

Abstract
Hippocampal kindling, a model of mesial temporal lobe epilepsy, is developed through repetitive stimulation of the hippocampus and leads to increased after-discharges as measured by EEG and an enduring seizure-prone state. Synthesis of new proteins is thought to form the basis for sustained seizure-induced physiological and/or pathological changes in synaptic reorganization and apoptotic/necrotic neuronal death. Here we examined the effect of kindling on stimulus-induced c-Jun N-terminal kinase (JNK) and p38 phosphorylation, events postulated to lie upstream of seizure-induced changes in gene transcription. We found that stimulus-induced phosphorylation of JNK, but not of p38, is significantly enhanced in kindled animals compared with their naive counterparts in the CA1 subregion of the hippocampus. Immunofluorescent staining confirmed this region-specific pattern of JNK activation and revealed that reactive astrocytes mediate this effect. Astrocyte proliferation and hypertrophy, as well as upregulation of vimentin protein levels, common markers of astrogliosis, were present after 4 d of kindling. Moreover, this reactive astrogliosis was associated with neuronal death as visualized with Fluoro-jade B and anti-active caspase-3 staining. Stimulus-induced phosphorylation of the JNK substrate paxillin was enhanced in kindled animals, but not that of c-Jun. Moreover, a pan-antibody against MAPK/CDK (mitogen-activated protein kinases/cyclin-dependent kinase) substrates indicated the presence of phosphorylated proteins in cytosolic, membrane, and nuclear fractions. The consequence of these phosphorylation events is not completely understood, but these findings suggest a selective astrocytic signaling response to aberrant synaptic activity, signaling that may modulate kindling progression and/or neuronal death.
Keywords: epileptogenesis, kindling, hippocampus, c-Jun N-terminal kinase, JNK, p38, reactive astrogliosis
Introduction
Hippocampal kindling is a model of temporal lobe epilepsy. (Goddard et al., 1969; Racine, 1972; Tu and Bazan, 2003). Several studies have validated it as a reliable predictor of anti-convulsant drug efficacy (Majkowski, 1999); however, the mechanisms underlying kindling epileptogenesis are incompletely understood.
During seizures, release of glutamate initiates the reorganization of neuronal connections favoring a permanent hyperexcitable state, often associated with plasticity (Sutula et al., 1989; Cavazos et al., 1994). Synthesis of proteins is thought to form the basis for sustained seizure induced in synaptic reorganization and apoptotic/necrotic neuronal death. We postulate that seizure-induced accumulation of bioactive lipid messengers, such as 1-O-alkyl-2-acetyl-glycero-3-phosphocholine [platelet-activating factor (PAF)], helps drive these changes in synaptic restructuring and neuronal death through the activation of kinase signaling, and consequently, modulation of gene transcription (Bazan et al., 2002).
We demonstrated that cyclooxygenase-2 (COX-2) mRNA and protein levels increase during kindling and play a role in its progression (Tu and Bazan, 2003). COX-2 levels are regulated by PAF, and in vitro data suggest that mitogen-activated protein kinase (MAPK) signaling cascades may mediate this effect in neurons (Marcheselli and Bazan, 1994; Marcheselli and Bazan, 1996; Mukherjee et al., 1999). This has led us to examine the effect of kindling on stimulus-induced activation of the MAP kinases, c-Jun N-terminal kinase (JNK), and p38, in the hippocampus and the neocortex.
JNK is fully activated after phosphorylation at Thr183 and Tyr185 by MAP kinase kinases (MKK4 or MKK7, respectively). The MKKs are activated by a group of upstream kinases known as the MAP kinase kinase kinases (MKKKs). This cascade is triggered under a variety of cellular stresses and has also been observed in the hippocampus after seizures elicited by diverse means (Yang et al., 1997; Herdegen et al., 1998; Brecht et al., 1999; Mielke et al., 1999; Jeon et al., 2000). Numerous studies have demonstrated the importance of JNK activation in promoting excitotoxic neuronal death in the CNS (Mattson and Bazan, 2006). The prodeath role of JNK is best illustrated by the protection afforded to JNK-3 knock-out mice in response to kainic acid injection (Yang et al., 1997).
JNK activation may have other functions aside from neuronal life-or-death decisions. For instance, there is some evidence that JNK may modulate synaptic plasticity and is also associated with the exploration of novel environments (Xu et al., 1997; Berman et al., 1998). In accord with a possible role in modulating synaptic plasticity, Curran et al. (2003) recently demonstrated that JNK activation is necessary for induction of long-term depression in the dentate gyrus. Nevertheless, the effect of kindling on stimulus-induced JNK activation remains unexplored.
Here, we demonstrate that stimulus-induced JNK phosphorylation is significantly enhanced in the CA1 of animals that undergo kindling for four consecutive days. Moreover, this enhanced JNK phosphorylation is mediated by astrocytes with a reactive phenotype and is associated with hippocampal neuronal death. Our data support a model whereby recurrent abnormal synaptic activity induces a specific astrocytic signaling response, which may ultimately modulate the progression of kindling and/or neuronal death.
Materials and Methods
Kindling.
Adult male Wistar rats weighing 175–200 g received intraperitoneal injections of a 1 ml/kg anesthetic mixture (ketamine 100 mg/ml and xylazine 10 mg/ml) and were surgically implanted with tripolar electrode units (Plastics One, Roanoke, VA). The electrode units contained a polymide-coated twisted pair (diameter 0.2 mm, tip separation ∼0.06–0.08 mm) that was stereotaxically implanted in the right hippocampus (anteroposterior, 4.0 mm; mediolateral, 2.0 mm; dorsoventral, 3.7 mm) and a ground electrode that was attached to a stainless-steel screw in the left frontal bone (Reibel et al., 2000). The electrode was secured further with three other stainless-steel screws and dental acrylic. After a 1 week recovery, animals underwent a rapid kindling protocol consisting of 12 daily electrical subconvulsive stimulations (10 s train containing 50 Hz biphasic pulses of 1 ms width and 400 μA amplitude) at 30 min intervals for four consecutive days. On the fifth day, animals received one stimulus and were killed by decapitation. The progression of kindling was verified by scoring seizures according to Racine’s scale: I, standing still, wet-dog shaking, mouth and facial movements; II, head nodding or jerking; III, forelimb clonus; IV, rearing; V, falling (Racine, 1972). The EEG was recorded through electrodes using Enhanced Graphics Acquisition for Analysis (version 3.63; RS Electronics, Santa Barbara, CA) and mean spike number was determined using Matlab (MathWorks, Natcik, MA). To determine spike number, Matlab software was configured to determine the baseline amplitude from EEG recordings taken 5 min before stimulus application. All animal-use procedures were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved beforehand by the Louisiana State University Health Sciences Center Institutional Animal Care and Use Committee.
Western blot analysis.
Rats that received one stimulus per day for 1, 2, or 4 d and naive rats were immediately decapitated after stimulation, and the dentate gyrus (DG), CA3, and CA1 subregions of the hippocampus as well as the neocortex were dissected out from the right brain hemisphere. To obtain whole-cell extracts, brain tissues were placed in ice-cold lysis buffer [20 mm Tris-HCl, pH 7.5, 1 mm of an extracellular calcium chelator (EGTA), 20 mm MgCl2, 20 mm β-glycerol phosphate, 10 mm NaF, 2 mm DTT, 10 mm NaVO3, 0.1 μm okadaic acid, 1 mm phenylmethyl sulfonyl fluoride, and 10 μl/ml HALT Protease Inhibitor cocktail (Pierce, Rockford, IL)] and sonicated. Alternatively, to separate brain tissue into cytosolic, membrane, and nuclear protein fractions, a compartmental extraction kit from BioChain (Hayward, CA) was used. Tissue was homogenized in a Dounce homogenizer with five strokes of the loose-fitting pestle and manipulated further according to manufacturer’s instructions. The final protein concentration of each sample was determined using Bio-Rad (Hercules, CA) protein assay and 5 μg protein from each sample was denatured and electrophoresed on a 10% nondenaturing SDS-PAGE gel. All gels were stained with Coomassie blue to verify equal loading among lanes. Proteins were then transferred to polyvinylidene difluoride membranes and probed with antibodies to total and Thr183- and Tyr185-phosphorylated JNK-1, -2, and -3 (Cell Signaling, Danvers, MA), JNK-1 (Santa Cruz Biotechnology, Santa Cruz, CA), vimentin (Santa Cruz Biotechnology), phosphorylated MAPK/cyclin-dependent kinase (CDK) substrate (Cell Signaling), Ser178-phosphorylated paxillin (Bethyl Laboratories, Montgomery, TX), glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Research Diagnostics, Concord MA), and Ser73-phosphorylated c-Jun (Cell Signaling). Bands were visualized by the addition of IRDye 800 (Rockland Immunochemicals, Gilbertsville, PA) and Alexa 680 (Invitrogen, Eugene, OR)-conjugated secondary antibodies and use of the Odyssey Infrared Imaging System from LI-COR (Lincoln, NE) . Relative band intensity was determined using Odyssey software version 1.0 (LI-COR).
Immunofluorescent staining and Fluoro-jade B staining.
After anesthesia, rats that received 12 trains of stimulation during the first day and 24 trains of stimulation during the second day of kindling, as well as fully kindled and naive rats were transcardially perfused with 4% paraformaldehyde. The brains were removed and postfixed in 4% paraformaldehyde for 1 d at 4°C and incubated overnight in 30% sucrose at 4°C. Brains were then embedded in Tissue-Tek OCT compound, frozen, and cut into 14-μm-thick sections. For staining of phosphorylated JNK, sections were permeabilized with 100% ice-cold methanol, blocked in 10% goat serum, 5% bovine serum albumin (BSA), and 0.1% Triton X-100, and incubated with anti-Thr186- and Tyr185-phosphorylated JNK-1, -2, and -3 monoclonal antibody overnight at 4°C. After incubation in Alexa 555-conjugated goat anti-mouse IgG (highly cross-adsorbed; Invitrogen), sections were rinsed, dried, and mounted with Vectashield mounting medium with 4,6-diamidino-2-phenylindole (DAPI; Vector Laboratories, Burlingame, CA). For active caspase-3 labeling, the procedure was identical to that described above except that sections were incubated with 1 μg/ml rabbit anti-active caspase-3 (17 and 12 kDa cleaved caspase-3 fragments; BD PharMingen, San Diego, CA) and an Alexa 488-conjugated goat anti-rabbit (highly cross-adsorbed; Invitrogen). For glial fibrillary acidic protein (GFAP), vimentin, and β tubulin III the procedure was the same except sections were incubated with rabbit anti-GFAP (Sigma, St. Louis, MO), rabbit anti-vimentin (Abcam, Cambridge, MA), and rabbit anti-β tubulin III (Sigma) antibodies at room temperature for 1 h. For double-labeling experiments, the above procedures were performed sequentially as described with the addition of a reblocking step in between individual protein-labeling steps. Immunofluorescent staining was visualized and images were recorded using a Zeiss (Oberkochen, Germany) Axioplan 2 imaging microscope system.
Fluoro-jade B (FJ) staining was conducted to detect degenerating neurons (Schmued and Hopkins, 2000). Sections were immersed in 100% ethanol for 3 min and 70% ethanol for 2 min. After a 2 min rinse in distilled H2O (dH2O), sections were incubated in 0.06% potassium permanganate for 10 min, rinsed in dH2O for 2 min, and stained with 0.0002% FJ (Chemicon) in 2% acetic acid for 20 min. After three 1 min rinses in dH2O, the slides were allowed to dry, cleared in xylene, and mounted with distrene plasticizer xylene (Sigma). Sections taken at least 200 μm from the electrode-insertion point (caudal and rostral to electrode) and obtained from control and kindled animals were visually inspected for FJ-positive cells. Sections taken at the electrode-insertion point and stained with FJ served as a positive control for damaged neurons.
Quantitation of GFAP immunofluorescent staining.
For quantitation of GFAP-positive cells, 14-μm-thick coronal brain sections caudal to the stimulating electrode and ∼6 mm from bregma were isolated from five implanted, nonstimulated (control) animals and four 4-d-kindled animals and processed for GFAP as described above. Images from fifteen random, nonoverlapping 40× magnification fields were recorded from the ipsilateral DG, hilus, CA3, and CA1. Because of the size of the hilus, images from 10 random, nonoverlapping magnification fields were recorded; however, these 10 40× fields effectively incorporated all areas of the hilus. The hilus was defined as the region bordered by the suprapyramidal and infrapyramidal blades of the granule cell layer and the beginning of the CA3. The CA3 region extended ventrally to an imaginary boundary extending from the rhinal fissure, perpendicular to the long axis of the hippocampus. The CA1 region extended from the tip of the blade of the DG and ended ventrally at the same approximate location as the CA3, using the rhinal fissure as a landmark. Using Slidebook software, masks were generated for all images, and the thresholding size was set to 1000 voxels. This thresholding value was chosen based on visual inspection of images with the intention of including staining associated with cell bodies but not processes. Astrocytic processes may span the thickness of the section and the corresponding cell body may or may not be contained within the same section. Also, in some cases there may be discontinuities in the processes because of the plane in which the section was taken. For these reasons, counting processes would yield unrealistic estimates, and therefore, only cell bodies were counted. The numbers of GFAP-positive cells and areas (voxels) of computer-generated masks were quantified (as a rough approximation of cellular area) and mean values from 10 or 15 fields were calculated for both parameters. This method was validated by comparing manual counts of forty randomly chosen images to Slidebook-generated counts. The mean counts obtained for the manually counted group were statistically equivalent to those generated by Slidebook (p = 0.8468, paired two-tailed t test).
Immunoprecipitation/kinase assay.
Approximately 600 μg of whole-cell extracts were incubated with 50 μg agarose-conjugated anti-JNK-1 antibody (Santa Cruz Biotechnology) or agarose-conjugated rabbit IgG (Santa Cruz Biotechnology) at 4°C for 2 h. Immunocomplexes were pelleted by centrifuging at 1000 rpm for 10 s. Pellets were resuspended in PBS (supplemented with 1× HALT protease inhibitor cocktail and 10 mm NaVO3) and recentrifuged. For assay of JNK-1 activity, pellets were resuspended in kinase assay buffer [containing the following (in mm): 25 Tris-HCl, pH 7.5, 5 β-glycerol phosphate, 2 DTT, 0.1 NaVO3, 10 MgCl2], 200 μm ATP, and 4 μg c-Jun fusion protein (Cell Signaling) and incubated at 30°C for 30 min. To terminate the reaction, 5× SDS sample buffer was added and reaction mixtures were placed on ice. Reaction mixtures were then subjected to Western analysis as described.
Statistical analyses.
Comparison of multiple groups was performed using one-way ANOVA with Student–Newman–Keuls post-tests. A value of p < 0.05 was accepted as significant.
Results
Kindling results in increased seizure scores and after-discharge spikes
The rapid kindling protocol used here is described in greater detail previously (Tu and Bazan, 2003). As expected, all 12 animals used in the current study exhibited seizure-score profiles that were characterized by a gradual increase in seizure severity, culminating in stage-V seizures (Fig. 1A). Individual animals progressed through kindling at slightly different rates, such that at the end of day four, three animals had experienced two stage-V seizures, two animals had experienced four or five, two animals had experienced eight, and the remaining five animals had experienced 10 to 16 stage-V seizures. Electrophysiological data correlated well with the behavioral changes: all animals exhibited a significantly higher number of after-discharge spikes at the termination of kindling (data not shown).
Figure 1.

Rapid hippocampal kindling increases the number of after-discharge spikes and seizure intensity. A, Kindling progression was also verified electrophysiologically by recording after-discharges. Examples of recordings taken after one stimulation and after the 48th stimulation are shown. Note the increase in mean spike number. A more detailed characterization of the after-discharge profile of this kindling protocol has been described previously (Tu and Bazan, 2003). B, Seizure intensities for all 12 animals included in the current study were scored according to Racine’s scale and the average values for the indicated number of stimulations are plotted ±SEM. As expected, there was a progressive, reproducible increase in stimulus-evoked seizure intensity with each day of kindling (12 stimulations/day). Although all animals in the current study progressed at slightly different rates, all animals experienced a stage-V seizure in response to the stimulation administered on day 5, before they were killed.
Stimulus-induced JNK phosphorylation is enhanced in kindled animals
Western blot analysis of phosphorylated JNK-1, -2, and-3 was used as an indication of JNK activation state and stimulus-induced JNK phosphorylation was compared among rats stimulated for 1 or 2 d, fully kindled, and naive animals. Total JNK-1, -2, and -3 protein levels were also examined, and these levels remained unchanged in all stimulated animals compared with their naive counterparts in all brain regions examined (data not shown). Because of this, total JNK (p54 and p46 bands) was used as an internal loading control, and data were expressed as a ratio of the relative intensity of phosphorylated JNK to that of total JNK (Fig. 2). The fraction of phosphorylated JNK in the total JNK population was not statistically different between naive, 1- or 2-d-stimulated, and fully kindled animals in the DG, CA3, or neocortex. In contrast, there was a statistically significant increase in the CA1 of kindled animals as compared with their naive counterparts (p < 0.05, one-way ANOVA). A similar trend was seen in the CA3, although this fell short of statistical significance. There was no apparent relationship between the degree of JNK phosphorylation and the number of stage-V seizures an animal had experienced. Correlation analysis (Pearson) of these variables yielded r values of 0.4645, −0.5995, −0.2479, and −0.4305 for the DG, CA3, CA1, and neocortex, respectively.
Figure 2.

Kindling enhances stimulus-induced JNK phosphorylation. A, C, E, Western blot analysis for Thr283- and Tyr185-phosphorylated JNK (red) and for total JNK (green) was performed on whole-cell lysates collected from the indicated brain regions 15 min after the administration of one electrical stimulus to naive or kindled animals. Representative Western blots are shown, and the p54 and p46 JNK isoforms are indicated. B, D, F, The relative intensity of bands corresponding to phosphorylated p54 and p46 (combined) were determined and this value was divided by that obtained for total JNK. The means of four independent trials ± SEM are plotted. All groups are n ≥ 6. There was a statistically significant increase in the proportion of phosphorylated JNK in the CA1 of kindled animals compared their naive counterparts, and also in animals stimulated for 1 or 2 d (*p < 0.05, paired ANOVA, Student–Newman–Keuls post-tests). Note that increases in phosphorylated JNK were occasionally observed in the CA3 and neocortex (as seen in the Western blot above), but they were not consistent among all animals.
To determine whether this kindling-induced response was specific for JNK or whether this observation could be extended to other MAP kinase family members, Western blot analysis was performed for total and phosphorylated p38. As with total JNK levels, total p38 levels did not differ between naive and kindled animals in any region examined (data not shown). Moreover, there were no statistically significant differences in the fraction of phosphorylated p38 in any of the hippocampal subregions or in the neocortex. It is worth mentioning that a trend toward an increased p38 phosphorylation in CA3 and CA1 was apparent: these were the same regions that exhibited a tendency toward enhanced JNK phosphorylation; however, this effect was not consistent in our sampling and therefore did not attain statistical significance.
Kindling leads to increased stimulus-induced substrate phosphorylation
To understand the downstream effects of kindling-induced enhanced JNK responsiveness, and its downstream intercompartmental effects, hippocampal cytosolic, membrane, and nuclear protein fractions were separated by SDS-PAGE and probed with a pan-antibody that recognizes proteins containing the motifs PXS*P and S*PXR/K (S*, phosphorylated serine), or more simply, MAPK/CDK substrates. As can be seen in Figure 3A, several band intensities increased in kindled animals in all three protein fractions examined, indicating the presence of several kindling-responsive substrates in each cellular compartment. Interestingly, analysis of JNK-1 in these extracts indicated an accumulation of this protein in nuclear fractions as a result of kindling (Fig. 3B). Phosphorylation of the well known JNK substrates c-Jun and paxillin was also examined. There was a clear increase in the Ser178-phosphorylated species of paxillin, but not of Ser73-phosphorylated c-Jun or Ser100-JunD (Fig. 3B). Because enhanced c-Jun phosphorylation was not observed in vivo, JNK-1 was immunoprecipitated from whole-cell hippocampal extracts and its ability to phosphorylate a c-Jun fusion protein was assayed in vitro. JNK-1 from hippocampal extracts isolated from kindled animals was, in fact, better able to phosphorylate c-Jun in vitro, as seen by the Western blot shown in Figure 3C.
Figure 3.
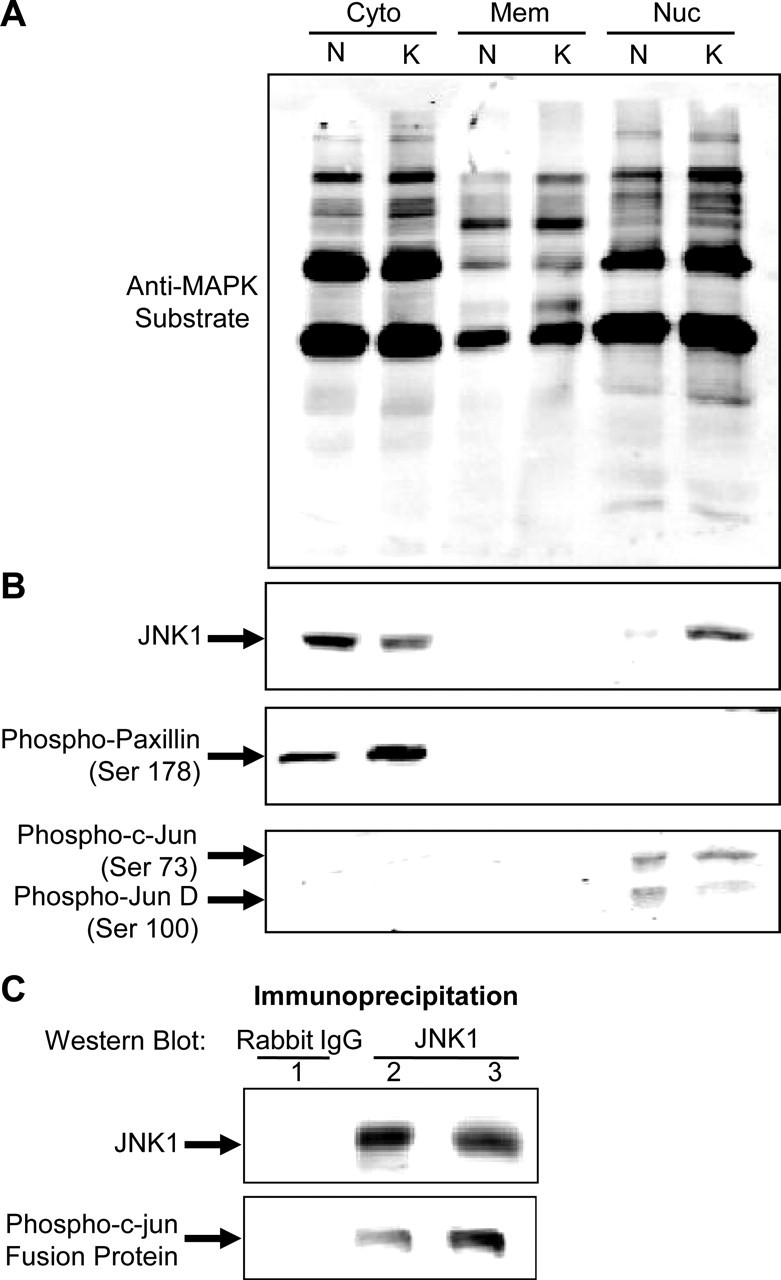
Kindling leads to increased stimulus-induced MAPK substrate phosphorylation. A, Hippocampi ipsilateral to the stimulating electrode were collected from naive and kindled animals 15 min after the administration of one stimulus. Hippocampal tissues were separated into cytoplasmic, membrane, and nuclear protein fractions and subjected to Western blot analysis. Membranes were probed with anti-MAPK/CDK substrate antibody. This antibody recognizes proteins containing the following consensus sequences: PXS*P or S*PXR/K, where S* represents a phosphorylated serine. B, Membranes containing cytoplasmic and nuclear fractions were probed with anti-JNK-1, anti-phosphopaxillin (ser178), and anti-phospho-c-Jun (ser73), respectively. C, JNK-1 immunoprecipitated from whole-cell extracts isolated from kindled hippocampus and an immunocomplex kinase assay was performed followed by Western analysis of phosphorylated substrate (c-Jun fusion protein). An immunoprecipitation using normal rabbit IgG was included as a negative control. Western analysis of the total JNK-1 immunoprecipitate is shown along with that for phospho-c-Jun (Ser 73). These results demonstrate that kindling increase MAPK sustrates in hippocampal cellular compartments whereas the accumulation of JNK-1 in the nuclear fraction suggests possible modulatory action at gene expression level.
Enhanced JNK phosphorylation is mediated by astrocytes
Immunofluorescent staining was performed not only to confirm region-specific localization of phosphorylated JNK as determined by Western blot analysis, but also to characterize its cell type-specific distribution. Sections both rostral and caudal to the stimulating electrode were examined. Because mechanical implantation of the electrode could confound interpretation of results, care was taken to examine sections that were at least 200 μm either rostral or caudal to the electrode-insertion point. Four kindled animals and five naive animals were examined for phospho-JNK staining. In contrast to sections obtained from naive animals, which exhibited diffuse staining throughout the hippocampus with slightly more intense staining in the principal cell layers, sections from kindled animals exhibited a phospho-JNK staining pattern that was confined to well demarcated, star-shaped silhouettes, very closely resembling astrocytes (Fig. 4). The omission of the primary antibody or the addition of blocking peptides completely abolished this pattern of staining (Fig. 4I,J). In all four kindled animals examined, this star-shaped pattern of staining was present throughout the hippocampus, being most concentrated in the CA3 and even more so in the CA1. There was very sparse, only occasional staining in the DG and hilus. No staining was observed in the somatosensory cortex. Although phospho-JNK was present in all layers of the CA1, it was most intense in the more superficial portions of the hippocampus, the stratum radiatum, and the stratum lacunosum-moleculare (Fig. 4E–H). At higher magnification, the majority of phospho-JNK label appeared as tiny punctae, largely present in the perikarya and stellae with a smaller amount of stain in the nucleus as visualized by the blue nuclear counterstain, DAPI. To confirm that this phospho-JNK label was present in astrocytes, a double immunofluorescent stain for the glia-specific intermediate filament, GFAP, and phospho-JNK were performed. Both labels overlapped perfectly. In contrast, double labeling with the neuronal marker β-tubulin III revealed no overlap between this label and that of phospho-JNK.
Figure 4.

Kindling induces JNK phosphorylation in all layers of the CA1. Immunofluorescent phospho-JNK staining (red) was performed on frozen brain sections isolated from five naive and four kindled animals 15 min after the administration of one stimulus. Sections were counterstained with the nuclear dye DAPI (blue). In contrast to sections obtained from naive animals, sections from all four kindled animals exhibited a star-like staining pattern that was consistently present in the CA1 subregion. Representative images from phospho-JNK staining in the four layers of the CA1 from a naive (A, C, E, G) and a kindled animal (B, D, F, H) are shown. Although phospho-JNK staining was present in all layers (A, B, stratum oriens; C, D, pyramidal cell layer; E, F, stratum radiatum; G, H, stratum lacunosum moleculare), it was frequently most intense in the more superficial portions of the hippocampus, the stratum radiatum and stratum lacunosum-moleculare. The omission of the primary anti-phospho-JNK antibody (I) and the addition of blocking peptides (J) completely abolished this pattern of staining (high-exposure images of the stratum radiatum are shown in I and J). Dual immunofluorescent staining for phospho-JNK (red) and either the glial marker, GFAP (green), or the neuronal marker, β-tubulin III (green), was performed on frozen sections from kindled animals (K and L, respectively). Phospho-JNK staining overlapped with that of GFAP but not that of β-tubulin III. Scale bar, 30 μm. There were no immunoreactive phospho-JNK-related patterns in pyramidal cell layers from animals stimulated for 1 (M) or 2 d (N), nor in the molecular layers (O, P).
Kindling induces reactive astrogliosis and reactive astrocytes mediate stimulus-induced JNK phosphorylation
“Reactive astrogliosis” is the term used to describe the morphological transformation of astrocytes in response to a wide variety of neurotoxic stimuli. These changes include the proliferation and hypertrophy of astrocytes, as well as the upregulation of cytoskeletal proteins such as GFAP and vimentin (Khurgel et al., 1992; Khurgel and Ivy, 1996). Reactive gliosis is an early event associated with kindling and more recent data suggest that reactive astrocytes may mediate responses to abnormal seizure activity (Khurgel et al., 1992; Adams et al., 1998; Aronica et al., 2000; Gorter et al., 2002). Therefore, we set out to determine whether markers of reactive astrogliosis were present in our kindled animals and whether these reactive glia were responsible for the enhanced stimulus-induced JNK phosphorylation after kindling. GFAP immunofluorescent staining was performed on sections from control and kindled animals and the number and area (approximated by computer-generated masks) of GFAP-positive cells were quantified. There was a clear increase in both parameters in kindled animals compared with their naive counterparts in the CA1 as well in rats stimulated for 1 or 2 d (p < 0.05, ANOVA). In addition, Western blot analysis of vimentin protein levels consistently revealed a robust increase in the levels of this protein as a result of 4 d of kindling (Fig. 5C). This increase in vimentin protein levels was also clearly seen using immunofluorescent staining (data not shown). To determine whether astrocytes with a reactive phenotype were responsible for enhanced JNK phosphorylation, costaining for vimentin and phospho-JNK was performed and revealed that all vimentin-positive cells were also positive for phospho-JNK (Fig. 5D).
Figure 5.
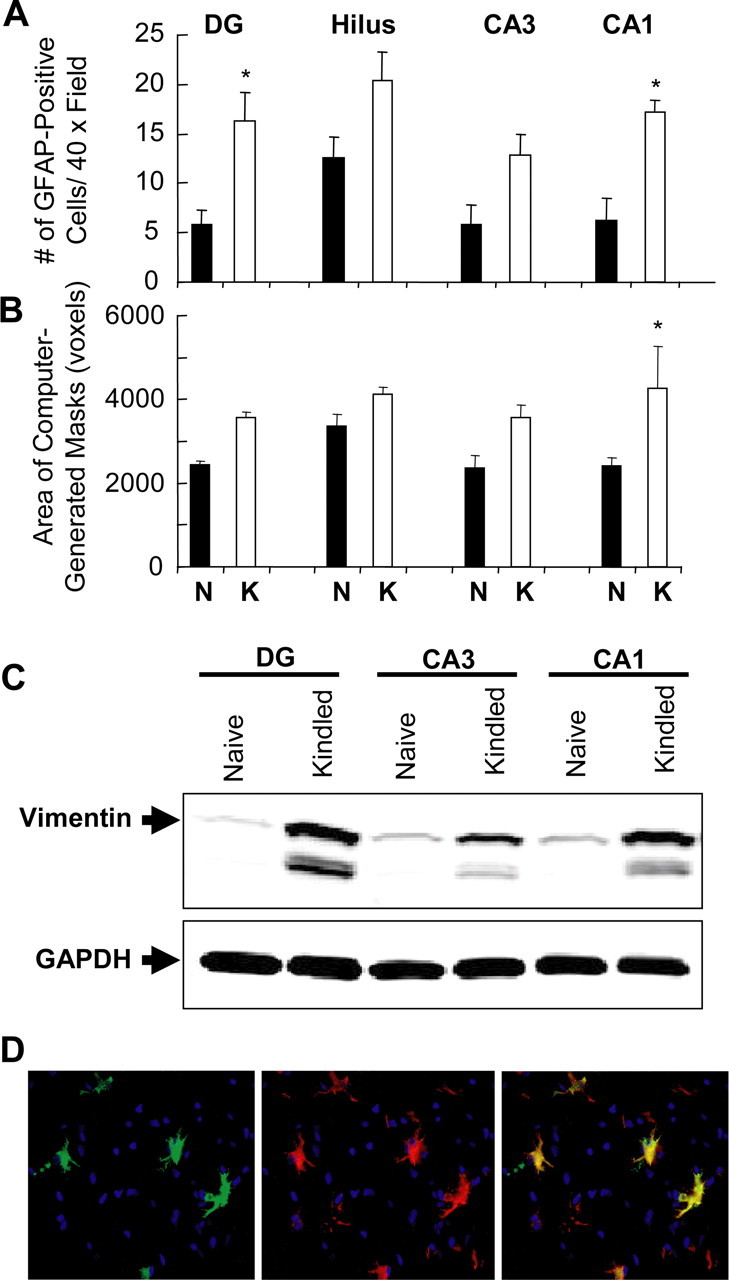
Kindling induces reactive astrogliosis and reactive astrocytes mediate JNK phosphorylation. A, B, Immunofluorescent GFAP staining was performed on frozen brain sections isolated from five naive and four kindled animals. The numbers (A) and areas (B) of GFAP-positive cells were quantified in different regions of the ipsilateral hippocampus (DG, hilus, CA3, and CA1). There was a significant increase in the numbers of GFAP-positive cells and areas of these cells (approximated as computer-generated masks) in the CA1 (*p < 0.05, one-way ANOVA, Student–Newman–Keuls post-tests). C, Western blot analysis for vimentin was performed on whole-cell lysates collected from the indicated brain regions from either naive or kindled animals, and a representative Western blot is shown. Kindling consistently induced a clear increase in vimentin protein levels in the DG, CA3, and CA1 subregions of the hippocampus in all animals examined. The same blot was reprocessed with anti-GAPDH antibody to show that each lane received approximately the same amount of protein. D, Frozen sections from kindled animals were stained for both phospho-JNK (red) and vimentin (green) and counterstained with the nuclear stain DAPI (blue). Images show both stains alone and in combination with one another. There is clear overlap between both stains, indicating that reactive astrocytes are responsible for the enhanced JNK phosphorylation observed during kindling. Original magnification, 400×. Error bars indicate SEM.
Four days of kindling is associated with neuronal death
Cavazos et al. (1994) have demonstrated previously that as few as three generalized tonic-clonic seizures leads to detectable death in the hilus and CA1 as quantified by stereological techniques. Therefore, we set out not to quantify kindling-induced neuronal death, as has already been done by others, but to verify the presence of neuronal death, particularly in the regions of the hippocampus that exhibited enhanced JNK phosphorylation. To do this, sections obtained from four control animals and three kindled animals (animals had experienced four, eight, and 16 stage-V seizures) were stained with FJ, a marker for degenerating neurons, and immunostained for active caspase-3 and visually inspected for cells positive for these labels in the hippocampus and cortex. Staining in sections from all three animals revealed numerous intensely staining cells dispersed throughout the DG, hilus, CA3, CA1, and very infrequently, in lateral aspects of the somatosensory cortex (Fig. 6B,G,L). FJ-positive cells were also present, although they were very sparse, in sections obtained from naive animals (Fig. 6A,F). On closer inspection of sections obtained from kindled animals, it was apparent that some FJ-positive cells had a neuronal morphology and some had an astrocytic morphology (Fig. 6B, inset, K). This is consistent with the work of others demonstrating that reactive astrocytes are labeled with FJ (Colombo and Puissant, 2002).
Figure 6.

Four days of rapid kindling results in neuronal death. Frozen brain sections isolated from four naive and three kindled animals were either stained with Fluoro-jade B, a marker for degenerating neurons (A, B, F, G, K), or immunostained for active caspase-3 (C–E, H–J, M–O). Representative images of Fluoro-jade staining from the ipsilateral hippocampus are shown [A, F (naive); B, G (kindled; four stage-V seizures)]. The region of the hippocampus from which the image is obtained is indicated to the left. Note the increase in staining in kindled animals. This increase is partly attributable to staining of neurons as shown in the inset in B and reactive astrocytes (taken from the hilus) as shown in K. FJ staining was also occasionally detected in the somatosensory cortex of a kindled animal (four stage-V seizures; L). No caspase-3 staining was detected in the animal experiencing four stage-V seizures (C, H). In contrast, a punctate pattern of staining was present in animals experiencing eight (D, I) and 16 (E, J) stage-V seizures. The significance of these puncta is currently unknown. Large circular blebs, consistent with the appearance of apoptotic bodies, are particularly abundant in the CA1 and hilus in the latter animal (E, J, arrowheads). K, Higher-magnification image of large blebs indicated by the arrow in E (blue, DAPI counterstain). M, A few active caspase-3 positive cells were present in the somatosensory cortex (only in the animal experiencing 16 stage-V seizures; blue, DAPI counterstain). N, O, Double immunofluorescent staining for C3 (green) and the neuronal marker NeuN (red); blue, DAPI counterstain. The image was taken from the granule cell layer border. Scale bars: (in A) A–E, 300 μm; (in F) F–J, 300 μm; K, N, O, 60 μm; L, M, 30 μm.
Previous work has shown that kindling-induced neuronal death has both necrotic and apoptotic components (Pretel et al., 1997). It has also been demonstrated that neuronal nuclei in the DG stain positive for TUNEL (terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling) after only one subconvulsive stimulus, suggesting that kindling stimulations may readily activate apoptotic signaling (Bengzon et al., 1997). In an attempt to understand whether these apoptotic pathways may be activated in our kindled animals, we also examined sections for active caspase-3. Processing of the proenzyme form of caspase-3 results in 17 and 12 kDa fragments that heterodimerize to form a functional protease that cleaves various cytosolic and nuclear substrates during the final stages of apoptosis. As shown in Figure 6, active caspase-3 was detected in the hippocampus of the animals that had experienced eight and 16 stage-V seizures, but not in the animal that had experienced four stage-V seizures (Fig. 6C–E, H–J). The main cell layers of the DG, CA3, and CA1, as well as the intervening layers, all exhibited a very fine punctate pattern of staining in these animals. The significance of this staining is unknown. Occasionally, in sections obtained from the animal that experienced eight stage-V seizures, and more frequently in sections from the one that experienced 16, there were large, intensely staining circular blebs interposed between these puncta that, on closer inspection, had typical attributes of apoptotic bodies (Fig. 6N). In both animals, these “blebs” were present in the DG, hilus, CA3, and CA1. Notably, these blebs were abundant in the hilus and CA1 in the animal that had experienced 16 stage-V seizures (Fig. 6E, arrow). This staining was specific as it was completely abolished by omission of the primary antibody (data not shown). As with FJ staining, a small amount of active caspase-3 labeling was also detected in the somatosensory cortex, but only in the animal that experienced 16 stage-V seizures. Double labeling for active caspase-3 and the neuronal marker NeuN revealed that both labels did overlap, although a few cells that stained positive for active caspase-3 did not label for NeuN (Fig. 6M,O).
Discussion
Kindling results in an increase in after-discharge spikes as measured by EEG, as well as a gradual increase in seizure intensity, culminating in stage-V seizures. The rapid hippocampal kindling protocol used here has been characterized in greater detail previously (Tu and Bazan, 2003). Although animals experienced variable numbers of stage-V seizures (at least two to 16), all animals experienced (stage-V) generalized tonic-clonic seizures in response to the electrical stimulus administered on the fifth day (before they were killed). Thus, despite their variable seizure history, this group of animals was homogenous with regard to their stimulus-induced seizure susceptibility.
Western blot analysis of Thr183/Tyr185-phosphorylated JNK was used as an indication of JNK activation state and revealed that stimulus-induced JNK activation was significantly enhanced in the CA1 subregion of the hippocampus. The permanence of enhanced stimulus-induced JNK phosphorylation (i.e., whether it lasts weeks or months after kindling) awaits additional experimentation.
Reactive gliosis, characterized by proliferation and hypertrophy of glia as well as by an upregulation of vimentin protein levels, is also present after 4 d of kindling and reactive glia mediate enhanced stimulus-induced JNK phosphorylation. Although to our knowledge this is the first study examining changes in JNK activity during kindling, Kashihara et al. (1997) showed that AP-1 binding activity, an event downstream of JNK activation, was enhanced in amygdala-kindled rats compared with nonkindled rats. The small (less than twofold) increase in phosphorylated JNK seen here is in keeping with what others have observed. The CNS has high basal levels of phosphorylated JNK compared with other tissues, and these high levels of phosphorylated JNK are thought to mask stress-induced JNK activation (Coffey et al., 2000). Other studies have shown that JNK translocates to the nucleus when activated, agreeing well with the nuclear accumulation of JNK1 seen here. Despite this accumulation, no increased phosphorylation of c-Jun could be detected. This does not, however, rule out the possibility that the time course of c-Jun phosphorylation precludes detection of increased phosphorylation at this time point (15 min after the stimulus); nor does it rule out the possibility that kindling-induced JNK activation may affect other nuclear targets. Indeed, Western analysis with an MAPK/CDK substrate antibody indicated the presence of several phosphorylated proteins in the nucleus, suggesting that kindling-induced enhanced JNK responsiveness might lead to changes in gene expression.
The enhanced stimulus-induced JNK phosphorylation seen as a result of kindling could not be explained by either an increase in JNK protein levels or a change in protein levels of the JNK-regulating protein, JNK-interacting protein-1 (data not shown). These results may point to a more complex mechanism of JNK regulation under these conditions. Interestingly, the stimulus-induced JNK response observed here was not consistently paralleled by the other MAP kinase, p38. Phosphorylation of the prosurvival kinase Akt1 was also examined and found to remain unchanged (data not shown). The selective activation of JNK, but not p38 or Akt1, under these conditions may indicate different time courses of activation for these kinases, or possibly a selective role for JNK in mediating astrocyte responses to abnormal synaptic activity.
The consequences of augmented stimulus-induced JNK activation in reactive astrocytes caused by kindling are not completely understood. Historically, the role of astrocytes in the CNS was regarded simply as a supportive one, providing energy and structural support to neurons. However, more recent studies challenge this view and suggest that astrocytes actively contribute to synaptic plasticity, both during development and in response to abnormal synaptic activity associated with seizures (Ullian et al., 2004). Recent data suggest that reactive astrocytes, in particular, may mediate responses to abnormal seizure activity. Gorter et al. (2002) have shown that reactive astrocytes upregulate sodium channel β1-subunit protein levels after status epilepticus. Upregulation of the metabotropic glutamate receptors 3 and 5 by these cells also occurs after status epilepticus (Aronica et al., 2000). In both cases, these changes were hypothesized to result in altered glial-neuronal connectivity and communication, thereby altering neuronal activity under these conditions. The current finding, that kindling enhances stimulus-induced phosphorylation of integral membrane proteins, suggests that the activity of various ion channels, transporters, and receptors may be regulated by the MAPK members. This finding, together with the data of others, may indicate a selective signaling response to abnormal neuronal activity, as opposed to a general response to neuronal damage normally associated with reactive glia, which alters the extracellular milieu (ion and neurotransmitter concentrations) and modulates synaptic activity.
Augmented stimulus-induced JNK activation in reactive astrocytes may also portend the demise of neurons during kindling. It is well documented that temporal lobe epilepsy is associated with significant neuronal death in the CA1, and this neuronal loss can be reproduced in kindling (Cavazos et al., 1994). Regions of the hippocampus exhibit different sensitivities to stimulus-induced neuronal death, with the hilus and the CA1 being the most vulnerable regions (Cavazos et al., 1994). In accord with this, we detected degenerating neurons, via Fluoro-jade B staining and active caspase-3 labeling, in these regions. Our data are consistent with the findings of others showing that kindling-induced neuronal death is attributable, at least in part, to apoptosis in these areas. Given the fact that JNK is a well established mediator of excitotoxic neuronal death, significantly enhanced JNK phosphorylation in the hippocampus, specifically the CA1 subregion, may explain the marked neuronal death observed in these regions in kindled animals.
Activated JNK is frequently found in astrocytes under a variety of neuropathological circumstances (Migheli et al., 1997; Ma and Quirion, 2002; Ferrer et al., 2003a,b; Shin et al., 2003). Indeed, there is accumulating evidence that MAP kinase activation mediates astroglial production of a host of inflammatory mediators under these conditions, which contribute to neuronal death (Migheli et al., 1997; Falsig et al., 2004). Using a special neuron-glia coculture system, Xie et al. (2004) demonstrated recently that lipopolysaccharide-induced activation in glia triggers neuronal death. The authors postulated that JNK activation may be part of a glial signaling pathway that leads to the release of diffusible neurotoxic molecules such as NO. Importantly, there is evidence that NO and other inflammatory mediators increase as a result of ongoing seizure activity (Kaneko et al., 2002). Increased interleukin-6 (IL-6) protein levels have been observed in the CSF of epileptic patients (Peltola et al., 1998). Also, temporal lobe tissue from epileptic patients exhibits increased IL-1β immunoreactivity (Sheng et al., 1994). In accord with these data, increased transcription of IL-1β, IL-6, and tumor necrosis factor α has been reported in experimental models of epilepsy (Minami et al., 1991; Plata-Salaman et al., 2000; Kaneko et al., 2002; Vezzani et al., 2002). Vezzani et al. (2002) demonstrated that upregulation of these cytokines after limbic seizures is attributable to astroglia. In vitro studies suggest that the MAP kinases mediate astroglial induction of some of these molecules. For example, JNK mediates upregulation of the chemokine RANTES/CCL5 (regulated upon activation, normal T-cell expressed and secreted), as well as inducible NO synthase in astroglial cultures treated with IL-1 (Hua et al., 2002; Kim et al., 2004). These data, together with our current finding that kindling increases astroglial JNK activation responses, may indicate a role for these cells in contributing to events leading to neuronal death. Additional study of astroglial signaling pathways during kindling will clarify the relationship between MAP kinase activation, the release of neurotoxic factors, and seizure-induced neuronal damage and death.
Footnotes
This work was supported by United States Public Health Service Grants R01NS23002 from the National Institute of Neurological Disorders and Stroke and P20RR16816 from the National Center for Research Resources (COBRE Program), National Institutes of Health (Bethesda, MD).
References
- Adams B, Von Ling E, Vaccarella L, Ivy GO, Fahnestock M, Racine RJ (1998). Time course for kindling-induced changes in the hilar area of the dentate gyrus: reactive gliosis as a potential mechanism. Brain Res 804:331–336. [DOI] [PubMed] [Google Scholar]
- Aronica E, van Vliet EA, Mayboroda OA, Troost D, da Silva FH, Gorter JA (2000). Upregulation of metabotropic glutamate receptor subtype mGluR3 and mGluR5 in reactive astrocytes in a rat model of mesial temporal lobe epilepsy. Eur J Neurosci 12:2333–2344. [DOI] [PubMed] [Google Scholar]
- Bazan NG, Tu B, Rodriguez de Turco EB (2002). What synaptic lipid signaling tells us about seizure-induced damage and epileptogenesis. Prog Brain Res 135:175–185. [DOI] [PubMed] [Google Scholar]
- Bengzon J, Kokaia Z, Elmer E, Nanobashvili A, Kokaia M, Lindvall O (1997). Apoptosis and proliferation of dentate gyrus neurons after single and intermittent limbic seizures. Proc Natl Acad Sci USA 94:10432–10437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman DE, Hazvi S, Rosenblum K, Seger R, Dudai Y (1998). Specific and differential activation of mitogen-activated protein kinase cascades by unfamiliar taste in the insular cortex of the behaving rat. J Neurosci 18:10037–10044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brecht S, Simler S, Vergnes M, Mielke K, Marescaux C, Herdegen T (1999). Repetitive electroconvulsive seizures induce activity of c-Jun N-terminal kinase and compartment-specific desensitization of c-Jun phosphorylation in the rat brain. Brain Res Mol Brain Res 68:101–108. [DOI] [PubMed] [Google Scholar]
- Cavazos JE, Das I, Sutula TP (1994). Neuronal loss induced in limbic pathways by kindling: evidence for induction of hippocampal sclerosis by repeated brief seizures. J Neurosci 14:3106–3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffey ET, Hongisto V, Dickens M, Davis RJ, Courtney MJ (2000). Dual roles for c-Jun N-terminal kinase in developmental and stress responses in cerebellar granule neurons. J Neurosci 20:7602–7613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo JA, Puissant VI (2002). Fluoro Jade stains early and reactive astroglia in the primate cerebral cortex. J Histochem Cytochem 50:1135–1137. [DOI] [PubMed] [Google Scholar]
- Curran BP, Murray HJ, O’Connor JJ (2003). A role for c-Jun N-terminal kinase in the inhibition of long-term potentiation by interleukin-1beta and long-term depression in the rat dentate gyrus in vitro. Neuroscience 118:347–357. [DOI] [PubMed] [Google Scholar]
- Falsig J, Porzgen P, Lotharius J, Leist M (2004). Specific modulation of astrocyte inflammation by inhibition of mixed lineage kinases with CEP-1347. J Immunol 173:2762–2770. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Barrachina M, Tolnay M, Rey MJ, Vidal N, Carmona M, Blanco R, Puig B (2003a). Phosphorylated protein kinases associated with neuronal and glial tau deposits in argyrophilic grain disease. Brain Pathol 13:62–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, Pastor P, Rey MJ, Munoz E, Puig B, Pastor E, Oliva R, Tolosa E (2003b). Tau phosphorylation and kinase activation in familial tauopathy linked to deln296 mutation. Neuropathol Appl Neurobiol 29:23–34. [DOI] [PubMed] [Google Scholar]
- Goddard GV, McIntyre DC, Leech CK (1969). A permanent change in brain function resulting from daily electrical stimulation. Exp Neurol 25:295–330. [DOI] [PubMed] [Google Scholar]
- Gorter JA, van Vliet EA, Lopes da Silva FH, Isom LL, Aronica E (2002). Sodium channel beta1-subunit expression is increased in reactive astrocytes in a rat model for mesial temporal lobe epilepsy. Eur J Neurosci 16:360–364. [DOI] [PubMed] [Google Scholar]
- Herdegen T, Claret FX, Kallunki T, Martin-Villalba A, Winter C, Hunter T, Karin M (1998). Lasting N-terminal phosphorylation of c-Jun and activation of c-Jun N-terminal kinases after neuronal injury. J Neurosci 18:5124–5135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua LL, Zhao ML, Cosenza M, Kim MO, Huang H, Tanowitz HB, Brosnan CF, Lee SC (2002). Role of mitogen-activated protein kinases in inducible nitric oxide synthase and TNFalpha expression in human fetal astrocytes. J Neuroimmunol 126:180–189. [DOI] [PubMed] [Google Scholar]
- Jeon SH, Kim YS, Bae CD, Park JB (2000). Activation of JNK and p38 in rat hippocampus after kainic acid induced seizure. Exp Mol Med 32:227–230. [DOI] [PubMed] [Google Scholar]
- Kaneko K, Itoh K, Berliner LJ, Miyasaka K, Fujii H (2002). Consequences of nitric oxide generation in epileptic-seizure rodent models as studied by in vivo EPR. Magn Reson Med 48:1051–1056. [DOI] [PubMed] [Google Scholar]
- Kashihara K, Sato K, Akiyama K, Okada S, Ishihara T, Hayabara T, Shomori T (1997). Temporal pattern of AP-1 DNA-binding activity in the rat hippocampus following a kindled seizure. Neuroscience 80:753–761. [DOI] [PubMed] [Google Scholar]
- Khurgel M, Ivy GO (1996). Astrocytes in kindling: relevance to epileptogenesis. Epilepsy Res 26:163–175. [DOI] [PubMed] [Google Scholar]
- Khurgel M, Racine RJ, Ivy GO (1992). Kindling causes changes in the composition of the astrocytic cytoskeleton. Brain Res 592:338–342. [DOI] [PubMed] [Google Scholar]
- Kim MO, Suh HS, Brosnan CF, Lee SC (2004). Regulation of RANTES/CCL5 expression in human astrocytes by interleukin-1 and interferon-beta. J Neurochem 90:297–308. [DOI] [PubMed] [Google Scholar]
- Ma W, Quirion R (2002). Partial sciatic nerve ligation induces increase in the phosphorylation of extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) in astrocytes in the lumbar spinal dorsal horn and the gracile nucleus. Pain 99:175–184. [DOI] [PubMed] [Google Scholar]
- Majkowski J (1999). Kindling: clinical relevance for epileptogenicity in humans. Adv Neurol 81:105–113. [PubMed] [Google Scholar]
- Marcheselli VL, Bazan NG (1994). Platelet-activating factor is a messenger in the electroconvulsive shock-induced transcriptional activation of c-fos and zif-268 in hippocampus. J Neurosci Res 37:54–61. [DOI] [PubMed] [Google Scholar]
- Marcheselli VL, Bazan NG (1996). Sustained induction of prostaglandin endoperoxide synthase-2 by seizures in hippocampus. Inhibition by a platelet-activating factor antagonist. J Biol Chem 271:24794–24799. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Bazan NG (2006). Apoptosis and necrosis. In: Basic neurochemistry (Siegel G, Albers RW, Brady S, Price D, eds) Ed 7 in press London: Elsevier.
- Mielke K, Brecht S, Dorst A, Herdegen T (1999). Activity and expression of JNK1, p38 and ERK kinases, c-Jun N-terminal phosphorylation, and c-jun promoter binding in the adult rat brain following kainate-induced seizures. Neuroscience 91:471–483. [DOI] [PubMed] [Google Scholar]
- Migheli A, Piva R, Atzori C, Troost D, Schiffer D (1997). c-Jun, JNK/SAPK kinases and transcription factor NF-kappa B are selectively activated in astrocytes, but not motor neurons, in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 56:1314–1322. [DOI] [PubMed] [Google Scholar]
- Minami M, Kuraishi Y, Satoh M (1991). Effects of kainic acid on messenger RNA levels of IL-1 beta, IL-6, TNF alpha and LIF in the rat brain. Biochem Biophys Res Commun 176:593–598. [DOI] [PubMed] [Google Scholar]
- Mukherjee PK, DeCoster MA, Campbell FZ, Davis RJ, Bazan NG (1999). Glutamate receptor signaling interplay modulates stress-sensitive mitogen-activated protein kinases and neuronal cell death. J Biol Chem 274:6493–6498. [DOI] [PubMed] [Google Scholar]
- Peltola J, Hurme M, Miettinen A, Keranen T (1998). Elevated levels of interleukin-6 may occur in cerebrospinal fluid from patients with recent epileptic seizures. Epilepsy Res 31:129–133. [DOI] [PubMed] [Google Scholar]
- Plata-Salaman CR, Ilyin SE, Turrin NP, Gayle D, Flynn MC, Romanovitch AE, Kelly ME, Bureau Y, Anisman H, McIntyre DC (2000). Kindling modulates the IL-1beta system, TNF-alpha, TGF-beta1, and neuropeptide mRNAs in specific brain regions. Brain Res Mol Brain Res 75:248–258. [DOI] [PubMed] [Google Scholar]
- Pretel S, Applegate CD, Piekut D (1997). Apoptotic and necrotic cell death following kindling induced seizures. Acta Histochem 99:71–79. [DOI] [PubMed] [Google Scholar]
- Racine RJ (1972). Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol 32:281–294. [DOI] [PubMed] [Google Scholar]
- Reibel S, Larmet Y, Le BT, Carnahan J, Marescaux C, Depaulis A (2000). Brain-derived neurotrophic factor delays hippocampal kindling in the rat. Neuroscience 100:777–788. [DOI] [PubMed] [Google Scholar]
- Schmued LC, Hopkins KJ (2000). Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res 874:123–130. [DOI] [PubMed] [Google Scholar]
- Sheng JG, Boop FA, Mrak RE, Griffin WS (1994). Increased neuronal beta-amyloid precursor protein expression in human temporal lobe epilepsy: association with interleukin-1 alpha immunoreactivity. J Neurochem 63:1872–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin T, Ahn M, Jung K, Heo S, Kim D, Jee Y, Lim YK, Yeo EJ (2003). Activation of mitogen-activated protein kinases in experimental autoimmune encephalomyelitis. J Neuroimmunol 140:118–125. [DOI] [PubMed] [Google Scholar]
- Sutula T, Cascino G, Cavazos J, Parada I, Ramirez L (1989). Mossy fiber synaptic reorganization in the epileptic human temporal lobe. Ann Neurol 26:321–330. [DOI] [PubMed] [Google Scholar]
- Tu B, Bazan NG (2003). Hippocampal kindling epileptogenesis upregulates neuronal cyclooxygenase-2 expression in neocortex. Exp Neurol 179:167–175. [DOI] [PubMed] [Google Scholar]
- Ullian EM, Christopherson KS, Barres BA (2004). Role for glia in synaptogenesis. Glia 47:209–216. [DOI] [PubMed] [Google Scholar]
- Vezzani A, Moneta D, Richichi C, Aliprandi M, Burrows SJ, Ravizza T, Perego C, De Simoni MG (2002). Functional role of inflammatory cytokines and antiinflammatory molecules in seizures and epileptogenesis. Epilepsia 43:Suppl 5, 30–35. [DOI] [PubMed] [Google Scholar]
- Xie Z, Smith CJ, Van Eldik LJ (2004). Activated glia induce neuron death via MAP kinase signaling pathways involving JNK and p38. Glia 45:170–179. [DOI] [PubMed] [Google Scholar]
- Xu X, Raber J, Yang D, Su B, Mucke L (1997). Dynamic regulation of c-Jun N-terminal kinase activity in mouse brain by environmental stimuli. Proc Natl Acad Sci USA 94:12655–12660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang DD, Kuan CY, Whitmarsh AJ, Rincon M, Zheng TS, Davis RJ, Rakic P, Flavell RA (1997). Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature 389:865–870. [DOI] [PubMed] [Google Scholar]