

Abstract
Brain-derived neurotrophic factor (BDNF) is a member of the nerve growth factor family of neurotrophins and plays a vital role in synaptic plasticity. This study investigated the involvement of the amygdaloid BDNF system in molecular mechanisms underlying anxiety and alcohol-drinking behaviors. Male Sprague Dawley rats were cannulated targeting central amygdala (CeA), medial amygdala (MeA), or basolateral amygdala (BLA), and BDNF expression was manipulated using an antisense oligodeoxynucleotide (ODN) strategy. Anxiety-like and alcohol-drinking behaviors were measured after infusion of BDNF sense and antisense ODNs with or without BDNF coinfusion, using the elevated plus-maze test and two-bottle free-choice paradigm, respectively. Here we report that BDNF antisense ODN infusions into the CeA and MeA, but not BLA, provoked anxiety-like behaviors in rats, which were rescued by BDNF coinfusion. The levels of BDNF, p-ERK1/2 (phosphorylated extracellular signal-regulated kinases 1/2), and p-CREB (phosphorylated cAMP responsive-element binding protein) were decreased by BDNF antisense, but not by sense, ODN infusions, which were restored to normal after BDNF coinfusions. Furthermore, BDNF antisense ODN infusions into the CeA or MeA, but not into BLA, increased alcohol intake, which was attenuated by BDNF coinfusions. These novel results suggest that decreased BDNF levels in the CeA and MeA, but not in the BLA, are crucial in regulating alcohol-drinking and anxiety-like behaviors in rats.
Keywords: BDNF, CREB, amygdala, anxiety, alcohol preference, alcohol addiction
Introduction
Brain-derived neurotrophic factor (BDNF) is a member of the nerve growth factor family of neurotrophic factors that is highly expressed in various brain regions and is involved in neuronal survival and differentiation and synaptic strength (Thoenen, 1995, 2000; Bibel and Barde, 2000; Poo, 2001; Carter et al., 2002; Monteggia et al., 2004). The action of BDNF is mediated by binding to tyrosine kinase B (TrkB) receptors and subsequently the activation of the MAP kinase (mitogen-activated protein kinase), phospholipase C-γ (PLC-γ), and phosphoinositide 3-kinase (PI 3-kinase) signal transduction pathways (Thoenen, 1995; Impey et al., 1999; Ying et al., 2002; Pandey, 2004). BDNF and TrkB are cAMP-inducible genes and are regulated by the cAMP responsive-element binding protein (CREB) gene transcription factor (Shieh et al., 1998; Deogracias et al., 2004; Duman, 2004; Pandey et al., 2004).
The role of BDNF has been implicated in several neurological and psychiatric conditions, such as learning and memory, depression, stress, and drug addiction (Siuciak et al., 1997; Horger et al., 1999; Manji et al., 2001; Poo, 2001; Nestler et al., 2002; Shirayama et al., 2002; Duman, 2004). However, the role of the BDNF gene in anxiety is less clear. For example, BDNF-haplodeficient mice do not display anxiety-like behaviors, whereas BDNF conditional knock-out mice are prone to anxiety-like behaviors (Montkowski and Holsboer, 1997; MacQueen et al., 2001; Rios et al., 2001). Previous studies indicate that haplodeficiency in BDNF promotes alcohol intake in mice (Hensler et al., 2003; McGough et al., 2004). However, these studies do not identify the involvement of the amygdaloid BDNF system in anxiety or alcohol-drinking behaviors. The amygdaloid structures have been shown to be centers for emotion and anxiety and also play a role in motivational aspects of alcohol-drinking behaviors (Davis, 1997; McBride, 2002; Koob, 2003; Pandey, 2004). We recently found that decreased CREB function in the central amygdala (CeA) may be involved in anxiety and alcohol-drinking behaviors (Pandey et al., 2003, 2005). In addition, it was found that CREB-deficient mice display anxiety-like behaviors and also have higher alcohol preference. Interestingly, these mice have lower expression of BDNF in several brain regions, including amygdala (Pandey et al., 2004). These results suggest the possibility that decreased CREB levels may regulate alcohol-drinking and anxiety-like behaviors via the reduction of BDNF levels in amygdaloid structures.
To explore the direct association of BDNF, in the specific neurocircuitry of amygdala, with alcohol-drinking and anxiety-like behaviors, the present study was designed to alter the expression of BDNF by infusion of BDNF antisense oligodeoxynucleotides (ODNs) in various amygdaloid structures and then examine the phenotypes of alcohol preference and anxiety-like behaviors. We used this approach to test the hypothesis that decreased BDNF in the amygdaloid structures may be one of the molecular mechanisms for regulating alcohol-drinking and anxiety-like behaviors. The antisense ODN strategy circumvents the global deletion of a gene, as in knock-out mice, and allows brain region-specific reduction in BDNF gene expression. BDNF acts via TrkB receptors, which regulate the activation of extracellular signal-regulated kinases 1/2 (ERK1/2), finally converging onto CREB phosphorylation at serine-133 (Impey et al., 1999); we therefore also studied the phosphorylation status of CREB and ERK1/2 after blocking BDNF expression as a functional measure of BDNF in the amygdaloid structures.
Materials and Methods
Animals and implantation of cannulas.
All experiments were conducted in accordance with the National Institutes of Health Guidelines for Care and Use of Laboratory Animals and were approved by the Institutional Animal Care Committee. Adult male Sprague Dawley rats (225–250 g at the beginning of the experiment) were used in this study. All rats were housed under a 12 h light/dark cycle and had ad libitum access to water and food. Rats were anesthetized with sodium pentobarbital (50 mg/kg, i.p.) and placed in a stereotaxic apparatus. Rats were implanted bilaterally with CMA/11 guided cannulas (CMA Microdialysis, North Chelmsford, MA) targeted 3 mm above the central amygdala (CeA), medial amygdala (MeA), or basolateral amygdala (BLA). Cannulas were secured to the skull using dental cement and screws. The coordinates for CeA were 2.5 mm posterior and ±4.2 mm lateral to bregma and 5.1 mm ventral from the point of entry at the skull surface; for MeA: 2.56 mm posterior and ±3.2 mm lateral to bregma and 6.0 mm ventral from the point of entry at the skull surface; and for BLA: 2.56 mm posterior and ±4.8 mm lateral to bregma and 5.4 mm ventral from the point of entry at the skull surface. Cannulas were covered with guided caps (CMA Microdialysis).
Measurement of anxiety-like behaviors by elevated plus-maze test.
One week after surgery, rats were subjected to BDNF sense/antisense ODN infusions. Rats (n = 8 for CeA; n = 8–9 for MeA; n = 6–12 for BLA) were infused with phosphorothioate-modified BDNF sense (5′-TGAGAAGAGTGATGACAA-3′)/antisense (5′-TTGTCATCACTCTTCTCA-3′) 18-mer ODNs. This sequence is based on the literature in which BDNF antisense ODN infusion into hippocampus modified long-term potentiation (Ma et al., 1998). The phosphorothioate-modified ODNs were synthesized by Integrated DNA Technologies (Coralville, IA). One group of rats was also infused with BDNF protein (98% recombinant; Sigma-Aldrich, St. Louis, MO) using a microdialysis probe (CMA Microdialysis) connected to an automatic infusion pump. The microdialysis probe extended 3 mm beyond the cannula into the CeA, MeA, and BLA. The BDNF sense/antisense ODNs and BDNF were dissolved in artificial CSF (aCSF). The following groups were generated for infusions into CeA, MeA, and BLA: (1) aCSF (0.5 μl); (2) BDNF antisense ODNs (0.5 μl of 0.1 mm); (3) BDNF sense ODNs (0.5 μl of 0.1 mm); (4) BDNF protein (50 ng in 0.5 μl); and (5) BDNF protein (50 ng in 0.5 μl; 15 min before BDNF antisense ODN infusion) plus BDNF antisense ODNs (0.5 μl of 0.1 mm). BDNF sense or antisense ODNs were infused once (over a period of 2 min) 12 h before anxiety-like behavior measurement, whereas BDNF was infused twice: 12 h and 1 h before elevated plus-maze (EPM) testing in both the BDNF and BDNF plus BDNF antisense ODN infusion groups.
The EPM procedure was the same as that described by us previously (Pandey et al., 1999, 2003, 2005). The apparatus consists of two open arms and two closed arms, which are arranged directly adjacent to each other and connected to a central platform (Lafayette Instrument, Lafayette, IN). Rats were taken to the test room individually in a transport cage and were allowed a 5 min habituation period before testing. Each rat was observed for open- and closed-arms exploration for a 5 min test. The number of entries made to each type of arm (open or closed) and time spent on each arm was recorded. EPM test results were expressed as the mean ± SEM of the percentage of open-arm entries and the mean percentage of time spent on the open arms (open-arm activity). The locomotor activity of rats was measured by the total number of closed-arm entries as reported by other investigators (File, 1993; Rodgers and Johnson, 1995; Schulteis et al., 1998). Immediately after behavioral testing, rats were perfused, and brains were collected and used for histochemical studies of various proteins as described below.
Gold immunolabeling of BDNF, phosphorylated CREB, phosphorylated ERK1/2, and neuron-specific neuronal proteins in rat brain.
The cellular expression of various proteins was determined by the gold-immunolabeling histochemical procedure as described previously by us (Pandey et al., 2001, 2003, 2004). Briefly, the procedure was as follows: brain sections (20 μm) were first blocked and then incubated with anti-phosphorylated CREB (p-CREB), phosphorylated ERK1/2 (p-ERK1/2), BDNF, or neuron-specific neuronal (NeuN) marker primary antibodies [1:500 dilution for p-CREB (Millipore, Billerica, MA) and a 1:200 dilution for NeuN (Millipore), BDNF (Santa Cruz Biotechnology, Santa Cruz, CA), and p-ERK1/2 (Cell Signaling Technology, Beverly, MA)] in 1% BSA prepared in 0.01 m PBS containing 0.25% Triton X-100 for 18 h at room temperature. After two 10 min washes with PBS and two 10 min washes with 1% BSA in PBS, sections were incubated with gold particle (1–1.4 nm; Nanoprobes, Yaphank, NY) conjugated anti-rabbit or anti-mouse secondary antibody (1:200 dilution in 1% BSA in PBS) for 1 h at room temperature. Sections were further rinsed several times in 1% BSA in PBS, followed by rinsing in double-distilled water. The gold immunolabeling was then silver enhanced (Ted Pella, Redding, CA) for 15–25 min and washed several times with tap water. Sections were then mounted on slides and examined under a light microscope. The quantification of gold-immunolabeled proteins was performed using an image analysis system (Loats Associates, Westminster, MD) connected to a light microscope that calculated the number of gold particles per 100 μm2 area of amygdaloid brain structure at high magnification (100×). The threshold of each image was set up in such a way that an area without staining should give zero counts. Under this condition, immunogold particles in the CeA, MeA, and BLA of three adjacent brain sections (three fields in each brain structure per section) of each rat were counted, and then values were averaged for each rat.
In situ RT-PCR for BDNF mRNA measurement in brain. Rat brain sections (40 μm) were used to determine the mRNA levels of BDNF using in situ reverse transcription-PCR as reported previously by us (Pandey et al., 2004). BDNF-positive cell bodies were detected by using alkaline phosphatase-conjugated anti-digoxigenin antibody and subsequent staining of the complex with specific substrate nitro blue tetrazolium chloride/BCIP (5-bromo-4-chloro-3-indolylphosphate; Roche Diagnostics, Indianapolis, IN). The optical density (OD) of positive cell bodies was calculated with an image analyzer (Loats Associates) connected to a light microscope (20× magnification). The OD from negative brain sections was subtracted from the OD of positive brain sections. The mean OD in the CeA, MeA, and BLA structures of three adjacent brain sections (three fields in each brain structure per section) of each rat was calculated, and then values were averaged for each rat. The results were represented as mean OD per 100 pixels of area for BDNF mRNA levels.
Ethanol preference procedure.
Ethanol preference was measured by the two-bottle free-choice paradigm used previously by us (Pandey et al., 2003, 2004, 2005). Rats were bilaterally implanted with cannulas targeted 3 mm above the CeA (n = 6), MeA (n = 6), or BLA (n = 6–8), as described above. After a recovery period of 1 week, rats were habituated to drinking water from two bottles. Once they started drinking water equally from either bottle, rats were infused with 0.5 μl of aCSF, 0.5 μl of 0.1 mm BDNF antisense ODNs, 0.5 μl of 0.1 mm BDNF sense ODNs, 50 ng in 0.5 μl of BDNF, or BDNF plus BDNF antisense ODNs, using a microdialysis probe attached to an automatic pump. The microdialysis probe extended 3 mm beyond the guide cannula into the CeA, MeA, or BLA. The groups for infusions into CeA, MeA, and BLA were similar to the groups described above for anxiety measurements. BDNF sense/antisense ODNs were infused (over a period of 2 min) once (5:00–6:00 P.M.), and rats were given 7% (v/v) ethanol in one bottle and water in the other bottle every day for 4 d. In BDNF groups, rats were infused with BDNF 15 min before the infusion with BDNF antisense ODNs or aCSF on the first day, and then on the second day, only BDNF or aCSF was infused. Consumption of ethanol and water was measured daily at 6:00 P.M., and fresh water and 7% (v/v) ethanol solution in water were provided every day for 4 d. The mean percentage of ethanol intake and percentage of water intake were calculated from their total fluid intake (in milliliters) for each day. The rats were perfused, and brains were processed for histochemistry to check the position of cannulas or tissue damage using cresyl violet staining.
Statistical analyses.
The differences between the various groups for anxiety-like behaviors and neurochemical data were evaluated by a one-way ANOVA or Kruskal–Wallis test. The alcohol intake data were evaluated by repeated-measure ANOVA with day as the within-subject factor and experimental groups as the between-subject factor. Post hoc comparisons were performed using Tukey’s test.
Results
Effects of intra-CeA infusion of BDNF antisense ODNs on anxiety-like behaviors
To examine the association between decreased expression of BDNF in the CeA and anxiety-like behaviors, we implanted bilateral cannulas targeting the CeA (Fig. 1A). After 1 week of recovery, rats were infused with 0.5 μl of aCSF, BDNF sense ODNs, BDNF antisense ODNs, BDNF, or BDNF plus BDNF antisense ODNs. The open- and closed-arm activities were measured on the EPM test. It was found that intra-CeA infusion of BDNF antisense ODNs produced a significant reduction in percentage of open-arm entries (F(4,35) = 35.96; p < 0.001) and percentage of time (F(4,35) = 8.84; p < 0.001) spent on open arms in rats (Fig. 1B). Post hoc comparisons revealed that BDNF antisense ODN infusions into CeA significantly decreased the percentage of open-arm entries (p < 0.001) and time spent (p = 0.007) on open arms compared with aCSF-infused rats (Fig. 1B). Coinfusion of BDNF prevented the BDNF antisense ODN-induced reduction in percentage of open-arm entries and time spent on open arms. Rats infused with BDNF sense ODNs or BDNF alone, however, did not show any significant alterations in anxiety-like behaviors compared with aCSF-infused rats. The total number of entries (closed and open arms) was not significantly different among the groups. However, the total number of closed-arm entries was significantly (df = 4; H = 19.17; p < 0.001) different among groups, but post hoc comparisons revealed that closed-arm entries of BDNF antisense ODNs, sense ODNs, or BDNF-infused rats were not significantly different from aCSF-infused rats. Thus, the locomotor activity was not modulated by infusions of these agents into the CeA of rats (Fig. 1B). These results indicate that BDNF antisense ODN infusion into the CeA provoked anxiety-like effects in rats.
Figure 1.

A, Low-magnification view of Nissl staining of the amygdaloid structures of rats. This photomicrograph indicates the position of the cannula, which is just above the targeted area of central amygdala. Scale bar, 200 μm. B, Effect of CeA infusion of aCSF and BDNF sense and antisense ODNs (with and without BDNF coinfusion) on open- and closed-arm activities of the EPM. Total number of entries represents the total numbers of closed- and open-arm entries in EPM. Values are the mean ± SEM of eight rats in each group. Asterisks indicate significant (p < 0.01–0.001) difference from aCSF-infused rats.
Cellular changes in the CeA after infusions of BDNF antisense ODNs and BDNF
Immediately after the measurement of anxiety-like behaviors, the brains were used to examine the protein and mRNA levels of BDNF as well as the cellular expressions of p-CREB, p-ERK1/2, and NeuN proteins. Figure 2 shows the expression of BDNF protein and mRNA along with gold immunolabeling of p-CREB, p-ERK1/2, and NeuN in the CeA after BDNF manipulations. BDNF antisense ODN infusion into the CeA significantly decreased both the protein (F(4,30) = 13.99; p < 0.001) and mRNA (F(4,25) = 31.56; p < 0.001) levels of BDNF only in the CeA, not in the MeA and BLA of rats (Figs. 2, 3). Infusion of BDNF along with BDNF antisense ODNs prevented the decrease in BDNF protein and mRNA levels in the CeA. Infusions of BDNF sense ODNs or BDNF alone into the CeA did not produce any significant changes in the protein or mRNA levels of BDNF in CeA, MeA, or BLA compared with the aCSF-infused group (Figs. 2, 3).
Figure 2.
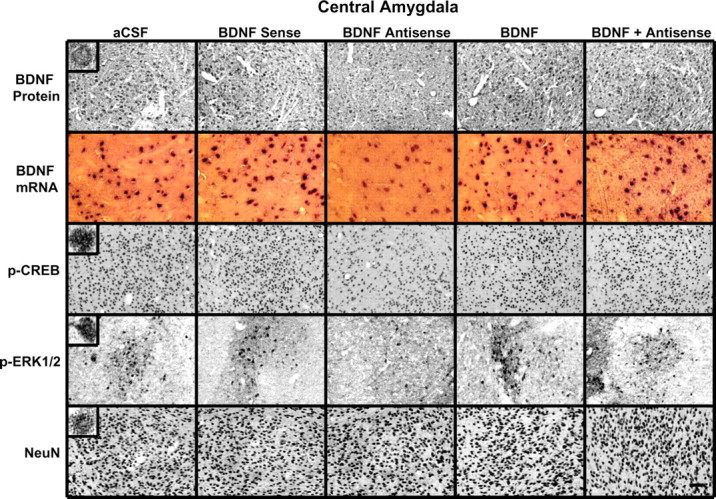
Representative photomicrographs at low magnification showing BDNF (top row), p-CREB (third row), p-ERK1/2 (fourth row), and NeuN (bottom row) gold-immunolabeling and mRNA levels of BDNF (second row) in the CeA brain structures of rats infused with aCSF, BDNF sense, and antisense ODNs (with and without BDNF coinfusion) into the CeA. Scale bar, 30 μm. Insets in the aCSF column indicate immunogold particles within a single cell body or single nucleus at high magnification (100×).
Figure 3.
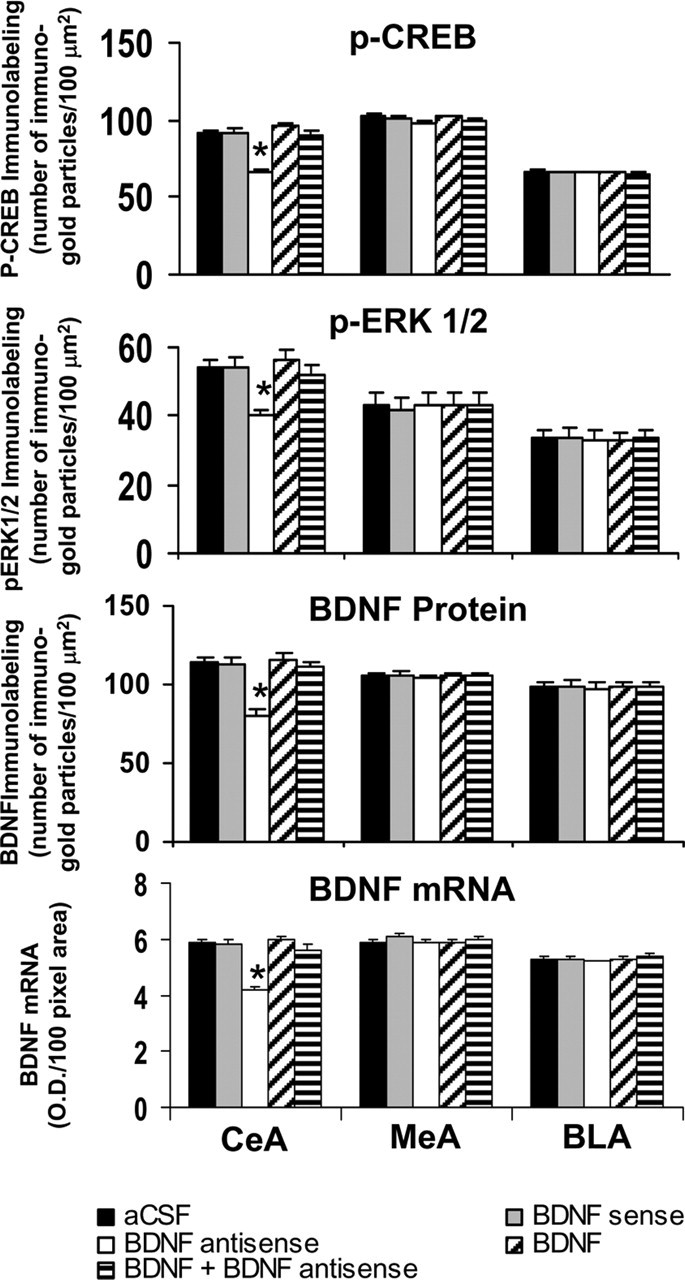
Quantitation of BDNF, p-CREB, and p-ERK1/2 protein levels (number of immunogold particles per 100 μm2 area) and mRNA levels of BDNF in the amygdaloid structures of rats infused with aCSF, BDNF sense, and antisense ODNs (with and without BDNF coinfusion) into the CeA. Values are the mean ± SEM of five to seven rats in each group. The asterisks indicate significant (p < 0.001) difference from aCSF-infused rats.
We also studied the functional consequences of decreased levels of BDNF in the CeA by measuring the protein levels of p-ERK1/2 and p-CREB. It was found that BDNF antisense ODN infusion into the CeA significantly decreased the protein levels of p-ERK1/2 (F(4,20) = 4.93; p = 0.006) and p-CREB (F(4,20) = 35.28; p < 0.001) in the CeA, which was prevented by coinfusion with BDNF (Figs. 2, 3). Post hoc comparisons revealed that BDNF antisense ODN infusion into CeA significantly reduced the protein levels of p-ERK1/2 (p < 0.001) and p-CREB (p < 0.001) in CeA, but not in MeA and BLA, compared with CeA aCSF-infused rats. Infusions of BDNF sense ODNs or BDNF protein alone in the CeA did not produce any significant changes in the protein levels of p-ERK1/2 or p-CREB in CeA, MeA, or BLA compared with the aCSF-infused rats. BDNF sense and antisense ODN infusions did not cause any neuronal loss, because the levels of NeuN were not significantly altered among the treatment groups (Fig. 2, bottom) (mean data not shown). These results indicate that BDNF antisense or sense ODNs or BDNF infusions into CeA do not produce any neurotoxicity.
Relationship between decreased BDNF levels in the CeA and alcohol preference
We next examined the association between decreased BDNF levels in CeA and alcohol preference. We implanted bilateral cannulas targeting the CeA, and after 1 week of recovery, rats were habituated to drinking water from two bottles. When rats started drinking water equally from either bottle, bilateral infusions of pharmacological agents into the CeA were performed. Rats were provided with 7% (v/v) ethanol solution in one of the bottles and water in the other bottle for 4 d. The mean percentage of ethanol or water intake of their total fluid intake (in milliliters) for each day was calculated. A 5 × 4 (group × day) repeated-measure ANOVA was performed on changes in alcohol intake that yielded a significant group effect (F(4,75) = 14.93; p < 0.001), a significant day effect (F(3,75) = 8.50; p < 0.001), and a significant interaction (F(12,75) = 8.48; p < 0.001) of these variables in rats. A 5 × 4 (group × day) repeated-measure ANOVA was performed on changes in water intake that yielded a significant group effect (F(4,75) = 15.56; p < 0.001), a significant day effect (F(3,75) = 7.49; p < 0.001), and a significant interaction (F(12,75) = 7.70; p < 0.001) of these variables in rats. It was found that BDNF antisense ODN-infused rats, but not the sense ODN-infused rats, consumed significantly higher (p < 0.001) ethanol solution and lower water (p < 0.001) on days 1 and 2 of the postinfusion period than aCSF-infused rats. Interestingly, the alcohol and water preferences in the BDNF antisense ODN-infused rats, on the third and fourth days of the postinfusion period, were similar to the aCSF group of rats (Fig. 4). It was found that BDNF (50 ng) infusion (once daily for 2 d, 24 h interval) alone into the CeA produced a nonsignificant decrease in alcohol preference and increase in water preference, but coinfusing with BDNF antisense ODNs significantly antagonized the increase in alcohol preference in rats (Fig. 4). The total fluid intake (in milliliters per day) was similar in all groups at each day of the postinfusion period (Fig. 4). The body weights were also not significantly different among the groups. These results indicate that in the CeA, decreased BDNF levels increase preference to alcohol over water without modulating total fluid intake. These results also indicate that the effects of BDNF antisense ODNs on alcohol intake are reversible. Furthermore, these results indicate that rescue of the decreased BDNF levels in the CeA via coinfusion with BDNF attenuated the BDNF antisense ODN-induced increases in alcohol intake in rats.
Figure 4.
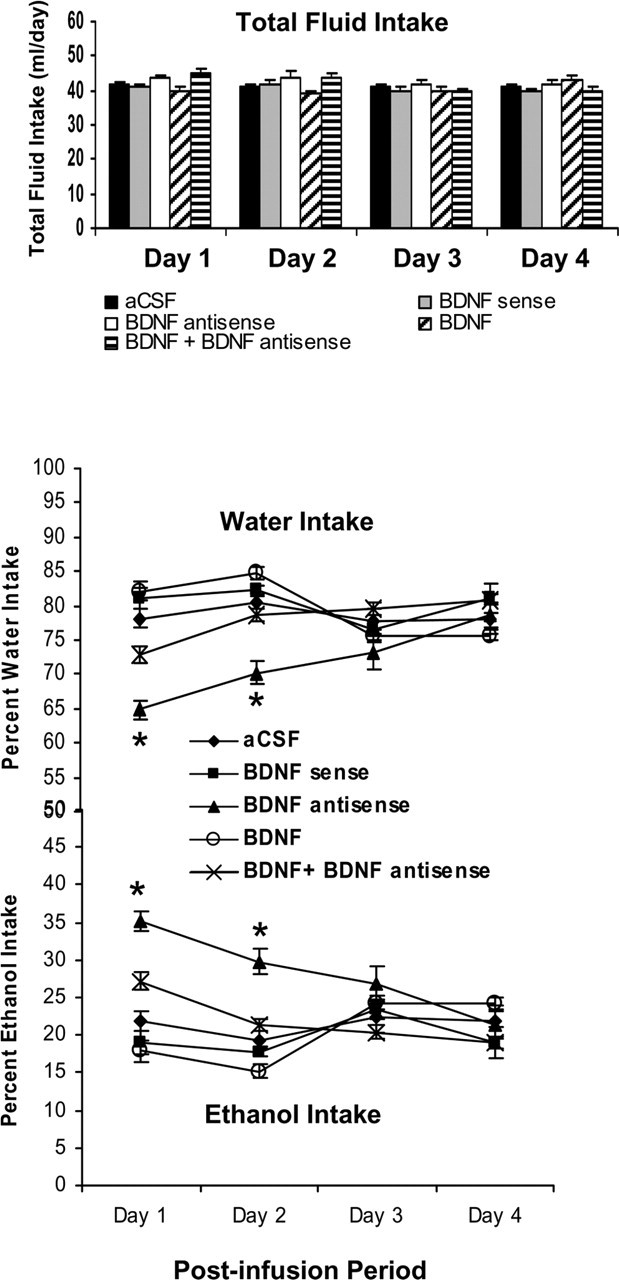
Effect of CeA infusion (once at day 1) of BDNF sense and antisense ODNs and/or BDNF (twice; first infusion at day 1 and second infusion at day 2) on alcohol preference as measured by the two-bottle free-choice paradigm. Results represent the percentage of 7% v/v (ethanol in water solution) ethanol intake and percentage of water intake of total fluid intake (milliliters per day). Values are the mean ± SEM of six rats in each group, and alcohol intake was measured for 4 d in the same group of rats. Asterisks indicate significant (p < 0.001) difference from aCSF-infused rats.
Effects of intra-MeA infusion of BDNF antisense and sense ODNs on anxiety-like behaviors in rats
To examine whether BDNF manipulations in the MeA also affected anxiety-like behaviors, we implanted bilateral cannulas targeting the MeA (Fig. 5A). After 1 week of recovery, rats were injected with 0.5 μl of aCSF, BDNF sense ODNs, BDNF antisense ODNs, BDNF, or BDNF plus BDNF antisense ODNs as described above for the CeA. The open- and closed-arm activities were measured on the EPM. It was found that intra-MeA infusion of BDNF antisense ODNs produced a significant reduction of percentage of open-arm entries (F(4,37) = 18.52; p < 0.001) and percentage of time (F(4,37) = 9.10; p < 0.001) spent on open arms in rats. Post hoc comparisons revealed that BDNF antisense ODN infusions into MeA significantly decreased the percentage of open-arm entries (p < 0.001) and time spent (p < 0.001) on open arms compared with aCSF-infused rats (Fig. 5B). Coinfusion of BDNF prevented the development of BDNF antisense ODN-induced anxiety-like behaviors. Rats infused with BDNF sense ODNs or BDNF alone, however, did not show any significant alterations in anxiety-like behaviors (changes in open-arm activities) compared with aCSF-infused rats. The total number of entries (closed and open arms) or total number of closed-arm entries was not significantly different among the groups. Thus, locomotor activity was not modulated by infusions of these agents into the MeA of rats (Fig. 5B). These results indicate that BDNF antisense ODN infusion into the MeA provoked an anxiety-like effect in rats.
Figure 5.

A, Low-magnification view of Nissl staining of the amygdaloid structures of rats. This photomicrograph indicates the position of the cannula, which is just above the targeted area of medial amygdala. Scale bar, 200 μm. B, Effect of MeA infusion of aCSF and BDNF sense and antisense ODNs (with and without BDNF coinfusion) on open- and closed-arm activities of the elevated plus maze. Total number of entries represents the total numbers of closed- and open-arm entries in EPM. Values are the mean ± SEM of eight to nine rats in each group. Asterisks indicate significant (p < 0.001) difference from aCSF-infused rats.
Cellular changes in the MeA after infusions of BDNF antisense ODNs and BDNF
Immediately after the measurement of anxiety-like behavior, rats were perfused, and the brains were used to examine the protein and mRNA levels of BDNF as well as the protein levels of p-CREB, p-ERK1/2, and NeuN. Fig. 6 shows the expression of BDNF protein and mRNA along with gold immunolabeling of p-CREB, p-ERK1/2, and NeuN in the MeA after BDNF manipulations. BDNF antisense ODN infusion in the MeA significantly decreased both the protein (F(4,30) = 127.22; p < 0.001) and mRNA (F(4,25) = 68.79; p < 0.001) levels of BDNF in the MeA of rats. Post hoc comparisons revealed that BDNF antisense ODN infusion into MeA significantly decreased mRNA (p < 0.001) and protein (p < 0.001) levels of BDNF specifically in MeA but not in the CeA or BLA compared with the aCSF-infused rats. Infusion of BDNF along with BDNF antisense ODNs prevented the decrease in protein and mRNA levels of BDNF in the MeA. Infusions of BDNF sense ODNs or BDNF alone in the MeA did not produce any significant changes in the protein or mRNA levels of BDNF in CeA, MeA, or BLA compared with the aCSF-infused group (Figs. 6, 7).
Figure 6.
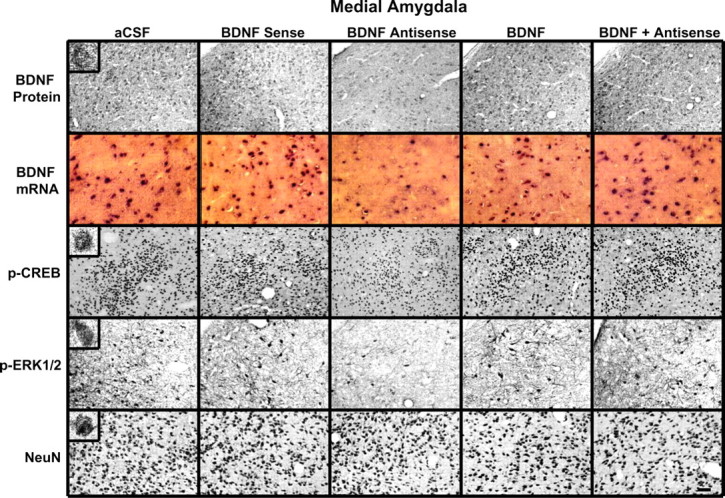
Representative photomicrographs at low magnification showing BDNF (top row), p-CREB (third row), p-ERK1/2 (fourth row), and NeuN (bottom row) gold immunolabeling and mRNA levels of BDNF (second row) in the MeA brain structures of rats infused with aCSF, BDNF sense, and antisense ODNs (with and without BDNF coinfusion) into the MeA. Scale bar, 30 μm. Insets in the aCSF column indicate immunogold particles within a single cell body or single nucleus at high magnification (100×).
Figure 7.
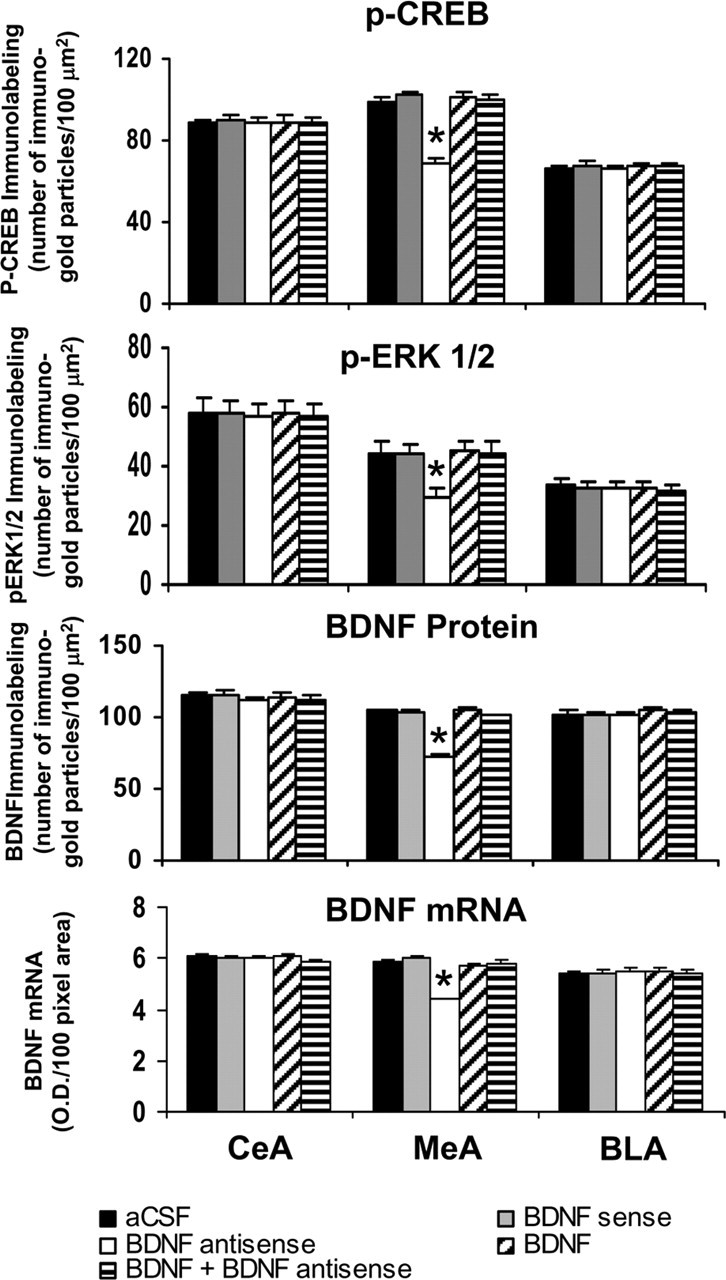
Quantitation of BDNF, p-CREB, and p-ERK1/2 protein levels (number of immunogold particles per 100 μm2 area) and mRNA levels of BDNF in the amygdaloid structures of rats infused in the MeA with aCSF and BDNF sense and antisense ODNs (with and without BDNF coinfusion). Values are the mean ± SEM of five to seven rats in each group. Asterisks indicate significant (p < 0.001) difference from aCSF-infused rats.
We also found that BDNF antisense ODN infusion into the MeA significantly decreased the phosphorylation of ERK1/2 (df = 4; H = 11.13; p = 0.025) and CREB (F(4,20) = 41.43; p < 0.001) specifically in the MeA, which was prevented by coinfusion with BDNF. Infusions of BDNF sense ODNs or BDNF alone in the MeA did not produce any significant changes in the protein levels of p-ERK1/2 and p-CREB in CeA, MeA, or BLA compared with the aCSF-infused group (Figs. 6, 7). Similarly to CeA infusion, BDNF sense and antisense ODN infusions into MeA did not cause any neuronal loss in this region of amygdala, because the levels of NeuN were not significantly altered among the treatment groups (Fig. 6, bottom) (mean data not shown).
Relationship between decreased BDNF expression in the MeA and alcohol preference
To determine the relationship between BDNF levels in MeA and alcohol preference, we also measured alcohol intake after manipulating the BDNF levels in the MeA. We implanted bilateral cannulas targeting the MeA, and after 1 week of recovery, rats were habituated to drinking water from two bottles. When rats started drinking water equally from either bottle, bilateral infusions of pharmacological agents into the MeA were performed. Rats were provided with 7% (v/v) ethanol solution in one of the bottles and water in the other bottle for 4 d. The mean percentage of ethanol or water intake of their total fluid intake for each day was calculated. A 5 × 4 (group × day) repeated-measure ANOVA was performed on changes in alcohol intake that yielded a significant group effect (F(4,75) = 31.20; p < 0.001), a significant day effect (F(3,75) = 7.09; p < 0.001), and a significant interaction (F(12,75) = 14.56; p < 0.001) of these variables in rats. A 5 × 4 (group × day) repeated-measure ANOVA was performed on changes in water intake that also yielded a significant group effect (F(4,75) = 31.36; p < 0.001), a significant day effect (F(3,75) = 6.66; p < 0.001), and a significant interaction (F(12,75) = 14.64; p < 0.001) of these variables in rats (Fig. 8). It was found that intra-MeA infusions of BDNF antisense, but not sense, ODNs in rats significantly increase ethanol preference (p < 0.001) and decrease water preference (p < 0.001) on days 1 and 2 of the postinfusion period compared with aCSF-infused rats. Interestingly, alcohol intake on postinfusion days 3 and 4 returned to levels similar to the aCSF group of rats (Fig. 8). It was found that BDNF (50 ng) infusion (once daily for 2 d) alone into MeA significantly (p < 0.001) decreased alcohol intake, and coinfusion of BDNF with BDNF antisense ODNs significantly antagonized the increase in alcohol intake of rats. The total fluid intake (in milliliters per day) was similar for each group at each day (Fig. 8). There were no significant differences in body weights among the groups. These results indicate that the decreased BDNF levels in the MeA are also associated with higher alcohol preference. Furthermore, these results indicate that rescue of the decreased BDNF levels in the MeA attenuated the BDNF antisense ODN-induced increases in alcohol intake in rats.
Figure 8.
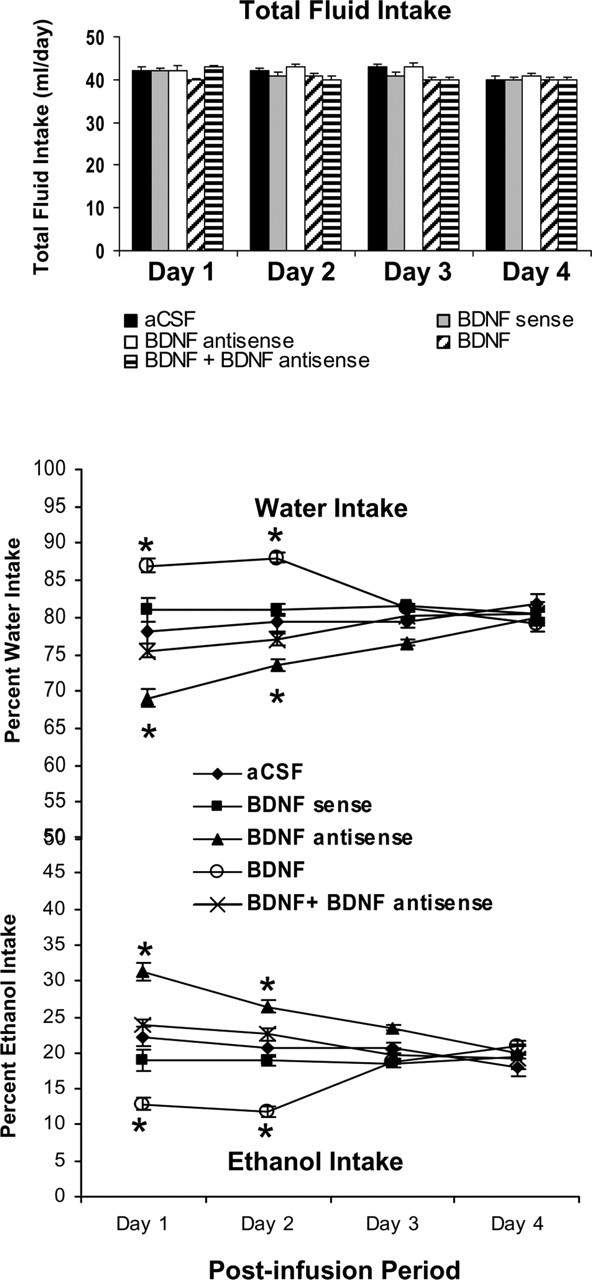
Effect of MeA infusion (once at day 1) of BDNF sense and antisense ODNs and/or BDNF (twice; first infusion at day 1 and second infusion at day 2) on alcohol preference as measured by the two-bottle free-choice paradigm. Results represent the percentage of 7% v/v (ethanol in water solution) ethanol intake and percentage of water intake of total fluid intake (milliliters per day). Values are the mean ± SEM of six rats in each group, and alcohol intake was measured for 4 d in the same group of rats. Asterisks indicate significant (p < 0.001) difference from aCSF-infused rats.
Effects of intra-BLA infusion of BDNF antisense ODNs on anxiety-like behaviors
We also examined the effects of BDNF manipulations in the BLA on anxiety-like behaviors. We implanted bilateral cannulas targeting the BLA (Fig. 9A). After 1 week of recovery, rats were infused with 0.5 μl of aCSF, BDNF sense, antisense ODNs, and BDNF. Twelve hours after infusion, open- and closed-arm activities were measured on the EPM. Unlike intra-CeA or -MeA infusions, it was found that intra-BLA infusion of BDNF antisense ODNs produced no significant changes in percentage of open-arm entries and percentage of time spent on open arm compared with aCSF-infused rats (Fig. 9B). Rats infused with BDNF sense ODNs or BDNF also did not show any changes in open- or closed-arm activities. The total number of entries (closed and open arms) or total number of closed-arm entries was not significantly different among the groups. Thus, locomotor activity was not modulated by infusions of these agents into BLA of rats (Fig. 9B). These results indicate that BDNF antisense ODN infusions into BLA have no effects on anxiety-like behaviors in rats.
Figure 9.

A, Low-magnification view of Nissl staining of amygdaloid structures of rats. This photomicrograph indicates the position of the cannula, which is just above the targeted area of basolateral amygdala. Scale bar, 200 μm. B, Effect of BLA infusion of aCSF and BDNF sense and antisense ODNs (with and without BDNF) on open- and closed-arm activities of the elevated plus maze. Total number of entries represents the total numbers of closed- and open-arm entries in EPM. Values are the mean ± SEM of 6–12 rats in each group.
Cellular changes in the BLA after infusions of BDNF antisense ODNs and BDNF
Immediately after EPM test, brains were used to examine the protein and mRNA levels of BDNF as well as the protein levels of p-CREB, p-ERK1/2, and NeuN in the CeA, MeA, and BLA. Figure 10 shows the expression of BDNF protein and mRNA, along with gold immunolabeling of p-CREB, p-ERK1/2, and NeuN. It was found that BDNF antisense ODN infusion in the BLA significantly decreased both the protein (F(4,43) = 11.66; p < 0.001) and mRNA (F(4,34) = 37.83; p < 0.001) levels of BDNF in the BLA of rats (Fig. 11). Post hoc comparisons revealed that BDNF antisense ODN infusions into BLA significantly decreased mRNA (p < 0.001) and protein (p < 0.001) levels of BDNF in the BLA but not in MeA and CeA compared with aCSF-infused rats. Coinfusion of BDNF with BDNF antisense ODNs significantly attenuated the reductions in mRNA and protein levels of BDNF in the BLA of rats. Infusion of BDNF sense ODNs or BDNF alone in the BLA did not produce any significant changes in the protein or mRNA levels of BDNF in CeA, MeA, or BLA compared with the aCSF-infused rats (Figs. 10, 11).
Figure 10.
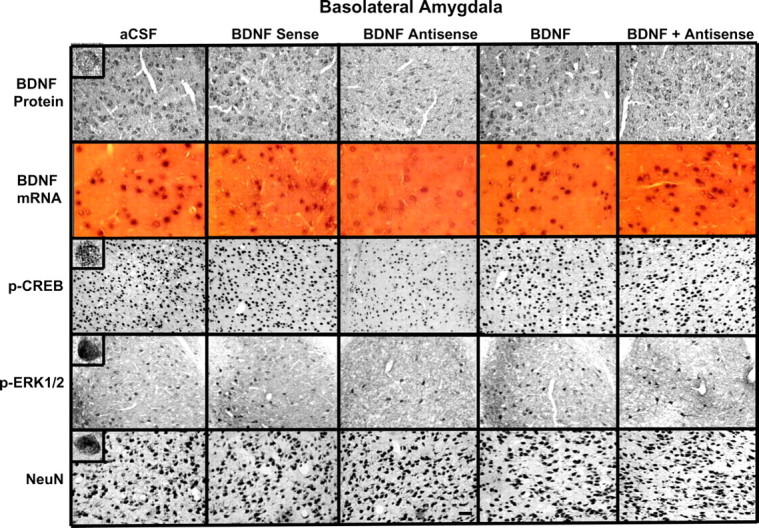
Representative photomicrographs at low magnification showing BDNF (top row), p-CREB (third row), p-ERK1/2 (fourth row), and NeuN (bottom row) gold immunolabeling and mRNA levels of BDNF (second row) in the BLA brain structures of rats infused with aCSF and BDNF sense and antisense ODNs (with or without BDNF infusion) into the BLA. Scale bar, 30 μm. Insets in the aCSF column indicate immunogold particles within a single cell body or single nucleus at high magnification (100×).
Figure 11.
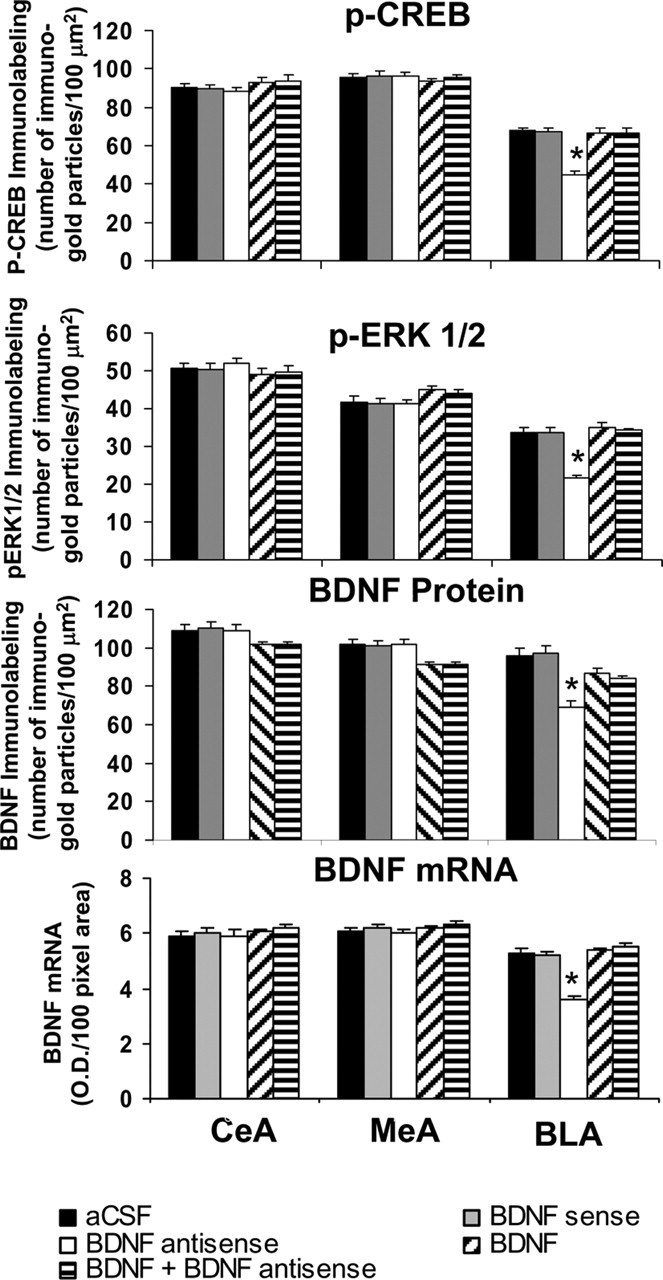
Quantitation of BDNF, p-CREB, and p-ERK1/2 protein levels (number of immunogold particles per 100 μm2 area) and mRNA levels of BDNF in the amygdaloid structures of rats infused in the BLA with aCSF and BDNF sense and antisense ODNs (with or without BDNF coinfusion). Values are the mean ± SEM of 6–12 rats in each group. Asterisks indicate significant (p < 0.001) difference from aCSF-infused rats.
We also studied the effects of BDNF manipulations in the BLA on the protein levels of p-ERK1/2 and p-CREB. It was found that BDNF antisense ODN infusion into the BLA significantly decreased the protein levels of p-ERK1/2 (F(4,34) = 27.63; p < 0.001) and p-CREB (F(4,34) = 29.91; p < 0.001) specifically in the BLA, which was normalized by coinfusion with BDNF (Fig. 11). Infusion of BDNF sense ODNs or BDNF alone in the BLA did not produce any significant changes in the protein levels of p-ERK1/2 and p-CREB in CeA, MeA, or BLA compared with the aCSF-infused rats. Similar to CeA and MeA, BDNF sense, antisense ODNs, and BDNF infusions into BLA did not produce any neuronal loss in this region of amygdala, because the levels of NeuN were not significantly altered among the treatment groups (Fig. 10, bottom) (mean data not shown).
Relationship between decreased BDNF levels in the BLA and alcohol preference
We also examined the association between decreased BDNF levels in BLA and alcohol preference. We implanted bilateral cannulas targeting the BLA, and after 1 week of recovery, rats were habituated to drinking water from two bottles. When rats started drinking water equally from either bottle, bilateral infusions of pharmacological agents into the BLA were performed. Rats were provided with 7% (v/v) ethanol solution in one of the bottles and water in the other bottle for 4 d. The mean percentage of ethanol or water intake of their total fluid intake for each day was calculated. A 5 × 4 (group × day) repeated-measure ANOVA was performed on changes in alcohol and water intake that yielded no significant group or day effect and also no significant interaction of these variables in rats. It was found that infusion of BDNF antisense and sense ODNs into BLA does not alter the consumption of water or ethanol compared with aCSF-infused rats. It was found that BDNF (50 ng) infusion (once daily for 2 d) into BLA had no significant effects on alcohol and water intake of rats (Fig. 12). The total fluid intake (in milliliters per day) was similar for each group at each day. There were no significant differences in body weights among the groups. These results indicate that in the BLA, decreased BDNF levels have no effects on alcohol intake.
Figure 12.
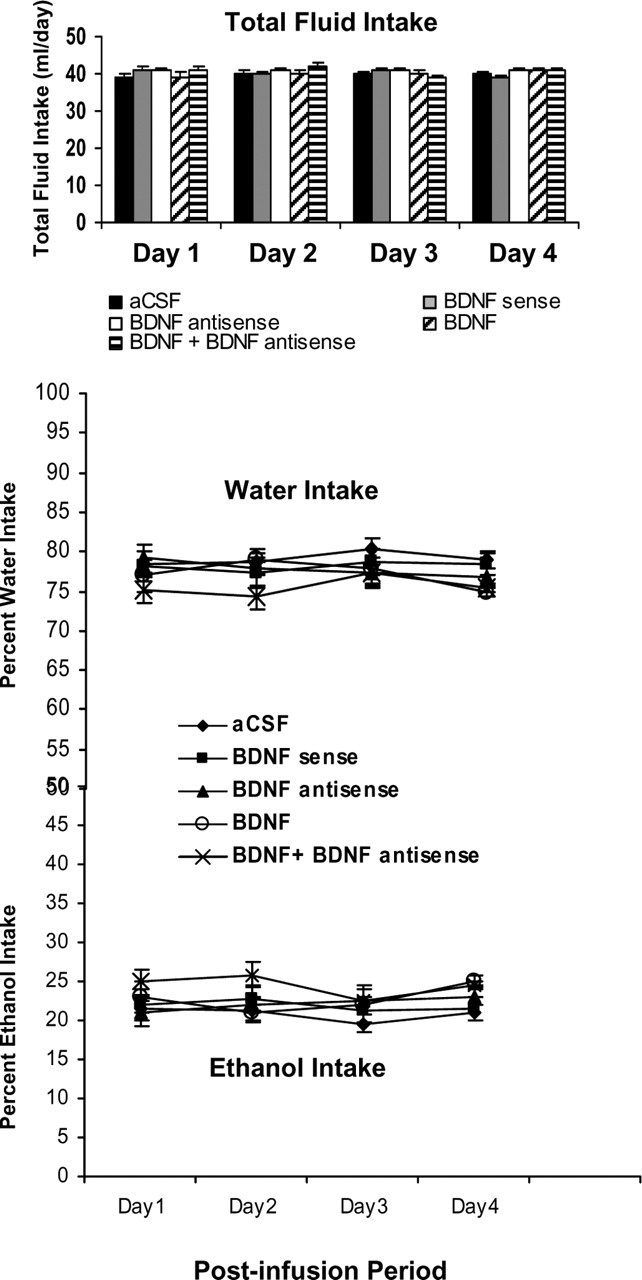
Effect of BLA infusion of BDNF sense and antisense ODNs (once on day 1) with and without BDNF (twice; first infusion on day 1 and second infusion on day 2) on alcohol preference as measured by the two-bottle free-choice paradigm. Results represent the percentage of 7% v/v (ethanol in water solution) ethanol intake and percentage of water intake of total fluid intake (milliliters per day). Values are the mean ± SEM of six to eight rats in each group, and alcohol intake was measured for 4 d in the same group of rats.
Discussion
The data presented here provide the first evidence that a deficiency of BDNF in the neurocircuitry of CeA and MeA but not BLA is involved in alcohol-drinking and anxiety-like behaviors in rats. It has been shown recently that BDNF may be an important molecule in the hippocampus and striatal brain regions to mediate some of the behavioral consequences of ethanol exposure (McGough et al., 2004). Using knock-out strategy, it has been shown that a deficiency in BDNF promotes alcohol intake in mice (Hensler et al., 2003; McGough et al., 2004). Here we extended these studies and used an antisense ODN strategy to establish the direct role of CeA, MeA, and BLA BDNF signaling in alcohol-drinking behaviors. It was found that a deficiency in BDNF in CeA and MeA but not in BLA is associated with higher anxiety-like and alcohol-drinking behaviors. Furthermore, the alcohol intake and anxiety-like behaviors were rescued via coinfusion of BDNF with BDNF antisense ODNs in the CeA or MeA. It was found that infusion of BDNF sense ODNs into CeA, MeA, or BLA does not produce any effects on anxiety-like and alcohol-drinking behaviors or on BDNF levels and related signaling, suggesting that effects of BDNF antisense ODNs are specific. These results clearly indicate that a deficient BDNF system in the CeA and MeA may be a part of the molecular processes that regulate both alcohol-drinking and anxiety-like behaviors.
Several studies have shown that a BDNF gene polymorphism is associated with personality traits of anxiety, depression, and alcoholism (Uhl et al., 2001; Matsushita et al., 2004; Jiang et al., 2005; Lang et al., 2005). Preexisting higher anxiety levels appear to play an important role in the initiation and maintenance of alcohol-drinking behaviors in some alcoholics (Koob, 2003; Pandey, 2003). Because various nuclei of amygdala play an important role in alcohol intake and anxiety behavior (Davis, 1997; McBride, 2002; Koob, 2003), we asked whether decreased BDNF expression in CeA, MeA, and BLA neurocircuitries acts as a common molecular marker for these behaviors. Here, we report that depleting BDNF levels in the CeA and MeA leads to development of anxiety-like behaviors and also promotes alcohol intake, suggesting a correlation between drinking and anxiety-like behaviors. It has been shown that acute ethanol exposure increases the BDNF levels in hippocampus, whereas chronic treatment either decreases or increases the BDNF levels in forebrain regions (MacLennan et al., 1995; Miller et al., 2002; McGough et al., 2004). We found that ethanol withdrawal after chronic ethanol exposure decreases the protein levels of BDNF in cortex as well as mRNA and protein levels of BDNF in CeA and MeA but not in BLA of rats (Pandey et al., 1999; Zhang and Pandey, 2003). Ethanol-withdrawn rats behaviorally display anxiety-like behaviors (Pandey et al., 1999, 2003). Because the results from the present study indicate that decreased BDNF levels in CeA and MeA are associated with anxiety-like behaviors and promote alcohol intake, it is possible that decreased BDNF levels in the CeA and MeA may be involved in anxiety related to ethanol withdrawal, and this may maintain the continued consumption of ethanol. Future studies are needed to explore this possibility to demonstrate the role of BDNF in relapse to alcohol-drinking behaviors.
BDNF acts via TrkB to activate intracellular signaling pathways, which include PLC-γ, ERK1/2, and PI 3-kinase signaling pathway (Impey et al., 1999). It has been shown that higher expression of TrkB decreases anxiety-like behaviors in mice (Koponen et al., 2004). Furthermore, it was also reported that acute social defeat decreases BDNF levels in various brain structures of mice (Pizarro et al., 2004). These findings support the current results that decreased BDNF function in the CeA and MeA is associated with anxiety-like behavior in rats. We found a decrease in CREB phosphorylation after BDNF antisense ODN infusion, and this decrease may be attributable to decreased ERK1/2 activity. Interestingly, when BDNF was coinfused with BDNF antisense ODNs, it prevented the development of anxiety-like behaviors and attenuated alcohol intake. The reductions in phosphorylation of ERK1/2 and CREB and in protein and mRNA levels of BDNF were also normalized after BDNF coinfusion. It is possible that decreased CREB phosphorylation, attributable to BDNF antisense ODN infusion in the CeA, MeA, or BLA, may also lead to a decreased expression of TrkB. Thus, overall BDNF signaling may be deficient because of depletion of BDNF. The pattern of cellular expression of p-CREB, BDNF, and p-ERK1/2 in the CeA, MeA, and BLA indicate that the number of p-CREB-positive cells is higher than the numbers of BDNF- and p-ERK1/2-positive cells. This differential expression may be related to the fact that CREB may be expressed in all cells, whereas BDNF and p-ERK1/2 may be expressed in certain kinds of cells in the brain (Irving et al., 2000; Riley et al., 2004). We reported previously that CREB-haplodeficient mice display higher anxiety-like behaviors and have a higher alcohol preference. These mice have lower levels of BDNF in several brain regions, including the amygdala, because of deletion of CREB (Pandey et al., 2004). In agreement with these findings, it appears that decreased CREB phosphorylation in the CeA and MeA, caused by depletion of BDNF, provokes anxiety-like behaviors and promotes alcohol intake in rats, which is prevented by BDNF coinfusion.
The next question that arises is how lower BDNF levels in the CeA and MeA regulate anxiety-like and alcohol-drinking behaviors. One possibility is that BDNF may directly regulate these behaviors by regulating synaptic plasticity via altering the synaptic strength and dendritic spines in the CeA and MeA (Agassandian et al., 2006). Another possibility is that decreased BDNF in CeA and MeA may modulate the function of GABAergic, glutamatergic, serotonergic, and/or the neuropeptide Y (NPY) system and thereby regulate anxiety and alcohol-drinking behaviors (Mamounas et al., 1995; Barnea and Roberts, 2001; Hensler et al., 2003; Matsumoto et al., 2006). In fact, it has been shown that NPY in the CeA is anxiolytic and that decreased NPY levels in CeA are involved in alcohol-drinking behaviors (Pandey et al., 2003, 2005; Heilig, 2004). Several studies indicate that decreased BDNF levels are associated with decreased NPY levels in the brain, whereas infusion of BDNF in brain or treatment of culture cells with BDNF increases the levels of NPY (Jones et al., 1994; Nawa et al., 1994; Barnea and Roberts, 2001). Thus it is possible that BDNF antisense ODN infusion into CeA and MeA may decrease NPY expression via decreased CREB phosphorylation, which in turn may modulate anxiety and alcohol-drinking behaviors. It is important to mention that infusion of BDNF alone into CeA produced a trend toward decreased alcohol intake, whereas infusion into MeA significantly decreased the alcohol intake; however, infusion into BLA had no effects on alcohol intake. In alcohol preference studies, we infused a low dose of BDNF twice at 24 h intervals, and under this condition we did not measure the BDNF signaling. However, when we infused a low dose of BDNF twice within 12 h and measured the anxiety-like behaviors 1 h after the last infusion, BDNF alone had no effects on anxiety-like behaviors and BDNF signaling but significantly attenuated the BDNF antisense ODN-induced anxiety-like behaviors and the reduction in BDNF signaling. Although more studies are needed to establish the dose and time course of the action of BDNF in the CeA and MeA on alcohol-drinking and anxiety-like behaviors, these data suggest that neurons in the MeA and possibly the CeA are more sensitive to a low dose of BDNF in relation to alcohol-drinking behaviors.
The BLA structures also play a role in anxiety (Davis, 1997; Davis and Whalen, 2001), but we found that CREB phosphorylation and BDNF protein levels were not altered in this amygdaloid structure of rats during ethanol withdrawal after chronic ethanol exposure, suggesting the possibility that BDNF in the BLA may not be mediating anxiety or alcohol-drinking behaviors (Pandey et al., 2003; Zhang and Pandey, 2003). The levels of various proteins linked to BDNF signaling were also reduced in the BLA after BDNF antisense ODN infusion; however, this did not induce anxiety-like behaviors or promote alcohol intake. Therefore, it is possible that decreased BDNF signaling in this brain region does not play a role in anxiety or alcohol-drinking behaviors. In contrast, it has been shown recently that overexpression of CREB in BLA produces anxiety-like behavior (Wallace et al., 2004). Furthermore, BDNF signaling in BLA plays a role in fear conditioning (Rattiner et al., 2004). However, our previous as well as current studies suggest that decreased CREB phosphorylation and BDNF expression in the BLA does not provoke anxiety-like behaviors and also does not modulate alcohol intake (Pandey et al., 2003). In fact, it has been shown that ERK signaling in the CeA, but not in BLA, is involved in cocaine craving in rats (Lu et al., 2005). Nonetheless, our results clearly suggest that decreased BDNF levels in the CeA and MeA, but not in the BLA, produce higher alcohol intake and anxiety-like behaviors, and this is correlated with decreased phosphorylation of ERK1/2 and CREB.
Conclusions
The present investigation provides a common molecular mechanism that implicates that decreased BDNF function in the CeA and MeA, but not BLA, may be responsible for the predisposition to anxiety and alcohol-drinking behaviors. Because BDNF is highly implicated in synaptic plasticity (Thoenen, 2000; Poo, 2001), decreased expression of BDNF in CeA and MeA may alter the synaptic plasticity within these amygdaloid nuclei, which may provoke anxiety and alcohol-drinking behaviors.
Footnotes
This study was supported by National Institute on Alcohol Abuse and Alcoholism Grants AA-10005 and AA13341 and by the Department of Veterans Affairs (Merit Review Grant and Research Career Scientist Award to S.C.P.).
References
- Agassandian K, Gedney M, Cassell MD (2006). Neurotrophic factors in the central nucleus of amygdala may be organized to provide substrates for associative learning. Brain Res 1076:78–86. [DOI] [PubMed] [Google Scholar]
- Barnea A, Roberts J (2001). Induction of functional and morphological expression of neuropeptide Y (NPY) in cortical cultures by brain-derived neurotrophic factor (BDNF): evidence for a requirement for extracellular-regulated kinase (ERK)-dependent and ERK-independent mechanisms. Brain Res 919:57–69. [DOI] [PubMed] [Google Scholar]
- Bibel M, Barde YA (2000). Neurotrophins: key regulators of cell fate and cell shape in the vertebrate nervous system. Genes Dev 14:2919–2937. [DOI] [PubMed] [Google Scholar]
- Carter AR, Chen C, Schwartz PM, Segal RA (2002). Brain-derived neurotrophic factor modulates cerebellar plasticity and synaptic ultrastructure. J Neurosci 22:1316–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M (1997). Neurobiology of fear responses: the role of the amygdala. J Neuropsychiatry Clin Neurosci 9:382–402. [DOI] [PubMed] [Google Scholar]
- Davis M, Whalen PJ (2001). The amygdala: vigilance and emotion. Mol Psychiatry 6:13–34. [DOI] [PubMed] [Google Scholar]
- Deogracias R, Espliguero G, Iglesias T, Rodriguez-Pena A (2004). Expression of neurotrophin receptor trkB is regulated by cAMP/CREB pathway in neurons. Mol Cell Neurosci 26:470–480. [DOI] [PubMed] [Google Scholar]
- Duman RS (2004). Role of neurotrophic factors in the etiology and treatment of mood disorders. Neuromolecular Med 5:11–25. [DOI] [PubMed] [Google Scholar]
- File SE (1993). The interplay of learning and anxiety in the elevated plus-maze. Behav Brain Res 58:199–202. [DOI] [PubMed] [Google Scholar]
- Heilig M (2004). The NPY system in stress, anxiety, and depression. Neuropeptides 38:213–224. [DOI] [PubMed] [Google Scholar]
- Hensler JG, Ladenheim EE, Lyons WE (2003). Ethanol consumption and serotonin-1A (5-HT1A) receptor function in heterozygous BDNF (+/−) mice. J Neurochem 85:1139–1147. [DOI] [PubMed] [Google Scholar]
- Horger BA, Iyasere CA, Berhow MT, Messer CJ, Nestler EJ, Taylor JR (1999). Enhancement of locomotor activity and conditioned reward to cocaine by brain-derived neurotrophic factor. J Neurosci 19:4110–4122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Impey S, Obrietan K, Storm DR (1999). Making new connections: role of ERK/MAP kinase signaling in neuronal plasticity. Neuron 23:11–14. [DOI] [PubMed] [Google Scholar]
- Irving EA, Barone FC, Reith AD, Hadingham SJ, Parsons AA (2000). Differential activation of MAPK/ERK and p38/SAPK in neurons and glia following focal cerebral ischaemia in the rat. Mol Brain Res 77:65–75. [DOI] [PubMed] [Google Scholar]
- Jiang X, Xu K, Hoberman J, Tian F, Marko AJ, Waheed JF, Harris CR, Marini AM, Enoch MA, Lipsky RH (2005). BDNF variation and mood disorders: a novel functional promoter polymorphism and Val 66Met are associated with anxiety but have opposing effects. Neuropsychopharmacology 30:1353–1361. [DOI] [PubMed] [Google Scholar]
- Jones KR, Farinas I, Backus C, Reichardt LF (1994). Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Cell 76:989–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF (2003). Alcoholism: allostasis and beyond. Alcohol Clin Exp Res 27:232–243. [DOI] [PubMed] [Google Scholar]
- Koponen E, Voikar V, Riekki R, Saarelainen T, Rauramaa T, Rauvala H, Taira T, Castren E (2004). Transgenic mice overexpressing the full-length neurotrophin receptor trkB exhibit increased activation of the trkB-PLC gamma pathway, reduced anxiety, and facilitated learning. Mol Cell Neurosci 26:166–181. [DOI] [PubMed] [Google Scholar]
- Lang UE, Hellweg R, Kalus P, Bajbouj M, Lenzen KP, Sander T, Kunz D, Gallinat J (2005). Association of a functional BDNF polymorphism and anxiety-related personality traits. Psychopharmacology 180:95–99. [DOI] [PubMed] [Google Scholar]
- Lu L, Hope BT, Dempsey J, Liu SY, Bossert JM, Shaham Y (2005). Central amygdala ERK signaling pathway is critical to incubation of cocaine craving. Nat Neurosci 8:212–219. [DOI] [PubMed] [Google Scholar]
- Ma YL, Wang HL, Wu HC, Wei CL, Lee EH (1998). Brain-derived neurotrophic factor antisense oligonucleotide impairs memory retention and inhibits long term potentiation in rats. Neuroscience 82:957–967. [DOI] [PubMed] [Google Scholar]
- MacLennan AJ, Lee N, Walker DW (1995). Chronic ethanol administration decreases brain-derived neurotrophic factor gene expression in rat hippocampus. Neurosci Lett 197:105–108. [DOI] [PubMed] [Google Scholar]
- MacQueen GM, Ramakrishnan K, Croll SD, Siuciak JA, Yu G, Young LT, Fahnestock M (2001). Performance of heterozygous brain-derived neurotrophic factor knockout mice on behavioral analogues of anxiety, nociception, and depression. Behav Neurosci 115:1145–1153. [DOI] [PubMed] [Google Scholar]
- Mamounas LA, Blue ME, Siuciak JA, Altar CA (1995). Brain-derived neurotrophic factor promotes the survival and sprouting of serotonergic axons in rat brain. J Neurosci 15:7929–7939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manji HK, Drevets WC, Charney DS (2001). The cellular neurobiology of depression. Nat Med 7:541–547. [DOI] [PubMed] [Google Scholar]
- Matsumoto T, Numakawa T, Yokomaku D, Adachi N, Yamagishi S, Numakawa Y, Kunugi H, Taguchi T (2006). Brain-derived neurotrophic factor-induced potentiation of glutamate and GABA release: different dependency on signaling pathways and neuronal activity. Mol Cell Neurosci 31:70–84. [DOI] [PubMed] [Google Scholar]
- Matsushita S, Kimura M, Miyakawa T, Yoshino A, Murayama M, Masaki T, Higuchi S (2004). Association study of brain-derived neurotrophic factor gene polymorphism and alcoholism. Alcohol Clin Exp Res 28:1609–1612. [DOI] [PubMed] [Google Scholar]
- McBride WJ (2002). Central nucleus of the amygdala and the effects of alcohol and alcohol drinking behavior in rodents. Pharmacol Biochem Behav 71:509–515. [DOI] [PubMed] [Google Scholar]
- McGough NN, He DY, Logrip ML, Jeanblanc J, Phamluong K, Luong K, Kharazia V, Janak PH, Ron D (2004). RACK1 and brain-derived neurotrophic factor: a homeostatic pathway that regulates alcohol addiction. J Neurosci 24:10542–10552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller R, King MA, Heaton MB, Walker DW (2002). The effects of chronic ethanol consumption on neurotrophins and their receptors in the rat hippocampus and basal forebrain. Brain Res 950:137–147. [DOI] [PubMed] [Google Scholar]
- Monteggia LM, Barrot M, Powell CM, Berton O, Galanis V, Gemelli T, Meuth S, Nagy A, Greene RW, Nestler EJ (2004). Essential role of brain-derived neurotrophic factor in adult hippocampal function. Proc Natl Acad Sci USA 29:10827–10832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montkowski A, Holsboer F (1997). Intact spatial learning and memory in transgenic mice with reduced BDNF. Neuroreport 8:779–782. [DOI] [PubMed] [Google Scholar]
- Nawa H, Pelleymounter MA, Carnahan J (1994). Intraventricular administration of BDNF increases neuropeptide expression in newborn rat brain. J Neurosci 14:3751–3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM (2002). Neurobiology of depression. Neuron 34:13–25. [DOI] [PubMed] [Google Scholar]
- Pandey SC (2003). Anxiety and alcohol abuse disorders: a common role for CREB and its target, the neuropeptide Y gene. Trends Pharmacol 24:456–460. [DOI] [PubMed] [Google Scholar]
- Pandey SC (2004). The gene transcription factor cyclic AMP responsive-element binding protein: role in positive and negative affective states of alcohol addiction. Pharmacol Ther 104:47–58. [DOI] [PubMed] [Google Scholar]
- Pandey SC, Zhang D, Mittal N, Nayyar D (1999). Potential role of the gene transcription factor cyclic AMP-responsive element binding protein in ethanol withdrawal-related anxiety. J Pharmacol Exp Ther 288:866–878. [PubMed] [Google Scholar]
- Pandey SC, Roy A, Mittal N (2001). Effects of chronic ethanol intake and its withdrawal on the expression and phosphorylation of the CREB gene transcription factor in rat cortex. J Pharmacol Exp Ther 296:857–868. [PubMed] [Google Scholar]
- Pandey SC, Roy A, Zhang H (2003). The decreased phosphorylation of cyclic adenosine monophosphate (cAMP) response element binding (CREB) protein in the central amygdala acts as a molecular substrate for anxiety related to ethanol withdrawal in rats. Alcohol Clin Exp Res 27:396–409. [DOI] [PubMed] [Google Scholar]
- Pandey SC, Roy A, Zhang H, Xu T (2004). Partial deletion of the cAMP response element-binding protein gene promotes alcohol-drinking behaviors. J Neurosci 24:5022–5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey SC, Zhang H, Roy A, Xu T (2005). Deficits in amygdaloid cAMP responsive-element binding protein signaling play a role in genetic predisposition to anxiety and alcoholism. J Clin Invest 115:2762–2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizarro JM, Lumley LA, Medina W, Robison CL, Chang WE, Alagappan A, Bah MJ, Dawood MY, Shah JD, Mark B, Kendall N, Smith MA, Saviolakis GA, Meyerhoff JL (2004). Acute social defeat reduces neurotrophin expression in brain cortical and subcortical areas in mice. Brain Res 1025:10–20. [DOI] [PubMed] [Google Scholar]
- Poo MM (2001). Neurotrophins as synaptic modulators. Nat Rev Neurosci 2:24–32. [DOI] [PubMed] [Google Scholar]
- Rattiner LM, Davis M, French CT, Ressler KJ (2004). Brain-derived neurotrophic factor and tyrosine kinase receptor B involvement in amygdala-dependent fear conditioning. J Neurosci 24:4796–4806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley CP, Cope TC, Buck CR (2004). CNS neurotrophins are biologically active and expressed by multiple cell types. J Mol Histol 35:771–783. [DOI] [PubMed] [Google Scholar]
- Rios M, Fan G, Fekete C, Kelly J, Bates B, Kuehn R, Lechan RM, Jaenisch R (2001). Conditional deletion of brain-derived neurotrophic factor in the postnatal brain leads to obesity and hyperactivity. Mol Endocrinol 15:1748–1757. [DOI] [PubMed] [Google Scholar]
- Rodgers RJ, Johnson NJT (1995). Factor analysis of spatiotemporal and ethological measures in the murine elevated plus-maze test of anxiety. Pharmacol Biochem Behav 52:297–303. [DOI] [PubMed] [Google Scholar]
- Schulteis G, Yackey M, Risbrough V, Koob GF (1998). Anxiogenic-like effects of spontaneous and naloxone-precipitated opiate withdrawal in the elevated-plus maze. Pharmacol Biochem Behav 60:727–731. [DOI] [PubMed] [Google Scholar]
- Shieh PB, Hu SC, Bobb K, Timmusk T, Ghosh A (1998). Identification of a signaling pathway involved in calcium regulation of BDNF expression. Neuron 20:727–740. [DOI] [PubMed] [Google Scholar]
- Shirayama Y, Chen AC, Nakagawa S, Russell DS, Duman RS (2002). Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J Neurosci 22:3251–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siuciak JA, Lewis DR, Wiegand SJ, Lindsay RM (1997). Antidepressant-like effect of brain-derived neurotrophic factor (BDNF). Pharmacol Biochem Behav 56:131–137. [DOI] [PubMed] [Google Scholar]
- Thoenen H (1995). Neurotrophins and neuronal plasticity. Science 270:593–598. [DOI] [PubMed] [Google Scholar]
- Thoenen H (2000). Neurotrophins and activity-dependent plasticity. Prog Brain Res 128:183–191. [DOI] [PubMed] [Google Scholar]
- Uhl GR, Liu QR, Walther D, Hess J, Naiman D (2001). Polysubstance abuse-vulnerability genes: genome scans for association, using 1,004 subjects and 1,494 single-nucleotide polymorphisms. Am J Hum Genet 69:1290–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace TL, Stellitano KE, Neve RL, Duman RS (2004). Effects of cyclic adenosine monophosphate response element binding protein overexpression in the basolateral amygdala on behavioral models of depression and anxiety. Biol Psychiatry 56:151–160. [DOI] [PubMed] [Google Scholar]
- Ying SW, Futter M, Rosenblum K, Webber MJ, Hunt SP, Bliss TV, Bramham CR (2002). Brain-derived neurotrophic factor induces long-term potentiation in intact adult hippocampus: requirement for ERK activation coupled to CREB and upregulation of Arc synthesis. J Neurosci 22:1532–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Pandey SC (2003). Effects of chronic ethanol exposure and withdrawal on the expression and function of brain-derived neurotrophic factor in rat amygdaloid structures. Alcohol Clin Exp Res [Suppl] 27:87A. [Google Scholar]