

Abstract
Activity-dependent regulation of synaptic AMPA receptor (AMPAR) number is critical to NMDA receptor (NMDAR)-dependent synaptic plasticity. Using quantitative high-resolution in situ hybridization, we show that mRNAs encoding the AMPA-type glutamate receptor subunits (GluRs) 1 and 2 are localized to dendrites of hippocampal neurons and are regulated by paradigms that alter synaptic efficacy. A substantial fraction of synaptic sites contain AMPAR mRNA, consistent with strategic positioning and availability for “on-site” protein synthesis. NMDAR activation depletes dendritic levels of AMPAR mRNAs. The decrease in mRNA occurs via rise in intracellular Ca2+, activation of extracellular signal-regulated kinase/mitogen-activated protein kinase signaling, and transcriptional arrest at the level of the nucleus. The decrease in mRNA is accompanied by a long-lasting reduction in synaptic AMPAR number, consistent with reduced synaptic efficacy. In contrast, group I metabotropic GluR signaling promotes microtubule-based trafficking of existing AMPAR mRNAs from the soma to dendrites. Bidirectional regulation of dendritic mRNA abundance represents a potentially powerful means to effect long-lasting changes in synaptic strength.
Keywords: messenger RNA, transcription, mRNA trafficking, dendrites, AMPA receptors, synaptic plasticity
Introduction
Activity-dependent regulation of synaptic AMPA receptor (AMPAR) number is critical to enduring forms of synaptic plasticity such as NMDA receptor (NMDAR)-dependent long-term depression (LTD). The precise events between synaptic activation and long-lasting modification of individual synapses remain unclear. Whereas regulated synaptic delivery and retrieval of AMPARs are mechanisms critical to the early phase of NMDAR-dependent synaptic plasticity (Carroll et al., 2001; Barry and Ziff, 2002; Malinow and Malenka, 2002; Sheng and Kim, 2002; Song and Huganir, 2002; Collingridge et al., 2004), the mechanisms underlying the expression phase are less clear. Local protein synthesis is an attractive mechanism that could underlie long-lasting, changes in synaptic efficacy (Eberwine et al., 2001; Steward and Schuman, 2003; Martin and Zukin, 2006). Activity-dependent mRNA trafficking and local protein synthesis in dendrites have recently gained widespread acceptance as mechanisms fundamental to synaptic plasticity. By spatially restricting gene expression within neurons, localized mRNAs endow synapses with the capacity to regulate morphology and efficacy in response to specific stimuli.
Studies involving in situ hybridization demonstrate the presence of mRNAs encoding cytosolic, cytoskeletal, and integral membrane proteins in dendritic layers of hippocampus and in dendrites of hippocampal neurons in vivo and in vitro. These include mRNAs encoding microtubule associated protein 2 (MAP2), the α subunit of Ca2+/calmodulin-dependent protein kinase II (CaMKII-α), brain-derived neurotrophic factor, activity-regulated cytoskeleton-associated protein (Arc), TrkB receptor, IP3 receptor, the atypical protein kinase Mzeta, the NMDAR NR1 subunit, and glycine receptor α subunit (for review, see Steward and Schuman, 2003). More recently, unbiased approaches to amplify mRNAs from dendritic and/or synaptic compartments indicate as many as 400 localized mRNAs (Eberwine et al., 2001). Wide acceptance of the dendritic localization of such a large number of mRNAs, however, has been delayed by the lack of in situ hybridization data documenting the presence of mRNAs in dendrites.
Dendritic RNA transport is specific and rapid and can be regulated by neuronal activity. Synaptic stimulation in vivo delivers Arc mRNA to segments of dendrites with activated synapses and promotes “on-site” synthesis of Arc protein (Steward and Worley, 2001). Neuronal activity in vitro promotes movement of CaMKIIα mRNA in dendrites (Rook et al., 2000) and transport of β-actin mRNA into spines (Tiruchinapalli et al., 2003). Dopamine receptor activation promotes local translation of AMPAR mRNAs (Smith et al., 2005). These findings support a model in which activated or “tagged” synapses recruit mRNAs, enabling local regulation of translation and synaptic efficacy (Martin and Zukin, 2006).
Using quantitative fluorescence in situ hybridization (Q-FISH) and Northern blot analysis, we show localization of AMPAR mRNAs to dendrites of hippocampal neurons and regulation of mRNA abundance by glutamatergic signaling. NMDAR activation, a signaling paradigm that induces endocytosis of AMPARs in neurons in culture (Beattie et al., 2000; Ehlers, 2000; Lee et al., 2004) and chemical LTD in hippocampal slices (Lee et al., 1998; Kamal et al., 1999; van Dam et al., 2002; Li et al., 2004), depletes dendritic AMPAR mRNAs. NMDA triggers a rise in intracellular Ca2+, activation of extracellular signal-regulated kinase (ERK)/mitogen-activated protein kinase (MAPK) and transcriptional arrest, leading to decreased mRNA and synaptic AMPAR number. In contrast, group I metabotropic glutamate receptor (mGluR) activation elevates dendritic AMPAR mRNA via microtubule-based, anterograde RNA transport. Bidirectional regulation of dendritic AMPAR mRNAs is a novel mechanism for long-lasting modifications of receptor composition and number, mechanisms relevant to synaptic plasticity.
Materials and Methods
Cell culture.
Primary cultures of hippocampal neurons were prepared from embryonic day 18 (E18) Sprague Dawley rat brains as described previously (Goslin et al., 1988). Dissociated cells were plated on 18 mm poly-l-lysine-coated coverslips at low density (100,000 cells per dish), inverted over a glial feeder layer, and maintained in Neurobasal medium supplemented with B27 medium (Invitrogen, Carlsbad, CA) with once per week feedings for 14–21 d in vitro (DIV). Primary cultures of cortical neurons were prepared from E18 rats and plated at high density (one brain hemisphere per 100 mm dish). Cortical cultures were maintained in Neurobasal medium supplemented with B27 (Invitrogen) with once per week feedings for 10–14 DIV. B27 suppresses glial cell growth to <0.5%, thus ensuring a nearly pure neuronal population (Brewer et al., 1993).
Drug treatments.
Hippocampal neurons at 14–21 DIV were stimulated for 1 min (Q-FISH) or 2 min (immunocytochemistry) with 50 μm NMDA (plus 100 μm CNQX to block AMPARs) or for 15 min with 25 μm (RS)-3,5-dihydroxyphenylglycine (DHPG) (plus d-APV and CNQX to block NMDARs and AMPARs) and further incubated for a total of 30 min at 37°C in conditioned medium containing d-APV (50 μm) and CNQX (100 μm). Control cells were incubated for a total of 30 min at 37°C in conditioned medium containing d-APV (50 μm) and CNQX (100 μm). In addition, to assess a possible effect of d-APV plus CNQX, a minimum of two coverslips per experiment were incubated with conditioned medium in the absence of added drugs (“untreated controls”). BAPTA-AM (10 μm) was applied 30 min before NMDA for a total of 60 min. PD98059 [2-(2-amino-3-methyoxyphenyl)-4H-1-benzopyran-4-one] (50 μm), SB203580 [4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)-1H-imidazole] (10 μm), actinomycin D (30 μm), colchicine (10 μm), and emetine (20 μm) were applied 15 min before NMDA for a total of 45 min. After drug treatment, cells were washed in ice-cold PBS two times, fixed with 4% paraformaldehyde/5 mm MgCl2, and processed for in situ hybridization and/or immunolabeling. Within this range of ages, we observed no detectable effect of age on the abundance per unit area and/or localization of mRNAs or on the ability of drugs to elicit alterations in mRNA abundance and/or localization.
Oligonucleotide probes.
A mixture of four oligonucleotides (1:1:1:1, 50 bases each), complementary to nucleotide positions 136–185, 532–595, 787–836, and 1168–1218 of rat GluR2 cDNA (Keinanen et al., 1990), four oligonucleotides complementary to nucleotide positions 609–658, 711–760, 811–860, and 911–960 of the rat GluR1 cDNA, and four oligonucleotides complementary to nucleotide positions 166–215, 1184–1233, 1750–1794, and 2107–2156 of the rat NR1 cDNA were reacted with digoxigenin succinimide ester (Roche, Indianapolis, IN) at five amino-modified thymidine residues per oligonucleotide as described previously (Zhang et al., 2001). For control experiments, mixtures of three oligonucleotide probes were made to each of the following RNAs: γ-actin mRNA, β-galactosidase mRNA, and adult rat 18S ribosomal RNA as described previously (Bassell et al., 1998). In addition, scrambled probes (probes of the same size and base content but in a scrambled order) were prepared. In all cases, a BLAST (basic local alignment search tool) search revealed that the sequences of oligonucleotide probes and scrambled probes were not homologous to any other sequences within the database. To assess background staining, fluorescence and alkaline phosphatase in situ hybridization were performed with the omission of oligonucleotide probes. An advantage of oligonucleotide probes over RNA probes is that probe length, guanine–cytosine content, and hapten-labeling efficiency can be standardized, permitting direct analysis of differences in signal distribution.
Quantitative fluorescence in situ hybridization. Digoxigenin-labeled oligonucleotide probe mixtures (5 ng/oligonucleotide) were dried with Escherichia coli tRNA (10 mg/ml) and salmon sperm DNA (10 mg/ml) and resuspended in hybridization buffer [40% formamide/10% dextran sulfate (Sigma, St.
Louis, MO)/SSC/0.2% bovine serum albumin (BSA)/50 mm sodium phosphate buffer, pH 7.0/ribonucleoside vanadyl complex (Sigma)]. Cells were fixed by incubation in 4% paraformaldehyde/PBS/5 mm MgCl2 for 20 min at room temperature, rinsed three times in 1× PBS/5 mm MgCl2 and equilibrated in 40% formamide containing 20 mm sodium phosphate buffer, pH 7.0. Fixed cells were hybridized in hybridization buffer containing probe overnight at 39°C. After hybridization, cells were incubated in 40% formamide in 1× SSC for 15 min at 37°C and washed three times with 1× SSC and three times with Tris-buffered saline (TBS) (50 mm Tris/HCl buffer/150 mm NaCl, pH 7.4) at room temperature. Cells were then permeabilized by incubation with 0.3% Triton X-100 for 5 min at room temperature, washed three times in Tris–glycine buffer (0.1 m Tris-HCl/0.1 m glycine, pH 7.4), and blocked by incubation with 2% BSA/2% fetal bovine serum/0.1% Triton X-100 for 1 h at room temperature. Digoxigenin-labeled oligonucleotide probes were detected by immunocytochemistry with cyanine 3 (Cy3)-conjugated mouse monoclonal anti-digoxigenin antibody (Jackson ImmunoResearch, West Grove, PA) in conjunction with Cy3-conjugated anti-mouse antibody (Jackson ImmunoResearch) as described previously (Zhang et al., 2001). Dendritic structures and synapses were detected with rabbit polyclonal anti-MAP2 antibody (dendrites; Boehringer Mannheim, Mannheim, Germany) and rabbit polyclonal anti-synapsin-1 antibody (synapses; Chemicon, Temecula, CA) after permeabilization of cells with Triton X-100 in conjunction with Cy5-conjugated donkey anti-rabbit IgG (Jackson ImmunoResearch).
Standard nonradioactive in situ hybridization. Neurons were fixed by incubation in 4% paraformaldehyde/4% sucrose/1× PBS, acetylated with 0.1 m triethanolamine-HCl, pH 8.0, containing 0.2% acetic anhydride, rinsed with 1× PBS, and permeabilized by incubation in 0.2N HCl.
Cells were rinsed two times with 1× PBS and incubated in 40% formamide/1× SSC/0.01 m sodium phosphate buffer, pH 7.0, for 10 min at room temperature. Probes were dried with a mixture of 10 μg of E. coli tRNA and 10 μg of salmon sperm DNA and resuspended in hybridization buffer (40% formamide/10% dextran sulfate/1× SSC/0.2% BSA/15 mm sodium phosphate buffer, pH 7.0). Cells were hybridized with probe overnight at 37°C. After hybridization, cells were rinsed for 5 min at 37°C once with 40% formamide/SSC, three times with 1×SSC, and once with 1× TBS and then blocked by incubation in 1× TBS/10% BSA for 30 min at room temperature. Digoxigenin-labeled oligonucleotides were detected by incubation with an anti-digoxigenin Fab fragment conjugated to alkaline phosphatase (1:1000; Boehringer Mannheim) in 1× TBS containing 10% BSA for 1 h at 37°C. Cells were rinsed once in TBS and incubated with a solution containing 5-bromo-4-chloro-3-indolyl-phosphate and 3-nitro blue tetrazolium chloride (Boehringer Mannheim) overnight at 4°C according to the directions of the manufacturer. The colorimetric reaction was quenched by rinsing in 100 mm Tris-HCl, pH 8.5/1 mm EDTA. Coverslips were mounted on glass slides with glycerol/H20.
Fluorescence microscopy and quantitative digital imaging.
Neurons that appeared healthy and morphologically intact were selected by an investigator blind to the drug treatment under phase optics and imaged by fluorescence microscopy. Fluorescence labeling was imaged with a Nikon (Tokyo, Japan) Eclipse inverted microscope equipped with a 60× Plan-Neofluar or a 60× Plan-Apochromat objective, phase optics, 100 W mercury arc lamp, and HiQ bandpass filters (Chroma Technology, Brattleboro, VT). Images were captured with a cooled CCD camera (Hamamatsu, Bridgewater, NJ) using an electronically controlled illumination shutter (Sutter Instruments, Novato, CA) as described previously (Zhang et al., 1999). In all experiments, images for all experimental conditions were collected blind to the treatment and were processed and imaged in parallel. Comparisons were made between control and stimulated cells from the same culture preparation. For experiments involving Q-FISH and immunolabeling, minimal bleed-through between channels was confirmed by imaging single-labeled specimens. Images of control and stimulated cells from a given experiment were analyzed using identical acquisition parameters. Exposure times were kept constant and below grayscale saturation. Pixel size was 100 nm in x and y.
For quantitation of hybridization signal, cell bodies and all neurites of each neuron were delineated from the phase image and transferred to the fluorescent image for analysis. The total fluorescence intensity, integrated intensity, and area of the region of interest (all neurites of a given neuron) were analyzed using MetaMorph software (Universal Imaging Corporation, West Chester, PA). To determine average signal intensity, the total fluorescence intensity of all neurites emanating from the cell soma up to and including the fourth branch point (∼150 μm from the soma) was normalized to the total area of the compartment. To ensure that fluorescence was measured only in dendrites emanating from a single neuron, identifying hallmarks were verified in MAP2-labeled cells like those in Figure 2. Images were thresholded to subtract the average background fluorescence in cells labeled with negative control probes (randomized GluR2 or β-galactosidase oligonucleotides). Fluorescence intensity data for drug-treated cells are reported as the average integrated intensity/area of the region of interest of all cells after normalization to the average of the vehicle-treated, control cells. To assess synaptic localization of receptor mRNA, GluR2 mRNA and synapsin images were merged. Puncta were identified as discrete regions of fluorescence more than twofold of background. Positive colocalization was scored as puncta either overlapping or directly apposed (within two pixels) using Adobe Photoshop software (Adobe Systems, San Jose, CA).
Figure 2.

GluR2 mRNA clusters localize to dendritic shaft and postsynaptic structures. GluR2 mRNA (a) and MAP2 label (b) fill the shafts of dendritic processes; GluR2 mRNA clusters decorate proximal and distal segments of MAP2-filled dendrites (merge, c). Some GluR2 mRNA clusters (but not MAP2 label) appear to emanate from the dendritic shaft and localize to the heads of spine-like protrusions (a, c, arrows). d, Phase-contrast image of a cultured hippocampal neuron. e, Merge of GluR2 mRNA (red) and synapsin-1 protein (green) for neuron shown under phase optics in d. GluR2 mRNA clusters colocalize with synapsin-1 puncta, indicating the presence of abundant GluR2 mRNA at and near to synaptic sites. Inset, Enlarged view of boxed segment in e. A number of GluR2 mRNA clusters localize in close proximity with synapsin puncta, indicating localization opposite presynaptic terminals (arrows). f, Quantitation of juxtaposition of GluR2 mRNA clusters and synapsin-1 puncta. Approximately 26% of synapses (marked by synapsin-1) contain GluR2 mRNA clusters (n = 29 cells from 5 independent experiments). Approximately 12% of GluR2 mRNA clusters localize opposite synapsin puncta (n = 29 cells from 5 independent experiments). Scale bars: a–c, 20 μm; insets, 2 μm; d, e, 25 μm; inset, 5 μm.
To estimate the relative volumes of the soma versus dendrites for typical hippocampal pyramidal neurons in culture, confocal images were acquired on an Olympus (Optical Thornwood, NY) Fluoview imaging system. The pinhole was adjusted to give ∼1 μm depth of field, and a z stack of images were taken every 1 μm through the whole cell. Background-subtracted images were thresholded in MetaMorph to highlight positive GluR2 signal. To determine average signal intensity, the total fluorescence intensity of the cell soma and of all neurites emanating from the cell soma up to and including the fourth branch point (∼150 μm from the soma) was measured in each confocal plane. To estimate the relative volumes of the two compartments, the sum of these signals was calculated for all of the planes (total integrated signal). The fluorescent signal in the dendrites was expressed as a percentage of the total fluorescent signal contained in the soma and all neurites.
Except when otherwise noted, the n value refers to the number of independent experiments, each involving analysis of two coverslips per treatment group and a minimum of 10 cells per coverslip. All errors reported are as SEMs. Statistical analysis was assessed by means of a Student’s two-tailed t test.
Northern blot analysis.
To examine drug-induced alterations in mRNA abundance, primary cultures of cortical neurons were maintained in Neurobasal medium with B27 medium for 10–14 DIV (Brewer et al., 1993). On the day of the experiment, neurons (three 100 mm dishes per treatment group) were stimulated with NMDA in the absence or presence of actinomycin D as described above. Control cultures were treated with conditioned medium. Immediately after stimulation, cells were harvested and mRNA was extracted with the FastTrack system (Invitrogen) according to the instructions of the manufacturer. Northern blot analysis was as described previously (Oguro et al., 2001). In brief, samples (1 μg of mRNA with 0.3 μg/ml ethidium bromide) were loaded on a 1% agarose gel and subjected to gel electrophoresis. The contents of the gel were capillary blotted onto Hybond-N nylon membranes (Amersham Biosciences, Arlington Heights, IL) and fixed by exposure to ultraviolet light. For preparation of probes, GluR1, GluR2 (gift from Dr. Stephen Heinemann, Salk Institute, La Jolla CA), NR1 (cloned by our laboratory), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (loading control; Ambion, Austin, TX) cDNAs were denatured and end labeled with the RadPrime DNA labeling system (Invitrogen) in the presence of 32P-dCTP (Amersham Biosciences) and purified by elution on NICK columns (Amersham Biosciences). For detection of mRNA, membranes were prehybridized for 1 h at 68°C in ULTRAHyb solution (Ambion) and then hybridized for 2 h at 68°C in ULTRAHyb solution containing 32P-labeled-GluR1, GluR2, or NR1 cDNA. The membranes were washed twice in 2× SSC buffer containing 0.1% SDS at room temperature and two times with 0.1× SSC buffer containing 0.1% SDS at 60°C before exposure to film. Blots were reprobed with 32P-labeled-GAPDH cDNA to confirm the equivalent loading of mRNA samples. Exposure time was 72 h for GluR1, GluR2, and NR1 and 12 h for GAPDH. For quantitation of mRNA, Northern blots were analyzed with a Scan Jet 4-C computing densitometer using NIH Image 1.61 image-processing and analysis software. After background subtraction, mean optical densities of bands were corrected for differences in loading, and SEMs were computed for a minimum of four independent experiments. Statistical significance was assessed by means of an unpaired Student’s t test.
Immunocytochemistry.
Immunolabeling of cellular GluR2 or NR1 protein was performed as described previously (Lissin et al., 1999). Surface AMPARs were visualized in 18–21 DIV cultures by using an antibody to the N terminus of GluR2 after treatment with vehicle or drugs. Neurons were chilled on ice and washed three times with 1× PBS before fixation under nonpermeabilizing conditions with 4% paraformaldehyde/1× PBS for 10 min at 22°C. Fixed cells were washed three times in 1× PBS for 10 min, blocked with 2% BSA/1× TBS, and incubated with 1× TBS containing mouse monoclonal anti-GluR2 (Chemicon) to detect AMPARs or mouse monoclonal anti-NR1 antibody (Chemicon) to detect NMDARs for 1 h at room temperature. After washing, antibody-labeled surface receptors were visualized with Cy3-conjugated secondary antibody (goat anti-mouse IgG; Jackson ImmunoResearch). To verify that cells fixed under nonpermeabilizing conditions were indeed nonpermeabilized, we labeled neurons with an antibody to the intracellular protein PICK1 (protein interacting with C kinase-1). Whereas PICK1 immunoreactivity was detected in permeabilized cells, it was not detected under these conditions (n = 4) (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). To label total receptors, fixed neurons were washed three times in 1× TBS, permeabilized with 0.1% Triton X-100/1× PBS, washed three times in 1×TBS, and blocked with 2% BSA/1× TBS. Fixed, permeabilized neurons were then incubated with anti-GluR2 or anti-NR1 antibody as above. After washing, total receptors were visualized with Cy3-conjugated secondary antibody (affinity-purified donkey anti-mouse). For experiments involving double immunolabeling, receptors were labeled under nonpermeabilizing conditions as above; synapses were visualized after permeabilization of cells with 0.1% Triton X-100 by incubation with rabbit anti-synapsin-1 antibody (1:200; Chemicon) in conjunction with FITC-conjugated secondary (donkey anti-rabbit IgG) antibody (Jackson ImmunoResearch). Images were acquired and analyzed as described above for Q-FISH. Fluorescence intensity values for experimental cells were normalized to values for control cells at each time point.
Results
GluR2 and GluR1 mRNA granules localize to dendrites of hippocampal neurons
To examine the spatial distribution of GluR2 mRNA, we performed high-resolution Q-FISH and digital imaging analysis of hippocampal neurons at 14–21 DIV (Bassell et al., 1998; Zhang et al., 1999). Q-FISH afforded striking visual documentation of GluR2 mRNA within cell somata (Fig. 1b) (for lower-intensity images, see supplemental Fig. S2, available at www.jneurosci.org as supplemental material) and neuronal processes, in which it appeared as clusters of fluorescence. The clusters resembled RNA granules or “packets,” implicated in transport of RNA in neurons (Krichevsky and Kosik, 2001). Within dendrites, GluR2 mRNA granules extended to distal structures >50 μm from the cell soma (Fig. 1b) and were detected in spine-like protrusions (Fig. 1c, arrows). GluR2 mRNA fluorescence was not detectable in axons marked by neurofilament-H (supplemental Fig. S3a–f, available at www.jneurosci.org as supplemental material) or in MAP2-negative processes (see below). GluR1 mRNA fluorescence exhibited a pattern similar to that of GluR2 (Fig. 1d–f). In contrast, γ-actin mRNA in neurons at 9–10 DIV was confined primarily to the soma (Fig. 1g,h). These results demonstrate the presence of AMPAR mRNAs in dendrites and show that mRNAs exhibit distinct distributions within the neuron.
Figure 1.

GluR2 mRNA is highly clustered in proximal and distal dendrites of neurons. a–c, Localization of GluR2 mRNA in hippocampal neurons visualized by high-resolution FISH. a, Phase-contrast image of a cultured hippocampal neuron at 21 DIV. b, GluR2 mRNA appeared as fluorescent clusters extending into proximal and distal processes. The mRNA clusters resembled RNA granules or packets, implicated in RNA transport in neurons. c, Enlarged view of boxed segment in b; arrows denote spine-like protrusions. d, Phase-contrast image of a cultured hippocampal neuron. e, GluR1 exhibited a granular organization similar to that observed for GluR2 mRNA. f, Enlarged view of boxed segment in e; arrows denote spine-like protrusions. g, Phase-contrast image of a cultured hippocampal neuron. h, γ-Actin mRNA fluorescence in the same neuron as in g was confined to cell somata, with little labeling in neuronal processes. i, Phase-contrast image of a cultured hippocampal neuron. j, Hybridization with a randomized GluR2 probe of the same neuron as in i showed no detectable labeling in the soma or dendrites. Scale bars: a, b, 25 μm; c, 5 μm; g, i, 20 μm.
A number of observations document the specificity of the mRNA labeling technique. First, no detectable labeling was observed in neurons hybridized with scrambled GluR2 probe (Fig. 1i,j). Second, GluR2 mRNA fluorescence was not detectable in astrocytes (supplemental Fig. S3g–i, available at www.jneurosci.org as supplemental material) or cardiac myocytes (data not shown). Third, GluR2 mRNA distribution in neurons assessed by an alkaline phosphatase-based in situ hybridization protocol in combination with a specific GluR2 probe (supplemental Fig. S4a,b, available at www.jneurosci.org as supplemental material) was essentially identical to that obtained by Q-FISH (Fig. 1c). In contrast, labeling by the alkaline phosphatase method with a scrambled GluR2 probe (supplemental Fig. S4c,d, available at www.jneurosci.org as supplemental material) or a specific probe to LacZ mRNA (supplemental Fig. S4e,f, available at www.jneurosci.org as supplemental material) was undetectable.
To evaluate the relative proportion of total GluR2 mRNA that is present in dendrites, we performed two experiments. First, we evaluated the relative volumes of the dendrites and soma of typical hippocampal neurons at 14–21 DIV. Toward this end, we imaged dye-labeled neurons by confocal microscopy and acquired a z stack. Analysis of signal in the two compartments indicated that the dendrites comprise 88.9 ± 9.1% of the cell volume and the soma comprises 11.2 ± 1.4% of the cell volume (n = 40). Second, we measured distribution of mRNA fluorescence between the compartments by confocal microscopy. Analysis of mRNA fluorescence signal indicated that ∼50% of GluR2 mRNA is in the dendrites and ∼50% in the soma. Because we did not measure signal in dendrites beyond the third branch point, the dendritic value may represent an underestimate and the soma an overestimate of the actual volumes. Thus, dendritic mRNA comprises a rather sizable proportion of total mRNA.
GluR2 mRNA is targeted to apparent synaptic sites
We next performed double labeling for GluR2 mRNA and the known dendritic marker MAP2 (Fig. 2a–c). GluR2 mRNA fluorescence was prominent in MAP2-positive processes or dendrites. Within dendrites, GluR2 mRNA granules extended beyond the third or fourth branch points (Fig. 2c, merge). Whereas MAP2 label was confined within dendritic shafts, GluR2 mRNA fluorescence appeared to emanate from the shaft and localize to spine-like protrusions (Fig. 2a,c, arrows). GluR2 mRNA fluorescence was not detected in MAP2-negative processes (n = 40 cells in 4 independent experiments). To characterize more rigorously localization of GluR2 mRNA to synapses, we performed double labeling for GluR2 mRNA and the presynaptic marker synapsin-1 (Fig. 2d,e). A subset of GluR2 mRNA clusters was juxtaposed to synapsin puncta (Fig. 2e, inset, arrows). Altogether, 12.5 ± 1.4% of GluR2 mRNA clusters localized opposite synapsin (n = 5), and 25.8 ± 2.5% of synapsin puncta localized with a GluR2 mRNA cluster (n = 5) (Fig. 2f). Thus, GluR2 mRNA targets to apparent synaptic sites, consistent with possible local (on-site) synthesis of AMPARs.
NMDAR activation promotes loss of AMPAR mRNAs from dendrites
Dendritic RNA transport can be regulated in response to neuronal activity (Kiebler and DesGroseillers, 2000; Steward and Schuman, 2001; Farina and Singer, 2002). We next examined the impact of NMDAR activation on localization of AMPAR mRNAs in dendrites. In control cells, GluR2 mRNA fluorescence was abundant in cell soma and dendrites (Fig. 3a). Application of NMDA (50 μm) in the presence of the AMPAR antagonist CNQX (100 μm) for 1 min, followed by additional incubation for a total of 30 min in the presence of d-APV and CNQX (“NMDA”), induced a marked decrease in GluR2 mRNA fluorescence in dendrites relative to that of control cells incubated for 30 min in the presence of d-APV and CNQX (“control”) (decrease by 34.8 ± 2.9%; n = 12; p < 0.001) (Fig. 3b,g), with little or no effect on fluorescence in soma (Fig. 3g). The NMDAR effect was blocked by the NMDAR antagonist d-APV (50 μm; data not shown), indicating a critical role for NMDAR activation. Similarly, NMDA decreased GluR1 mRNA fluorescence in dendrites relative to that of control neurons (reduction by 35.3 ± 4.8%; n = 5; p < 0.001) (Fig. 3c,d,g). The NMDA effect was specific in that it did not alter NR1 mRNA fluorescence (increase by 21.30 ± 8.59; n = 4; NS) (Fig. 3e,f,g) or number of synapses, marked by synapsin-1 puncta (control, 16 ± 2.2 puncta/10 μm dendrite, n = 4; NMDA, 15.1 ± 1.1 puncta/10 μm dendrite, n = 4; NS). Moreover, the impact of NMDAR activation on somatic and dendritic mRNA abundance was confirmed by confocal microscopy and analysis of z stacks of images. Moreover, application of d-APV (50 μm) and CNQX (100 μm) for 30 min in the absence of NMDA did not detectably alter GluR2 mRNA fluorescence intensity in dendrites relative to that of control cells incubated in conditioned medium in the absence of added drugs (untreated control; n = 5; NS; data not shown). These findings indicate that NMDAR activation promotes a selective decrease in AMPAR mRNA abundance in dendrites. Although less likely, we cannot rule out the possibility that the observed decrease in signal is attributable, at least in part, to masking of the mRNA by RNA binding protein(s), rendering the mRNA less accessible to the probe (cf. Tiruchinapalli et al., 2003). Several factors, however, argue against a prominent contribution of RNA masking in this experiment. First, Q-FISH is performed with a mixture of five oligonucleotides directed to different sequences within the mRNA; it is unlikely that accessibility of all of the sequences is similarly affected. Second, whereas RNA binding proteins typically recognize sequences in the 3′ untranslated region (UTR), our probes target sequences in the coding region. Third, the decrease in GluR2 mRNA signal assessed by Q-FISH was confirmed by Northern blots (see Fig. 5).
Figure 3.

NMDAR activation promotes loss of dendritic GluR1 and GluR2 (but not NR1) mRNAs. a, GluR2 mRNA localization in the soma and dendrites of a control (vehicle-treated) hippocampal neuron at 21 DIV. b, Application of 50 μm NMDA (plus 100 μm CNQX, 1 min), followed by additional incubation for a total of 30 min in the presence of d-APV and CNQX, caused a prominent loss of GluR2 mRNA fluorescence (intensity and number of GluR2 mRNA clusters) in proximal and distal dendrites, assessed by Q-FISH at 30 min after initiation of drug treatment, with little or no change in soma. c, GluR1 mRNA localization in the soma and dendrites of a control hippocampal neuron from the same batch as in a and b. d, NMDA produced a marked decrease in GluR1 mRNA fluorescence in dendrites, assessed by Q-FISH at 30 min after initiation of drug treatment. e, NR1 localization in the soma and dendrites of a control hippocampal neuron. NR1 mRNA clusters were abundant in cell somata and throughout shafts of proximal and distal dendrites of a control (vehicle-treated) hippocampal neuron at 14 DIV. f, NMDA did not significantly alter NR1 mRNA fluorescence in dendrites of a hippocampal neuron, assessed at 30 min. g, Quantitation of data from images like those illustrated in a–f. Data represent the percentage change in the mean integrated intensity values per designated area of interest. Error bars represent SEM (***p ≤ 0.001). Scale bars: a–d, 25 μm; e, f, 15 μm; insets, 2 μm.
Figure 5.

NMDAR activation regulates total GluR2 mRNA abundance but does alter mRNA stability. a, Representative Northern blot analysis of mRNA samples from control (cntrl) and NMDA-treated cortical neurons (see Materials and Methods). b, Quantitation of data. Brief stimulation with NMDA (50 μm, 60 s) in the presence of CNQX reduced cellular GluR1 and GluR2, but not NR1 or GAPDH, mRNA content in neurons, assessed at 30 min after initiation of drug application. c–e, Representative Northern blots and decay rates of GluR2 (c), NR1 (d), and GAPDH (e) mRNAs in the presence of the transcriptional inhibitor actinomycin D or after NMDA treatment in the presence of actinomycin D. c, In the presence of actinomycin D, GluR2 mRNA exhibited a relatively rapid rate of constitutive decay relative to that of NR1 (d) or GAPDH (e) mRNA. NMDA did not significantly alter the constitutive decay of GluR2, NR1, or GAPDH mRNA observed in the presence of actinomycin D, indicating no detectable effect on mRNA stability. Error bars represent SEMs for four independent experiments (**p ≤ 0.01).
The NMDA-induced decrease in dendritic GluR2 mRNA requires Ca2+ and ERK/MAPK signaling
Ca2+ is a crucial second messenger implicated in the molecular transduction of synaptic activity into changes in gene expression. Ca2+ influx via NMDARs activates an array of intracellular signaling cascades. To examine a possible role for Ca2+ signaling, we loaded cells with the cell-permeable Ca2+ chelator BAPTA-AM (10 μm). Because local protein synthesis is implicated in NMDA-dependent synaptic plasticity and AMPARs in principal hippocampal neurons are GluR2 containing, we focused on NMDA regulation of GluR2 mRNA. BAPTA-AM produced a slight increase in dendritic GluR2 mRNA (increase by 14.9 ± 8.6%; n = 4) (Fig. 4, compare c with a) and abolished the NMDA-induced loss of dendritic GluR2 mRNA (NMDA plus BAPTA, increase by 10.0 ± 11.1%; n = 5; p < 0.001 vs NMDA) (Fig. 4, compare d with b). Thus, a rise in intracellular Ca2+ is critical to regulation of GluR2 mRNA by NMDA.
Figure 4.
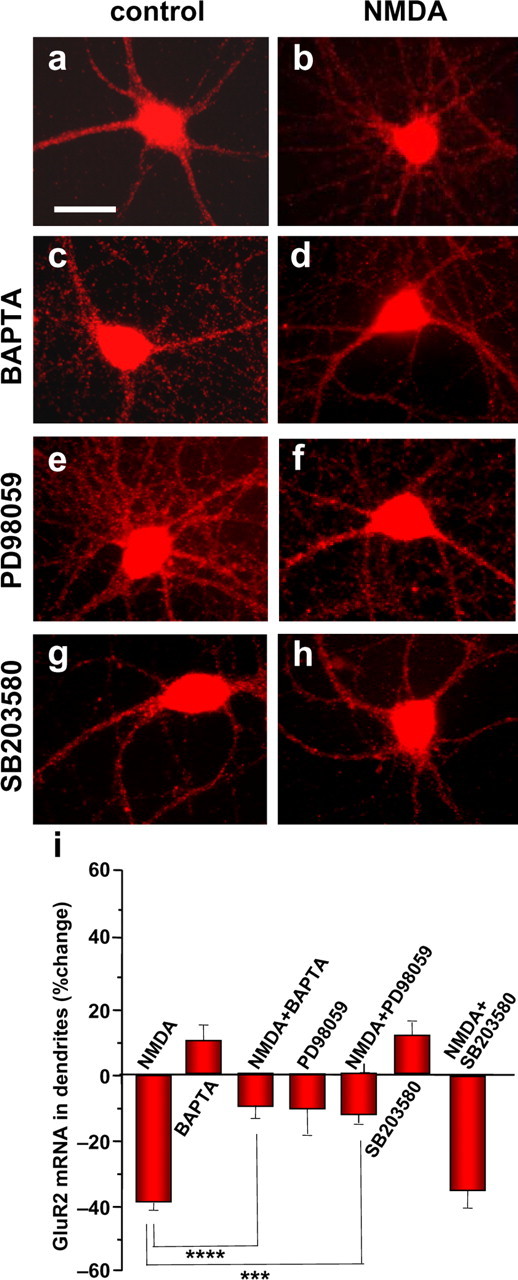
NMDA-induced loss of dendritic GluR2 mRNA requires Ca2+ and MAPK signaling. a, Representative images showing control (a, c, e, g) and NMDA-treated (b, d, f, h) neurons preincubated with vehicle (a, b), the cell-permeant Ca2+ chelator BAPTA-AM (c, d), the ERK/MAPK inhibitor PD98059 (e, f), and the p38 MAPK inhibitor SB203580 (g, h). a, b, NMDA (30 s) promoted loss/removal of GluR2 mRNA fluorescence in dendrites, as assessed by Q-FISH at 30 min after initiation of drug treatment. c–h, The NMDA-induced loss of GluR2 mRNA was markedly inhibited by BAPTA-AM (c, d) and PD98059 (e, f) but not by SB203580 (g, h), indicating that an elevation of intracellular Ca2+ and ERK/MAPK (but not p38 MAPK) signaling are critical to the NMDA effect. BAPTA-AM (c), PD98059 (e), and SB203580 (g) alone did not significantly alter GluR2 mRNA levels in dendrites or soma. i, Quantitation of data from images like those illustrated in a–h. NMDA treatment was as described in the legend to Figure 3. Drug treatments were as described in Materials and Methods. Error bars represent SEMs for 5–11 independent experiments (***p ≤ 0.001; ****p < 0.0005). Scale bar, 20 μm.
Ca2+ influx via NMDARs activates Ca2+-dependent signaling cascades such as the ERK/MAPK cascade, a well characterized signaling cascade implicated in regulation of gene expression (Pearson et al., 2001; Sweatt, 2004; Thomas and Huganir, 2004). To examine a possible role for ERK/MAPK signaling, we treated neurons with the selective, membrane-permeant ERK inhibitor PD98059 (50 μm). PD98059 did not significantly alter GluR2 mRNA abundance (reduction by 12 ± 19.9%; n = 6; NS) but inhibited the NMDA-induced loss of GluR2 mRNA from dendrites (NMDA plus PD98059, reduction by 13.5 ± 5.6%; n = 6; p < 0.01 vs NMDA alone) (Fig. 4e–i). The inhibition by PD98059 was ERK specific in that SB203580 (10 μm), a selective p38 MAPK inhibitor, was without effect (NMDA plus SB203580, reduction by 27.2 ± 4.1%; n = 5; NS vs NMDA alone) (Fig. 4g–l). These findings indicate that ERK/MAPK activation is required for NMDA regulation of GluR2 mRNA.
NMDA signaling inhibits GluR2 transcription
The results thus far indicate that NMDA signaling promotes a reduction in dendritic GluR2 mRNA via NMDAR activation, a rise in intracellular Ca2+, and activation of MAPK but do not distinguish between a loss of cellular GluR2 mRNA versus redistribution (regulated trafficking) of mRNA from dendrites to cell soma, with no change in abundance. To address this issue, we treated neurons with NMDA (50 μm, 1 min), isolated mRNA, and monitored cellular GluR2 mRNA abundance by Northern blot analysis under high stringency conditions (Fig. 5a,b). GluR2 mRNA exhibited two bands, 5.9 and 3.9 kb, corresponding to two splice variants that arise by alternative RNA splicing within the 3′ UTR of mouse (Kohler et al., 1994) and rat GluR2 mRNA (R. Dingledine, personal communication). NMDA induced a marked reduction in cellular GluR2 mRNA (reduction to 0.69 ± 0.10% of the control value; n = 15; p < 0.01) (Fig. 5a,b), with no effect on cell viability (supplemental Fig. S5, available at www.jneurosci.org as supplemental material). Similar results were observed for GluR1 mRNA (Fig. 5a,b), consistent with coregulation of GluR1 and GluR2 mRNAs. The effect was specific in that NMDA did not significantly alter NR1 or GAPDH mRNA (Fig. 5a,b). AMPA treatment also reduced dendritic levels of GluR2 mRNA (reduction to 68.1 ± 9.1% of the control value; n = 4; p < 0.05; data not shown), indicating that glutamatergic signaling specifically induces a loss of cellular AMPAR mRNAs.
The loss of cellular GluR2 mRNA could arise as a consequence of transcriptional regulation and/or regulation of mRNA stability. To examine a possible effect of NMDA on mRNA stability, we blocked RNA synthesis with the transcriptional inhibitor actinomycin D (30 μm) and monitored GluR2 mRNA by Northern blot analysis. The constitutive rate of GluR2 mRNA decay (measured in the presence of actinomycin D) was relatively rapid (reduction by ∼40% in 30 min) (Fig. 5c). The rates of constitutive decay of NR1 and GAPDH mRNA were slower than that of GluR2 (Fig. 5d,e). Application of NMDA (50 μm, 1 min) in the presence of actinomycin D did not further alter GluR2, NR1, or GAPDH mRNA (Fig. 5c–e), indicating that NMDA does not alter mRNA stability. These findings suggest that NMDA acts via transcriptional arrest to reduce dendritic GluR2 mRNA.
Actinomycin D mimics and occludes the NMDA-induced loss of dendritic GluR2 mRNA
To examine the mechanism by which NMDA regulates GluR2 mRNA in dendrites, we performed Q-FISH. Actinomycin D (30 μm, 45 min) reduced GluR2 mRNA in dendrites (reduction by 39.5 ± 4.6%; n = 12) (Fig. 6c,h), a degree of loss similar to that observed for NMDA (Fig. 6b,h). In the presence of actinomycin D, NMDA did not further alter dendritic GluR2 mRNA (NMDA plus actinomycin D, reduction by 33.3 ± 4.4%; n = 5; NS vs either NMDA or actinomycin D alone) (Fig. 6d,h). Thus, actinomycin D mimics and occludes the NMDA-induced loss of GluR2 mRNA in dendrites, consistent with a common mechanism of action. These findings strongly suggest that NMDA acts via transcriptional inhibition to decrease dendritic GluR2 mRNA.
Figure 6.

Actinomycin D mimics and occludes the NMDA-induced decrease in dendritic GluR2 mRNA. a–g, Representative images showing GluR2 mRNA in control (a, c, e, g) and NMDA-treated (b, d, f) neurons after preincubation with the transcription inhibitor actinomycin D (c, d), the microtubule- depolymerizing agent colchicine (e, f) or actinomycin D and colchicine (g), assessed by Q-FISH at 30 min after initiation of drug application. c, d, The transcriptional inhibitor actinomycin D decreased dendritic GluR2 mRNA to nearly the same extent as NMDA (b) and occluded additional reduction of GluR2 mRNA abundance by NMDA (d). Colchicine did not detectably alter GluR2 mRNA abundance in dendrites but strikingly inhibited the NMDA- and actinomycin D-induced loss/removal of GluR2 mRNA from dendrites (f). h, Quantitation of data. Drug treatments were as described in Materials and Methods. Error bars represent SEMs for 4–12 independent experiments (**p ≤ 0.01). Scale bars: a, 20 μm; inset, 2 μm.
NMDA-induced loss of dendritic GluR2 mRNA requires microtubule-based transport
Trafficking of localized neuronal mRNAs is mediated by microtubules (Knowles et al., 1996; Bassell et al., 1998; Zhang et al., 1999; Shan et al., 2003; Richter, 2004). We reasoned that, if NMDA signaling shuts off GluR2 transcription, it might act via a “synapse-to-nucleus signal.” Moreover, under conditions of transcriptional block, RNA is removed by constitutive degradation. Microtubules could mediate transport of a synapse-to-nucleus signal and/or removal of dendritic mRNA by constitutive degradation. To address this issue, we treated neurons with the microtubule destabilizing agent colchicine (10 μm). Colchicine did not alter GluR2 mRNA (increase by 8.7 ± 9.4%; n = 12; NS) (Fig. 6e,h) but markedly inhibited the NMDA-induced decrease in dendritic GluR2 mRNA (increase of 3.8 ± 14.8%; n = 4; p < 0.001 vs NMDA alone) (Fig. 6f,h). These findings indicate that the NMDA-induced loss of dendritic GluR2 mRNA is microtubule dependent.
To distinguish between a role for microtubules in transport of a synapse-to-nucleus signal versus a role in constitutive RNA degradation, we treated neurons with actinomycin D in the presence of colchicine. Because actinomycin D acts directly in the nucleus to inhibit active transcription, it does not require a synapse-to-nucleus signal. Actinomycin D reduced GluR2 mRNA abundance in dendrites by ∼40% (Fig. 6c,h). Colchicine itself did not alter GluR2 mRNA abundance (Fig. 6e,h) but markedly inhibited the actinomycin D-induced loss of GluR2 mRNA from proximal and distal dendrites (reduction by 10.3 ± 11.5%; n = 4; p < 0.05 vs actinomycin D alone) (Fig. 6g,h). These findings indicate that microtubules are required for constitutive degradation of dendritic mRNA and implicate microtubule-based transport in access of the degradative machinery to the mRNA. Microtubules are also critical for transport of RNA granules to and from dendrites (for review, see Hirokawa and Takemura, 2005). Thus, colchicine has two effects: it blocks local degradation and it blocks export of RNA to dendrites, thus maintaining dendritic levels mRNA.
Group I mGluR1 activation promotes transport of AMPAR mRNAs into dendrites
Activation of group I mGluRs promotes dendritic synthesis of proteins such as GluR1 and GluR2 (Weiler and Greenough, 1993; Kacharmina et al., 2000; Ju et al., 2004). We hypothesized that mGluR activation might also promote AMPAR mRNA transport into dendrites. Application of the group I mGluR agonist DHPG (25 μm, 15 min) increased the abundance of GluR2 mRNA in dendrites (increase by 52.2 ± 11.6%; n = 10 independent experiments; p < 0.001), with little or no effect in soma (Fig. 7a–f). DHPG produced a similar increase in dendritic GluR1 mRNA (increase by 40 ± 14.3%; n = 3 independent experiments; p < 0.01) (Fig. 7g–l). The DHPG effect was specific in that NR1 mRNA was unchanged (data not shown).
Figure 7.
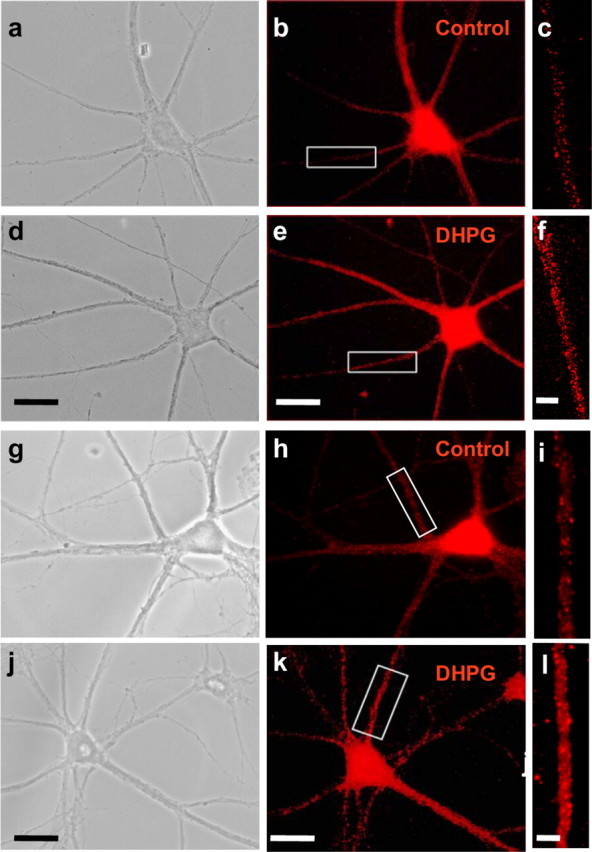
Group I mGluR1 activation enhances levels of GluR2 and GluR1 mRNA in dendrites. a–f, Representative images showing GluR2 mRNA in control (b, c) and DHPG-treated (e, f) neurons, as assessed by Q-FISH at 30 min after initiation of drug treatment. DHPG (25 μm plus 50 μm d-APV and 100 μm CNQX) was applied for 15 min. b, e, GluR2 mRNA fluorescence is prominent in the soma and dendrites of the same neurons shown under phase optics in a and d, respectively. c, f, Enlarged view of areas indicated in boxes in b and e, respectively. g–l, Representative images showing GluR1 mRNA fluorescence in a control (g–i) and DHPG-treated (j–l) neuron, as assessed by Q-FISH at 30 min after initiation of drug treatment. h, k, GluR1 mRNA fluorescence in the soma and dendrites of the same neurons shown under phase optics in g and j, respectively. i, l, Enlarged view of areas indicated in boxes in h and k, respectively. Activation of group I mGluRs markedly enhanced GluR2 and GluR1 mRNA fluorescence intensity in proximal and distal dendrites of hippocampal neurons, with no significant change in cell soma. Error bars represent SEMs for 10 (GluR2 mRNA) and 3 (GluR1 mRNA) independent experiments, each involving a minimum of 20 cells per treatment group (**p ≤ 0.01). Scale bars: b, e, h, k, 20 μm; c, f, i, l, 2 μm.
The increase in GluR2 mRNA could arise as a result of enhanced transcription, a decrease in RNA degradation, and/or regulated mRNA transport. To examine a possible role for transcriptional regulation, we treated neurons with actinomycin D. Although actinomycin D reduced dendritic GluR2 mRNA (reduction by 33.7 ± 7.2%; n = 4; p < 0.01 vs control) (Fig. 8c,g), DHPG increased dendritic GluR2 mRNA even in the presence of actinomycin D (DHPG plus actinomycin D, decrease by 4.0 ± 12.6%; n = 4; p < 0.01 vs actinomycin D or DHPG alone) (Fig. 8d,g). Moreover, DHPG did not significantly alter cellular GluR2 mRNA content, assessed by Northern blot analysis (increase to 1.18 ± 0.09%; n = 6; NS) (Fig. 8h,i). These findings indicate that the DHPG-induced increase in dendritic mRNA does not require active transcription. Thus, glutamatergic signaling bidirectionally regulates AMPAR mRNA abundance in dendrites.
Figure 8.
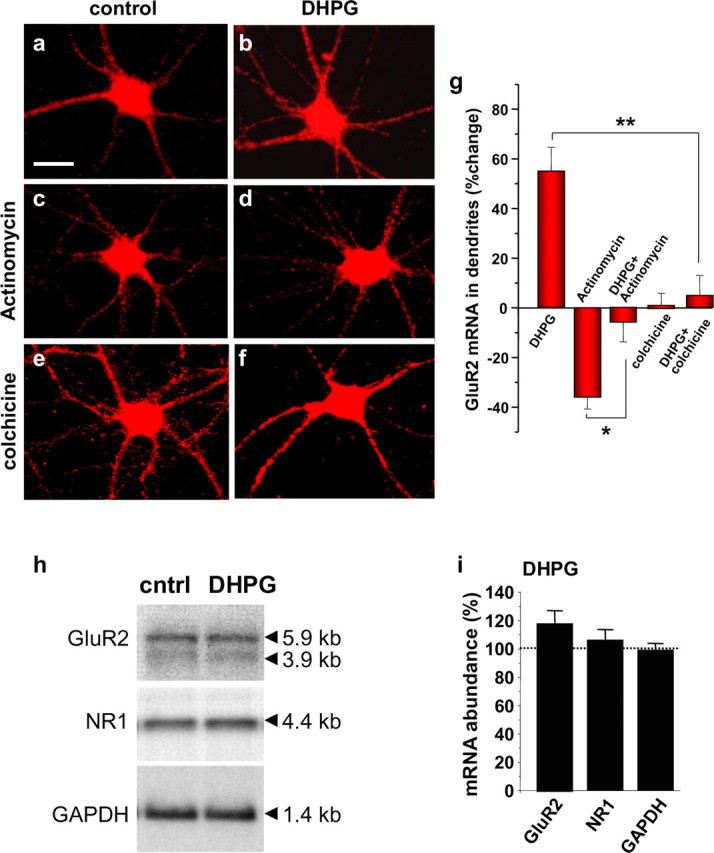
mGluR-induced increase in dendritic GluR2 mRNA involves regulated mRNA transport. a–f, Representative images showing GluR2 mRNA in control (a, c, e) and DHPG-treated (b, d, f) neurons pretreated with vehicle (a, b), the transcriptional inhibitor actinomycin D (c, d) and the microtubule-depolymerizing agent colchicine (e, f). g, Quantitation of data. Drug treatments were as described in Materials and Methods. b, DHPG (15 min) markedly enhanced GluR2 mRNA fluorescence in dendrites relative to that of control neurons (a), as assessed by Q-FISH at 30 min after initiation of DHPG application. c, Actinomycin D reduced fluorescence signal in dendrites. d, DHPG markedly increased GluR2 mRNA fluorescence in dendrites relative to that observed in the presence of actinomycin D alone. e, f, Colchicine did not detectably alter dendritic GluR2 mRNA fluorescence in dendrites or soma but markedly inhibited the DHPG-induced increase in GluR2 mRNA in dendrites. g, Quantitation of data. Error bars represent SEMs for four to six independent experiments. Scale bar, 20 μm. h, Representative Northern blot analysis of mRNA samples from control (cntrl) and DHPG-treated cortical neurons (see Materials and Methods). i, Quantitation of data. Stimulation with DHPG (25 μm, 15 min) in the presence of 50 μm d-APV and 100 μm CNQX did not significantly alter cellular GluR2, NR1, or GAPDH mRNA content in neurons, assessed at 30 min after initiation of drug application. Error bars represent SEMs for four independent experiments. *p < 0.05; **p < 0.01. Scale bar (in a), 25 μm.
We next examined whether the increase in mRNA requires microtubules. Colchicine did not detectably alter dendritic GluR2 mRNA but markedly inhibited the DHPG-induced increase in dendritic GluR2 mRNA (Fig. 8e–g). This finding suggests that mGluR1/5 signaling promotes transport of existing GluR transcripts from the cell body to dendrites via microtubules.
Regulation of dendritic mRNAs alters synaptic AMPAR number
Local protein synthesis in dendrites is critical to plasticity at several synapses (Steward and Schuman, 2003; Kanai et al., 2004; Richter, 2004; Martin and Zukin, 2006). We therefore investigated whether regulation of dendritic mRNAs is associated with alterations in AMPAR number. Toward this end, we fixed neurons at times after pharmacological stimulation and performed double immunolabeling for GluR2 and synapsin-1. In control neurons, GluR2 surface expression was prominent in proximal and distal dendrites (Fig. 9a). Brief stimulation with NMDA (50 μm, 30 s) reduced surface GluR2 surface expression in dendrites (decrease by 45 ± 9% at 4 h; n = 6 independent experiments; p < 0.001 at 4 h) (Fig. 9b,e), with no change in soma (decrease by 12 ± 5% at 4 h; n = 6; NS at 4 h) (Fig. 9b). The NMDA effect was blocked by d-APV (50 μm; data not shown), indicating a critical role for NMDAR activation. In contrast, NMDA did not significantly reduce dendritic or somatic NR1 protein, assessed as late as 24 h (increase by 2.51 ± 0.76% in dendrites, n = 4, NS; increase by 1.35 ± 0.13% in soma, n = 4, NS; data not shown). Moreover, NMDA did not detectably alter the number of synapses (control neurons, 8.9 ± 0.6 puncta/10 μm dendrite; n = 10; NMDA neurons, 8.1 ± 0.9 puncta/10 μm dendrite; n = 10; NS), indicating the specificity of the NMDA effect on AMPAR number.
Figure 9.

NMDAR activation induces a long-lasting decrease in synaptic AMPAR number. a–d, Representative images showing surface GluR2 (red) and synapsin-1 (green) protein immunolabeling in neurons at 4 h after treatment with vehicle(a), NMDA (b), actinomycin D (c), and NMDA (d) in the presence of actinomycin D. e, Time course of surface and total GluR2 protein in dendrites after treatment with NMDA. f, Quantitative analysis of total GluR2 protein abundance in dendrites at 4 h after NMDA, actinomycin D, NMDA plus actinomycin D, emetine, and NMDA plus emetine. g, Quantitative analysis of synaptic GluR2 assessed in images like those in a–d. NMDAR stimulation (30 s) markedly reduced surface and total GluR2 protein abundance in dendrites, with little or no change in soma (b, e, f). NMDA did not detectably alter NR1 protein fluorescence in dendrites (data not shown). Emetine and actinomycin D mimicked and occluded the NMDA-induced loss of GluR2 protein in dendrites (f) and synaptic sites (g). Drug treatments were as described in Materials and Methods. Data represent the percentage change in the mean integrated intensity values per designated area of interest for a minimum of four independent experiments. Error bars represent SEMs (**p ≤ 0.01). Scale bars: a–d, 10 μm; insets, 2 μm.
The findings thus far show that NMDAR signaling produces a reduction in surface dendritic GluR2 protein but do not distinguish between regulated receptor endocytosis versus a reduction in protein abundance. To distinguish between these possibilities, we assessed the impact of NMDA on total GluR2 protein in dendrites. NMDA reduced total GluR2 protein in dendrites (decrease by 46.0 ± 9% at 4 h; n = 4; p < 0.01 vs control) (Fig. 9e,f). The decrease in dendritic GluR2 was sustained for as long as 24 h (data not shown). The finding of a comparable decrease in surface and total GluR2 protein indicates that regulated receptor endocytosis alone cannot account for the altered receptor number. A reduction in protein could arise as a result of reduced protein synthesis and/or enhanced degradation. To distinguish between these possibilities, we treated cells with the irreversible protein synthesis inhibitor emetine (20 μm, applied 15 min before NMDA) (Linden, 1996); this protocol affords a measure of constitutive receptor degradation in the absence of protein synthesis. In the presence of emetine, the levels of dendritic GluR2 protein declined (decrease by 39.6 ± 7.5% at 4 h; n = 4; p < 0.01) (Fig. 9f). In the presence of emetine, NMDA did not further reduce GluR2 in dendrites (decrease by 41 ± 9.7%; n = 4; NS vs emetine alone) (Fig. 9f). These findings indicate that NMDA does not regulate protein stability.
The observation of a reduction in surface and total GluR2 protein in dendrites, with little or no change in protein stability, suggests a role for translational and/or transcriptional regulation. To address this issue, we treated neurons with actinomycin D (30 μm, applied 15 min before NMDA) and examined dendritic AMPAR numbers by immunocytochemistry. Actinomycin D reduced dendritic GluR2 (decrease by 42 ± 10%; n = 4; p < 0.01 vs control) (Fig. 9c,f). In the presence of actinomycin D, NMDA did not further alter dendritic GluR2 (decrease by 37 ± 12%; n = 4; NS vs NMDA or actinomycin D alone) (Fig. 9d,f). Thus, the two effects occluded one another, consistent with a common mechanism of action. Neither treatment altered the number of synapses (actinomycin D neurons, 8.2 ± 0.4 puncta/10 μm dendrite, n = 10, NS; NMDA plus actinomycin D neurons, 8.7 ± 0.8 puncta/10 μm dendrite, n = 10, NS), indicating specificity of the drug effects.
The results thus far show that NMDA reduces surface AMPAR expression in dendrites but do not distinguish between synaptic and extrasynaptic receptors. To address this issue, we assessed synaptic AMPAR numbers, scored as GluR2 puncta that colocalize with synapsin-1 puncta. In control neurons, many GluR2 puncta were juxtaposed to synapsin-1 puncta, indicating localization of AMPARs at synaptic sites (Fig. 9a). Altogether, 72.5 ± 1.8% of synapses contained GluR2 (n = 10 cells, 4 neurites per cell) (Fig. 9g). NMDA (30 s) reduced the percentage of synapses with GluR2 (decrease to 13.6 ± 2.8% at 4 h; n = 10; p < 0.001) (Fig. 9b,g). Actinomycin D produced a similar decrease in percentage of synapses with GluR2 (decrease to 13.9 ± 2.4% synapses with GluR2; n = 10; p < 0.001 vs control) (Fig. 9c,g). Moreover, in the presence of actinomycin D, NMDA did not further alter the number of synaptic AMPARs (decrease to 16.9 ± 2.3% synapses with GluR2; n = 10; NS vs actinomycin D alone) (Fig. 9d,g). Thus, the two effects occluded one another, consistent with a common mechanism of action. These findings suggest, but do not prove, a causal relationship between regulated dendritic mRNA abundance and loss of synaptic AMPARs and implicate regulated dendritic mRNA abundance as a mechanism relevant to synaptic efficacy. We cannot, however, rule out the possibility that the loss of receptors arises as a result of a loss of somatic mRNA, reduced translation centrally, and reduced transport of GluR2 subunits to synaptic sites.
Discussion
Activity-dependent alterations in AMPAR number are critical to many forms of synaptic plasticity. Whereas regulated insertion and removal of AMPARs from synaptic sites are critical to the early phase of NMDAR-dependent plasticity, regulated translation of dendritic mRNAs is well suited to the late, expression phase of long-term potentiation (LTP) and LTD. Here we show the novel observation that endogenous mRNAs encoding the AMPA receptor subunits GluR1 and GluR2 are localized to proximal and distal dendrites of hippocampal neurons. A substantial fraction of synaptic sites contain GluR2 mRNA clusters, consistent with their strategic positioning and availability for on-site protein synthesis. We demonstrate that glutamatergic signaling regulates GluR1 and GluR2 (but not NR1) mRNA localization in dendrites of hippocampal neurons. The finding that NR1 mRNA abundance is not altered underscores the specificity of the effect. NMDAR activation induces a loss of cellular GluR1 and GluR2 (but not NR1 or GAPDH) mRNA content. The NMDA-induced decrease in dendritic GluR2 mRNA occurs via a rise in intracellular Ca2+, activation of ERK/MAPK (but not p38 MAPK) signaling, and transcriptional arrest at the level of the nucleus, consistent with a synapse-to-nucleus signal.
We further show that regulated AMPAR mRNA localization has functional consequences. The NMDAR-induced decrease in levels of dendritic AMPAR mRNAs is associated with a long-lasting decrease in synaptic AMPAR number. Experiments involving emetine indicate that NMDA does not promote receptor degradation but rather inhibits protein synthesis. Experiments involving actinomycin D indicate a causal relationship between loss of dendritic mRNA and loss of synaptic GluR2 protein. A possible scenario is that NMDAR activation inhibits transcription, thereby reducing dendritically localized GluR2 mRNA and local translation, leading to a decrease in dendritic and synaptic AMPAR numbers. The decrease in synaptic AMPAR number is consistent with NMDA-induced LTD (Lee et al., 1998; Kamal et al., 1999; Beattie et al., 2000; van Dam et al., 2002; Li et al., 2004). It should, however, be noted that, although locally translated in dendrites, integral membrane proteins may undergo long-range transport to Golgi apparatus in the soma, followed by transport back to synaptic sites (Horton and Ehlers, 2003). Moreover, only a small minority of dendrites contain Golgi outposts (Horton et al., 2005). Thus, the abundance of dendritic mRNA encoding membrane proteins would not necessarily predict abundance of the fully processed protein product.
Whereas NMDAR signaling inhibits transcription and causes a decrease in GluR1 and GluR2 mRNA abundance in dendrites, group I mGluR signaling promotes anterograde trafficking of GluR1 and GluR2 mRNA to dendrites. The mGluR-induced increase in dendritic GluR2 mRNA requires microtubule-based mRNA transport but not active transcription, consistent with transport of an available pool of mRNA. This same signaling paradigm drives translation of exogenously expressed AMPAR mRNAs and synaptic insertion of AMPARs in severed dendrites, demonstrating the presence of key translational machinery in dendrites and regulation of AMPAR synthesis by neuronal activity (Kacharmina et al., 2000; Ju et al., 2004). The presence of endogenous AMPAR mRNAs in dendrites together with key elements of translational machinery suggests an important relationship between AMPAR synthesis and activity-dependent synaptic and structural plasticity. Consistent with this concept, recent findings link AMPAR GluR2 expression to remodeling of dendritic spines and structural plasticity (Passafaro et al., 2003). The present study extends previous studies in that it shows for the first time that endogenous AMPAR mRNAs are localized to dendrites and that mRNA abundance is bidirectionally regulated by neuronal activity.
Localization of mRNAs and regulated translation in dendrites have recently gained widespread acceptance as mechanisms fundamental to synaptic plasticity (Eberwine, 2001; Steward and Schuman, 2003; Martin and Zukin, 2006). By spatially restricting gene expression within neurons, dendritically localized mRNAs endow synapses with the capacity to autonomously regulate morphology and efficacy. Bidirectional regulation of AMPAR mRNA localization in dendrites represents a novel and potentially powerful mechanism for altering synaptic structure and efficacy. Although not addressed by the present study, our findings suggest that AMPAR mRNAs may also be present in the dendrites of hippocampal neurons in situ. Consistent with this, pilocarpine-induced seizures promote translocation of GluR2 mRNA to dendrites of hippocampal neurons in situ, as assessed by laser capture of the CA1 dendritic layer (R. Dingledine, personal communication). Nevertheless, we cannot rule out the possibility that AMPAR mRNAs are primarily localized to dendrites of immature neurons in culture.
Ca2+ and MAPK signaling are critical to regulation of dendritic GluR2 mRNA abundance
Our study has begun to define the signaling mechanisms that couple NMDAR activation to changes in dendritic levels of AMPAR mRNAs in dendrites. We show that a rise in intracellular Ca2+ and ERK/MAPK signaling is critical to NMDAR-regulated levels of dendritic GluR2 mRNA. We further show that NMDA acts via ERK/MAPK signaling to arrest GluR2 transcription and thereby depress GluR2 mRNA in dendrites. These findings are consistent with the concept that Ca2+ influx via NMDARs serves as an “on switch” for ERK-dependent synapse-to-nucleus communication (Hardingham et al., 2001). The ERK/MAPK cascade is a well characterized Ca2+-dependent signaling cascade implicated in regulation of gene expression (Sweatt, 2004). Once activated, ERK assembles with a downstream kinase such as pp90 ribosomal protein S6 kinase (Rsk); the MAPK/Rsk complex translocates to the nucleus in which it regulates transcription of genes involved in synaptic plasticity. Although ERK signaling is well known to activate transcription in neurons, it can inhibit transcription via phosphorylation and activation of repressors (Allen et al., 2000; Lo et al., 2001).
Regulated trafficking of AMPAR mRNAs in dendrites
Whereas NMDA signaling induces transcriptional inhibition, group I mGluR signaling promotes trafficking of mRNA from the cell soma into dendrites by microtubule-based transport. RNAs are transported into developing axons and dendrites in the form of large granules containing RNA, RNA binding proteins, ribosomes, and translational factors (RNA-containing granules); RNA transport in neuronal processes is rapid (average speed of 0.1 μm/s), bidirectional, and microtubule dependent (for review, see Hirokawa and Takemura, 2005). Recent studies have begun to reveal the underpinnings of RNA transport in dendrites. In a tour de force, Kanai et al. (2004) used affinity chromatography and proteomics to identify 42 components of large RNA-containing granules and showed that kinesin family member KIF5 is a motor critical to transport of granules into distal dendrites (Kanai et al., 2004). Our finding that mGluR signaling regulates AMPAR (but not NMDAR) mRNA trafficking into dendrites is consistent with the notion that RNA transport is rapid and specific and can be regulated by neuronal activity. Of binding proteins known to mediate mRNA trafficking in dendrites, perhaps the best characterized is cytoplasmic polyadenylation element binding protein (CPEB) (Huang et al., 2003). CPEB facilitates mRNA targeting to dendrites (Huang et al., 2003) and triggers on-site translation in response to external cues (Shin et al., 2004). The 3′ UTR of the long splice form of GluR2 mRNA contains putative CPE consensus sequences, which might subserve regulated trafficking in dendrites.
Based on these findings, we propose a model whereby activity bidirectionally regulates AMPAR mRNA abundance in dendrites (supplemental Fig. S6, available at www.jneurosci.org as supplemental material). GluR1 and GluR2 mRNAs are localized in the soma and dendrites of hippocampal neurons. NMDAR activation produces a rise in intracellular Ca2+ (localized primarily to spines) and activation of the Ca2+-dependent MAPK signaling cascade. ERK, the putative synapse-to-nucleus signal, translocates to the nucleus and inhibits transcription of GluR1 and GluR2. Under conditions of transcriptional block, dendritic levels of AMPAR mRNA are decreased. Microtubule-based transport is required for the decrease in dendritic mRNA, consistent with a role for microtubules in RNA degradation (Cleveland and Havercroft, 1983; Gong and Brandhorst, 1988; Aronov et al., 1999; Tadros et al., 2003). A possible scenario is that GluR2 mRNA undergoes microtubule-based transport from synaptic sites to processing- (or P-) bodies, organized sites in which decapping and degradation take place (Sheth and Parker, 2003). Future studies will be required to determine whether P-bodies are present in neurons or localized to dendrites. In contrast, the mGluR-mediated effect requires RNA transport but not transcription. The finding that mGluR signaling produces a different and opposing action to NMDARs is not surprising given that activation of mGluRs and NMDARs elicit Ca2+ signals with differing spatiotemporal dynamics.
Physiological significance of regulated trafficking of AMPAR mRNAs in dendrites
The present study provides the first evidence of regulated AMPAR abundance and trafficking in dendrites. Regulated endocytosis of synaptic AMPARs is a mechanism critical to the early phase of NMDAR-dependent LTD at hippocampal synapses (cf. Carroll et al., 2001). We propose that, whereas regulated endocytosis produces a change in surface (but not total) AMPARs and lasts on the order of minutes, regulated mRNA in dendrites can produce alterations in receptor number that endure for many hours. We show that NMDAR activation promotes transcriptional arrest of AMPAR mRNAs, decreased dendritic mRNA, and decreased receptor number, mechanisms that may be relevant to long-lasting LTD. In contrast, mGluR activation drives trafficking of GluR mRNAs into dendrites (present study) and promotes local on-site protein synthesis (Kacharmina et al., 2000; Ju et al., 2004). Whereas mGluR activation promotes AMPAR endocytosis and LTD on the order of minutes, mGluR LTD is followed by priming of LTP, a state in which persistent enhanced membrane excitability facilitates LTP induction on the order of hours (Cohen and Abraham, 1996; Cohen et al., 1999; Raymond et al., 2000; Miura et al., 2002). We propose that enhanced levels of GluR mRNAs in dendrites may represent a mechanism relevant to priming. Thus, regulation of AMPAR mRNA abundance and trafficking by glutamatergic signaling provides a potentially powerful means to effect long-lasting changes in synaptic efficacy.
Footnotes
*S.Y.G., K.-M.N., and R.R. contributed equally to this work.
†R.C.C. and R.S.Z. contributed equally to this work.
S. Y. Grooms’ present address: AstraZeneca, Columbia, SC 29223.
G. J. Bassell’s present address: Departments of Cell Biology and Neurology, Emory University School of Medicine, Atlanta, GA 30322.
This work was supported by National Institutes of Health Grants NS51127 (G.J.B.), NS049661 (R.C.C.), and NS20752 (R.S.Z.) and the Howard Hughes Medical Institute–Research Resources Program for Medical Schools (R.C.C.). We thank Dr. Oswald Steward for helpful comments on this manuscript and Aleksandra Fayer, Rownak Afroz, and Maritza Martinez for technical assistance.
References
- Allen MP, Xu M, Zeng C, Tobet SA, Wierman ME (2000). Myocyte enhancer factors-2B and -2C are required for adhesion related kinase repression of neuronal gonadotropin releasing hormone gene expression. J Biol Chem 275:39662–39670. [DOI] [PubMed] [Google Scholar]
- Aronov S, Marx R, Ginzburg I (1999). Identification of 3′UTR region implicated in tau mRNA stabilization in neuronal cells. J Mol Neurosci 12:131–145. [DOI] [PubMed] [Google Scholar]
- Barry MF, Ziff EB (2002). Receptor trafficking and the plasticity of excitatory synapses. Curr Opin Neurobiol 12:279–286. [DOI] [PubMed] [Google Scholar]
- Bassell GJ, Zhang H, Byrd AL, Femino AM, Singer RH, Taneja KL, Lifshitz LM, Herman IM, Kosik KS (1998). Sorting of β-actin mRNA and protein to neurites and growth cones in culture. J Neurosci 18:251–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beattie EC, Carroll RC, Yu X, Morishita W, Yasuda H, von Zastrow M, Malenka RC (2000). Regulation of AMPA receptor endocytosis by a signaling mechanism shared with LTD. Nat Neurosci 3:1291–1300. [DOI] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR, Evege EK, Price PJ (1993). Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res 35:567–576. [DOI] [PubMed] [Google Scholar]
- Carroll RC, Beattie EC, von Zastrow M, Malenka RC (2001). Role of AMPA receptor endocytosis in synaptic plasticity. Nat Rev Neurosci 2:315–324. [DOI] [PubMed] [Google Scholar]
- Cleveland DW, Havercroft JC (1983). Is apparent autoregulatory control of tubulin synthesis nontranscriptionally regulated? J Cell Biol 97:919–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen AS, Abraham WC (1996). Facilitation of long-term potentiation by prior activation of metabotropic glutamate receptors. J Neurophysiol 76:953–962. [DOI] [PubMed] [Google Scholar]
- Cohen AS, Coussens CM, Raymond CR, Abraham WC (1999). Long-lasting increase in cellular excitability associated with the priming of LTP induction in rat hippocampus. J Neurophysiol 82:3139–3148. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Isaac JT, Wang YT (2004). Receptor trafficking and synaptic plasticity. Nat Rev Neurosci 5:952–962. [DOI] [PubMed] [Google Scholar]
- Eberwine J (2001). Single-cell molecular biology. Nat Neurosci [Suppl] 4:1155–1156. [DOI] [PubMed] [Google Scholar]
- Eberwine J, Miyashiro K, Kacharmina JE, Job C (2001). Local translation of classes of mRNAs that are targeted to neuronal dendrites. Proc Natl Acad Sci USA 98:7080–7085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD (2000). Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron 28:511–525. [DOI] [PubMed] [Google Scholar]
- Farina KL, Singer RH (2002). The nuclear connection in RNA transport and localization. Trends Cell Biol 12:466–472. [DOI] [PubMed] [Google Scholar]
- Gong ZY, Brandhorst B (1988). Autogenous regulation of tubulin synthesis via RNA stability during sea urchin embryogenesis. Development 102:31–43. [DOI] [PubMed] [Google Scholar]
- Goslin K, Schreyer DJ, Skene JH, Banker G (1988). Development of neuronal polarity: GAP-43 distinguishes axonal from dendritic growth cones. Nature 336:672–674. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Arnold FJ, Bading H (2001). A calcium microdomain near NMDA receptors: on switch for ERK-dependent synapse-to-nucleus communication. Nat Neurosci 4:565–566. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Takemura R (2005). Molecular motors and mechanisms of directional transport in neurons. Nat Rev Neurosci 6:201–214. [DOI] [PubMed] [Google Scholar]
- Horton AC, Ehlers MD (2003). Dual modes of endoplasmic reticulum-to-Golgi transport in dendrites revealed by live-cell imaging. J Neurosci 23:6188–6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton AC, Racz B, Monson EE, Lin AL, Weinberg RJ, Ehlers MD (2005). Polarized secretory trafficking directs cargo for asymmetric dendrite growth and morphogenesis. Neuron 48:757–771. [DOI] [PubMed] [Google Scholar]
- Huang YS, Carson JH, Barbarese E, Richter JD (2003). Facilitation of dendritic mRNA transport by CPEB. Genes Dev 17:638–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju W, Morishita W, Tsui J, Gaietta G, Deerinck TJ, Adams SR, Garner CC, Tsien RY, Ellisman MH, Malenka RC (2004). Activity-dependent regulation of dendritic synthesis and trafficking of AMPA receptors. Nat Neurosci 7:244–253. [DOI] [PubMed] [Google Scholar]
- Kacharmina JE, Job C, Crino P, Eberwine J (2000). Stimulation of glutamate receptor protein synthesis and membrane insertion within isolated neuronal dendrites. Proc Natl Acad Sci USA 97:11545–11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamal A, Ramakers GM, Urban IJ, de Graan PN, Gispen WH (1999). Chemical LTD in the CA1 field of the hippocampus from young and mature rats. Eur J Neurosci 11:3512–3516. [DOI] [PubMed] [Google Scholar]
- Kanai Y, Dohmae N, Hirokawa N (2004). Kinesin transports RNA: isolation and characterization of an RNA-transporting granule. Neuron 43:513–525. [DOI] [PubMed] [Google Scholar]
- Keinanen K, Wisden W, Sommer B, Werner P, Herb A, Verdoorn TA, Sakmann B, Seeburg PH (1990). A family of AMPA-selective glutamate receptors. Science 249:556–560. [DOI] [PubMed] [Google Scholar]
- Kiebler MA, DesGroseillers L (2000). Molecular insights into mRNA transport and local translation in the mammalian nervous system. Neuron 25:19–28. [DOI] [PubMed] [Google Scholar]
- Knowles RB, Sabry JH, Martone ME, Deerinck TJ, Ellisman MH, Bassell GJ, Kosik KS (1996). Translocation of RNA granules in living neurons. J Neurosci 16:7812–7820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler M, Kornau HC, Seeburg PH (1994). The organization of the gene for the functionally dominant alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor subunit GluR-B. J Biol Chem 269:17367–17370. [PubMed] [Google Scholar]
- Krichevsky AM, Kosik KS (2001). Neuronal RNA granules: a link between RNA localization and stimulation-dependent translation. Neuron 32:683–696. [DOI] [PubMed] [Google Scholar]
- Lee HK, Kameyama K, Huganir RL, Bear MF (1998). NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron 21:1151–1162. [DOI] [PubMed] [Google Scholar]
- Lee SH, Simonetta A, Sheng M (2004). Subunit rules governing the sorting of internalized AMPA receptors in hippocampal neurons. Neuron 43:221–236. [DOI] [PubMed] [Google Scholar]
- Li R, Dozmorov M, Hellberg F, Tian Y, Jilderos B, Wigstrom H (2004). Characterization of NMDA induced depression in rat hippocampus: involvement of AMPA and NMDA receptors. Neurosci Lett 357:87–90. [DOI] [PubMed] [Google Scholar]
- Linden DJ (1996). A protein synthesis-dependent late phase of cerebellar long-term depression. Neuron 17:483–490. [DOI] [PubMed] [Google Scholar]
- Lissin DV, Carroll RC, Nicoll RA, Malenka RC, von Zastrow M (1999). Rapid, activation-induced redistribution of ionotropic glutamate receptors in cultured hippocampal neurons. J Neurosci 19:1263–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo RS, Wotton D, Massague J (2001). Epidermal growth factor signaling via Ras controls the Smad transcriptional co-repressor TGIF. EMBO J 20:128–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinow R, Malenka RC (2002). AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci 25:103–126. [DOI] [PubMed] [Google Scholar]
- Martin KC, Zukin RS (2006). RNA trafficking and local protein synthesis in dendrites: an overview. J Neurosci 26:7131–7134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura M, Watanabe M, Offermanns S, Simon MI, Kano M (2002). Group I metabotropic glutamate receptor signaling via Gαq/Gα11 secures the induction of long-term potentiation in the hippocampal area CA1. J Neurosci 22:8379–8390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oguro K, Jover T, Tanaka H, Lin Y, Kojima T, Oguro N, Grooms SY, Bennett MV, Zukin RS (2001). Global ischemia-induced increases in the gap junctional proteins connexin 32 (Cx32) and Cx36 in hippocampus and enhanced vulnerability of Cx32 knock-out mice. J Neurosci 21:7534–7542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passafaro M, Nakagawa T, Sala C, Sheng M (2003). Induction of dendritic spines by an extracellular domain of AMPA receptor subunit GluR2. Nature 424:677–681. [DOI] [PubMed] [Google Scholar]
- Pearson G, Robinson F, Beers GT, Xu BE, Karandikar M, Berman K, Cobb MH (2001). Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev 22:153–183. [DOI] [PubMed] [Google Scholar]
- Raymond CR, Thompson VL, Tate WP, Abraham WC (2000). Metabotropic glutamate receptors trigger homosynaptic protein synthesis to prolong long-term potentiation. J Neurosci 20:969–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter JD (2004). RNA transport (partly) revealed!. Neuron 43:442–443. [DOI] [PubMed] [Google Scholar]
- Rook MS, Lu M, Kosik KS (2000). CaMKIIα 3′ untranslated region-directed mRNA translocation in living neurons: visualization by GFP linkage. J Neurosci 20:6385–6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan J, Munro TP, Barbarese E, Carson JH, Smith R (2003). A molecular mechanism for mRNA trafficking in neuronal dendrites. J Neurosci 23:8859–8866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Kim MJ (2002). Postsynaptic signaling and plasticity mechanisms. Science 298:776–780. [DOI] [PubMed] [Google Scholar]
- Sheth U, Parker R (2003). Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science 300:805–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin CY, Kundel M, Wells DG (2004). Rapid, activity-induced increase in tissue plasminogen activator is mediated by metabotropic glutamate receptor-dependent mRNA translation. J Neurosci 24:9425–9433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith WB, Starck SR, Roberts RW, Schuman EM (2005). Dopaminergic stimulation of local protein synthesis enhances surface expression of GluR1 and synaptic transmission in hippocampal neurons. Neuron 45:765–779. [DOI] [PubMed] [Google Scholar]
- Song I, Huganir RL (2002). Regulation of AMPA receptors during synaptic plasticity. Trends Neurosci 25:578–588. [DOI] [PubMed] [Google Scholar]
- Steward O, Schuman EM (2001). Protein synthesis at synaptic sites on dendrites. Annu Rev Neurosci 24:299–325. [DOI] [PubMed] [Google Scholar]
- Steward O, Schuman EM (2003). Compartmentalized synthesis and degradation of proteins in neurons. Neuron 40:347–359. [DOI] [PubMed] [Google Scholar]
- Steward O, Worley PF (2001). Selective targeting of newly synthesized Arc mRNA to active synapses requires NMDA receptor activation. Neuron 30:227–240. [DOI] [PubMed] [Google Scholar]
- Sweatt JD (2004). Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol 14:311–317. [DOI] [PubMed] [Google Scholar]
- Tadros W, Houston SA, Bashirullah A, Cooperstock RL, Semotok JL, Reed BH, Lipshitz HD (2003). Regulation of maternal transcript destabilization during egg activation in Drosophila. Genetics 164:989–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GM, Huganir RL (2004). MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci 5:173–183. [DOI] [PubMed] [Google Scholar]
- Tiruchinapalli DM, Oleynikov Y, Kelic S, Shenoy SM, Hartley A, Stanton PK, Singer RH, Bassell GJ (2003). Activity-dependent trafficking and dynamic localization of zipcode binding protein 1 and beta-actin mRNA in dendrites and spines of hippocampal neurons. J Neurosci 23:3251–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dam EJ, Ruiter B, Kamal A, Ramakers GM, Gispen WH, de Graan PN (2002). N-methyl-d-aspartate-induced long-term depression is associated with a decrease in postsynaptic protein kinase C substrate phosphorylation in rat hippocampal slices. Neurosci Lett 320:129–132. [DOI] [PubMed] [Google Scholar]
- Weiler IJ, Greenough WT (1993). Metabotropic glutamate receptors trigger postsynaptic protein synthesis. Proc Natl Acad Sci USA 90:7168–7171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HL, Eom T, Oleynikov Y, Shenoy SM, Liebelt DA, Dictenberg JB, Singer RH, Bassell GJ (2001). Neurotrophin-induced transport of a beta-actin mRNP complex increases beta-actin levels and stimulates growth cone motility. Neuron 31:261–275. [DOI] [PubMed] [Google Scholar]
- Zhang HL, Singer RH, Bassell GJ (1999). Neurotrophin regulation of beta-actin mRNA and protein localization within growth cones. J Cell Biol 147:59–70. [DOI] [PMC free article] [PubMed] [Google Scholar]