ghosh, unpublished observations). It remains to be determined whether the morphology of neurons in layer 4 is affected in conditional lmo4 null mice. This would be interesting to investigate, given that axon segregation is affected in that layer.
Discussion
In this study, we identify LMO4 as a novel mediator of calcium-dependent transcription that plays an important role in the patterning of connections in barrel cortex. LMO proteins originally were discovered from studies of translocations in human acute T-cell leukemia (Boehm et al., 1988; Royer-Pokora et al., 1991) and are expressed throughout the adult body as well as the nervous system, but little is known about their function (Grutz et al., 1998; Sugihara et al., 1998; Chen et al., 2002). It recently was reported that LMO4 is expressed asymmetrically in the developing human cerebral cortex, suggesting that it might contribute to the emergence of brain asymmetry (Sun et al., 2005). Our observations suggest that observed asymmetries in brain LMO4 expression well may translate into differences in connectivity in the two hemispheres.
LMO4-mediated transcription is responsive to calcium influx through both L-VSCCs and NMDARs. Because nifedipine blocks only ∼15–30% of the KCl-induced somatic calcium influx, these results suggest that the source of calcium influx plays an important role in LMO4 activation (Redmond et al., 2002). Previous studies have shown that L-VSCCs have a preferential nuclear signaling capability over other members of the high-voltage-activated calcium channel family (Murphy et al., 1991; Shieh et al., 1998; Dolmetsch et al., 2001). One explanation for this capability is the close association of the intracellular domain of the channel with calmodulin (Erickson et al., 2001). A recent study has shown that mutation of the L-VSCC calmodulin-binding domain prevents activation of a Cre-dependent reporter via the Ras-MAPK pathway (Dolmetsch et al., 2001). This activation depends on the calmodulin-binding properties of the cytoplasmic tail of the α-1C subunit of the channel (Dolmetsch et al., 2001; Erickson et al., 2001). A similar mechanism may be involved in the VSCC-dependent activation of LMO4.
To determine whether LMO4 can drive transcription in response to synaptic stimulation, we performed experiments in mature cortical cultures that were treated with bicuculline. Bicuculline-induced increase in activity can drive LMO4-mediated transcription and is blocked by APV, indicating that NMDAR activation is necessary for synaptic activation of LMO4-mediated transcription. Interestingly, nifedipine also blocks synaptic activation of LMO4. This suggests that both NMDARs and L-VSCCs are necessary for synaptic activation of LMO4. It is possible that NMDAR-mediated calcium influx activates second messengers that directly drive LMO4-mediated transcription. Another possibility is that NMDARs provide the depolarizing current necessary for the opening of L-VSCCs, which then activate LMO4. These two models are not mutually exclusive, and it may be that both the calcium influx and voltage change resulting from NMDAR activation are necessary for LMO4-mediated transcription.
The effect of calcium influx on the activation of LMO4 is mediated principally by CaMKIV, which is both necessary and sufficient for LMO4 activation. Inhibitors of the MAPK pathway also block LMO4-mediated transcription, indicating that signaling via MAPK also is required for activation of LMO4. The observation that the Ras-MAPK pathway acts downstream of both L-VSCCs (Dolmetsch et al., 2001) and NMDARs (Hardingham et al., 2001) is consistent with this model.
LMO4 contains two closely spaced LIM domains that are implicated in a number of protein–protein interactions (Sugihara et al., 1998; Sum et al., 2002). Because LMO4 has not been shown to bind DNA directly, it seems likely that LMO4 exerts its effects on nervous system development by interacting with DNA-binding transcription factors and acting as a mediator of activity-dependent transcription, perhaps in a manner analogous to CREB-binding protein (CBP) (Hu et al., 1999). In this regard the interaction of LMO4 with CREB and the effects of LMO4 mutations on CREB-mediated transcription are very interesting, because they suggest that LMO4 contributes to calcium activation of the CREB complex. We should note, however, that the in vivo interaction of CREB and LMO4 remains to be confirmed. Our attempts at exploring the interaction of the endogenous proteins in vivo were confounded by the poor quality of commercial LMO4 antibodies, which did not allow us to obtain definitive coimmunoprecipitation data on the endogenous proteins. Future experiments are needed to show that this complex does, in fact, assemble in neurons. It also will be important to determine whether the CREB–LMO4–CLIM complex is distinct from the CREB–CBP complex or whether all of the proteins exist in one large transcription complex.
Our results show that conditional deletion of lmo4 in the cortex disrupts the normal formation of barrels in the somatosensory cortex. Specifically, we find that thalamocortical afferents fail to segregate into distinct patches in lmo4 mutant mice and that the barrels are smaller in cross-sectional area when they do form. These defects are similar to defects reported in conditional deletion of the NR1 subunit in mouse cortex (Iwasato et al., 2000). Given our observation that NMDAR activation regulates LMO4-dependent transcription, it is likely that the effects of NMDAR activation on barrel formation are mediated, at least in part, via LMO4-dependent transcription. We propose that activation of NMDARs at thalamocortical synapses leads to LMO4-dependent expression of genes that are important for the segregation of thalamic afferents into cortical barrels. Although the identity of genes regulated by LMO4 remains to be determined, in preliminary experiments we found that LMO4 expression enhances calcium-dependent expression of a BDNF reporter gene. In summary, our findings suggest that activity-dependent regulation of LMO4-mediated transcription plays an important role in the patterning of thalamocortical connections.
Footnotes
This work was supported by National Institutes of Health Grants MH60598 and MH68578 and a National Alliance for Research on Schizophrenia and Depression Independent Investigator Award (A.G.). We thank Jordan Buchman, Kathryn Bobb, and Shu-Ching Hu for their contributions to this project, Dr. Gordon Gill (University of California, San Diego, La Jolla, CA) for his generous contribution of LMO4 antibody, and members of the Ghosh laboratory for discussions.
References
- Ahn S, Riccio A, Ginty DD (2000). Spatial considerations for stimulus-dependent transcription in neurons. Annu Rev Physiol 62:803–823. [DOI] [PubMed] [Google Scholar]
- Aizawa H, Hu SC, Bobb K, Balakrishnan K, Ince G, Gurevich I, Cowan M, Ghosh A (2004). Dendrite development regulated by CREST, a calcium-regulated transcriptional activator. Science 303:197–202. [DOI] [PubMed] [Google Scholar]
- Bading H, Ginty DD, Greenberg ME (1993). Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science 260:181–186. [DOI] [PubMed] [Google Scholar]
- Boehm T, Baer R, Lavenir I, Forster A, Waters JJ, Nacheva E, Rabbitts TH (1988). The mechanism of chromosomal translocation t(11;14) involving the T-cell receptor Cδ locus on human chromosome 14q11 and a transcribed region of chromosome 11p15. EMBO J 7:385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulchand S, Subramanian L, Tole S (2003). Dynamic spatiotemporal expression of LIM genes and cofactors in the embryonic and postnatal cerebral cortex. Dev Dyn 226:460–469. [DOI] [PubMed] [Google Scholar]
- Chen HH, Yip JW, Stewart AF, Frank E (2002). Differential expression of a transcription regulatory factor, the LIM domain only 4 protein LMO4, in muscle sensory neurons. Development 129:4879–4889. [DOI] [PubMed] [Google Scholar]
- Constantine-Paton M, Cline HT, Debski E (1990). Patterned activity, synaptic convergence, and the NMDA receptor in developing visual pathways. Annu Rev Neurosci 13:129–154. [DOI] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M, Cohen P (2000). Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J 351:95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deisseroth K, Bito H, Tsien RW (1996). Signaling from synapse to nucleus: postsynaptic CREB phosphorylation during multiple forms of hippocampal synaptic plasticity. Neuron 16:89–101. [DOI] [PubMed] [Google Scholar]
- Dolmetsch RE, Pajvani U, Fife K, Spotts JM, Greenberg ME (2001). Signaling to the nucleus by an L-type calcium channel–calmodulin complex through the MAP kinase pathway. Science 294:333–339. [DOI] [PubMed] [Google Scholar]
- Erickson MG, Alseikhan BA, Peterson BZ, Yue DT (2001). Preassociation of calmodulin with voltage-gated Ca2+ channels revealed by FRET in single living cells. Neuron 31:973–985. [DOI] [PubMed] [Google Scholar]
- Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL, Scherle PA, Trzaskos JM (1998). Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem 273:18623–18632. [DOI] [PubMed] [Google Scholar]
- Feuerstein R, Wang X, Song D, Cooke NE, Liebhaber SA (1994). The LIM/double zinc-finger motif functions as a protein dimerization domain. Proc Natl Acad Sci USA 91:10655–10659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fimia GM, De Cesare D, Sassone-Corsi P (2000). A family of LIM-only transcriptional coactivators: tissue-specific expression and selective activation of CREB and CREM. Mol Cell Biol 20:8613–8622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox K, Schlaggar BL, Glazewski S, O’Leary DD (1996). Glutamate receptor blockade at cortical synapses disrupts development of thalamocortical and columnar organization in somatosensory cortex. Proc Natl Acad Sci USA 93:5584–5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Greenberg ME (1995). Calcium signaling in neurons: molecular mechanisms and cellular consequences. Science 268:239–247. [DOI] [PubMed] [Google Scholar]
- Goodman CS, Shatz CJ (1993). Developmental mechanisms that generate precise patterns of neuronal connectivity. Cell 72:[Suppl], 77–98. [DOI] [PubMed] [Google Scholar]
- Grutz G, Forster A, Rabbitts TH (1998). Identification of the LMO4 gene encoding an interaction partner of the LIM-binding protein LDB1/NLI1: a candidate for displacement by LMO proteins in T-cell acute leukemia. Oncogene 17:2799–2803. [DOI] [PubMed] [Google Scholar]
- Hahm K, Sum EY, Fujiwara Y, Lindeman GJ, Visvader JE, Orkin SH (2004). Defective neural tube closure and anteroposterior patterning in mice lacking the LIM protein LMO4 or its interacting partner Deaf-1. Mol Cell Biol 24:2074–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannan AJ, Blakemore C, Katsnelson A, Vitalis T, Huber KM, Bear M, Roder J, Kim D, Shin HS, Kind PC (2001). PLC-β1, activated via mGluRs, mediates activity-dependent differentiation in cerebral cortex. Nat Neurosci 4:282–288. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Arnold FJ, Bading H (2001). A calcium microdomain near NMDA receptors: on switch for ERK-dependent synapse-to-nucleus communication. Nat Neurosci 4:565–566. [DOI] [PubMed] [Google Scholar]
- Hu SC, Chrivia J, Ghosh A (1999). Regulation of CBP-mediated transcription by neuronal calcium signaling. Neuron 22:799–808. [DOI] [PubMed] [Google Scholar]
- Iwasato T, Erzurumlu RS, Huerta PT, Chen DF, Sasaoka T, Ulupinar E, Tonegawa S (1997). NMDA receptor-dependent refinement of somatotopic maps. Neuron 19:1201–1210. [DOI] [PubMed] [Google Scholar]
- Iwasato T, Datwani A, Wolf AM, Nishiyama H, Taguchi Y, Tonegawa S, Knopfel T, Erzurumlu RS, Itohara S (2000). Cortex-restricted disruption of NMDAR1 impairs neuronal patterns in the barrel cortex. Nature 406:726–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz LC, Shatz CJ (1996). Synaptic activity and the construction of cortical circuits. Science 274:1133–1138. [DOI] [PubMed] [Google Scholar]
- Kenny DA, Jurata LW, Saga Y, Gill GN (1998). Identification and characterization of LMO4, an LMO gene with a novel pattern of expression during embryogenesis. Proc Natl Acad Sci USA 95:11257–11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebrand C, Cases O, Adelbrecht C, Doye A, Alvarez C, El Mestikawy S, Seif I, Gaspar P (1996). Transient uptake and storage of serotonin in developing neurons. Neuron 17:823–835. [DOI] [PubMed] [Google Scholar]
- Lee SK, Jurata LW, Nowak R, Lettieri K, Kenny DA, Pfaff SL, Gill GN (2005). The LIM domain-only protein LMO4 is required for neural tube closure. Mol Cell Neurosci 28:205–214. [DOI] [PubMed] [Google Scholar]
- Lu HC, She WC, Plas DT, Neumann PE, Janz R, Crair MC (2003). Adenylyl cyclase I regulates AMPA receptor trafficking during mouse cortical “barrel” map development. Nat Neurosci 6:939–947. [DOI] [PubMed] [Google Scholar]
- Mitrovic N, Mohajeri H, Schachner M (1996). Effects of NMDA receptor blockade in the developing rat somatosensory cortex on the expression of the glia-derived extracellular matrix glycoprotein tenascin-C. Eur J Neurosci 8:1793–1802. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Worley PF, Baraban JM (1991). L-type voltage-sensitive calcium channels mediate synaptic activation of immediate early genes. Neuron 7:625–635. [DOI] [PubMed] [Google Scholar]
- O’Leary DD, Ruff NL, Dyck RH (1994). Development, critical period plasticity, and adult reorganizations of mammalian somatosensory systems. Curr Opin Neurobiol 4:535–544. [DOI] [PubMed] [Google Scholar]
- Redmond L, Kashani AH, Ghosh A (2002). Calcium regulation of dendritic growth via CaM kinase IV and CREB-mediated transcription. Neuron 34:999–1010. [DOI] [PubMed] [Google Scholar]
- Royer-Pokora B, Loos U, Ludwig WD (1991). TTG-2, a new gene encoding a cysteine-rich protein with the LIM motif, is overexpressed in acute T-cell leukaemia with the t(11;14)(p13;q11). Oncogene 6:1887–1893. [PubMed] [Google Scholar]
- Schlaggar BL, Fox K, O’Leary DD (1993). Postsynaptic control of plasticity in developing somatosensory cortex. Nature 364:623–626. [DOI] [PubMed] [Google Scholar]
- Schwab MH, Druffel-Augustin S, Gass P, Jung M, Klugmann M, Bartholomae A, Rossner MJ, Nave KA (1998). Neuronal basic helix–loop–helix proteins (NEX, Neuro D, NDRF): spatiotemporal expression and targeted disruption of the NEX gene in transgenic mice. J Neurosci 18:1408–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab MH, Bartholomae A, Heimrich B, Feldmeyer D, Druffel-Augustin S, Goebbels S, Naya FJ, Zhao S, Frotscher M, Tsai MJ, Nave KA (2000). Neuronal basic helix–loop–helix proteins (NEX and BETA2/Neuro D) regulate terminal granule cell differentiation in the hippocampus. J Neurosci 20:3714–3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaywitz AJ, Greenberg ME (1999). CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem 68:821–861. [DOI] [PubMed] [Google Scholar]
- Shieh PB, Hu SC, Bobb K, Timmusk T, Ghosh A (1998). Identification of a signaling pathway involved in calcium regulation of BDNF expression. Neuron 20:727–740. [DOI] [PubMed] [Google Scholar]
- Sugihara TM, Bach I, Kioussi C, Rosenfeld MG, Andersen B (1998). Mouse deformed epidermal autoregulatory factor 1 recruits a LIM domain factor, LMO-4, and CLIM coregulators. Proc Natl Acad Sci USA 95:15418–15423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sum EY, Peng B, Yu X, Chen J, Byrne J, Lindeman GJ, Visvader JE (2002). The LIM domain protein LMO4 interacts with the cofactor CtIP and the tumor suppressor BRCA1 and inhibits BRCA1 activity. J Biol Chem 277:7849–7856. [DOI] [PubMed] [Google Scholar]
- Sun P, Enslen H, Myung PS, Maurer RA (1994). Differential activation of CREB by Ca2+/calmodulin-dependent protein kinases type II and type IV involves phosphorylation of a site that negatively regulates activity. Genes Dev 8:2527–2539. [DOI] [PubMed] [Google Scholar]
- Sun T, Patoine C, Abu-Khalil A, Visvader J, Sum E, Cherry TJ, Orkin SH, Geschwind DH, Walsh CA (2005). Early asymmetry of gene transcription in embryonic human left and right cerebral cortex. Science 308:1794–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Threadgill R, Bobb K, Ghosh A (1997). Regulation of dendritic growth and remodeling by Rho, Rac, and Cdc42. Neuron 19:625–634. [DOI] [PubMed] [Google Scholar]
- Tse E, Smith AJ, Hunt S, Lavenir I, Forster A, Warren AJ, Grutz G, Foroni L, Carlton MB, Colledge WH, Boehm T, Rabbitts TH (2004). Null mutation of the Lmo4 gene or a combined null mutation of the Lmo1/Lmo3 genes causes perinatal lethality, and Lmo4 controls neural tube development in mice. Mol Cell Biol 24:2063–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Loos H, Woolsey TA (1973). Somatosensory cortex: structural alterations following early injury to sense organs. Science 179:395–398. [DOI] [PubMed] [Google Scholar]
- West AE, Chen WG, Dalva MB, Dolmetsch RE, Kornhauser JM, Shaywitz AJ, Takasu MA, Tao X, Greenberg ME (2001). Calcium regulation of neuronal gene expression. Proc Natl Acad Sci USA 98:11024–11031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong-Riley M (1979). Changes in the visual system of monocularly sutured or enucleated cats demonstrable with cytochrome oxidase histochemistry. Brain Res 171:11–28. [DOI] [PubMed] [Google Scholar]
- Wu GY, Deisseroth K, Tsien RW (2001). Activity-dependent CREB phosphorylation: convergence of a fast, sensitive calmodulin kinase pathway and a slow, less-sensitive mitogen-activated protein kinase pathway. Proc Natl Acad Sci USA 98:2808–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SX, Goebbels S, Nakamura K, Nakamura K, Kometani K, Minato N, Kaneko T, Nave KA, Tamamaki N (2005). Pyramidal neurons of upper cortical layers generated by NEX-positive progenitor cells in the subventricular zone. Proc Natl Acad Sci USA 102:17172–17177. [DOI] [PMC free article] [PubMed] [Google Scholar]