

Abstract
Endocannabinoid signaling has been demonstrated to mediate depolarization-induced suppression of excitation at climbing fiber (CF) and parallel fiber (PF) synapses onto cerebellar Purkinje cells. Here, we show that CF-evoked release of cannabinoids (CBs) additionally suppresses a presynaptic form of long-term potentiation (LTP) at PF synapses. PF-LTP can be induced by 8 Hz PF tetanization but is blocked when the PF tetanization is paired with 4 or 1 Hz CF coactivation. CF activity can be substituted for by bath application of the CB receptor agonist WIN55,212-2 [R(+)-[2,3-dihydro-5-methyl-3-[(morpholinyl)methyl]pyrrolo[1,2,3-de]-1,4-benzoxazinyl]-(1-naphthalenyl) methanone]. In the presence of the CB1 receptor antagonist AM251 [N-1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-1-piperidinyl-1H-pyrazole-3-carboxamide], CF activity no longer suppresses PF-LTP. Presynaptic potentiation can also be obtained by the adenylyl cyclase activator forskolin. WIN55,212-2 blocked this forskolin-mediated enhancement, showing that CB1 receptor activation interferes with the adenylyl cyclase–protein kinase A cascade, which participates in LTP induction. CF activity has been described to promote the induction of postsynaptic PF-long-term depression (LTD) and to impair postsynaptic PF-LTP. Our observation that CF activity blocks the induction of presynaptic LTP suggests that the CF input controls all forms of presynaptic and postsynaptic PF plasticity and that CF activity provides a “safety lock” to prevent an enhancement of transmitter release while postsynaptic AMPA receptor function is downregulated during LTD.
Keywords: cannabinoids, cerebellum, long-term depression, long-term potentiation, parallel fiber, Purkinje cell
Introduction
Long-term depression (LTD) at cerebellar parallel fiber (PF)–Purkinje cell (PC) synapses is considered a cellular correlate of cerebellar motor learning (Hansel et al., 2001; Ito, 2001). Whereas PF-LTD induction requires the simultaneous activity of the PF and the climbing fiber (CF) inputs onto PCs at low frequencies (e.g., 1 Hz for 5 min), PF stimulation alone leads to the induction of long-term potentiation (LTP) (Lev-Ram et al., 2002; Coesmans et al., 2004). The CF acts as a heterosynaptic control switch for postynaptic PF plasticity: CF-evoked complex spikes evoke large dendritic calcium transients that trigger LTD induction, whereas LTP results from smaller calcium transients in the absence of complex spikes (Coesmans et al., 2004). PF stimulation alone can also elicit a presynaptically expressed form of PF-LTP when applied for short durations at high frequencies (e.g., 8 Hz for 15 s) (Salin et al., 1996). Induction of presynaptic PF-LTP depends on the activation of calcium/calmodulin-sensitive adenylyl cyclase I and the subsequent activation of cAMP-dependent kinase [protein kinase A (PKA)] (Salin et al., 1996; Chen and Regehr, 1997; Linden, 1997; Storm et al., 1998; Jacoby et al., 2001).
In postsynaptic PF plasticity, CF activity determines whether LTD or LTP is induced (Lev-Ram et al., 2002; Coesmans et al., 2004). So far, it has not been examined whether CF activity can suppress presynaptic LTP. We reasoned that such a “safety lock” might exist to prevent the induction of presynaptic LTP while LTD is expressed postsynaptically, because the postsynaptic downregulation of response amplitudes might otherwise be accompanied by an increase in transmitter release. Retrograde endocannabinoid signaling might provide a possible link between CF activity and the suppression of presynaptic PF-LTP. At cerebellar and hippocampal synapses, dendritically released endocannabinoids can bind to presynaptically located CB1 receptors (Diana et al., 2002) and cause depolarization-induced suppression of inhibition (Llano et al., 1991; Pitler and Alger, 1992; Wilson et al., 2001), or its equivalent at excitatory synapses, depolarization-induced suppression of excitation (DSE) (Kreitzer and Regehr; 2001). The release of endocannaboids involved in DSE at PF–PC synapses can be activated by CF activity (Brenowitz and Regehr, 2005), suggesting that complex spike-associated calcium transients in PC dendrites are sufficient to initiate the release process.
There have been contradictory reports on the effects of retrograde endocannabinoid signaling on synaptic plasticity in the cerebellum. It has been demonstrated that cannabinoids impair PF-LTD induction (Levenes et al., 1998), but also that cannabinoid signaling is required for PF-LTD induction (Safo and Regehr, 2005). The suppressive effect was explained by a reduction in transmitter release during tetanization, and the permissive effect was explained by a CB1 receptor-mediated release of nitric oxide (NO) from PF terminals. Here, we demonstrate that endocannabinoid signaling can suppress presynaptic PF-LTP. We show that this retrograde signaling mechanism is recruited by CF activity to suppress presynaptic PF-LTP and that this effect can be attributed to the CB1 receptor-mediated blockade of adenylyl cyclase. This CB1 receptor-mediated suppression of presynaptic LTP provides an alternative explanation for the permissive effect of retrograde endocannabinoid signaling in PF-LTD induction.
Materials and Methods
Slice preparation
Sagittal slices of the cerebellar vermis (200–250 μm thick) were prepared from postnatal day 18–25 Sprague Dawley rats in ice-cold artificial CSF (ACSF). The slices were kept in ACSF containing the following (in mm): 124 NaCl, 5 KCl, 1.25 Na2HPO4, 2 MgSO4, 2 CaCl2, 26 NaHCO3, and 10 d-glucose bubbled with 95% O2 and 5% CO2. The ACSF used for perfusion was supplemented with 20 μm bicuculline methiodide to block GABAA receptors. Whole-cell patch-clamp recordings were performed at room temperature using an EPC-9 amplifier (HEKA Elektronik, Lambrecht/Pfalz, Germany). Recording electrodes were filled with a solution containing the following (in mm): 9 KCl, 10 KOH, 108 K-gluconate, 3.48 MgCl2, 10 HEPES, 4 NaCl, 4 Na2ATP, 0.4 Na3GTP, 5 BAPTA (K4+ salt), and 17.5 sucrose, pH 7.25. When the BAPTA concentration was raised to 30 mm (see Fig. 1B,D), 15 mm CaCl2 was added to maintain the resting calcium concentration. The K-gluconate concentration was lowered to maintain the desired osmolarity and ionic strength. All drugs were purchased from Sigma (St. Louis, MO), except for BAPTA (Invitrogen, Eugene, OR), AM251 [N-1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-1-piperidinyl-1H-pyrazole-3-carboxamide; Tocris, Bristol, UK], and PKC [19-36] (Calbiochem, La Jolla, CA). AM251, WIN55,212-2 [R(+)-[2,3-dihydro-5-methyl-3-[(morpholinyl)methyl]pyrrolo[1,2,3-de]-1,4-benzoxazinyl]-(1-naphthalenyl) methanone], and forskolin were dissolved in DMSO and kept as stock solution at −20°C.
Figure 1.
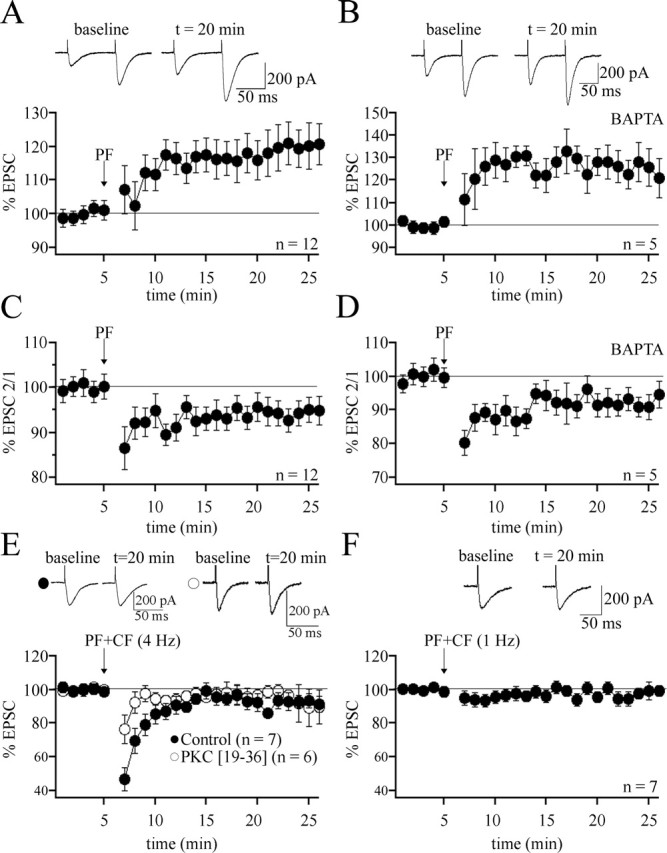
CF activity suppresses the induction of presynaptic PF-LTP. A, PF-LTP can be induced by PF stimulation at 8 Hz for 15 s (n = 12). Each data point represents the average of three successive test responses evoked at 0.05 Hz. The traces on top show EPSCs before and after LTP induction. The arrow indicates the onset of tetanization. B, PF-LTP can be induced when BAPTA (30 mm) is added to the internal saline (n = 5). C, PPF ratio (EPSC 2/EPSC 1) from the LTP group shown in A. D, PPF ratio from the BAPTA group shown in B. E, PF-LTP is abolished when 8 Hz PF stimulation is paired with 4 Hz CF stimulation (n = 7). LTP suppression using paired CF stimulation can also be observed when PKC [19-36] (100 μm) is added to the internal saline (n = 6). F, PF-LTP is blocked when the 8 Hz PF stimulation is paired with CF stimulation at 1 Hz (n = 7). Error bars indicate SEM.
Electrophysiology
Currents were filtered at 3 kHz, digitized at 8 kHz, and acquired using PULSE software (HEKA Elektronik). During voltage-clamp experiments, PCs were held at potentials in the range of −60 to −70 mV. For extracellular stimulation, standard patch pipettes were filled with external saline. The CF input was activated in the granule cell layer, and the PF input was activated in the molecular layer. Test responses (typically ∼200 pA) were evoked at a frequency of 0.05 Hz using 0.5–2 μA pulses. To evoke EPSCs, paired pulses (100 ms interpulse interval) were applied to the PF input. In all experiments, cells were switched to current-clamp mode during tetanization. The protocol for inducing presynaptic LTP consisted of 120 pulses at 8 Hz to the PFs. In experiments in which the CF was coactivated, the presence of a CF response was confirmed in the beginning of the recording, but CF stimulation was subsequently only resumed for tetanization. Recordings were excluded from the study if the series or the input resistance varied by >15% over the course of the experiments. All values are shown as percentage of baseline ± SEM. For statistical analysis, we used the paired Student’s t test and the Mann–Whitney U test where appropriate.
Results
To characterize the effect of CF-evoked cannabinoid signaling on presynaptic PF-LTP, we performed whole-cell patch-clamp recordings from PCs in rat cerebellar slices. During the test periods before and after tetanization, EPSCs were monitored in voltage-clamp mode. For tetanization, recordings were switched to current-clamp mode. LTP was observed after 8 Hz PF tetanization for 15 s. This manipulation resulted in a potentiation of EPSC amplitudes (120.2 ± 6.3% of baseline; n = 12; last 5 min) (Fig. 1A) that reached statistical significance (p < 0.01; paired Student’s t test). The LTP protocol used has been described to induce a presynaptically expressed form of PF-LTP (Salin et al., 1996). The presynaptic origin of the potentiation was confirmed by activating PF–EPSC pairs at an interpulse interval of 100 ms and monitoring the paired-pulse facilitation (PPF) ratio. The LTP observed after tetanization was associated with a modest reduction in the PPF ratio (94.3 ± 2.9% of baseline; n = 12; last 5 min) (Fig. 1C), which reached statistical significance (p < 0.01; paired Student’s t test), indicating a significant presynaptic component of the potentiation. In subsequent experimental groups (see below), we restricted measurements of the PPF ratio to test for the efficacies of drugs known to act presynaptically. The reason is that the PPF ratio is a simple, but rather unreliable, indicator of the expression site of synaptic gain changes. To obtain independent proof of the presynaptic origin of this form of LTP, we applied the 8 Hz PF tetanization protocol when the calcium chelator BAPTA (30 mm) was added to the internal saline. At this concentration, BAPTA blocks the induction of postsynaptic LTP (Coesmans et al., 2004). In the presence of BAPTA, EPSC amplitudes were still potentiated (124.7 ± 7.9% of baseline; n = 5; last 5 min; p < 0.05) (Fig. 1B), and the PPF ratio was reduced (92.2 ± 3.4%; n = 5; last 5 min; p < 0.05) (Fig. 1D). Neither the changes in the EPSC amplitudes nor the PPF alterations differed between the control group and the BAPTA group (p > 0.05; Mann–Whitney U test). These results suggest that the LTP obtained with 8 Hz PF tetanization is indeed presynaptically expressed.
To examine whether paired CF activity impairs the probability to induce LTP, we costimulated the PF and CF inputs. The same PF tetanization protocol was applied as described above, but now the CF input was coactivated with every second PF stimulation pulse, effectively resulting in a 4 Hz stimulation for 15 s. Although the PF activation pattern was the same, application of this pairing protocol did not result in PF-LTP induction (92.9 ± 4.6% of baseline; n = 7; last 5 min; p > 0.05) (Fig. 1E). This difference in LTP observed in the absence of paired CF stimulation was statistically significant (p < 0.01, Mann–Whitney U test). To be able to exclude the possibility that paired PF and CF stimulation led to the induction of postsynaptic LTD, which might simply mask presynaptic LTP, we applied the same stimulus protocol when LTD induction was blocked by adding the PKC inhibitory peptide PKC [19-36] (100 μm) to the internal saline (Hansel et al., 2001). In the presence of PKC [19-36], paired stimulation still did not elicit LTP (93.4 ± 7.9% of baseline; n = 6; last 5 min; p > 0.05) (Fig. 1E), indicating that LTD did not mask presynaptic LTP.
Paired PF and CF stimulation also blocks the induction of presynaptic LTP when the frequency of CF stimulation is reduced to 1 Hz, which is in the range of spontaneous complex spike activity observed in vivo (98.4 ± 3.8% of baseline; n = 7; last 5 min) (Fig. 1F). However, the transient EPSC depression seen after pairing with CF stimulation at 4 Hz was not observed after 1 Hz CF stimulation. Thus, it seems that this short-term depression depends on the level of CF activity, but it might not transfer into LTD because of the short duration of tetanization applied here.
Retrograde endocannabinoid signaling can provide a link between CF activity and inhibition of transmitter release at PF terminals (Brenowitz and Regehr, 2005). To test whether the CF-mediated suppression of PF-LTP involves the activation of CB1 receptors, we applied paired 8 Hz PF and 4 Hz CF tetanization, while the CB1 receptor antagonist AM251 was present in the bath (5 μm). In the presence of AM251, PF-LTP was rescued (127.0 ± 6.8% of baseline; n = 12; last 5 min; p < 0.01) (Fig. 2A). This potentiation was significantly different from the EPSC changes resulting from PF and CF stimulation in the absence of the drug (p < 0.01, Mann–Whitney U test). In the absence of tetanization, AM251 did not alter EPSC amplitudes (100.9 ± 3.3%; n = 6; last 5 min; p > 0.05) (Fig. 2B). The opposite strategy is to test whether PF tetanization alone at 8 Hz for 15 s can still elicit LTP when CB receptors are tonically activated. Bath application of the CB receptor agonist WIN55,212-2 (2 μm) caused a pronounced reduction in EPSC amplitudes (37.2 ± 5.1% of baseline; n = 7; last 5 min; p < 0.01) (Fig. 3A), which was associated with a significant increase in the PPF ratio (123.8 ± 8.5%; n = 7; last 5 min; p < 0.01) (Fig. 3B), indicating that the drug acts on presynaptic CB receptors to reduce transmitter release. When WIN55,212-2 was present in the bath, application of the otherwise LTP-inducing PF tetanization protocol did not cause a potentiation (96.1 ± 3.5%; n = 17; last 5 min; p > 0.05) (Fig. 3C). This blockade was significantly different from the LTP seen under control conditions (p < 0.01, Mann–Whitney U test). Together, the results obtained from the CB1 receptor antagonist (AM251) and the CB receptor agonist (WIN55,212-2) experiments suggest that activation of presynaptic CB1 receptors blocks the induction of presynaptic PF-LTP and that this pathway can be recruited by CF activity.
Figure 2.

The CB1 receptor antagonist AM251 rescues PF-LTP. A, PF-LTP is observed when PF and CF synapses are coactivated in the presence of AM251 (5 μm; n = 12). B, Bath application of AM251 does not alter EPSC amplitudes (n = 6). The horizontal bar indicates the period in which AM251 was bath applied. Error bars indicate SEM.
Figure 3.

The CB receptor agonist WIN55,212-2 blocks PF-LTP. A, Bath application of WIN55,212-2 (2 μm) depresses PF-EPSCs (n = 7). B, PPF ratio (EPSC 2/EPSC 1) from the group shown in A. The horizontal bar indicates the presence of WIN55,212-2 in the bath. C, PF-LTP induced by 8 Hz PF stimulation for 15 s is abolished in the presence of WIN55,212-2 (n = 17). Error bars indicate SEM.
CB1 receptor activation can inhibit adenylyl cyclase activity and can modify K- and Ca-selective channels (Ameri, 1999). It has been shown that the induction of presynaptic PF-LTP involves the activation of adenylyl cyclase (Salin et al., 1996; Storm et al., 1998). Therefore, we next examined whether, under our experimental conditions, receptor activation would interfere with the adenylyl cyclase pathway. To do so, we bath applied the adenylyl cyclase activator forskolin (50 μm). Forskolin application resulted in a strong potentiation of EPSCs (178.4 ± 17.7% of baseline; n = 6; last 5 min; p < 0.01) (Fig. 4A), which was associated with a significant reduction in the PPF ratio (89.2 ± 4.7%; n = 6; last 5 min; p < 0.01) (Fig. 4B). In contrast, when forskolin was added to the bath in the presence of WIN55,212-2, no subsequent enhancement of EPSC amplitudes could be observed (99.9 ± 3.8%; n = 6; last 5 min) (Fig. 4C). This effect was significantly different from the potentiation seen in the absence of WIN55,212-2 (p < 0.01; Mann–Whitney U test). As forskolin activates adenylyl cyclase and thus acts downstream of K- or Ca-selective ion channels, the blockade of the forskolin-mediated EPSC potentiation by WIN55,212-2 suggests that the CB receptor agonist indeed interferes at the level of adenylyl cyclase activation.
Figure 4.
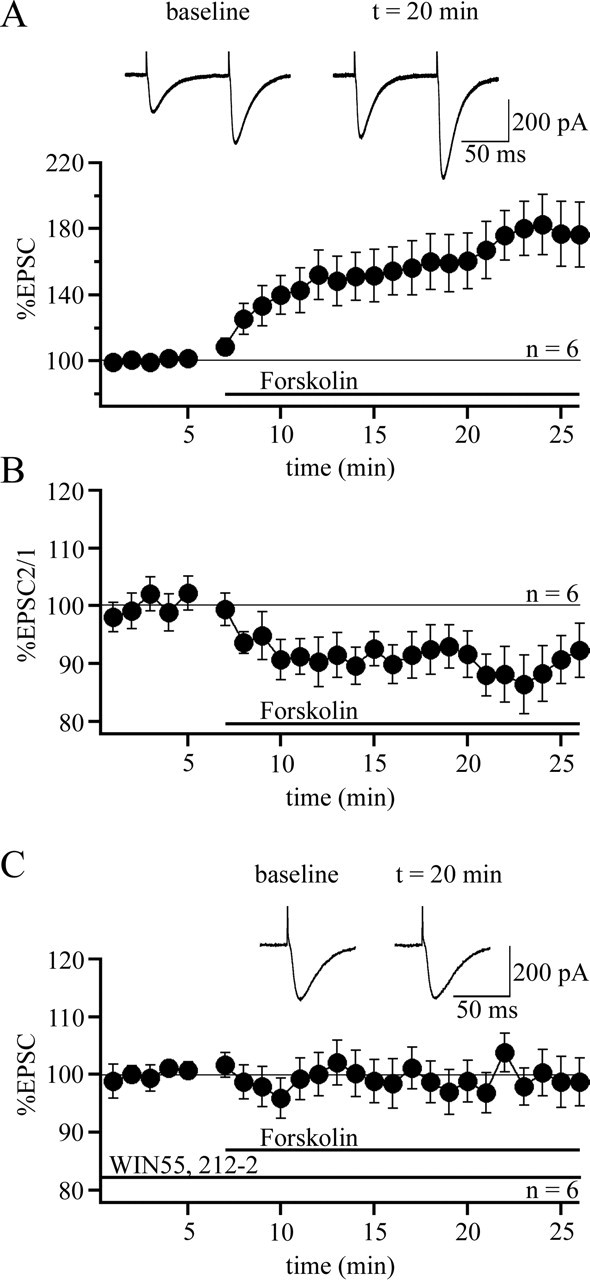
WIN55,212-2 prevents the activation of adenylyl cyclases by forskolin. A, Bath application of the adenylyl cyclase activator forskolin (50 μm) enhances PF-EPSCs (n = 6). B, PPF ratio (EPSC 2/EPSC 1) from the group shown in A. C, WIN55,212-2 (2 μm) abolishes the forskolin-mediated enhancement of PF-EPSCs (n = 6). The horizontal bars indicate the presence of WIN55,212-2 and forskolin, respectively, in the bath. Error bars indicate SEM.
Discussion
The main finding of this study is that activity of the heterosynaptic CF input suppresses the induction of a presynaptically expressed form of PF-LTP through retrograde cannabinoid signaling. Derivatives of the cannabinoid Δ9-tetrahydrocannabinol, the primary psychoactive component of the cannabis plant, are powerful modulators of synaptic transmission. In the brain, the endocannabinoids anandamide and 2-arachidonylglycerol function as modulators of transmitter release by binding to G-protein-coupled CB1 receptors, which are located on synaptic terminals. CB1 receptor activation can both inhibit adenylyl cyclases and modify K- and Ca-selective channels (Ameri, 1999). The latter mode of action has been shown to be involved in the acute effects of CB1 receptor activation on PF-EPSPs (Daniel et al., 2004) and DSEs (Brown et al., 2004). The resulting downregulation of transmitter release has consequences for LTP/LTD induction: cannabinoids impair LTP as well as LTD induction at glutamatergic synapses (Levenes et al., 1998; Misner and Sullivan, 1999) but have also been reported to facilitate the induction of LTD (Gerdeman et al., 2002). Moreover, cannabinoids have been described to reduce GABA release (Chevaleyre and Castillo, 2003). This effect likely explains the observation that endocannabinoids can, through heterosynaptic interaction, also facilitate LTP induction (Carlson et al., 2002; Chevaleyre and Castillo, 2004). The effects of CB1 receptor activation on K- and Ca-selective ion channels and adenylyl cyclases might interfere with presynaptic forms of long-term plasticity as well. For example, it has been shown that presynaptic LTD at neocortical synapses requires CB1 receptor activation (Sjöström et al., 2003).
Here, we report that endocannabinoid signaling can be triggered by CF activity and that the subsequent activation of CB1 receptors impairs presynaptic PF-LTP. Endocannabinoid signaling is well suited to mediate this CF-evoked suppression because it can be triggered in a calcium-dependent way (Kreitzer and Regehr, 2001; Ohno-Shosaku et al., 2001; Wilson and Nicoll; 2001; Brenowitz and Regehr, 2003), and because CB1 receptors are located at PF terminals. Our data show that cannabinoids indeed act as mediators of the CF effect. When the PF and CF inputs were coactivated, bath application of the CB1 receptor antagonist AM251 could rescue PF-LTP. Moreover, effects of CF stimulation could be mimicked by tonically activating CB1 receptors with bath application of WIN55,212-2, which impaired LTP induction. Agonists of CB1 receptors have been reported previously to reduce transmitter release (Levenes et al., 1998; Takahashi and Linden, 2000; Daniel et al., 2004). Activation of CB1 receptors has several effects, which all result in a reduction of transmitter release: (1) it can lead to a downregulation of voltage-dependent calcium channels; (2) it can cause an upregulation of K channels; and (3) it can interfere with the activation of adenylyl cyclases and thus with the PF-LTP induction cascade. All three mechanisms could account for the observed effects. By showing that in the presence of the CB receptor agonist WIN55,212-2 the adenylyl cyclase activator forskolin no longer potentiates PF-EPSCs, we confirm that CB receptor activation indeed suppresses the activity of adenylyl cyclase. This finding does not exclude the possibility that Ca- or K-selective channels are also modified, but it suggests that during tetanization, the activation of CB1 receptors blocks the adenylyl cyclase–PKA signaling cascade that otherwise leads to the induction of presynaptic LTP through phosphorylation of the PKA substrate RIM1α (Castillo et al., 2002; Lonart et al., 2003).
The observed endocannabinoid-mediated suppression of presynaptic PF-LTP occurs under conditions that also favor the induction of postsynaptically expressed LTD (paired PF and CF activity). However, when LTD induction was blocked by adding the PKC inhibitory peptide PKC [19-36], CF coactivation was still able to suppress presynaptic LTP, suggesting that LTD does not simply mask LTP. Rather, the CF-evoked suppression of presynaptic LTP might provide a safety lock, which prevents that LTP is expressed presynaptically, while LTD is expressed postsynaptically. The signaling cascades involved partially overlap with those described for DSE; CF activity causes complex spike-associated calcium transients that trigger the release of endocannabinoids (Brenowitz and Regehr, 2005). It is remarkable that the CF apparently controls all forms of PF plasticity. Simultaneous CF activity postsynaptically promotes the induction of PF-LTD by providing a large calcium transient, whereas postsynaptically expressed PF-LTP is triggered by smaller calcium transients in the absence of CF activity (Coesmans et al., 2004). Postsynaptic PF-LTP depends on the activation of protein phosphatases PP1, PP2A, and PP2B (Belmeguenai and Hansel, 2005), whereas PF-LTD depends on the activation of PKC (for review, see Hansel et al., 2001). Thus, it can be argued that CF activity, by promoting the PKC cascade, has a tight grip on the polarity of postsynaptic PF plasticity. Our observation that CF activity additionally suppresses the induction of presynaptic PF-LTP supports the notion that the CF input heterosynaptically controls all forms of PF synaptic plasticity.
The suppressive effect of CF coactivation was not only observed with CF stimulation at 4 Hz, but also at 1 Hz, suggesting the possibility that presynaptic LTP is permanently suppressed under resting conditions. In our recordings, spike activity is prevented in PCs by constant current injection. CF stimulation at 1 Hz thus provides a transient increase in PC activity that might trigger endocannabinoid release only under these in vitro conditions. Even if endocannabinoids were released under resting conditions in vivo, there are physiological activity patterns favoring presynaptic LTP because complex spikes are not constantly fired at 1 Hz. Transient inhibition of CFs by elevated cerebellar activity has been shown to facilitate the extinction of conditioned eyelid responses (Medina et al., 2002) and might create time windows without complex spike activity during which LTP can be induced.
Previous reports have suggested that cannabinoids interfere with the induction of PF-LTD. A previous study has shown that cannabinoid signaling impairs PF-LTD induction, which was explained by reduced transmitter release during tetanization (Levenes et al., 1998). The opposite effect was reported recently, showing that cannabinoid signaling is permissive for PF-LTD induction (Safo and Regehr, 2005). The resulting paradox that a retrograde messenger modifies the induction probability of postsynaptic LTD was addressed by suggesting that CB1 receptor activation might trigger NO release from PF terminals. Because NO has been suggested to be required for LTD induction, CB1 receptor activation could promote LTD induction, despite an accompanying transient reduction in transmitter release. The study by Safo and Regehr (2005) and ours complement each other and, in fact, provide two sides of the same coin. Whereas these authors demonstrated that cannabinoid signaling is required for the induction of PF-LTD (which is postsynaptically expressed), we demonstrate here that cannabinoid signaling suppresses a presynaptic form of PF-LTP. This LTP suppression might well provide a better explanation for the endocannabinoid-mediated promotion of PF-LTD than NO signaling as suggested by Safo and Regehr (2005). First, it has recently been shown that NO might indeed be involved in LTD induction; however, it is not released from PF terminals, but rather from interneurons (Shin and Linden, 2005). Second, simultaneous induction of presynaptic LTP would counteract postsynaptic LTD. It is likely that the CF-evoked calcium transients in PC dendrites can postsynaptically promote LTD and, through the release of endocannabinoids, presynaptically suppress LTP at the same time. The latter effect would facilitate LTD induction and enhance the observed LTD magnitude.
Footnotes
*B.J.v.B. and R.Y.N. contributed equally to this work.
This work was supported by a grant from NWO-VIDI (The Netherlands Organization for Scientific Research) to C.H. We thank members of the Hansel laboratory for helpful discussions.
References
- Ameri A (1999). The effects of cannabinoids on the brain. Prog Neurobiol 58:315–348. [DOI] [PubMed] [Google Scholar]
- Belmeguenai A, Hansel C (2005). A role for protein phosphatases 1, 2A, and 2B in cerebellar long-term potentiation. J Neurosci 25:10768–10772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenowitz SD, Regehr WG (2003). Calcium dependence of retrograde inhibition by endocannabinoids at synapses onto Purkinje cells. J Neurosci 23:6373–6384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenowitz SD, Regehr WG (2005). Associative short-term synaptic plasticity mediated by endocannabinoids. Neuron 45:419–431. [DOI] [PubMed] [Google Scholar]
- Brown SP, Safo PK, Regehr WG (2004). Endocannabinoids inhibit transmission at granule cell to Purkinje cell synapses by modulating three types of presynaptic calcium channels. J Neurosci 24:5623–5631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson G, Wang Y, Alger BE (2002). Endocannabinoids facilitate the induction of LTP in the hippocampus. Nat Neurosci 5:723–724. [DOI] [PubMed] [Google Scholar]
- Castillo PE, Schoch S, Schmitz F, Südhof TC, Malenka RC (2002). RIM1α is required for presynaptic long-term potentiation. Nature 415:327–330. [DOI] [PubMed] [Google Scholar]
- Chen C, Regehr WG (1997). The mechanism of cAMP-mediated enhancement at a cerebellar synapse. J Neurosci 17:8687–8694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE (2003). Heterosynaptic LTD of hippocampal GABAergic synapses: a novel role of endocannabinoids in regulating excitability. Neuron 38:461–472. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE (2004). Endocannabinoid-mediated metaplasticity in the hippocampus. Neuron 43:871–881. [DOI] [PubMed] [Google Scholar]
- Coesmans M, Weber JT, De Zeeuw CI, Hansel C (2004). Bidirectional parallel fiber plasticity in the cerebellum under climbing fiber control. Neuron 44:691–700. [DOI] [PubMed] [Google Scholar]
- Daniel H, Rancillac A, Crepel F (2004). Mechanisms underlying cannabinoid inhibition of presynaptic Ca2+ influx at parallel fiber synapses of the rat cerebellum. J Physiol (Lond) 557:159–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diana MA, Levenes C, Mackie K, Marty A (2002). Short-term retrograde inhibition of GABAergic synaptic currents in rat Purkinje cells is mediated by endogenous cannabinoids. J Neurosci 22:200–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdeman GL, Ronesi J, Lovinger DM (2002). Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat Neurosci 5:446–451. [DOI] [PubMed] [Google Scholar]
- Hansel C, Linden DJ, D’Angelo E (2001). Beyond parallel fiber LTD: the diversity of synaptic and non-synaptic plasticity in the cerebellum. Nat Neurosci 4:467–475. [DOI] [PubMed] [Google Scholar]
- Ito M (2001). Cerebellar long-term depression: characterization, signal transduction, and functional roles. Physiol Rev 81:1143–1194. [DOI] [PubMed] [Google Scholar]
- Jacoby S, Sims RE, Hartell NA (2001). Nitric oxide is required for the induction and heterosynaptic spread of long-term potentiation in rat cerebellar slices. J Physiol (Lond) 535:825–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG (2001). Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron 29:717–727. [DOI] [PubMed] [Google Scholar]
- Levenes C, Daniel H, Soubrie P, Crepel F (1998). Cannabinoids decrease excitatory synaptic transmission and impair long-term depression in rat cerebellar Purkinje cells. J Physiol (Lond) 510:867–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lev-Ram V, Wong ST, Storm DR, Tsien RY (2002). A new form of cerebellar long-term potentiation is postsynaptic and depends on nitric oxide but not cAMP. Proc Natl Acad Sci USA 99:8389–8393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden DJ (1997). Long-term potentiation of glial synaptic currents in cerebellar culture. Neuron 18:983–994. [DOI] [PubMed] [Google Scholar]
- Llano I, Leresche N, Marty A (1991). Calcium entry increases the sensitivity of cerebellar Purkinje cells to applied GABA and decreases inhibitory synaptic currents. Neuron 6:565–574. [DOI] [PubMed] [Google Scholar]
- Lonart G, Schoch S, Kaeser PS, Larkin CJ, Südhof TC, Linden DJ (2003). Phosphorylation of RIM1α by PKA triggers presynaptic long-term potentiation at cerebellar parallel fiber synapses. Cell 115:49–60. [DOI] [PubMed] [Google Scholar]
- Medina JF, Nores WL, Mauk MD (2002). Inhibition of climbing fibres is a signal for the extinction of conditioned eyelid responses. Nature 416:330–333. [DOI] [PubMed] [Google Scholar]
- Misner DL, Sullivan JM (1999). Mechanism of cannabinoid effects on long-term potentiation and depression in hippocampal CA1 neurons. J Neurosci 19:6795–6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Maejima T, Kano M (2001). Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron 29:729–738. [DOI] [PubMed] [Google Scholar]
- Pitler TA, Alger BE (1992). Postsynaptic spike firing reduces synaptic GABAA responses in hippocampal pyramidal cells. J Neurosci 12:4122–4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safo PK, Regehr WG (2005). Endocannabinoids control the induction of cerebellar LTD. Neuron 48:647–659. [DOI] [PubMed] [Google Scholar]
- Salin PA, Malenka RC, Nicoll RA (1996). Cyclic AMP mediates a presynaptic form of LTP at cerebellar parallel fiber synapses. Neuron 16:797–803. [DOI] [PubMed] [Google Scholar]
- Shin JH, Linden DJ (2005). An NMDA receptor/nitric oxide cascade is involved in cerebellar LTD but is not localized to the parallel fiber terminal. J Neurophysiol 94:4281–4289. [DOI] [PubMed] [Google Scholar]
- Sjöström PJ, Turrigiano GG, Nelson SB (2003). Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron 39:641–654. [DOI] [PubMed] [Google Scholar]
- Storm DR, Hansel C, Hacker B, Parent A, Linden DJ (1998). Impaired cerebellar long-term potentiation in type I adenylyl cyclase mutant mice. Neuron 20:1199–1210. [DOI] [PubMed] [Google Scholar]
- Takahashi KA, Linden DJ (2000). Cannabinoid receptor modulation of synapses received by cerebellar Purkinje cells. J Neurophysiol 83:1167–1180. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA (2001). Endogenous cannabinoids mediate retrograde signaling at hippocampal synapses. Nature 410:588–592. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Kunos G, Nicoll RA (2001). Presynaptic specificity of endocannabinoid signaling in the hippocampus. Neuron 31:453–462. [DOI] [PubMed] [Google Scholar]