

Abstract
Deletion of synaptobrevin/vesicle-associated membrane protein, the major synaptic vesicle soluble N-ethylmaleimide-sensitive factor attachment protein receptor (R-SNARE), severely decreases but does not abolish spontaneous and evoked synaptic vesicle exocytosis. We now show that the closely related R-SNARE protein cellubrevin rescues synaptic transmission in synaptobrevin-deficient neurons but that deletion of both cellubrevin and synaptobrevin does not cause a more severe decrease in exocytosis than deletion of synaptobrevin alone. We then examined the structural requirements for synaptobrevin to function in exocytosis. We found that substituting glutamine for arginine in the zero-layer of the SNARE motif did not significantly impair synaptobrevin-dependent exocytosis, whereas insertion of 12 or 24 residues between the SNARE motif and transmembrane region abolished the ability of synaptobrevin to mediate Ca2+-evoked exocytosis. Surprisingly, however, synaptobrevin with the 12-residue but not the 24-residue insertion restored spontaneous release in synaptobrevin-deficient neurons. Our data suggest that synaptobrevin mediates Ca2+-triggered exocytosis by tight coupling of the SNARE motif to the transmembrane region and hence forcing the membranes into close proximity for fusion. Furthermore, the fusion reactions underlying evoked and spontaneous release differ mechanistically.
Keywords: exocytosis, endocytosis, synaptic vesicle recycling, SNARE, spontaneous fusion, hippocampus, synapse, whole-cell patch-clamp
Introduction
Synapses release neurotransmitter when an action potential (AP) induces Ca2+ influx into nerve terminals, and Ca2+ in turn triggers synaptic vesicle exocytosis. However, quantal vesicular release that is electrically indistinguishable from AP-driven release also occurs spontaneously in all synapses; moreover, under experimental conditions, release can be induced by hypertonicity. Although all of these forms of release require soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins (for review, see Jahn et al., 2003), it is currently unclear whether the different forms of release are mechanistically identical or are mediated by distinct types of fusion.
During synaptic vesicle fusion, the synaptic vesicle SNARE protein synaptobrevin 2 (Syb2)/vesicle-associated membrane protein 2 (VAMP2) forms a complex with the plasma membrane SNARE proteins syntaxin 1 and synaptosome-associated protein of 25 kDa (SNAP-25) (Sollner et al., 1993). All SNARE proteins contain one or two SNARE motifs; SNARE complexes are assembled as four-helical bundles composed of four SNARE motifs (in the case of synaptic SNARE complexes, a single SNARE motif each from synaptobrevin and syntaxin and two from SNAP-25) (Hanson et al., 1997). Assembly of SNARE complexes is thought to catalyze fusion by forcing membranes into close proximity (Weber et al., 1998). Although SNARE complex formation is primarily fueled by hydrophobic interactions, SNARE complexes contain a central “zero layer” composed of a hydrophilic electrostatic interaction mediated by three glutamine and one arginine residue (Sutton et al., 1998). As a result, SNARE proteins are classified based on their SNARE motif sequences into types of Q-SNAREs and R-SNAREs (Fasshauer et al., 1998).
Synaptobrevin 2 is a minimal SNARE that consists only of a short NH2-terminal sequence, a SNARE motif, and a COOH-terminal transmembrane region (see Fig. 1A). Initial evidence that synaptobrevin mediates fusion was obtained when synaptobrevin was identified as the target of tetanus toxin, which blocks exocytosis by proteolytic cleavage of synaptobrevin (Link et al., 1992; Schiavo et al., 1992). Deletion of synaptobrevin impairs all forms of neurotransmitter release, albeit to different degrees (Schoch et al., 2001). Synaptobrevin-deficient synapses show an ∼100-fold reduction in AP-induced neurotransmitter release but only a ∼10-fold reduction in spontaneous release or in release triggered by hypertonic sucrose. This disparity between evoked and spontaneous release was also observed in neurons lacking SNAP-25 (Washbourne et al., 2002). Interestingly, in synaptobrevin-deficient chromaffin cells, release was only decreased by ∼50%, whereas double-mutant mice that lack both synaptobrevin and cellubrevin, a closely related R-SNARE protein (McMahon et al., 1993), lacked all release (Borisovska et al., 2005). Thus, in chromaffin cells, synaptobrevin and cellubrevin are functionally redundant. To some degree, redundancy was also reported for SNAP-25 and SNAP-23 function in chromaffin cells (Sorensen et al., 2003).
Figure 1.

Both N- and C-terminally tagged synaptobrevin 2 rescues synaptic function in synaptobrevin 2 knock-out neurons. A, Schematic drawings for synaptobrevin 2 tagged either N terminally with eCFP (eCFP–Syb2) or C terminally with another GFP analog fluorescence protein called venus (Syb2–venus). TMR, Transmembrane region. B, Representative responses to field stimulation at 1 Hz from WT, Syb2 KO hippocampal neurons, or synaptobrevin 2 knock-out neurons infected with lentivirus containing eCFP–Syb2 or Syb2–venus. C, Bar graph depicts the amplitudes of average responses to the first 60 pulses (n = 6 for WT, Syb2 KO, and Syb2-venus infected cells; n = 5 for eCFP–Syb2 infected cells). Note that statistical significance is marked on this and following figures as follows: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Although synaptic SNARE proteins have been extensively studied biochemically and physiologically, little is known about the structure/function relationships of synaptobrevin. For example, the current model for how SNAREs catalyze fusion requires that the SNARE motif must be close to the transmembrane region, but this has only been examined in yeast fusion reactions (Wang et al., 2001) and in in vitro fusion assays (McNew et al., 1999; Parlati et al., 2000; Melia et al., 2002). Therefore, in the present study, we used rescue experiments in cultured synaptobrevin-deficient neurons to examine the structural requirements for synaptobrevin function in synaptic exocytosis.
Materials and Methods
Hippocampal cultures.
Double knock-out (KO) mice were bred by crossing heterozygous mutant synaptobrevin 2 KO mice (Schoch et al., 2001) that are also homozygous mutant for cellubrevin (Yang et al., 2001). Hippocampal neurons from embryonic day 18 littermate mice were dissociated by trypsin (5 mg/ml for 5 min at 37°C), triturated with a siliconized Pasteur pipette, and plated onto 12 mm coverslips coated with poly-l-lysine (approximately three coverslips per hippocampus). Neurons were cultured at 37°C in a humidified incubator with 95% air/5% CO2 in Minimal Essential Media containing 5 g/liter glucose, 0.1 g/liter transferrin, 0.25 g/liter insulin, 0.3 g/liter glutamine, 5–10% heat-inactivated FCS, 2% B-27 supplement, and 1–2 μm cytosine arabinoside, and used after 12–22 d in vitro.
Constructs.
The different v-SNARE constructs were assembled in litmus28 vector (New England Biolabs, Ipswich, MA) and tagged N terminally with enhanced cyan fluorescent protein (eCFP) lacking the stop codon localizing the fluorescent protein in the cytoplasm. The following primer pairs were used for creating the mutants by PCR reaction: DF0007(C GTA GAC AAG GTC CTC GAG CAG GAC CAG AAG CTG TCG G) and DF0008 (C CGA CAG CTT CTG GTC CTG CTC GAG GAC CTT GTC TAC G) for R56Q point mutation, and P1 (GCT TCG AAT TCT GCA GTC G) and P2 (AAA GGA TCC AGC GCT ACC GGC TGA ACC AGC GCT ACC GGC CTT GAG CTT GGC TGC ACT AGT TTC AAA CTG GG) for the 12 insertion after introducing a new BamHI site with the primer pair of P3 (AAA GGA TCC CGC AAA TAC TGG TGG AAA AAC C) and P4 (CGC GCT AGC TCC TCA GGG GTT TAA GAG C). For the 24-insertion mutant, a digestion with BamHI/NheI was performed on the 12-insertion mutant, and the PCR product for 12 insertion with P4 and P5 (CA AGA TCT GCC GGT AGC GCT GGT TCA GC) was digested with BglII and NheI and subcloned, resulting in an insertion doubled in size. Bacterial expression was performed in Escherichia coli strain BL21. Later, all constructs were transferred into the main lentivirus vector (FUW, technique of lentiviral system was adopted from Dittgen et al., 2004) using BsiWI and EcoRI sites, except for the synaptobrevin 2–24 insertion, which was inserted between XbaI and NheI sites. All constructs were verified by sequencing. For the 12- and 24-insertion constructs, we preferred AGS repeats because of their hydrophobicity and predicted inability to form special secondary structure.
Lentiviral infection.
Constructs were cotransfected with plasmids for viral envelope proteins and enzymes into HEK 293 cells using the FuGENE6 transfection system (Roche Molecular Biochemicals, Indianapolis, IN) according to the specifications of the manufacturer, and lentivirus-containing culture medium was harvested 3 d later, filtered through 0.45 μm pores, and immediately used for infection or frozen in liquid nitrogen and stored at −80°C. Hippocampal cultures were infected at day 4 in vitro by adding 400 μl of viral suspension to each well.
Fluorescence imaging.
Modified Tyrode's bath solution [in mm: 150 NaCl, 4 KCl, 2MgCl2, 10 glucose, 10 HEPES-NaOH, pH 7.4, 0.01 CNQX, 0.05 AP-5, and 2 CaCl2 (∼310 mOsm)] was used in all experiments. Synaptic boutons were loaded with N-(3-triethylammoniumpropyl)-4-(4-(diethylamino)styryl)pyridinium dibromide (FM2-10) (400 μm; Invitrogen, Carlsbad, CA) by a 90 s exposure to 47 mm K+/2 mm Ca2+; full destaining was triggered with a hyperkalemic bath solution containing 90 mm KCl substituted for NaCl. Images were taken after 10 min wash in dye-free, nominally Ca2+-free solution to minimize spontaneous release. In all experiments, isolated boutons (∼1 μm2) were selected for analysis. Synaptic vesicle fusion was induced by gravity perfusion of hyperkalemic solution onto the field of interest (1–2 ml/min) for 60 s, followed by nominal Ca2+-free Tyrode's perfusion for 60 s again. Images were acquired with a cooled CCD camera (Roper Scientific, Trenton, NJ) during illumination (1 Hz and 200 ms) at 480 ± 20 nm (505 dichroic long pass and 534 ± 25 bandpass) via an optical switch (Sutter Instruments, Novato, CA) and analyzed using Metafluor Software (Universal Imaging, Downingtown, PA). Background staining levels determined after five consecutive rounds of high K+ application were subtracted from all fluorescence images.
Electrophysiology.
Synaptic responses were recorded from pyramidal cells in modified Tyrode's bath solution in the whole-cell patch configuration. Data were acquired with an Axopatch 200B amplifier and Clampex 8.0 software (Molecular Devices, Union City, CA), filtered at 2 kHz, and sampled at 200 μs. The internal pipette solution included the following (in mm): 115 Cs-MeSO3, 10 CsCl, 5 NaCl, 0.1 CaCl2, 10 HEPES, 4 Cs-BAPTA, 20 tetraethylammonium-Cl, 4 Mg-ATP, 0.3 mm Na2-GTP, and 10 lidocaine N-ethyl-bromide, pH 7.35 (300 mOsm). A hypertonic solution, prepared by addition of 500 mm sucrose to the nominally Ca2+-free Tyrode's solution, was perfused directly into the close vicinity of the cell from which the recording was made. Field stimulations (24 mA pulses of 0.6 ms) were applied with parallel platinum electrodes immersed into the perfusion chamber.
Coimmunoprecipitation of recombinant synaptobrevin 2 mutants with syntaxin 1.
Neuronal cultures in a six-well plate were infected with different synaptobrevin 2 mutant constructs and harvested using 1 ml of a HEPES buffer (50 mm HEPES, pH 7.2, 100 mm NaCl, 2 mm MgCl2, 4 mm EGTA, 1 mm PMSF, 5 μg/ml leupeptin, 2 μg/ml aprotinin, and 1 mm DTT). Proteins were extracted using 1% Triton X-100 for 1.5 h with rotation at 4°C and centrifuged at 20,800 × g for 10 min, and supernatants were diluted with the same HEPES buffer without Triton X-100 to reduce Triton X-100 concentration to 0.5%. A total of 10 μl of rabbit serum (U6251) raised against rat syntaxin 1 residue 1–264 was added to 600 μl of 0.5% Triton X-100 cell lysate and incubated for 2 h at 4°C with rotation. After 2 h incubation with U6251, 20 μl of protein A-Sepharose beads were added and further incubated another 1 h with rotation at 4°C. Beads were washed five times with HEPES buffer containing 0.5% Triton X-100, and 35 μl of 2× SDS sample buffer was added, vortexed, and boiled for 10 min. Immunoprecipitation sample preparations (30 μl) were used for SDS-PAGE and Western blotting. A total of 25 μl of cell lysate was used for SDS-PAGE and Western blotting after trichloroacetic acid precipitation of total protein. Blots were performed using the following sera or ascites: mouse anti-green fluorescent protein (GFP), C163 from Zymed (South San Francisco, CA); mouse anti-Syb2, CL69.1; mouse anti-syntaxin 1, HPC-1; and mouse anti-SNAP-25, CL71.1.
Miscellaneous.
Paired Student's t test or variance analysis was used to determine significance, which is marked on the figures as asterisks: *p < 0.05, **p < 0.01, ***p < 0.001 levels of significance.
Results
N- or C-terminal GFP fusions of synaptobrevin do not impair its function
Because in synaptotagmin 1 fusion of the cytoplasmic C terminus with GFP inactivates function (Han et al., 2005), we tested whether fusion of synaptobrevin with a GFP derivative (to visualize expression) at either the N or C terminus impairs function. In these experiments, we used cultured hippocampal neurons from synaptobrevin-deficient mouse embryos and expressed exogenous synaptobrevin proteins using a lentiviral expression system (Dittgen et al., 2004). The main advantage of the lentiviral expression system is its high infection efficacy (>90%), thus allowing electrophysiological assessment of presynaptic function in combination with optical analysis of synaptic vesicle recycling using the fluorescent styryl dye FM1-43 [N-(3-triethylammoniumpropyl)-4-(4-(dibutylamino)styryl) pyridinium dibromide]. Expression of synaptobrevin containing an N-terminally fused or a C-terminally fused GFP derivative (Fig. 1A) fully rescued the amplitude of evoked responses in synaptobrevin-deficient neurons (Fig. 1B). Thus, the GFP moiety does not interfere with synaptobrevin function during exocytosis when attached to synaptobrevin either N or C terminally.
Cellubrevin fully rescues synaptic transmission in synaptobrevin-deficient neurons
The R-SNARE protein cellubrevin (VAMP3) is highly homologous to synaptobrevin 2 (McMahon et al., 1993), with only a single amino acid difference between their SNARE motifs, although their N- and C-terminal sequences differ significantly (Fig. 2A). We used eCFP-tagged cellubrevin to test whether cellubrevin can substitute functionally for synaptobrevin. In synaptobrevin-deficient neurons, eCFP-tagged cellubrevin was targeted to synapses and fully restored spontaneous neurotransmitter release, evoked release in response to hypertonic sucrose application, and evoked release in response to action potentials (Fig. 2) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Cellubrevin can thus mediate the function of synaptobrevin in exocytosis.
Figure 2.
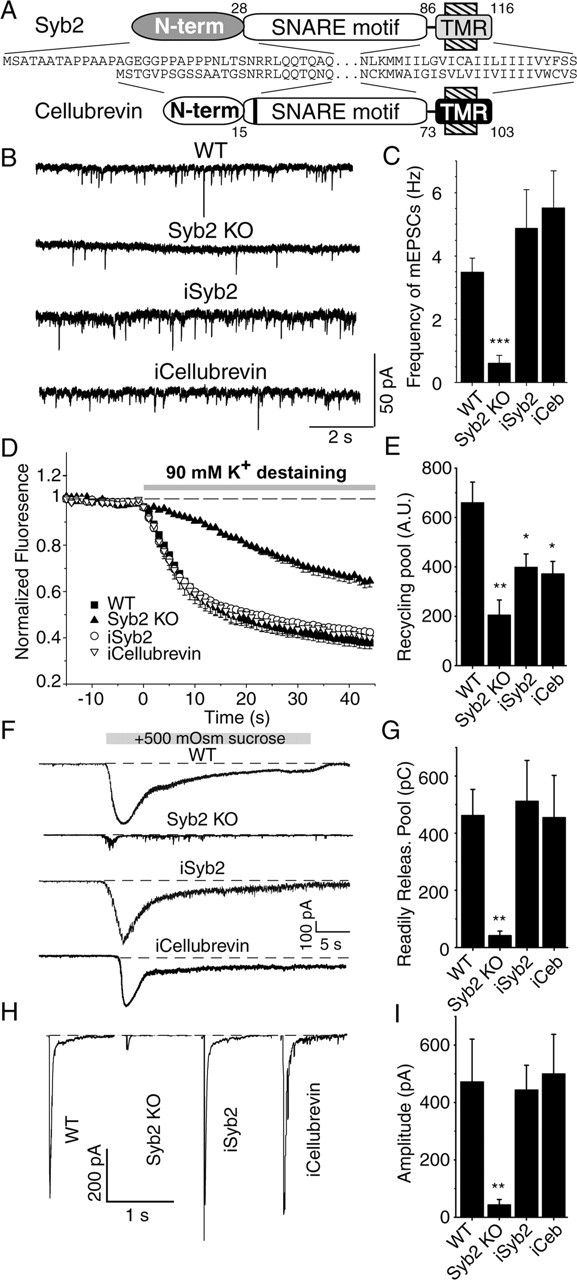
Rescue of synaptic transmission in synaptobrevin 2-deficient synapses by cellubrevin. Synaptic transmission was analyzed after infection of synaptobrevin 2-deficient hippocampal cultures with lentivirus containing either synaptobrevin 2 or cellubrevin. A, Schematic drawings compare the structure of synaptobrevin 2 and cellubrevin. TMR, Transmembrane region. B, Representative traces of spontaneous synaptic release at −70 mV in the presence of 1 μm tetrodotoxin and 100 μm picrotoxin. C, Graph depicts the average frequency and SEM of miniature EPSCs (mEPSCs). Note the markedly reduced average frequency of miniature EPSCs for synaptobrevin 2 knock-outs and its rescue by both synaptobrevin 2 and cellubrevin [n = 12 for WT; n = 6 for synaptobrevin 2 knock-out and cellubrevin infected (iCeb); n = 11 for synaptobrevin 2 infected (iSyb2)]. D, Synapses were loaded with 47 mm K+ for 90 s in the presence of 200 μm FM2-10, washed with calcium-free Tyrode's solution, and destained by applying 90 mm K+ for 60 s, which was repeated five times with a wash of 60 s after each. Averaged traces of FM2-10 fluorescence during the first round of 90 mm K+ destaining from wild-type (n = 7), Syb2 KO (n = 5), and synaptobrevin 2 knock-out synapses infected with synaptobrevin 2 (n = 7) or cellubrevin (n = 4). E, Bar graph depicts the recycling synaptic vesicle pool, which could be labeled by 47 mm K+ for 90 s and destained by five times application of 90 mm K+ for 1 min. F, Readily releasable pool recorded by addition of 500 mOsm sucrose to the bath solution. Representative traces recorded at −70 mV. G, Bar graph shows synaptic release rate estimated from the charge transfer rate [n = 4 for WT, synaptobrevin 2 KO, and synaptobrevin 2 infected (iSyb2); n = 5 for cellubrevin infected (iCeb)]. H, Representative responses to field stimulation at 1 Hz. I, Bar graphs depict the average responses to the first 60 pulses [n = 7 for WT and synaptobrevin 2 KO; n = 17 for synaptobrevin 2 infected (iSyb2); n = 5 for cellubrevin infected (iCeb)].
We next investigated whether cellubrevin can also rescue the synaptic vesicle endocytosis defect observed in synaptobrevin-deficient synapses (Deák et al., 2004). A hallmark of synaptobrevin-deficient synapses is a markedly reduced uptake of fluorescent styryl dyes such as FM2-10 and FM1-43 during a single round of high K+ stimulation (47 mm K+ for 90 s), which reflects a reduction in fast synaptic vesicle endocytosis (Deák et al., 2004). The N-terminal region of synaptobrevin could potentially mediate its role in fast endocytosis, for instance by triggering nucleation of an endocytic coat. Thus, the absence of this region in cellubrevin may result in diminished endocytic trafficking in cellubrevin-rescued synapses. In contrast to this expectation, the rescue of synaptic transmission by cellubrevin extended to styryl dye uptake and release (Fig. 2D,E). Cellubrevin-rescued synapses took up and released FM2-10 dye with the same efficiency as synaptobrevin-rescued synapses, suggesting that the N-terminal region of synaptobrevin (which is different from that of cellubrevin) (Fig. 2A) is not essential for endocytosis. In addition, in cellubrevin-rescued synapses, the rate of FM2-10 loss in response to 90 mm K+ stimulation was comparable with wild-type (WT) and synaptobrevin-rescued synapses, indicating full recovery of synaptic vesicle priming. Here, it is important to note that infection of synaptobrevin-deficient neurons with eCFP-tagged synaptobrevin 2 and cellubrevin constructs could only rescue the recycling pool size up to 60–70% of the wild type (Fig. 2E) (see Fig. 4E). There are two possibilities that may account for this observation. First, infections were performed 4 d after plating the cells, so it is likely that a significant portion of synaptic vesicles still lacked synaptobrevin 2 (or cellubrevin) at the time of the experiments. Second, all of these constructs had a N-terminal eCFP tag, which might interfere with other members of the release machinery by either masking a part of synaptobrevin 2 protein or weak dimerization.
Figure 4.
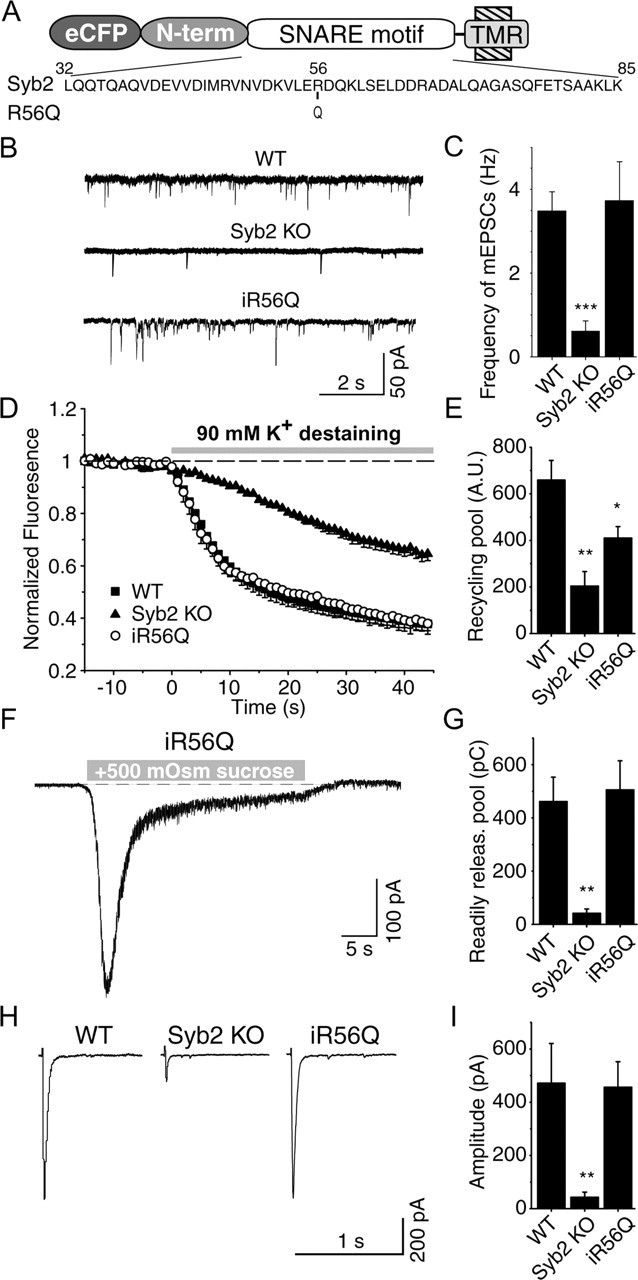
R56Q point mutation does not affect synaptobrevin 2 function. A, Diagram of the point mutation at the zero layer of SNARE motif, in which the 56arginine was mutated to glutamine. TMR, Transmembrane region. B, Representative traces of spontaneous synaptic events recorded from a cell infected with R56Q mutant form of synaptobrevin 2. C, Frequency of spontaneous synaptic events (n = 12 R56Q infected; n = 15 12-insertion infected KO cells; n = 10 24-insertion infected KO cells). D, Average destaining curves of FM2-10-loaded synapses during high potassium stimulation. E, Bar graphs depict size of recycling pool as mean ± SEM for WT (n = 16, 1146 synapses), synaptobrevin 2 KO (n = 11, 688 synapses) synapses, and KO synapses after infection with eCFP–synaptobrevin 2–R56Q (n = 3, 126 synapses). F, A representative recording of synaptic responses to hypertonic sucrose (+0.5 Osm) application in a synaptobrevin 2 KO culture rescued with the R56Q mutant. G, Summary graph of synaptic responses to hypertonic sucrose monitored in infected neurons. Abbreviations used are the same as in C. Responses are calculated as the cumulative charge transfer integrated over 1 s interval at the peak of the response (n = 6 R56Q infected). H, Representative traces from whole-cell recordings during 1 Hz field stimulations. I, Amplitudes of evoked responses (n = 7 WT and synaptobrevin 2 KO cells; n = 11 R56Q infected cells).
Additional deletion of cellubrevin does not aggravate the synaptobrevin knock-out phenotype
Because cellubrevin can substitute for synaptobrevin in synaptic exocytosis similar to chromaffin cell exocytosis (Borisovska et al., 2005), we tested whether low levels of cellubrevin, below the detection limit of the previous immunoblot analyses (Schoch et al., 2001), account for the residual fusion in synaptobrevin-deficient synapses. To address this question, we generated and analyzed double knock-out mice for synaptobrevin 2 and cellubrevin (see Materials and Methods). We found, however, that the amount of residual fusion present in synaptobrevin-deficient synapses was not decreased further in the double knock-out neurons (Fig. 3). We detected no additional decrease in mini frequency or amplitude (Fig. 3B,C) and no change in the synaptic vesicle pools monitored by loading synapses with the fluorescent dye FM2-10 (Fig. 3D,E). Moreover, the response to hypertonic sucrose (0.5 m sucrose in Tyrode's solution) was indistinguishable between synaptobrevin single knock-out neurons and synaptobrevin/cellubrevin double knock-out neurons (Fig. 3F,G). These data strongly argue that, in contrast to chromaffin cells (Borisovska et al., 2005), cellubrevin does not have a physiological role in exocytosis from hippocampal synapses.
Figure 3.
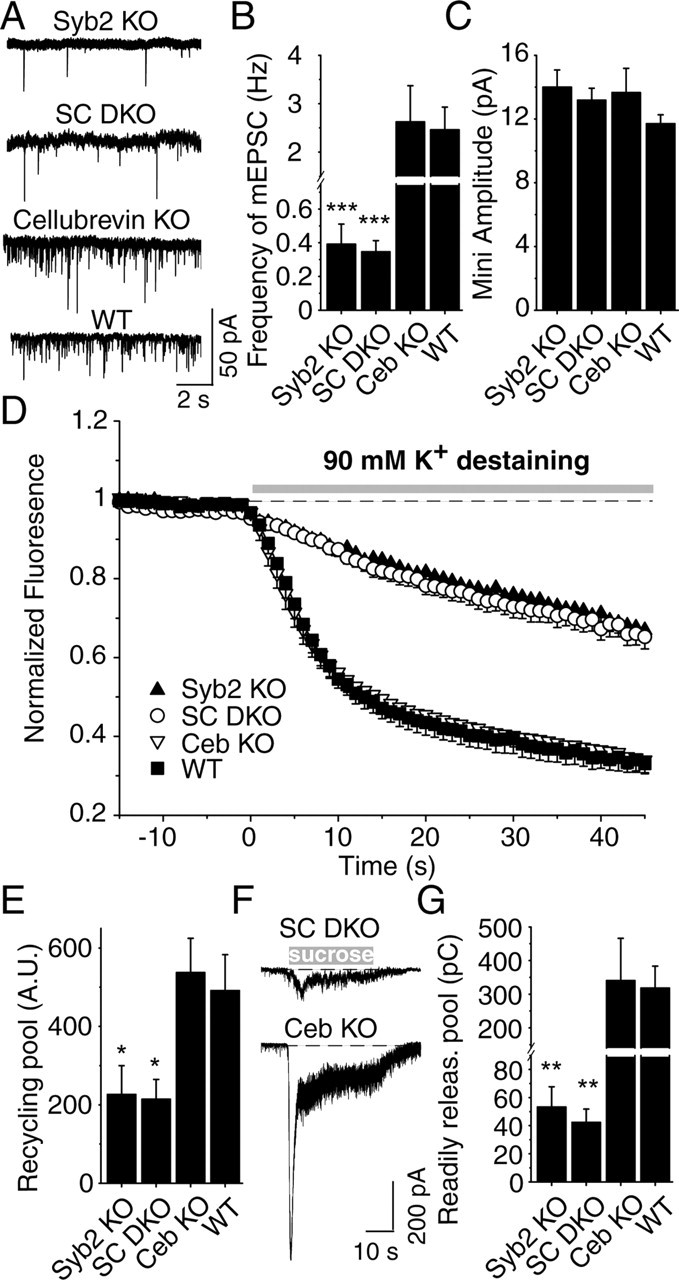
Analysis of synaptobrevin 2/cellubrevin double knock-outs. A, Spontaneous synaptic responses (“minis”) in cultured hippocampal neurons from mutant and control mice. mEPSCs, Miniature EPSCs. Frequency (B) and amplitude (C) of spontaneous synaptic events. All data shown are means ± SEMs [n = 9 Syb2 KO, WT, and cellubrevin KO (Ceb KO); n = 14 synaptobrevin/cellubrevin double KO mice (SC DKO)]; differences between synaptobrevin single KO neurons and synaptobrevin/cellubrevin double KO neurons were not significant. D, Average destaining curves for synaptobrevin 2 KO (n = 10, 632 synapses), SC double knock-out synapses (n = 7, 444 synapses), cellubrevin KO (n = 6, 467 synapses), and WT (n = 6, 435 synapses) neurons during the first 90 mm K+ application as shown on the top. E, Comparable pool size of functional vesicles in synaptobrevin 2 KO and SC double KO synapses. Graph depicts the recycling synaptic vesicle pool size loaded by a single round of 47 mm K+ stimulation. A.U., Arbitrary units. F, Synaptic responses to stimulation with hypertonic sucrose. Representative recordings of synaptic responses to hypertonic sucrose (+0.5 Osm). Traces were obtained during whole-cell recordings from neurons cultured from synaptobrevin 2/cellubrevin double KO mice and cellubrevin KO. G, Summary graph of synaptic responses to hypertonic sucrose application monitored in WT and various single and double KO neurons. Abbreviations used are the same as in A. Responses are calculated as the cumulative charge transfer integrated over 1 s interval at the peak of the response. Data shown are means ± SEMs (n = 8 for synaptobrevin 2 KO and WT; n = 7 for SC double KO; n = 5 for cellubrevin KO).
The Q/R substitution in the synaptobrevin SNARE motif does not impair neurotransmitter release
SNARE complex assembly requires the interaction of the three Q- and one R-SNARE motif, with the Q-SNAREs contributing glutamines and the R-SNARE contributing arginine to the central ionic zero layer of the complex. To test the functional significance of this interaction in the synapse, we infected synapses with synaptobrevin 2 mutants with a glutamine instead of an arginine in the central layer (R56Q), thus converting synaptobrevin 2 from an R-SNARE to a Q-SNARE (Fig. 4A). Expression of “Q-synaptobrevin” in synaptobrevin-deficient neurons fully rescued synaptic transmission, including spontaneous neurotransmitter release, release evoked in response to hypertonic sucrose application, and release evoked by APs (Fig. 4) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Thus, at this level of analysis, the zero layer Q/R interactions among SNARE proteins are not essential for SNARE function.
Effect of 12- and 24-residue insertions between the SNARE motif and transmembrane region of synaptobrevin
If SNARE complexes force membranes into close proximity, the distance between the SNARE motif and transmembrane region would be expected to be critical for fusion. To test this hypothesis, we examined whether insertions of 12 or 24 residues between the SNARE motif and transmembrane region of synaptobrevin impairs its ability to mediate synaptic vesicle exocytosis (Fig. 5A).
Figure 5.
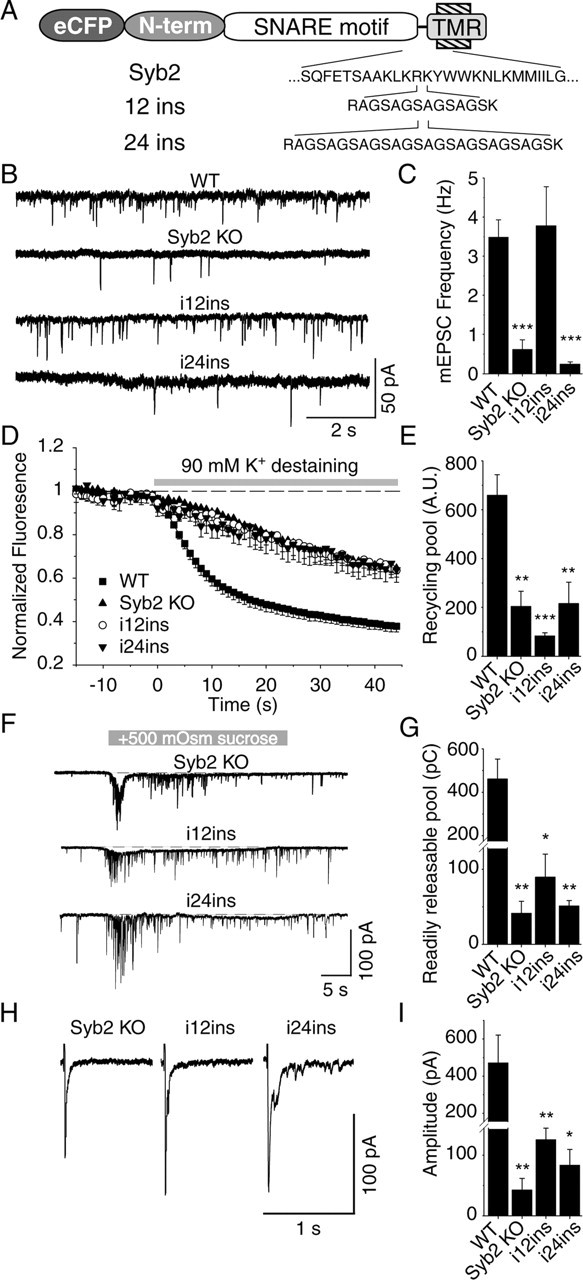
Membrane proximity is not an absolute requirement for spontaneous fusion. A, Diagram of constructs used for infection. B, Representative traces of spontaneous synaptic events recorded from a cell infected with 12- and 24-insertion mutants of synaptobrevin 2. C, Frequency of spontaneous synaptic events (n = 15 12-inserted infected KO cells; n = 10 24-insertion infected KO cells). Note that infection with 12 insertion increased the frequency of spontaneous synaptic events to the same level as WT. D, Average destaining curves of FM2-10-loaded synapses during high potassium stimulation. E, Bar graphs depict size of recycling pool as mean ± SEM for WT (n = 16, 1146 synapses), synaptobrevin 2 KO (n = 11, 688 synapses) synapses, and KO synapses after infection with 12 insertion (n = 5, 148 synapses) and 24 insertion (n = 4, 135 synapses). F, Representative traces of synaptic responses to sucrose monitored in infected neurons. G, Responses are calculated as the cumulative charge transfer integrated over 1 s interval at the peak of the response [n = 4 for WT and synaptobrevin 2 KO cells; n = 7 for 12-insertion mutant of synaptobrevin (12ins.) infected; n = 3 for 24-insertion (24ins.) infected KO cells]. H, Representative traces from whole-cell recordings during 1 Hz field stimulations. I, Amplitudes of evoked responses (n = 7 WT and synaptobrevin 2 KO cells; = 15 12-inserted infected KO cells; n = 9 24-inserted infected KO cells). Note that infection with 12 insertion significantly increased the amplitude of evoked responses (p < 0.05) in KO culture but that remained far less than in WT (p < 0.005).
We found that synaptobrevin carrying a 12-residue insertion between SNARE motif and transmembrane region fully rescued the decline in spontaneous release in synaptobrevin-deficient neurons (Fig. 5B,C). Strikingly, however, this mutant of synaptobrevin was unable to reverse the impairment of vesicle recycling observed in synaptobrevin-deficient neurons (Fig. 5D,E), rescue the decrease in hypertonic sucrose-evoked release (Fig. 5F,G), or fully restore evoked release (Fig. 5H,I) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). In contrast to synaptobrevin 2 with a 12-residue insertion, synaptobrevin with a 24-residue insertion was unable to rescue any fusion in synaptobrevin-deficient neurons, including spontaneous fusion (Fig. 5B–I). Direct comparison of the amount of release in synaptobrevin-deficient control synapses with synaptobrevin-deficient synapses expressing exogenous synaptobrevins containing a 12-residue or 24-residue insertion showed that the 12-residue insertion was not completely inactive but approximately doubled the amount of residual evoked release in synaptobrevin-deficient neurons (Fig. 5G,I) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). These findings suggest that the energetic constraints on spontaneous and evoked release differ and support a direct, energetic function of the SNARE complex in forcing the synaptic vesicle and the plasma membrane to fuse during evoked exocytosis. The small rescue effect of the 12-insertion mutant of synaptobrevin in evoked release indicates that the distance of the SNARE motif from the transmembrane region is barely too long in this mutant to still allow efficient fusion.
In a parallel set of experiments, we biochemically tested whether the synaptobrevin 2 with a 12-residue insertion (as well as the wild-type synaptobrevin 2) could participate in SNARE complex assembly. Lysates from hippocampal cultures infected with synaptobrevin 2 and the 12-insertion mutant (Fig. 6A) were incubated with a polyclonal antibody against rat syntaxin 1 residues 1–264, which recognizes assembled SNARE complexes (O.-H. Shin and T. C. Südhof, unpublished observations). The resulting immunoprecipitate was immunoblotted with antibodies against GFP, which recognizes the infected synaptobrevin 2 constructs (because an antibody raised against the N terminus of synaptobrevin 2 does not recognize these constructs because of the eCFP tag) as well as an antibody against SNAP-25 and a monoclonal antibody against syntaxin 1 (Fig. 6B). These immunoblots clearly documented that infected wild-type synaptobrevin 2 and synaptobrevin 2 with a 12-residue insertion could form SNARE complexes. This finding supports the above premise that the distance of the SNARE motif from the transmembrane region is a major determining factor for the ineffectiveness of this mutant in rescuing evoked fusion.
Figure 6.
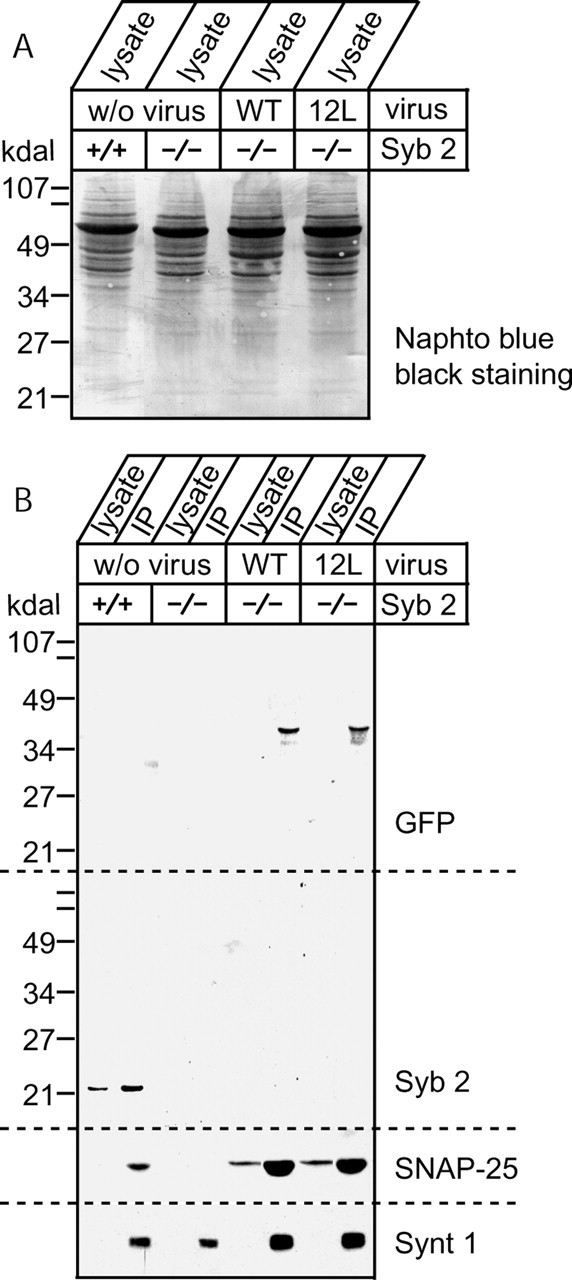
Infected wild-type and 12-insertion mutant of synaptobrevin 2 engage in assembled SNARE complexes. A, Cells from each six-well plate were harvested using 1 ml of buffer, and proteins were extracted using 1% Triton X-100. A total of 50 μl of cell lysate was used for SDS-PAGE and Naphto blue black staining. B, A total of 10 μl of rabbit serum raised against rat syntaxin 1 (U6251) and 20 μl of protein A-Sepharose beads were added to 1% Triton X-100 cell lysate expressing mutant synaptobrevin 2 to immunoprecipitate (IP) assembled SNARE complexes. After incubation, beads were washed five times, and proteins coimmunoprecipitated with syntaxin 1 were determined by Western blotting. Shown are Western blots of proteins coimmunoprecipitated with syntaxin 1. Blots were performed using the following sera or ascites: mouse anti-GFP, C163 from Zymed; mouse anti-Syb 2, CL69.1; mouse anti-syntaxin 1, HPC-1; and mouse anti-SNAP-25, CL71.1.
Functionally inactive synaptobrevin acts as a dominant negative in wild-type neurons
In a final set of experiments, we tested whether the synaptobrevin containing a 24-residue insertion that is unable to rescue the synaptobrevin knock-out phenotype alters synaptic vesicle exocytosis in wild-type synapses. When we monitored synaptic vesicle exocytosis and pool sizes in wild-type synapses expressing this construct using FM2-10 staining and destaining experiments, we found that exocytosis was severely depressed and the recycling vesicle pool size was decreased (Fig. 7). In contrast, wild-type synapses expressing either wild-type synaptobrevin or the mutant Q-synaptobrevin (both of which fully rescued the synaptobrevin deficiency phenotype in synapses) experienced no decline in fluorescent FM2-10 dye uptake and release, suggesting that only the synaptobrevin mutant that is inactive in exocytosis is dominant negative in inhibiting exocytosis. This observation further underlies the importance of the distance between the synaptobrevin SNARE motif and the transmembrane domain for SNARE core complex function in fusion.
Figure 7.

Dominant-negative action of the 24-insertion construct. A, High potassium induced destaining curves of FM2-10-loaded wild-type synapses infected with eCFP-tagged synaptobrevin2 (iwSyb2), the 24 insertion (iw24ins), and R56Q (iwR56Q) mutants. For comparison, average destaining curves from synaptobrevin 2 KO and wild-type neurons are also shown. B, Bar graphs depict the size of the recycling pool as mean ± SEM for WT (n = 7, 400 synapses), synaptobrevin 2 KO (n = 6, 364 synapses) neurons, and wild-type synapses after infection (iw24 ins, n = 5, 391 synapses; iwSyb2, n = 4, 409 synapses; iwR56Q, n = 3, 273 synapses).
Discussion
In this study, we used rescue experiments to examine how synaptobrevin acts in synaptic vesicle fusion. For this purpose, we tested mutant synaptobrevins and the closely related R-SNARE cellubrevin and used cultured hippocampal neurons from synaptobrevin-deficient mice in which we produced different proteins using a lentivirus expression system. We made five principal observations.
Fusion of synaptobrevin with GFP derivatives at either the N or the C terminus did not impair the ability of synaptobrevin to rescue synaptic neurotransmitter release in synaptobrevin-deficient synapses. This is significant because, in synaptotagmin 1, cytoplasmic C-terminal GFP fusions abolish function (Han et al., 2005). The fact that synaptobrevin function tolerates addition of a bulky GFP moiety at the N terminus close to the SNARE motif indicates that the N terminus in the SNARE complex is not present in a spatially constrained environment.
Cellubrevin expression successfully rescued spontaneous and evoked synaptic vesicle fusion as well as efficient synaptic vesicle recycling. In contrast, double knock-out of cellubrevin and synaptobrevin did not exhibit additional reduction in fusion efficacy below the reduction seen in synaptobrevin knock-out alone. These data show that, although synaptobrevin and cellubrevin are functionally interchangeable, the remaining fusion in synaptobrevin-deficient synapses is not mediated by cellubrevin.
Exchanging the arginine in the zero layer of the SNARE motif to glutamine (R56Q) in synaptobrevin 2 still resulted in full rescue of release; therefore, the four-Q combination is a functional core complex alternative.
Insertion of 12 and 24 residues between the SNARE motif and transmembrane region of synaptobrevin 2 severely impaired the ability of synaptobrevin to mediate evoked release, demonstrating that the physical distance between the SNARE motif and transmembrane region is critical for fusion. Interestingly, however, synaptobrevin containing the 12-residue insertion completely rescued spontaneous release, although evoked release was not recovered. This result suggests that the fusion reactions mediating evoked and spontaneous release differ.
Remarkably, synaptobrevin mutant with the insertion of 24 residues between the SNARE motif and the transmembrane region that was unable to rescue synaptic transmission in synaptobrevin-deficient synapses suppressed synaptic vesicle exocytosis in wild-type synapses.
Substitution of synaptobrevin function by cellubrevin
SNAREs were suggested as candidates for the target specificity of vesicular transport (Sollner et al., 1993). Later findings, however, challenged this view. Transgenic rescue experiments in Drosophila (Bhattacharya et al., 2002) argued for a functional interchangeability between nonsynaptic and synaptic synaptobrevin isoforms in Drosophila. Recently, cellubrevin was shown to be responsible for the remaining release observed in synaptobrevin-deficient chromaffin cells (Borisovska et al., 2005). We observed complete rescue of synaptic neurotransmitter release when cellubrevin was expressed in synaptobrevin-deficient neurons (Fig. 2). At the same time, we found that additional deletion of cellubrevin in synaptobrevin-deficient neurons did not further aggravate the impairment in exocytosis observed in these neurons (Fig. 3), consistent with our previous finding that cellubrevin is absent from hippocampal neurons (Schoch et al., 2001). We conclude that there is clear redundancy between the cellubrevin and synaptobrevin R-SNAREs role in fusion, despite their differences in N-terminal and C-terminal sequences, suggesting that these isoforms evolved as a means of diversifying expression patterns but not of diversifying SNARE functions.
The “four Q configuration” in the zero layer of the SNARE motif is functional
The lack of a major phenotype in synaptic exocytosis produced by the Q/R substitution agrees well with previous studies in yeast showing that similar substitutions did not abolish SNARE function (Katz and Brennwald, 2000; Ossig et al., 2000). Previous experiments in yeast, however, did not have the temporal resolution of synaptic exocytosis to detect changes in the kinetics or amplitude of exocytosis. Our present data thus extend the previous studies by showing that the four-Q SNARE complex is not only functional but appears to act with a similar kinetics in exocytosis as the three-Q/one-R SNARE complex. This is surprising given the tight interaction of the glutamines and arginine in the zero layer of the SNARE complex revealed in the crystal structure of the complex (Sutton et al., 1998). Recent experiments with different R–Q mutations in the zero layer of the yeast SNARE proteins ykt6 and sec22 found a developmental arrest in the mutants (Dilcher et al., 2001; Graf et al., 2005). These previous findings suggest that a subtle change in the SNARE function can lead to a cumulative defect in a longer time frame. These defects would be not detectable in relatively acute experimental settings such as ours. Furthermore, our system may not be optimal to detect the consequences of R-to-Q substitution in layer zero in exocytosis because synaptic vesicle fusion is less sensitive to intracellular fusion steps within a short time frame.
Differential requirement of membrane proximity in Ca2+-evoked and spontaneous synaptic release
It is assumed that the assembly of SNARE complexes provides the energy for membrane fusion (Rothman, 1994; Rothman and Warren, 1994). Our result with the 12- and 24-residue insertion motif constructs provides support for the notion that SNARE complex assembly drives approximation of membranes close together. Moreover, the observation that the 24-residue insertion mutant of synaptobrevin impaired fusion in wild-type synapses suggests that any inactive synaptobrevin, when expressed in a wild-type synapse, may impair synaptobrevin function. This result provides a possible explanation for the inactivation of synaptic exocytosis in wild-type synapses in which a synaptobrevin fusion protein is induced to multimerize by addition of a chemical dimerizer (Karpova et al., 2005). It is likely that, under these conditions, the multimerized synaptobrevin is inactivated, thereby having the same effect as the 24-residue insertion mutant described here.
Arguably the most striking finding of our experiments is that separating the SNARE motif and transmembrane region of synaptobrevin by sequence insertions has distinct effects on evoked and spontaneous fusion. The dramatic effect of the 12-residue insertion on evoked exocytosis confirms, to our knowledge for the first time for a physiological fusion reaction followed in real time, that tight coupling of the SNARE complex to the transmembrane region is essential for exocytosis. Curiously, however, the same insertion had no effect on the ability of synaptobrevin to rescue spontaneous release (Fig. 5B,C). This finding argues against the traditional notion that spontaneous release events arise from the random low-probability exocytosis of docked and fully primed vesicles in the readily releasable pool. According to this view, spontaneous fusion should possess the same structural requirements as evoked fusion. Our finding cannot be ascribed to a selective effect of synaptobrevin 2 in Ca2+ dependence of evoked fusion (Sakaba et al., 2005; Young, 2005) because synaptobrevin carrying the 12-residue insertion was also essentially unable to rescue hypertonic sucrose-evoked fusion, which is Ca2+ independent (Rosenmund and Stevens, 1996).
Our findings are consistent with the recent observation that the spontaneous neurotransmission originates, at least in part, from a different vesicle population than evoked neurotransmission (Sara et al., 2005). This recent study raised the question whether the release machinery for spontaneous fusion is identical to the one required for evoked fusion. Clearly, both forms of fusion are impaired in the absence of the same SNARE molecules, albeit to different degrees (Schoch et al., 2001; Washbourne et al., 2002). Together with the previous observations from the SNARE mutant studies, spontaneous fusion may require an alternative fusion complex (with a different R-SNARE) or the same complex formed with less stringency (Xu et al., 1999; Melia et al., 2002). Stronger dependency of evoked release on the close proximity of membranes is also more compatible with its strict Ca2+ dependence. Paradoxically, the mechanism of evoked release appears to be closely related to the evolutionally conserved exocytosis because yeast vacuolar fusion was similarly strictly dependent on membrane proximity and the length of R-SNAREs (McNew et al., 1999; Wang et al., 2001).
Footnotes
This work was supported by the National Institutes of Mental Health Grant MH066198 (E.T.K) and the Howard Hughes Medical Institute (F.D., T.C.S.). We are grateful to J. Pessin (Stony Brook, New York) for providing the cellubrevin knock-out animals. We thank, for the excellent technical assistance, Andrea Roth, Iza Kornblum, and Ewa Borowicz, and Nicky Hamlin and Jason Mitchell for the help with mouse husbandry. We thank the help and advice of Weiping Han, Peter Bronk, and Wen-Pin Chang with the lentivirus production.
References
- Bhattacharya S, Stewart BA, Niemeyer BA, Burgess RW, McCabe BD, Lin P, Boulianne G, O'Kane CJ, Schwarz TL (2002). Members of the synaptobrevin/vesicle-associated membrane protein (VAMP) family in Drosophila are functionally interchangeable in vivo for neurotransmitter release and cell viability. Proc Natl Acad Sci USA 99:13867–13872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borisovska M, Zhao Y, Tsytsyura Y, Glyvuk N, Takamori S, Matti U, Rettig J, Sudhof T, Bruns D (2005). v-SNAREs control exocytosis of vesicles from priming to fusion. EMBO J 24:2114–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deák F, Schoch S, Liu X, Sudhof TC, Kavalali ET (2004). Synaptobrevin is essential for fast synaptic-vesicle endocytosis. Nat Cell Biol 6:1102–1108. [DOI] [PubMed] [Google Scholar]
- Dilcher M, Kohler B, von Mollard GF (2001). Genetic interactions with the yeast Q-SNARE VTI1 reveal novel functions for the R-SNARE YKT6. J Biol Chem 276:34537–34544. [DOI] [PubMed] [Google Scholar]
- Dittgen T, Nimmerjahn A, Komai S, Licznerski P, Waters J, Margrie TW, Helmchen F, Denk W, Brecht M, Osten P (2004). Lentivirus-based genetic manipulations of cortical neurons and their optical and electrophysiological monitoring in vivo. Proc Natl Acad Sci USA 101:18206–18211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasshauer D, Sutton RB, Brunger AT, Jahn R (1998). Conserved structural features of the synaptic fusion complex: SNARE proteins reclassified as Q- and R-SNAREs. Proc Natl Acad Sci USA 95:15781–15786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf CT, Riedel D, Schmitt HD, Jahn R (2005). Identification of functionally interacting SNAREs by using complementary substitutions in the conserved “0” layer. Mol Biol Cell 16:2263–2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han W, Rhee JS, Maximov A, Lin W, Hammer RE, Rosenmund C, Sudhof TC (2005). C-terminal ECFP fusion impairs synaptotagmin 1 function: crowding out synaptotagmin 1. J Biol Chem 280:5089–5100. [DOI] [PubMed] [Google Scholar]
- Hanson PI, Roth R, Morisaki H, Jahn R, Heuser JE (1997). Structure and conformational changes in NSF and its membrane receptor complexes visualized by quick-freeze/deep-etch electron microscopy. Cell 90:523–535. [DOI] [PubMed] [Google Scholar]
- Jahn R, Lang T, Sudhof TC (2003). Membrane fusion. Cell 112:519–533. [DOI] [PubMed] [Google Scholar]
- Karpova AY, Tervo DGR, Gray NW, Svoboda K (2005). Rapid and reversible chemical inactivation of synaptic transmission in genetically targeted neurons. Neuron 48:727–735. [DOI] [PubMed] [Google Scholar]
- Katz L, Brennwald P (2000). Testing the 3Q:1R “rule”: mutational analysis of the ionic “zero” layer in the yeast exocytic SNARE complex reveals no requirement for arginine. Mol Biol Cell 11:3849–3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link E, Edelmann L, Chou JH, Binz T, Yamasaki S, Eisel U, Baumert M, Sudhof TC, Niemann H, Jahn R (1992). Tetanus toxin action: inhibition of neurotransmitter release linked to synaptobrevin proteolysis. Biochem Biophys Res Commun 189:1017–1023. [DOI] [PubMed] [Google Scholar]
- McMahon HT, Ushkaryov YA, Edelmann L, Link E, Binz T, Niemann H, Jahn R, Sudhof TC (1993). Cellubrevin is a ubiquitous tetanus-toxin substrate homologous to a putative synaptic vesicle fusion protein. Nature 364:346–349. [DOI] [PubMed] [Google Scholar]
- McNew JA, Weber T, Engelman DM, Sollner TH, Rothman JE (1999). The length of the flexible SNAREpin juxtamembrane region is a critical determinant of SNARE-dependent fusion. Mol Cell 4:415–421. [DOI] [PubMed] [Google Scholar]
- Melia TJ, Weber T, McNew JA, Fisher LE, Johnston RJ, Parlati F, Mahal LK, Sollner TH, Rothman JE (2002). Regulation of membrane fusion by the membrane-proximal coil of the t-SNARE during zippering of SNAREpins. J Cell Biol 158:929–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossig R, Schmitt HD, de Groot B, Riedel D, Keranen S, Ronne H, Grubmuller H, Jahn R (2000). Exocytosis requires asymmetry in the central layer of the SNARE complex. EMBO J 19:6000–6010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parlati F, McNew JA, Fukuda R, Miller R, Sollner TH, Rothman JE (2000). Topological restriction of SNARE-dependent membrane fusion. Nature 407:194–198. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF (1996). Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron 16:1197–1207. [DOI] [PubMed] [Google Scholar]
- Rothman JE (1994). Mechanisms of intracellular protein transport. Nature 372:55–63. [DOI] [PubMed] [Google Scholar]
- Rothman JE, Warren G (1994). Implications of the SNARE hypothesis for intracellular membrane topology and dynamics. Curr Biol 4:220–233. [DOI] [PubMed] [Google Scholar]
- Sakaba T, Stein A, Jahn R, Neher E (2005). Distinct kinetic changes in neurotransmitter release after SNARE protein cleavage. Science 309:491–494. [DOI] [PubMed] [Google Scholar]
- Sara Y, Virmani T, Deák F, Liu X, Kavalali ET (2005). An isolated pool of vesicles recycles at rest and drives spontaneous neurotransmission. Neuron 45:563–573. [DOI] [PubMed] [Google Scholar]
- Schiavo G, Benfenati F, Poulain B, Rossetto O, Polverino de Laureto P, DasGupta BR, Montecucco C (1992). Tetanus and botulinum-B neurotoxins block neurotransmitter release by proteolytic cleavage of synaptobrevin. Nature 359:832–835. [DOI] [PubMed] [Google Scholar]
- Schoch S, Deák F, Konigstorfer A, Mozhayeva M, Sara Y, Sudhof TC, Kavalali ET (2001). SNARE function analyzed in synaptobrevin/VAMP knockout mice. Science 294:1117–1122. [DOI] [PubMed] [Google Scholar]
- Sollner T, Whiteheart SW, Brunner M, Erdjument-Bromage H, Geromanos S, Tempst P, Rothman JE (1993). SNAP receptors implicated in vesicle targeting and fusion. Nature 362:318–324. [DOI] [PubMed] [Google Scholar]
- Sorensen JB, Nagy G, Varoqueaux F, Nehring RB, Brose N, Wilson MC, Neher E (2003). Differential control of the releasable vesicle pools by SNAP-25 splice variants and SNAP-23. Cell 114:75–86. [DOI] [PubMed] [Google Scholar]
- Sutton RB, Fasshauer D, Jahn R, Brunger AT (1998). Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature 395:347–353. [DOI] [PubMed] [Google Scholar]
- Wang Y, Dulubova I, Rizo J, Südhof TC (2001). Functional analysis of conserved structural elements in yeast syntaxin Vam3p. J Biol Chem 276:28598–28605. [DOI] [PubMed] [Google Scholar]
- Washbourne P, Thompson PM, Carta M, Costa ET, Mathews JR, Lopez-Bendito G, Molnar Z, Becher MW, Valenzuela CF, Partridge LD, Wilson MC (2002). Genetic ablation of the t-SNARE SNAP-25 distinguishes mechanisms of neuroexocytosis. Nat Neurosci 5:19–26. [DOI] [PubMed] [Google Scholar]
- Weber T, Zemelman BV, McNew JA, Westermann B, Gmachl M, Parlati F, Sollner TH, Rothman JE (1998). SNAREpins: minimal machinery for membrane fusion. Cell 92:759–772. [DOI] [PubMed] [Google Scholar]
- Xu T, Rammner B, Margittai M, Artalejo AR, Neher E, Jahn R (1999). Inhibition of SNARE complex assembly differentially affects kinetic components of exocytosis. Cell 99:713–722. [DOI] [PubMed] [Google Scholar]
- Yang C, Mora S, Ryder JW, Coker KJ, Hansen P, Allen LA, Pessin JE (2001). VAMP3 null mice display normal constitutive, insulin- and exercise-regulated vesicle trafficking. Mol Cell Biol 21:1573–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young SM Jr (2005). Proteolysis of SNARE proteins alters facilitation and depression in a specific way. Proc Natl Acad Sci USA 102:2614–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]