

Abstract
Multiple sclerosis is an inflammatory, neurodegenerative disease for which experimental autoimmune encephalomyelitis (EAE) is a model. Treatments with estrogens have been shown to decrease the severity of EAE through anti-inflammatory mechanisms. Here we investigated whether treatment with an estrogen receptor α (ERα) ligand could recapitulate the estrogen-mediated protection in clinical EAE. We then went on to examine both anti-inflammatory and neuroprotective mechanisms. EAE was induced in wild-type, ERα-, or ERβ-deficient mice, and each was treated with the highly selective ERα agonist, propyl pyrazole triol, to determine the effect on clinical outcomes, as well as on inflammatory and neurodegenerative changes. ERα ligand treatment ameliorated clinical disease in both wild-type and ERβ knock-out mice, but not in ERα knock-out mice, thereby demonstrating that the ERα ligand maintained ERα selectivity in vivo during disease. ERα ligand treatment also induced favorable changes in autoantigen-specific cytokine production in the peripheral immune system [decreased TNFα, interferon-γ, and interleukin-6, with increased interleukin-5] and decreased CNS white matter inflammation and demyelination. Interestingly, decreased neuronal staining [NeuN+ (neuronal-specific nuclear protein)/β3-tubulin+/Nissl], accompanied by increased immunolabeling of microglial/monocyte (Mac 3+) cells surrounding these abnormal neurons, was observed in gray matter of spinal cords of EAE mice at the earliest stage of clinical disease, 1–2 d after the onset of clinical signs. Treatment with either estradiol or the ERα ligand significantly reduced this gray matter pathology. In conclusion, treatment with an ERα ligand is highly selective in vivo, mediating both anti-inflammatory and neuroprotective effects in EAE.
Keywords: multiple sclerosis, experimental autoimmune encephalomyelitis, estrogen, cytokines, neuron, microglia
Introduction
In the most widely used animal model for multiple sclerosis (MS), experimental autoimmune encephalomyelitis (EAE), both estriol and estradiol (E2) were shown previously to be protective in both active and adoptive EAE in several strains of mice (Jansson et al., 1994; Kim et al., 1999; Bebo et al., 2001; Liu et al., 2003; Polanczyk et al., 2003; Palaszynski et al., 2004). Numerous anti-inflammatory mechanisms appear to underlie the protective effect of estrogen in EAE including downregulation of myelin protein-specific T helper 1 (Th1) immune responses, increased Th2 responses, decreased antigen presentation by dendritic cells, induction of CD4+CD25+ regulatory T cells and downregulation of chemokines in the CNS, all resulting in decreased inflammatory lesions in the CNS (Kim et al., 1999; Bebo et al., 2001; Ito et al., 2001; Liu et al., 2002; Zang et al., 2002; Liu et al., 2003; Subramanian et al., 2003; Xiao et al., 2004). Although a variety of anti-inflammatory mechanisms of estrogen treatment in EAE have been described, it is unknown whether estrogen treatment may be neuroprotective in EAE. A neuroprotective role of estrogens in EAE is possible because estrogens have been shown to be neuroprotective in a variety of animal models of neurodegenerative disease including Parkinson’s disease, spinal cord injury, cerebellar ataxia, Down’s syndrome, epilepsy, and some models of stroke and Alzheimer’s disease (Leranth et al., 2000; Veliskova et al., 2000; Dubal et al., 2001; Wise et al., 2001; Jover et al., 2002; Granholm et al., 2003; Rau et al., 2003; Sierra et al., 2003; Sribnick et al., 2003, 2005; Heikkinen et al., 2004; Green et al., 2005; van Groen and Kadish, 2005).
The actions of estrogen are mediated primarily by nuclear estrogen receptors ERα and ERβ, although nongenomic membrane effects have also been described previously (Weiss and Gurpide, 1988). Originally it was thought that ERα and ERβ would each have distinct tissue distributions, thereby providing a means through which use of selective estrogen receptor modifiers (SERMs) could improve tissue selectivity. However, the relationship between ERα and ERβ became complex, with most tissues expressing some detectable level of each of these receptors (Kuiper et al., 1998). The two receptors at times did, and at other times did not, colocalize to the same cells within a given tissue (Kuiper et al., 1997; Enmark and Gustafsson, 1999). Furthermore, in some tissues the two receptors were shown to act synergistically, whereas in other tissues they acted antagonistically. These tissue-specific differences in biologic outcomes are thought to be attributable in part to tissue-specific differences in transcription factors, which become activated after binding of each ER by ligand (Paech et al., 1997; Nilsson et al., 2001; Shang and Brown, 2002).
Our group and another have each used estrogen receptor knock-out (KO) mice to show that the protective effect of estrogen treatment (estradiol and estriol) in EAE is dependent on the presence of ERα (Liu et al., 2003; Polanczyk et al., 2003). However, whether targeted stimulation of ERα is both necessary and sufficient for the estrogen-mediated protection in EAE remains unknown. ERβ may act synergistically or antagonistically with ERα, in either the CNS or in the immune system, during estrogen treatment of EAE.
The purpose of this study was to use a highly selective ERα ligand, propyl pyrazole triol (PPT) (Harrington et al., 2003), to determine whether stimulation of ERα is sufficient for the estrogen-mediated protection in EAE. We report that treatment with this ERα-selective ligand is indeed sufficient for protection in EAE, mediating both anti-inflammatory and neuroprotective effects. This is the first report that treatment with an estrogen, or an ERα-selective ligand, is neuroprotective in EAE.
Materials and Methods
Animals.
Female C57BL/6 mice, 8 weeks of age, were purchased from Taconic (Germantown, NY). ERα KO mice backcrossed onto the C57BL/6 background for 16 generations were a generous gift from Dr. Dennis Lubahn (University of Missouri, Columbia, MO) (Lubahn et al., 1993). Wild-type littermates from F16 crosses served as ERα KO matched controls. ERβ KO mice, a generous gift from Dr. Jan Ake Gustafsson (Karolinska Institute, Stockholm, Sweden) (Krege et al., 1998), were backcrossed onto the C57BL/6 background for eight generations. Wild-type littermates from these crosses served as ERβ KO matched controls. Animals were housed under guidelines set by the National Institutes of Health, and experiments were conducted in accordance with the University of California, Los Angeles Chancellor’s Animal Research Committee and the Public Health Service Policy on Humane Care and Use of Laboratory Animals.
Reagents.
PPT was purchased from Tocris Bioscience (Ellisville, MO), and E2 was purchased from Sigma-Aldrich (St. Louis, MO). Miglyol 812 N, a thin liquid oil, was obtained from Sasol North America (Houston, TX). Myelin oligodendrocyte glycoprotein (MOG) peptide, amino acids 35–55, was synthesized to >98% purity by Chiron Mimotopes (San Diego, CA).
EAE.
Active EAE induction ensued with subcutaneous injection of an emulsion containing the autoantigen MOG peptide, amino acids 35–55 (300 μg/mouse) and Myobacterium tuberculosis (500 μg/mouse) in complete Freund’s adjuvant, as described previously (Suen et al., 1997; Liu et al., 2003). Mice underwent hormonal treatments as described below and were monitored daily for EAE disease severity using the standard EAE grading scale, as described previously (Pettinelli and McFarlin, 1981). Briefly, to determine the clinical score for each mouse on each day, each mouse was graded using the standard 0–5 scale: 0, unaffected; 1, tail limpness; 2, failure to right on attempt to roll over; 3, partial paralysis; 4, complete paralysis; and 5, moribund. On each day, the mean of the clinical scores of all mice within a given treatment group were determined, thereby yielding the mean clinical score for that treatment group. Some mice were followed clinically for up to 40 d after disease induction, and others were killed earlier for mechanistic studies, 1–2 d after the onset of clinical signs in the vehicle-treated group (day 16–19 after disease induction).
Treatments.
Isoflurane-anesthetized female mice were ovariectomized and allowed to recuperate for 10 d. Daily treatments of oil vehicle alone, estradiol, or PPT began 7 d before EAE immunization. Estradiol and PPT were dissolved in 10% ethanol and 90% oil to give the final proper concentration of 0.04 mg/kg/d estradiol (Jansson et al., 1994) and 10 mg/kg/d PPT per mouse (Harris et al., 2002). Estradiol, PPT or vehicle alone were given by daily subcutaneous injections along the midbackline and continued for the entire disease duration (up to 40 d after disease induction).
Perfusion.
Mice were deeply anesthetized with isoflurane and perfused transcardially with ice-cold 0.9% saline, followed by 10% formalin. Spinal cord columns were removed and postfixed overnight in 10% formalin and cryoprotected with 20% sucrose solution in PBS. Spinal cords were removed from the column, cut in three parts (cervical, thoracic, and lumbar), and embedded in gelatin/sucrose mix. Spinal cord regions in gelatin were further postfixed and stored in 20% sucrose. Free-floating sections (25 μm thick) were cut coronally with a sliding microtome and collected serially in PBS.
Uterine weights.
After the mice were killed, each uterus was extracted, and the fat, connective tissue, and excess fluid were removed to obtain each uterine weight, as described previously (Frasor et al., 2003).
Immune responses.
Spleens were harvested during deep anesthesia before perfusion. Splenocytes were stimulated with the autoantigen, MOG peptide 35–55, at 25 μg/ml. Supernatants were collected after 48 and 72 h, and levels of TNFα, interferon-γ (IFNγ), interleukin-6 (IL6), and IL5 were determined by cytometric bead array (BD Biosciences Pharmingen, San Diego, CA) as described previously (Liu et al., 2003).
Histopathology and immunohistochemistry.
Serial sections were mounted on slides and stained with hematoxylin and eosin (H&E), Nissl, or Luxol fast blue (LFB)–cresyl violet. Consecutive sections were also examined by immunohistochemistry. Briefly, 25 μm free-floating sections were permeabilized in 0.3% Triton X-100 in PBS and blocked with 10% normal goat serum. White matter immunostaining was enhanced by treating sections with 95% ethanol/5% acetic acid for 15 min before permeabilization and blocking. To detect specific cell types and structures, sections were preincubated with primary antibodies in PBS solution containing 2% NGS for 2 h at room temperature, and then overnight at 4°C. The following primary antibodies were used: anti-β3 tubulin and anti-neurofilament-NF200 [monoclonal (Chemicon, Temecula, CA); polyclonal (Sigma Biochemical)], anti-neuronal-specific nuclear protein (NeuN), anti-CD45 (Chemicon), anti-myelin basic protein (MBP; Chemicon) and anti-Mac 3 (BD Biosciences Pharmingen). The second antibody step was performed by labeling with antibodies conjugated to TRITC, FITC, and Cy5 [Vector Laboratories (Burlingame, CA) and Chemicon]. IgG control experiments were performed for all primary antibodies, and no staining was observed under these conditions. To assess the number of cells, a nuclear stain 4′,6′-diamidino-2-phenylindole dihydrochloride (DAPI; 2 ng/ml; Invitrogen, Eugene, OR) was added for 15 min before final washes after secondary antibody addition. The sections were mounted on slides, dried, and coverslipped in fluoromount G (Fisher Scientific, Hampton, NH).
Microscopy.
Stained sections were examined and photographed using a confocal microscope (TCS-SP; Leica, Mannheim, Germany) or a fluorescence microscope (BX51WI; Olympus, Tokyo, Japan) equipped with Plan Fluor objectives connected to a camera (DP70; Olympus). Digital images were collected and analyzed using Leica confocal and DP70 camera software. Images were assembled using Adobe Photoshop (Adobe Systems, San Jose, CA).
Quantification.
To quantify immunostaining results, sections from spinal cord levels T1–T5 were examined, six from each mouse, with n = 3 mice per treatment group, for a total of 18 sections per treatment group. Images were captured under microscope (4×, 10×, or 40×) using the DP70 Image software and a DP70 camera (both from Olympus). Identical light intensity and exposure times were applied to all photographs from each experimental set. Images from the same areas of spinal cord were compared (T1–T5) and were acquired separately from delineated whole gray and white matter regions. The middle region of the ventral horn was the focus for gray matter analysis, whereas the area lateral to the ventral horn was the focus for white matter analysis. Six gray matter and six white matter pictures were collected from the two sides of T1–T5 sections (100 μ m apart) from three animals in each treatment group. All images were converted to grayscale and then analyzed by density measurement with ImageJ version 1.29 (the Windows version of NIH Image), downloaded from http://rsb.info.nih.gov/ij. A fixed threshold range of 0–160 was chosen to highlight the staining signals in normal spinal cord sections, and the total area within this range was measured, averaged, and compared.
Increase in total number of infiltrating cells after induction of EAE was measured by density measurements of DAPI+ nuclei in the whole white matter. Neuronal cells were quantified by counting the NeuN+/β 3-tubulin+/DAPI+ cells per square millimeter in the whole gray matter. Both white and gray matter assessments occurred in the T1–T5 spinal cord sections. Laser-scanning confocal microscopic scans at 40× were performed on Mac 3+/β3-tubulin+ immunostained spinal cord sections corresponding to levels T1–T5 ventral horn. The results for each experimental condition were averaged from four unilateral levels per mouse (100 μm apart, three mice in each treatment group, total of 12 sections per treatment group) and were expressed as mean fold change compared with healthy matched controls.
Statistical analysis.
EAE disease severity was compared between groups using the Friedman test, histopathological changes were assessed using 1 × 4 ANOVAs, and uterine weights and cytokine levels were compared between treatment groups using Student’s t test, as described previously (Dalal et al., 1997).
Results
Treatment with an ERα ligand remains highly selective for ERα in vivo during EAE
We initially identified a dose of the ERα-selective ligand for use in our EAE experiments which could induce a known biological response on a control tissue (the uterus). Estrogen-induced increases in uterine weight had been shown previously to be mediated by ERα, and doses of the ERα ligand PPT needed for this in vivo treatment effect had been described (Frasor et al., 2003). When we gave daily subcutaneous injections of PPT, at a dose previously shown to increase uterine weight (10 mg/kg/d), we observed a significant increase in uterine weight in female C57BL/6 mice with EAE at day 40 after disease induction, Figure 1A. Sensitivity of this technique was shown by the decrease in uterine weight in ovariectomized compared with sham-operated, vehicle-treated mice. Treatment with injections of high doses of estradiol (to induce pregnancy levels in serum) served as a positive control, whereas treatment with injections of vehicle alone served as a negative control. To further demonstrate the in vivo selectivity of this dose of PPT, uterotrophic responses were also examined during PPT treatment of ERα or ERβ knock-out mice. Significant increases in uterine weight were observed in PPT-treated ERβ knock-out mice (Fig. 1B) but not in ERα knock-out mice (Fig. 1C). Together, these data demonstrated that our method of administration of the ERα ligand PPT induced an expected biological response in vivo on a positive control tissue.
Figure 1.
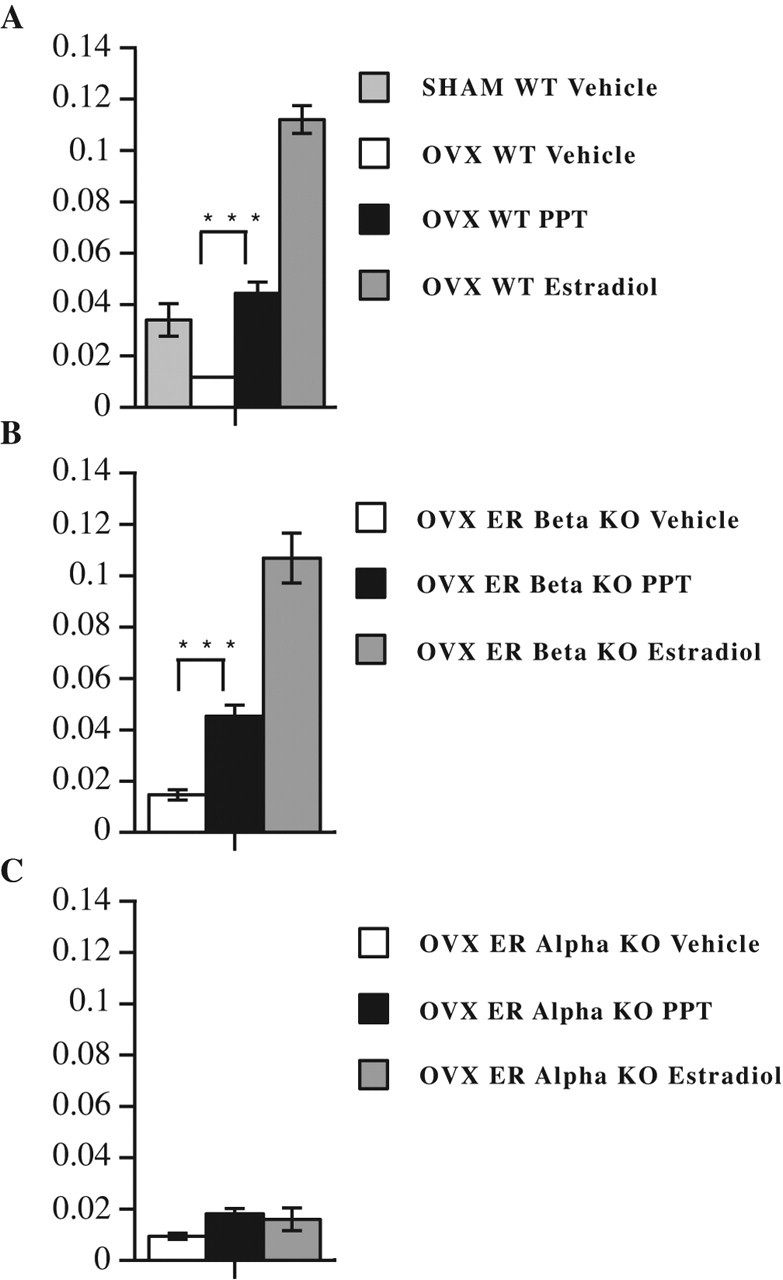
Treatment with an ERα-selective ligand is highly selective in vivo during EAE. A, Treatment with the ERα ligand PPT induced expected biological responses on uterine weight (y-axis = uterine weight in grams). Uterine weight was increased with PPT given as daily subcutaneous injections at 10 mg/kg/d. The decrease in uterine weight with ovariectomy compared with sham surgery demonstrated the sensitivity of the technique in detecting differences in uterine weights associated with differences in estrogen levels. Treatment with a dose of estradiol known to induce a late pregnancy level of estradiol was used as a positive control for an increase in uterine weight, whereas treatment with vehicle alone served as the negative control. The uteri were removed at day 35–40 during EAE treatment with the indicated hormone (sham vehicle, n = 6; OVX vehicle, n = 12; OVX estradiol, n = 18; OVX PPT, n = 18). OVX PPT and OVX Estradiol, each compared with OVX Vehicle, ∗∗∗p < 0.0001. WT, Wild type. B, Uterine weights were examined in ovariectomized ERβ knock-out mice as in A. Uterine weights were increased with PPT treatment in ERβ knock-out mice (OVX vehicle, n = 9; OVX estradiol, n = 12; OVX PPT, n = 12). OVX PPT and OVX Estradiol, each compared with OVX Vehicle, ∗∗∗p < 0.0001. C, Uterine weights were examined in ovariectomized ERα knock-out mice as in A. Uterine weights were not increased with PPT treatment in ERα knock-out mice (OVX vehicle, n = 6; OVX estradiol, n = 4; OVX PPT, n = 6).
Treatment with an ERα ligand reduces the clinical severity of EAE
Using the above dose and method of administration, PPT treatment was then assessed for its effect on the clinical course of EAE. Ovariectomized, C57BL/6 wild-type female mice with MOG 35–55 peptide-induced active EAE were treated with the ERα-selective ligand PPT. PPT treatment significantly reduced the clinical severity of EAE (Fig. 2A). Treatment with injections of estradiol served as a positive control, whereas treatment with injections of vehicle alone served as the negative control.
Figure 2.
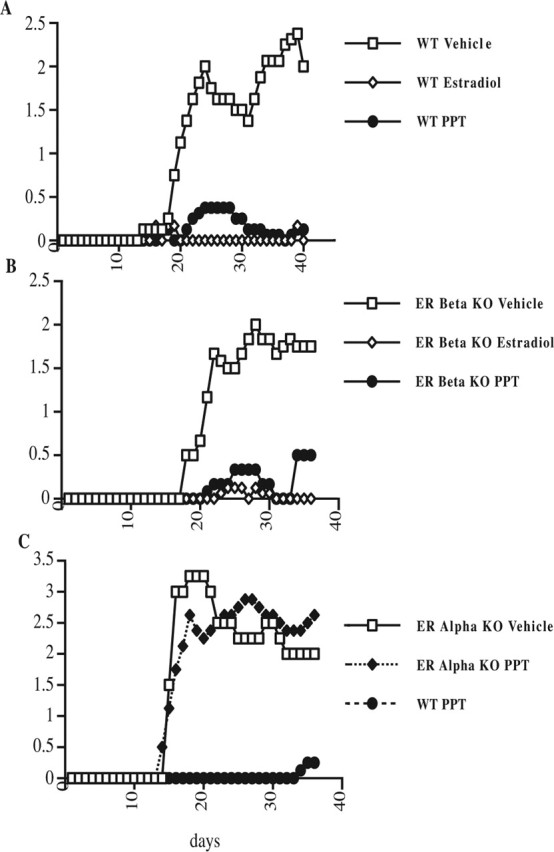
Treatment with an ERα-selective ligand is sufficient to reduce the clinical severity of EAE. A, EAE clinical severity was decreased in ovariectomized, wild-type (WT) C57BL/6 female mice treated with PPT. Daily treatments of ovariectomized mice with injections of vehicle (negative control), estradiol (positive control), or PPT (10 mg/kg/d) began, and then 7 d later, active EAE was induced with MOG 35–55 peptide. Mean clinical scores were significantly reduced in both estradiol- and PPT-treated mice compared with vehicle treated (p < 0.0001, Friedman test). Data are representative from experiments repeated a total of five times. B, The decrease in the mean clinical scores of EAE by PPT treatment was not dependent on the presence of ERβ. Ovariectomized, ERβ knock-out C57BL/6 female mice were treated with either PPT, estradiol, or vehicle as in A. Mean clinical scores were significantly reduced in both estradiol- and PPT-treated mice compared with vehicle treated (p < 0.0001, Friedman test). Data are representative from experiments repeated a total of three times. C, PPT treatment in vivo during EAE remains highly selective for ERα. Ovariectomized female ERα knock-out C57BL/6 mice were treated as in A. In ERα knock-out mice, mean clinical scores were not significantly different in PPT-treated compared with vehicle-treated. PPT-treated wild-type mice served as a positive control for a PPT treatment effect within the experiment. Data are representative from experiments repeated a total of three times. Error bars indicate variability of clinical scores between mice within a given treatment group. n = 5 mice per each treatment group.
We then addressed whether the selectivity of PPT remained specific for ERα during in vivo treatment of EAE, as opposed to being mediated in part through ERβ or nongenomic effects. When ovariectomized, ERβ knock-out C57BL/6 female mice were treated with PPT during active EAE, clinical disease severity was again significantly decreased (Fig. 2B). These data demonstrated that the presence of ERβ was not required for disease protection mediated by treatment with PPT. In contrast, when PPT was administered to ovariectomized ERα knock-out mice induced with active EAE, the disease-ameliorating effect of PPT treatment was abolished, as evidenced by the lack of a difference in mean clinical scores when comparing PPT-treated and vehicle-treated ERα knock-out mice (Fig. 2C). Similar results were obtained when castrated male mice were used instead of ovariectomized females (data not shown), consistent with a previous publication demonstrating that estrogen-mediated improvements in clinical EAE in castrated male mice were abrogated in the ERα knock-out (Liu et al., 2003). ERα knock-out female mice have high circulating estradiol levels; hence, estrogen unresponsiveness in this mouse could be attributable to the ERα genetic modification or the estrogen history of the mouse before ovariectomy at 4 weeks. Because male ERα knock-out mice do not have high circulating levels of estradiol, similar results in both the female and male ERα knock-outs make the ERα genetic modification, not the estrogen history of the mouse, most likely responsible for effects observed.
Together, these data demonstrated that the estrogen-mediated protection from EAE could be recapitulated by treatment with a highly selective ERα ligand, and that this protection was not dependent on an interaction with ERβ.
Treatment with an ERα ligand reduces autoantigen-specific proinflammatory cytokine production
Because it had been shown previously using ERα knock-out mice that both disease protection and a reduction in proinflammatory cytokines (TNFα and IFNγ) were dependent on ERα, we next determined whether treatment with an ERα ligand could reduce proinflammatory cytokine production. As demonstrated in Figure 3, PPT treatment significantly reduced TNFα, IFNγ, and IL6 production. Interestingly, we had shown previously that production of the Th2 cytokine IL5 was increased with estrogen treatment and that this was only partially, but not completely, abolished in the ERα knock-out (Liu et al., 2003). In the present study, when wild-type mice were treated with the ERα agonist PPT, treatment significantly increased IL5 production. Together, these data demonstrated that treatment with an ERα agonist induced changes in cytokine production during autoantigen-specific immune responses in the peripheral immune system that would be anti-inflammatory with respect to EAE immunopathogenesis.
Figure 3.

Treatment with an ERα ligand reduced proinflammatory cytokine production by peripheral immune cells in ovariectomized, wild-type C57BL/6 female mice with EAE. EAE was induced as in Figure 2, and at day 40 after disease induction, mice were killed, and cytokine production by MOG 35–55 stimulated splenocytes was determined. TNFα, IFNγ, and IL6 levels were each significantly reduced with PPT treatment, whereas IL5 levels were increased with PPT treatment. Error bars indicate variability of cytokine values for splenocytes between individual mice within a given treatment group, with n = 5 mice for each treatment group. Data are representative of experiments repeated three times. ∗p < 0.05.
Treatment with an ERα ligand reduces inflammation and demyelination in EAE
Because we had observed that treatment with the ERα ligand PPT recapitulated the protective effect of estrogen treatment on the clinical course of EAE and was anti-inflammatory with respect to the autoantigen-specific immune response in the periphery, we next ascertained the effect of treatment with PPT on inflammation and demyelination in the CNS of EAE mice. Spinal cord sections of ovariectomized, C57BL/6 mice at the acute phase of EAE (1–2 d after onset of clinical signs in vehicle-treated mice) were assessed for inflammation and demyelination. Mice from all treatment groups were killed at the same time point, to permit their examination in parallel. Compared with vehicle-treated EAE, both inflammation and demyelination were markedly reduced by treatment with the ERα ligand PPT or E2 (Fig. 4). H&E-stained vehicle-treated EAE mice, compared with normal healthy controls, had numerous multifocal to coalescing inflammatory cell infiltrates in the spinal cord. Infiltrates were present in the leptomeninges, around blood vessels in the leptomeninges, and in the parenchyma of the white matter (Fig. 4A). Inflammatory cell infiltrates were associated with pallor and vacuolation, consistent with demyelination. Quantification of white matter cell density by counting DAPI+ cells revealed a 60% increase in infiltrates of vehicle-treated EAE group. In contrast, both estradiol and PPT-treated mice had no detectable inflammation, with white matter cell densities similar to those in the normal control (Fig. 4D).
Figure 4.
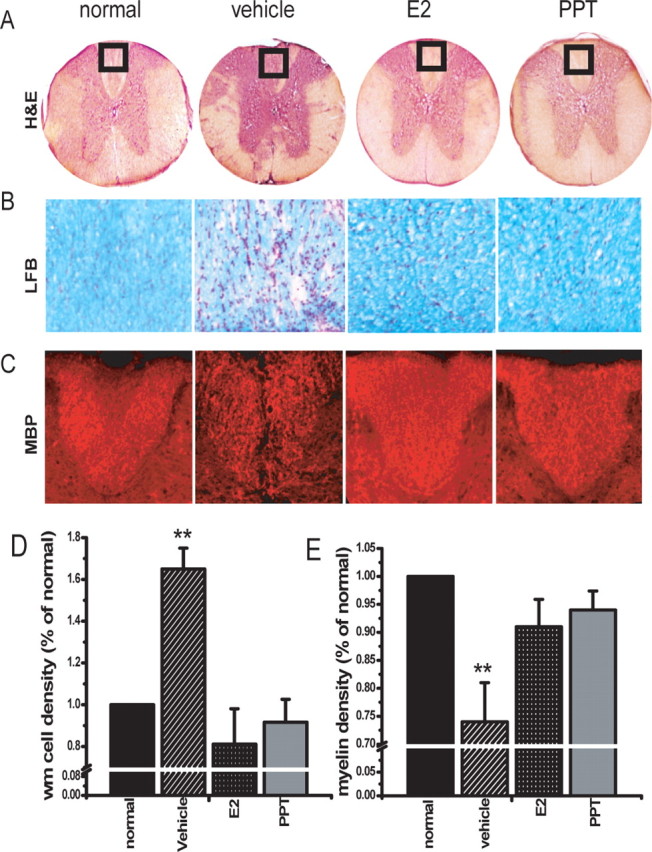
Treatment with an ERα ligand reduced inflammation and demyelination in spinal cords of mice with EAE. A, Representative H&E-stained thoracic spinal cord sections (4× magnification) from normal (healthy control), as well as vehicle-, E2-, and PPT-treated EAE mice. Vehicle-treated EAE spinal cord shows multifocal to coalescing areas of inflammation in the leptomeninges and white matter, around blood vessels, and in the parenchyma of the white matter (areas of inflammation shown by arrows). No inflammation was observed in either E2- or PPT-treated EAE spinal cords. B, Luxol fast blue-stained region of dorsal column (square in A) of spinal cords (40× magnification). Intense demyelination in the white matter is seen in vehicle-treated EAE sections only. C, Anti-MBP-immunostained dorsal column demonstrated demyelination in the white matter of vehicle-treated EAE sections only. D, Increase in total number of infiltrating cells after induction of EAE was semiquantified by counting DAPI+ cells in the entire delineated white matter (including dorsal, lateral, and ventral funiculi) and presented as percentage of normal. Vehicle-treated EAE mice had a significant increase in white matter cell density compared with healthy normal control, whereas E2-treated and the ERα ligand (PPT)-treated groups did not. E, The extent of demyelination was compared by staining thoracic spinal cord sections with Luxol fast blue. Myelin density is presented as percentage of normal. Vehicle-treated mice EAE mice had a significant decrease in myelin density in the entire delineated white matter as compared with normal control, whereas E2-treated and PPT-treated groups did not. Number of mice, three per treatment group; number of T1–T5 sections per mouse, six; total number of sections per treatment group, 18. ∗∗Statistically significant compared with normals (p < 0.001), 1 × 4 ANOVAs. Data are representative of experiments repeated in their entirety on another set of EAE mice with each of the treatments. Error bars represent SE of variability of the indicated measure between sections of mice within each treatment group.
The degree of myelin loss was assessed by Luxol fast blue and confirmed by MBP immunostaining. Luxol fast blue staining revealed demyelination at the sites of inflammatory cell infiltrates (Fig. 4B). Also, myelin staining of dorsal column regions of vehicle-treated spinal cord section had significantly less MBP immunostaining compared with normal control, E2-, and PPT-treated sections, Figure 4C. Quantification of demyelination by density analysis of Luxol fast blue-stained spinal cord sections revealed a 25% decrease in myelin density in vehicle-treated EAE mice. In contrast, both estradiol- and PPT-treated mice had much less demyelination, with myelin densities not significantly different from those in the normal control (Fig. 4E).
Treatment with an ERα ligand is neuroprotective in EAE
In light of the profound anti-inflammatory effect induced by PPT treatment of mice with EAE, we then ascertained whether this was associated with preservation of neuronal and axonal integrity. We used a combination of Nissl stain histology and anti-NeuN/β3-tubulin immunolabeling to identify and semiquantify neurons, and neurofilament antibody (anti-NF200) was used to identify axons. At the acute phase of EAE, 1–2 d after the onset of clinical signs in vehicle-treated mice, thoracic spinal cord sections of all treatment groups of EAE mice were assessed for NeuN+/β3 tubulin+ neurons in the gray matter and NF200+ axons in the white matter. A surprising decrease in neuronal staining (NeuN+/Nissl+) in gray matter occurred at this early time point in vehicle-treated EAE mice (Fig. 5B) compared with normal, healthy, age- and gender-matched control mice (Fig. 5A). This significant decrease in neuronal staining in gray matter of vehicle-treated EAE mice was not observed in EAE mice treated with either estradiol (Fig. 5C) or the ERα ligand (Fig. 5D). Quantification of NeuN+ cells in gray matter confirmed the significant loss in vehicle-treated EAE mice compared with normal controls, whereas estradiol- and PPT-treated mice had NeuN+ cell numbers that were no different from the normal control (Fig. 5E).
Figure 5.
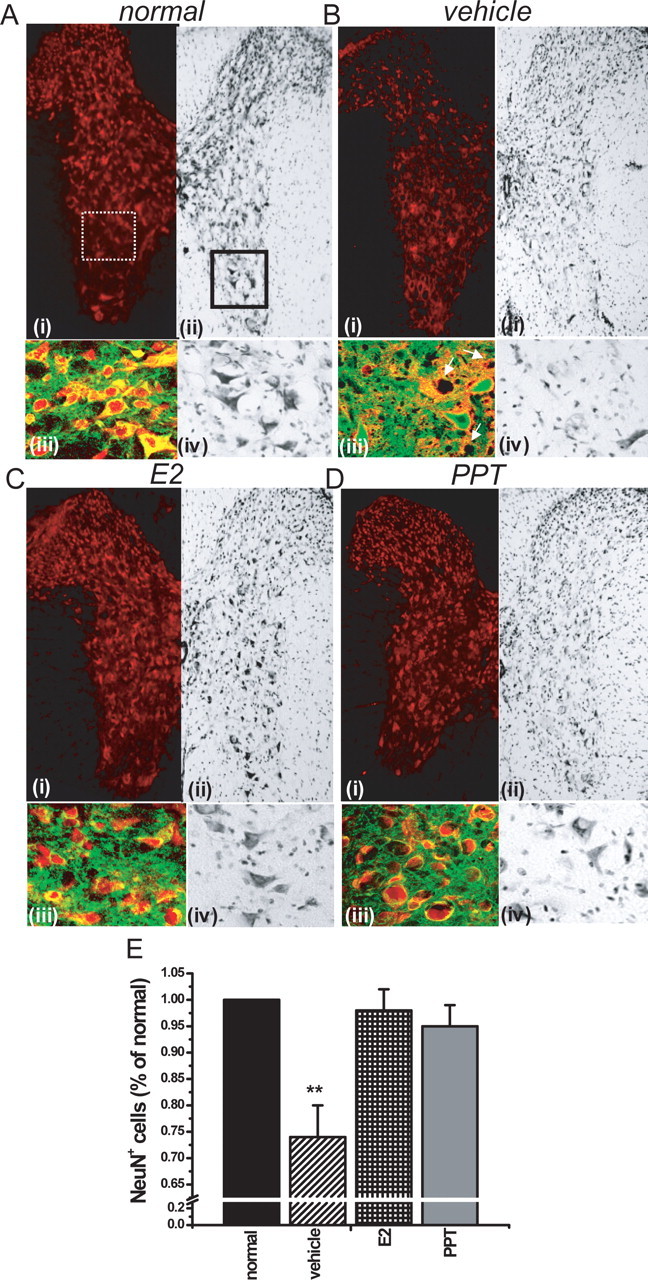
Treatment with an ERα ligand preserved neuronal staining in gray matter of spinal cords of mice with EAE. A–D, Split images of thoracic spinal cord sections stained with NeuN (red) in i and Nissl in ii at 4× magnification, derived from normal healthy control mice (A), vehicle-treated EAE (B), E2-treated EAE mice (C), and ERα ligand (PPT)-treated EAE mice (D), each killed very early during EAE, 1–2 d after the onset of clinical signs. iii, Merged confocal scan at 40× of NeuN+ (red) and β3-tubulin+ (green) colabeled neurons from an area represented by dotted white square area in i. iv, A 40× magnification of Nissl-stained area in solid black square in ii. A decrease in NeuN+ immunostaining and Nissl staining was observed in the dorsal horn, intermediate zone, and ventral horn of vehicle-treated EAE mice (B) compared with normal controls (A). White arrows in Biii denote loss of NeuN+ staining. In contrast, EAE mice treated with either estradiol (C) or PPT (D) had preserved NeuN and Nissl staining. E, After quantification of neurons in the entire delineated gray matter of T1–T5 sections, NeuN+ immunolabeled neurons were significantly decreased, by nearly 25%, in vehicle-treated EAE mice compared with normal controls, but E2- and PPT-treated EAE mice were not statistically different from normal controls. Number of mice, three per treatment group; number of T1–T5 sections per mouse, six; total number of sections per treatment group, 18. ∗∗Statistically significant compared with normals (p < 0.001); 1 × 4 ANOVAs. Data are representative of experiments repeated in their entirety on another set of EAE mice with each of the treatments. Error bars represent SE of variability of the indicated measure between sections of mice within each treatment group.
Immunostaining for neurofilament (NF200) resulted in clear identification of axons within the spinal cord of normal mice (Fig. 6A). A significant decrease in axonal NF200 staining (NF200+) in white matter occurred in vehicle-treated EAE mice compared with normal controls in areas positive for CD45 staining, consistent with previous observations of axonal transection within inflammatory white matter lesions in EAE (Wujek et al., 2002). EAE mice treated with either estradiol or the ERα ligand demonstrated no decrease in axonal NF200+ staining and only an occasional single cell positive for CD45 (Fig. 6A). Quantification of axon numbers in white matter confirmed the significant loss in vehicle-treated EAE mice, but no significant axonal loss occurred in EAE mice treated with either estradiol or the ERα ligand (Fig. 6C). These immunohistological data are consistent with our observation of markedly reduced inflammatory lesions by H&E in white matter with these treatments (Fig. 4A). Notably, at this early time point in EAE, there was no loss in axon numbers in white matter areas devoid of inflammatory lesions, even in the vehicle-treated EAE group, thereby providing no evidence for Wallerian degeneration of white matter tracts in these regions of the cord at this very early time point in EAE.
Figure 6.
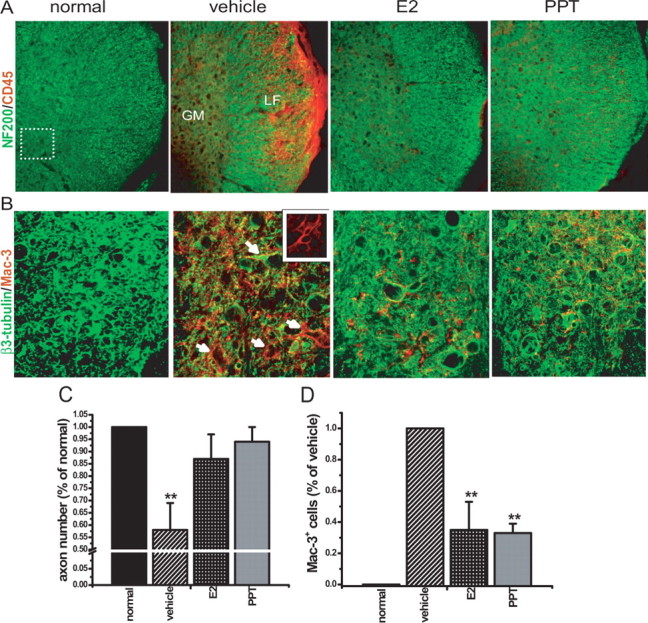
Treatment with an ERα ligand reduced CD45+ and Mac 3+ cells in white and gray matter of mice with EAE. A, Thoracic spinal cord sections from mice used in Figure 5 were coimmunostained with NF200 (green) and CD45 (red) at 10× magnification. Shown are partial images with white and gray matter from normal control, vehicle-treated EAE, E2-treated EAE, or ERα ligand (PPT)-treated EAE mice. LF, Lateral funiculus of white matter; GM, gray matter. The vehicle-treated EAE cords had large areas of CD45+ cells associated with reduced NF200 axonal staining in white matter compared with the normal control, whereas estradiol and ERα ligand-treated EAE mice had only occasional CD45 positivity, with intact NF200 axonal staining. B, Consecutive sections from the same mice were also coimmunostained with β3-tubulin (green) and Mac 3 (red), with the section of the ventral horn designated by the dotted line square area in A scanned at 40× magnification by confocal microscopy. Vehicle-treated EAE mice demonstrated markedly increased Mac 3 staining in ventral horn gray matter compared with normal control mice, with most of these Mac 3+ cells having the morphology of microglia (inset, 100× magnification). They were surrounding neuronal structures (white arrows). In contrast, E2- and ERα ligand (PPT)-treated EAE cord sections demonstrated less Mac 3 immunostaining compared with vehicle-treated EAE mice. C, After quantification, neurofilament-stained axon numbers in white matter were significantly lower in vehicle-treated EAE mice compared with normal mice, whereas E2- and PPT-treated EAE mice demonstrated no significant reduction in axon numbers. Axon number is presented as percentage of normal. ∗∗Statistically significant compared with normal (p < 0.001); 1 × 4 ANOVAs. D, Mac 3+ cells were analyzed by density measurements and represented as percentage of vehicle-treated groups. Compared with vehicle-treated EAE mice, both the E2-treated and PPT-treated had significantly lower Mac 3+ immunoreactivity in gray matter. Number of mice, three per treatment group; number of T1–T5 sections per mouse, four; total number of sections per treatment group, 12. ∗∗Statistically significant compared with normal (p < 0.001); 1 × 4 ANOVAs. Data are representative of experiments repeated in their entirety on another set of EAE mice with each of the treatments. Error bars represent SE of variability of the indicated measure between sections of mice within each treatment group.
Treatment with an ERα ligand reduces microglial/monocyte activation in white and gray matter of mice with EAE
Gray matter axonal pathology has been described in cortex of MS patients, which was characterized by activated microglia closely opposed to and ensheathing apical dendrites, neurites, and neuronal perikarya (Peterson et al., 2001). In light of our observation of a decrease in NeuN+/β3-tubulin+/Nissl+ neuronal staining in the gray matter of spinal cords in EAE, we next addressed the microglial reaction in this gray matter. Microglia/monocytes were stained for Mac 3, a lysosomal antigen equivalent to LAMP-2 (lysosomal-associated membrane protein 2)/CD107b, present on the surface of microglia and mature mononuclear phagocytes, and sections were coimmunolabeled with anti-β3-tubulin (Fig. 6B). Striking Mac 3+ reactivity was observed in gray matter of mice at this very early time point in EAE, only 1–2 d after the onset of clinical signs in the vehicle-treated group. Most of the Mac 3+ cells demonstrated a morphology similar to that of activated microglia (Fig. 6B, inset). They were in close vicinity to, and in direct contact with, gray matter neurons that had reduced and punctate β3-tubulin staining (Fig. 6B). In contrast, EAE mice treated with either the ERα ligand PPT, or estradiol, which were killed and examined in parallel, had some, but significantly less, Mac 3+ immunoreactivity (Fig. 6B). Quantification of Mac 3+ cells revealed an ∼65% decrease when E2- and PPT-treated spinal cords were compared with those from vehicle-treated EAE mice (Fig. 6D).
Discussion
Relapse rates in MS are decreased during late pregnancy, a time when circulating estrogen (estradiol and estriol) levels are increased (Abramsky, 1994; Confavreux et al., 1998). During late pregnancy, there is a downregulation of Th1 and an increase in Th2 immune responses, which may underlie disease amelioration during this time (Whitacre et al., 1999; Kruse et al., 2000; Tchorzewski et al., 2000; Elenkov et al., 2001; Voskuhl, 2003; Al-Shammri et al., 2004; Gilmore et al., 2004). In a pilot clinical trial, nonpregnant female MS patients were treated with estriol to induce a pregnancy level in serum. This treatment reduced the prototypic in vivo Th1 response, the delayed type hypersensitivity response, as well as reduced Th1 (TNFα, IFNγ) and increased Th2 (IL5, IL10) cytokine production by peripheral blood mononuclear cells (Sicotte et al., 2002; Soldan et al., 2003). Also, gadolinium-enhancing lesions on serial brain magnetic resonance images (MRIs) were reduced by >80% (Sicotte et al., 2002). Because enhancing lesion activity on brain MRI is a putative biomarker for relapses in MS, these reports together suggested that estriol treatment may recapitulate the anti-inflammatory effect of pregnancy in relapsing remitting MS (RRMS).
One must, however, consider the risk: benefit ratio of any estrogen treatment when considering its use in chronic disease. Estrogens in the form of hormone replacement therapy have been associated with side effects and therefore are not recommended for use in healthy menopausal women (Rossouw et al., 2002; Anderson et al., 2004). Although the risk/benefit ratio in a chronic disease such as MS is clearly different from the risk/benefit ratio in healthy individuals, optimizing efficacy and minimizing toxicity remains a major goal. Hence, determining which estrogen receptor mediates the protective effect of an estrogen in disease is of central importance. Our data demonstrate that despite the fact that both ERα and ERβ are expressed in both the immune system and the CNS (Kuiper et al., 1998; Enmark and Gustafsson, 1999; Erlandsson et al., 2001; Igarashi et al., 2001), treatment with an ERα ligand is sufficient to recapitulate the estrogen-mediated protection in EAE, the most widely used animal model of MS. Our data are consistent with a recent report by Elloso et al. (2005) that demonstrated that treatment with an ERα, but not an ERβ, ligand could reduce acute EAE disease severity in a relapsing remitting model of EAE in the SJL strain. This paper demonstrated immune modulation in splenocytes by estradiol and PPT treatment but did not address neuropathologic outcomes.
The degree of preservation of neuronal integrity in the gray matter of estradiol and ERα ligand-treated mice with EAE in our study was striking, and this has major implications for neurodegenerative changes that occur “beyond the lesion” in EAE and possibly MS. In both EAE and MS, inflammatory lesions classically occur in white matter, resulting in demyelination and axonal transection (Trapp et al., 1998, 1999). However, neuroimaging studies in MS have demonstrated that, although inflammatory lesions may serve as a biomarker for relapses, whole brain atrophy correlates much better with permanent disability (Rudick et al., 1999). Most recently, it has been shown that whole brain atrophy primarily involves the gray matter and that this can occur surprisingly early after MS onset (Chard et al., 2002; Dalton et al., 2004; Tiberio et al., 2005). The pathologic substrate of gray matter atrophy in MS remains unclear, but these imaging findings “beyond the lesion” in MS have prompted closer examination of gray matter abnormalities in MS and EAE. Pathological studies in cortical gray matter of MS patients initially detected axonal transection and microglial activation, with minimal inflammatory cell infiltrates (Bo et al., 2003). In a more recent study, cortical “lesions” in MS were characterized by demyelination, axonal sparing, and decreased neuronal density and were again not accompanied by an inflammatory infiltrate (Vercellino et al., 2005).
Gray matter abnormalities in EAE have included upregulation of expression of GFAP (Liedtke et al., 1998) and Mac-1 (Aharoni et al., 2005), thought to reflect activation of astrocytes and microglia, respectively, in the gray matter. Traumatic axotomy has been shown previously to induce decreased NeuN expression (McPhail et al., 2004), and recent studies have detected other abnormalities in neuronal staining in gray matter during EAE, ∼10 d after the initial onset of clinical signs (Aharoni et al., 2005; Aktas et al., 2005). In our study, decreases in NeuN and Nissl staining in gray matter, accompanied by increases in microglial/monocyte staining, were detected surprisingly early, within 1–2 d of the onset of clinical signs, but nevertheless were consistent with these previous observations. In pathologic studies in MS, it is difficult to ascertain the temporal relationship between gray matter abnormalities and disease onset. In EAE, we have detected gray matter abnormalities at the very earliest stages of clinical EAE.
Previously, estrogen treatment has been shown to decrease microglial activation in vitro (Drew and Chavis, 2000; Vegeto et al., 2001), but this is the first report that estrogen treatment can decrease activation in vivo. Whether microglial activation is a cause or consequence of neuronal pathology remains unknown. However, downregulation of microglial activation by either an ERα ligand (herein) or by antisense PARP-1 [poly (ADP-ribose)-polymerase-1] (Diestel et al., 2003) each resulted in decreased neuronal abnormalities in EAE. Therefore, it is tempting to speculate that activated microglia in gray matter during EAE may contribute to neuronal damage. This hypothesis is consistent with the observation that EAE clinical signs, CNS inflammation, and demyelination were each ameliorated in CD11b–HSVTK (herpes simplex virus thymidine kinase) transgenic mice, which were treated with gancyclovir and had bone marrow reconstituted from wild-type mice (Heppner et al., 2005). Unfortunately, neuronal abnormalities in gray matter were not assessed in these EAE mice with conditionally ablated activated microglia.
In addition to an effect on activated microglia, estrogen treatment may have other neuroprotective effects in EAE. Estrogen treatment has been shown previously to be neuroprotective in a variety of neurodegenerative disease models including Parkinson’s disease, cerebellar ataxia, stroke, and spinal cord injury (Leranth et al., 2000; Dubal et al., 2001; Wise et al., 2001; Jover et al., 2002; Rau et al., 2003; Sierra et al., 2003; Sribnick et al., 2003, 2005). Estrogens are lipophilic, readily traversing the blood–brain barrier, with the potential to be directly neuroprotective (Brinton, 2001; Garcia-Segura et al., 2001; Wise et al., 2001). Estrogen-mediated protection of neurons has been demonstrated in a variety of in vitro models of neurodegeneration including those induced by excitotoxicity and oxidative stress (Behl et al., 1995, 1997; Goodman et al., 1996; Harms et al., 2001). Estrogens have also been shown to decrease glutamate-induced apoptosis and preserve electrophysiologic function in primary cortical neurons (Sribnick et al., 2003, 2004). In addition, in vitro studies have demonstrated the ability of estrogen to modulate the astrocytic response to injury (Azcoitia et al., 1999; Garcia-Segura et al., 1999) and protect oligodendrocytes from cytotoxicity (Sur et al., 2003; Cantarella et al., 2004; Takao et al., 2004). Various combinations of these mechanisms could lead to our observed finding of a preservation of neuronal staining in the gray matter of spinal cords of estradiol- or ERα ligand-treated mice with EAE. Because treatment with either estradiol or the ERα ligand was each anti-inflammatory in the peripheral immune system, it is not possible to discern whether these treatments were directly neuroprotective, via one or more of the above mechanisms in the CNS, versus indirectly neuroprotective, via anti-inflammatory mechanisms in the peripheral immune system. However, it has been recently shown using irradiation bone marrow chimeras that the effect of estradiol treatment on clinical EAE and CNS inflammation was not dependent on ERα expression in the peripheral immune system but was conferred by ERα expression on cells resident to other tissues, such as the CNS (Garidou et al., 2004).
Direct and indirect neuroprotective mechanisms by estrogens in EAE are not mutually exclusive. The finding that estrogens are neuroprotective in EAE, regardless of mechanism, has relevance to estrogen treatment in MS, as well as pregnancy, a time when circulating estrogens are very high. Indeed, multiple pregnancies have been associated with a decrease in long-term disability accumulation in MS (Runmarker and Andersen, 1995; Damek and Shuster, 1997). Because it is known that up to 5 years of continuous treatment with immunomodulatory treatments are needed to impact disability in MS, a temporary anti-inflammatory effect of the third trimester of pregnancy would not be expected to impact long-term disability. A pregnancy-associated neuroprotective effect, combined with the temporary anti-inflammatory effect, could reconcile these findings.
Our data demonstrating that treatment with a highly selective ERα agonist can induce both anti-inflammatory and neuroprotective effects in EAE warrants additional study of this SERM as a novel treatment approach in MS and possibly other neurodegenerative diseases. Although a large study of estrogen replacement therapy in mild to moderate Alzheimer’s disease, with subjects averaging 75 years of age, yielded disappointing results (Mulnard et al., 2000), either the age of the subject population or the stage of disease may be critical to whether a neuroprotective effect of estrogen treatment can be appreciated (Mulnard et al., 2004). Adequate levels of estrogen receptors, or associated intracellular cofactors, may no longer be optimally expressed in either hormonally senescent, or significantly diseased, brains. Indeed, the effect of estrogen treatment on the blood–brain barrier has been shown to be quite different when comparing senescent versus younger rats (Bake and Sohrabji, 2004). Clinical reports also indicate differential effects of estrogen treatment depending on age and disease status. Estrogen replacement had no effect on cognition in elderly menopausal women (Binder et al., 2001), whereas it preserved cognition in younger women undergoing surgical hysterectomy (Sherwin, 1988; Verghese et al., 2000). In a large trial assessing the effects of estrogen treatment in stroke, treatment had no effect on cognition in women with stroke who had cognitive impairment at baseline, but treatment reduced the risk for cognitive decline in those with normal cognition at baseline (Viscoli et al., 2005). Finally, an improvement in cognitive function was observed with estriol treatment in early RRMS, but not in late secondary progressive MS (Sicotte et al., 2002). A “healthy cell bias of estrogen action” has been hypothesized to explain the disparity between promising results of estrogen treatment in animal models of neurodegenerative diseases versus disappointing results in estrogen treatment in some neurodegenerative disease trials (Brinton, 2005). Efficacy of estrogen treatment appears to depend critically on its administration early, as a preventative therapy, before neurodegeneration has occurred (Mulnard et al., 2000). Our data demonstrating neuronal abnormalities in gray matter very early in EAE, which were prevented by treatment with either estradiol or an ERα ligand, are consistent with these views. Together these data warrant additional study of treatments with ERα ligands, which consider the age and disease duration of the subjects, in MS and other neurodegenerative diseases.
Footnotes
This work was supported by National Institutes of Health–National Institute of Neurological Disorders and Stroke Grant NS45443, National Multiple Sclerosis Society Grants RD3407 and CA1028 (R.R.V.), the Sherak Foundation, and DirecTV.
References
- Abramsky O (1994). Pregnancy and multiple sclerosis. Ann Neurol 36:Suppl, S38–S41. [DOI] [PubMed] [Google Scholar]
- Aharoni R, Arnon R, Eilam R (2005). Neurogenesis and neuroprotection induced by peripheral immunomodulatory treatment of experimental autoimmune encephalomyelitis. J Neurosci 25:8217–8228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aktas O, Smorodchenko A, Brocke S, Infante-Duarte C, Topphoff US, Vogt J, Prozorovski T, Meier S, Osmanova V, Pohl E, Bechmann I, Nitsch R, Zipp F (2005). Neuronal damage in autoimmune neuroinflammation mediated by the death ligand TRAIL. Neuron 46:421–432. [DOI] [PubMed] [Google Scholar]
- Al-Shammri S, Rawoot P, Azizieh F, AbuQoora A, Hanna M, Saminathan TR, Raghupathy R (2004). Th1/Th2 cytokine patterns and clinical profiles during and after pregnancy in women with multiple sclerosis. J Neurol Sci 222:21–27. [DOI] [PubMed] [Google Scholar]
- Anderson GL, Limacher M, Assaf AR, Bassford T, Beresford SA, Black H, Bonds D, Brunner R, Brzyski R, Caan B, Chlebowski R, Curb D, Gass M, Hays J, Heiss G, Hendrix S, Howard BV, Hsia J, Hubbell A, Jackson R, et al (2004). Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women’s Health Initiative randomized controlled trial. JAMA 291:1701–1712. [DOI] [PubMed] [Google Scholar]
- Azcoitia I, Sierra A, Garcia-Segura LM (1999). Localization of estrogen receptor beta-immunoreactivity in astrocytes of the adult rat brain. Glia 26:260–267. [PubMed] [Google Scholar]
- Bake S, Sohrabji F (2004). 17beta-estradiol differentially regulates blood-brain barrier permeability in young and aging female rats. Endocrinology 145:5471–5475. [DOI] [PubMed] [Google Scholar]
- Bebo BF Jr, Fyfe-Johnson A, Adlard K, Beam AG, Vandenbark AA, Offner H (2001). Low-dose estrogen therapy ameliorates experimental autoimmune encephalomyelitis in two different inbred mouse strains. J Immunol 166:2080–2089. [DOI] [PubMed] [Google Scholar]
- Behl C, Widmann M, Trapp T, Holsboer F (1995). 17-beta estradiol protects neurons from oxidative stress-induced cell death in vitro. Biochem Biophys Res Commun 216:473–482. [DOI] [PubMed] [Google Scholar]
- Behl C, Skutella T, Lezoualc’h F, Post A, Widmann M, Newton CJ, Holsboer F (1997). Neuroprotection against oxidative stress by estrogens: structure-activity relationship. Mol Pharmacol 51:535–541. [PubMed] [Google Scholar]
- Binder EF, Schechtman KB, Birge SJ, Williams DB, Kohrt WM (2001). Effects of hormone replacement therapy on cognitive performance in elderly women. Maturitas 38:137–146. [DOI] [PubMed] [Google Scholar]
- Bo L, Vedeler CA, Nyland H, Trapp BD, Mork SJ (2003). Intracortical multiple sclerosis lesions are not associated with increased lymphocyte infiltration. Mult Scler 9:323–331. [DOI] [PubMed] [Google Scholar]
- Brinton RD (2001). Cellular and molecular mechanisms of estrogen regulation of memory function and neuroprotection against Alzheimer’s disease: recent insights and remaining challenges. Learn Mem 8:121–133. [DOI] [PubMed] [Google Scholar]
- Brinton RD (2005). Investigative models for determining hormone therapy-induced outcomes in brain: evidence in support of a healthy cell bias of estrogen action. Ann NY Acad Sci 1052:57–74. [DOI] [PubMed] [Google Scholar]
- Cantarella G, Risuglia N, Lombardo G, Lempereur L, Nicoletti F, Memo M, Bernardini R (2004). Protective effects of estradiol on TRAIL-induced apoptosis in a human oligodendrocytic cell line: evidence for multiple sites of interactions. Cell Death Differ 11:503–511. [DOI] [PubMed] [Google Scholar]
- Chard DT, Griffin CM, Parker GJ, Kapoor R, Thompson AJ, Miller DH (2002). Brain atrophy in clinically early relapsing-remitting multiple sclerosis. Brain 125:327–337. [DOI] [PubMed] [Google Scholar]
- Confavreux C, Hutchinson M, Hours MM, Cortinovis-Tourniaire P, Moreau T (1998). Rate of pregnancy-related relapse in multiple sclerosis. Pregnancy in Multiple Sclerosis Group. N Engl J Med 339:285–291. [DOI] [PubMed] [Google Scholar]
- Dalal M, Kim S, Voskuhl RR (1997). Testosterone therapy ameliorates experimental autoimmune encephalomyelitis and induces a T helper 2 bias in the autoantigen-specific T lymphocyte response. J Immunol 159:3–6. [PubMed] [Google Scholar]
- Dalton CM, Chard DT, Davies GR, Miszkiel KA, Altmann DR, Fernando K, Plant GT, Thompson AJ, Miller DH (2004). Early development of multiple sclerosis is associated with progressive grey matter atrophy in patients presenting with clinically isolated syndromes. Brain 127:1101–1107. [DOI] [PubMed] [Google Scholar]
- Damek DM, Shuster EA (1997). Pregnancy and multiple sclerosis. Mayo Clin Proc 72:977–989. [DOI] [PubMed] [Google Scholar]
- Diestel A, Aktas O, Hackel D, Hake I, Meier S, Raine CS, Nitsch R, Zipp F, Ullrich O (2003). Activation of microglial poly(ADP-ribose)-polymerase-1 by cholesterol breakdown products during neuroinflammation: a link between demyelination and neuronal damage. J Exp Med 198:1729–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew PD, Chavis JA (2000). Female sex steroids: effects upon microglial cell activation. J Neuroimmunol 111:77–85. [DOI] [PubMed] [Google Scholar]
- Dubal DB, Zhu H, Yu J, Rau SW, Shughrue PJ, Merchenthaler I, Kindy MS, Wise PM (2001). Estrogen receptor alpha, not beta, is a critical link in estradiol-mediated protection against brain injury. Proc Natl Acad Sci USA 98:1952–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elenkov IJ, Wilder RL, Bakalov VK, Link AA, Dimitrov MA, Fisher S, Crane M, Kanik KS, Chrousos GP (2001). IL-12, TNF-alpha, and hormonal changes during late pregnancy and early postpartum: implications for autoimmune disease activity during these times. J Clin Endocrinol Metab 86:4933–4938. [DOI] [PubMed] [Google Scholar]
- Elloso MM, Phiel K, Henderson RA, Harris HA, Adelman SJ (2005). Suppression of experimental autoimmune encephalomyelitis using estrogen receptor-selective ligands. J Endocrinol 185:243–252. [DOI] [PubMed] [Google Scholar]
- Enmark E, Gustafsson JA (1999). Oestrogen receptors–an overview. J Intern Med 246:133–138. [DOI] [PubMed] [Google Scholar]
- Erlandsson MC, Ohlsson C, Gustafsson JA, Carlsten H (2001). Role of oestrogen receptors alpha and beta in immune organ development and in oestrogen-mediated effects on thymus. Immunology 103:17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frasor J, Barnett DH, Danes JM, Hess R, Parlow AF, Katzenellenbogen BS (2003). Response-specific and ligand dose-dependent modulation of estrogen receptor (ER) alpha activity by ERbeta in the uterus. Endocrinology 144:3159–3166. [DOI] [PubMed] [Google Scholar]
- Garcia-Segura LM, Naftolin F, Hutchison JB, Azcoitia I, Chowen JA (1999). Role of astroglia in estrogen regulation of synaptic plasticity and brain repair. J Neurobiol 40:574–584. [PubMed] [Google Scholar]
- Garcia-Segura LM, Azcoitia I, DonCarlos LL (2001). Neuroprotection by estradiol. Prog Neurobiol 63:29–60. [DOI] [PubMed] [Google Scholar]
- Garidou L, Laffont S, Douin-Echinard V, Coureau C, Krust A, Chambon P, Guery JC (2004). Estrogen receptor alpha signaling in inflammatory leukocytes is dispensable for 17beta-estradiol-mediated inhibition of experimental autoimmune encephalomyelitis. J Immunol 173:2435–2442. [DOI] [PubMed] [Google Scholar]
- Gilmore W, Arias M, Stroud N, Stek A, McCarthy KA, Correale J (2004). Preliminary studies of cytokine secretion patterns associated with pregnancy in MS patients. J Neurol Sci 224:69–76. [DOI] [PubMed] [Google Scholar]
- Goodman Y, Bruce AJ, Cheng B, Mattson MP (1996). Estrogens attenuate and corticosterone exacerbates excitotoxicity, oxidative injury, and amyloid beta-peptide toxicity in hippocampal neurons. J Neurochem 66:1836–1844. [DOI] [PubMed] [Google Scholar]
- Granholm AC, Sanders L, Seo H, Lin L, Ford K, Isacson O (2003). Estrogen alters amyloid precursor protein as well as dendritic and cholinergic markers in a mouse model of Down syndrome. Hippocampus 13:905–914. [DOI] [PubMed] [Google Scholar]
- Green PS, Bales K, Paul S, Bu G (2005). Estrogen therapy fails to alter amyloid deposition in the PDAPP model of Alzheimer’s disease. Endocrinology 146:2774–2781. [DOI] [PubMed] [Google Scholar]
- Harms C, Lautenschlager M, Bergk A, Katchanov J, Freyer D, Kapinya K, Herwig U, Megow D, Dirnagl U, Weber JR, Hortnagl H (2001). Differential mechanisms of neuroprotection by 17 β-estradiol in apoptotic versus necrotic neurodegeneration. J Neurosci 21:2600–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington WR, Sheng S, Barnett DH, Petz LN, Katzenellenbogen JA, Katzenellenbogen BS (2003). Activities of estrogen receptor alpha- and beta-selective ligands at diverse estrogen responsive gene sites mediating transactivation or transrepression. Mol Cell Endocrinol 206:13–22. [DOI] [PubMed] [Google Scholar]
- Harris HA, Katzenellenbogen JA, Katzenellenbogen BS (2002). Characterization of the biological roles of the estrogen receptors, ERalpha and ERbeta, in estrogen target tissues in vivo through the use of an ERalpha-selective ligand. Endocrinology 143:4172–4177. [DOI] [PubMed] [Google Scholar]
- Heikkinen T, Kalesnykas G, Rissanen A, Tapiola T, Iivonen S, Wang J, Chaudhuri J, Tanila H, Miettinen R, Puolivali J (2004). Estrogen treatment improves spatial learning in APP + PS1 mice but does not affect beta amyloid accumulation and plaque formation. Exp Neurol 187:105–117. [DOI] [PubMed] [Google Scholar]
- Heppner FL, Greter M, Marino D, Falsig J, Raivich G, Hovelmeyer N, Waisman A, Rulicke T, Prinz M, Priller J, Becher B, Aguzzi A (2005). Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat Med 11:146–152. [DOI] [PubMed] [Google Scholar]
- Igarashi H, Kouro T, Yokota T, Comp PC, Kincade PW (2001). Age and stage dependency of estrogen receptor expression by lymphocyte precursors. Proc Natl Acad Sci USA 98:15131–15136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito A, Bebo BF Jr, Matejuk A, Zamora A, Silverman M, Fyfe-Johnson A, Offner H (2001). Estrogen treatment down-regulates TNF-alpha production and reduces the severity of experimental autoimmune encephalomyelitis in cytokine knockout mice. J Immunol 167:542–552. [DOI] [PubMed] [Google Scholar]
- Jansson L, Olsson T, Holmdahl R (1994). Estrogen induces a potent suppression of experimental autoimmune encephalomyelitis and collagen-induced arthritis in mice. J Neuroimmunol 53:203–207. [DOI] [PubMed] [Google Scholar]
- Jover T, Tanaka H, Calderone A, Oguro K, Bennett MV, Etgen AM, Zukin RS (2002). Estrogen protects against global ischemia-induced neuronal death and prevents activation of apoptotic signaling cascades in the hippocampal CA1. J Neurosci 22:2115–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Liva SM, Dalal MA, Verity MA, Voskuhl RR (1999). Estriol ameliorates autoimmune demyelinating disease: implications for multiple sclerosis. Neurology 52:1230–1238. [DOI] [PubMed] [Google Scholar]
- Krege JH, Hodgin JB, Couse JF, Enmark E, Warner M, Mahler JF, Sar M, Korach KS, Gustafsson JA, Smithies O (1998). Generation and reproductive phenotypes of mice lacking estrogen receptor beta. Proc Natl Acad Sci USA 95:15677–15682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse N, Greif M, Moriabadi NF, Marx L, Toyka KV, Rieckmann P (2000). Variations in cytokine mRNA expression during normal human pregnancy. Clin Exp Immunol 119:317–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuiper GG, Carlsson B, Grandien K, Enmark E, Haggblad J, Nilsson S, Gustafsson JA (1997). Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology 138:863–870. [DOI] [PubMed] [Google Scholar]
- Kuiper GG, Shughrue PJ, Merchenthaler I, Gustafsson JA (1998). The estrogen receptor beta subtype: a novel mediator of estrogen action in neuroendocrine systems. Front Neuroendocrinol 19:253–286. [DOI] [PubMed] [Google Scholar]
- Leranth C, Roth RH, Elswoth JD, Naftolin F, Horvath TL, Redmond DE Jr (2000). Estrogen is essential for maintaining nigrostriatal dopamine neurons in primates: implications for Parkinson’s disease and memory. J Neurosci 20:8604–8609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liedtke W, Edelmann W, Chiu FC, Kucherlapati R, Raine CS (1998). Experimental autoimmune encephalomyelitis in mice lacking glial fibrillary acidic protein is characterized by a more severe clinical course and an infiltrative central nervous system lesion. Am J Pathol 152:251–259. [PMC free article] [PubMed] [Google Scholar]
- Liu HB, Loo KK, Palaszynski K, Ashouri J, Lubahn DB, Voskuhl RR (2003). Estrogen receptor alpha mediates estrogen’s immune protection in autoimmune disease. J Immunol 171:6936–6940. [DOI] [PubMed] [Google Scholar]
- Liu HY, Buenafe AC, Matejuk A, Ito A, Zamora A, Dwyer J, Vandenbark AA, Offner H (2002). Estrogen inhibition of EAE involves effects on dendritic cell function. J Neurosci Res 70:238–248. [DOI] [PubMed] [Google Scholar]
- Lubahn DB, Moyer JS, Golding TS, Couse JF, Korach KS, Smithies O (1993). Alteration of reproductive function but not prenatal sexual development after insertional disruption of the mouse estrogen receptor gene. Proc Natl Acad Sci USA 90:11162–11166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPhail LT, McBride CB, McGraw J, Steeves JD, Tetzlaff W (2004). Axotomy abolishes NeuN expression in facial but not rubrospinal neurons. Exp Neurol 185:182–190. [DOI] [PubMed] [Google Scholar]
- Mulnard RA, Cotman CW, Kawas C, van Dyck CH, Sano M, Doody R, Koss E, Pfeiffer E, Jin S, Gamst A, Grundman M, Thomas R, Thal LJ (2000). Estrogen replacement therapy for treatment of mild to moderate Alzheimer disease: a randomized controlled trial. Alzheimer’s Disease Cooperative Study [see comments]. JAMA 283:1007–1015. [DOI] [PubMed] [Google Scholar]
- Mulnard RA, Corrada MM, Kawas CH (2004). Estrogen replacement therapy, Alzheimer’s disease, and mild cognitive impairment. Curr Neurol Neurosci Rep 4:368–373. [DOI] [PubMed] [Google Scholar]
- Nilsson S, Makela S, Treuter E, Tujague M, Thomsen J, Andersson G, Enmark E, Pettersson K, Warner M, Gustafsson JA (2001). Mechanisms of estrogen action. Physiol Rev 81:1535–1565. [DOI] [PubMed] [Google Scholar]
- Paech K, Webb P, Kuiper GG, Nilsson S, Gustafsson J, Kushner PJ, Scanlan TS (1997). Differential ligand activation of estrogen receptors ERalpha and ERbeta at AP1 sites. Science 277:1508–1510. [DOI] [PubMed] [Google Scholar]
- Palaszynski KM, Liu H, Loo KK, Voskuhl RR (2004). Estriol treatment ameliorates disease in males with experimental autoimmune encephalomyelitis: implications for multiple sclerosis. J Neuroimmunol 149:84–89. [DOI] [PubMed] [Google Scholar]
- Peterson JW, Bo L, Mork S, Chang A, Trapp BD (2001). Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann Neurol 50:389–400. [DOI] [PubMed] [Google Scholar]
- Pettinelli CB, McFarlin DE (1981). Adoptive transfer of experimental allergic encephalomyelitis in SJL/J mice after in vitro activation of lymph node cells by myelin basic protein: requirement for Lyt 1+ 2- T lymphocytes. J Immunol 127:1420–1423. [PubMed] [Google Scholar]
- Polanczyk M, Zamora A, Subramanian S, Matejuk A, Hess DL, Blankenhorn EP, Teuscher C, Vandenbark AA, Offner H (2003). The protective effect of 17beta-estradiol on experimental autoimmune encephalomyelitis is mediated through estrogen receptor-alpha. Am J Pathol 163:1599–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rau SW, Dubal DB, Bottner M, Gerhold LM, Wise PM (2003). Estradiol attenuates programmed cell death after stroke-like injury. J Neurosci 23:11420–11426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, Jackson RD, Beresford SA, Howard BV, Johnson KC, Kotchen JM, Ockene J (2002). Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women’s Health Initiative randomized controlled trial. JAMA 288:321–333. [DOI] [PubMed] [Google Scholar]
- Rudick RA, Fisher E, Lee JC, Simon J, Jacobs L (1999). Use of the brain parenchymal fraction to measure whole brain atrophy in relapsing-remitting MS. Multiple Sclerosis Collaborative Research Group. Neurology 53:1698–1704. [DOI] [PubMed] [Google Scholar]
- Runmarker B, Andersen O (1995). Pregnancy is associated with a lower risk of onset and a better prognosis in multiple sclerosis [see comments]. Brain 118:253–261. [DOI] [PubMed] [Google Scholar]
- Shang Y, Brown M (2002). Molecular determinants for the tissue specificity of SERMs. Science 295:2465–2468. [DOI] [PubMed] [Google Scholar]
- Sherwin BB (1988). Estrogen and/or androgen replacement therapy and cognitive functioning in surgically menopausal women. Psychoneuroendocrinology 13:345–357. [DOI] [PubMed] [Google Scholar]
- Sicotte NL, Liva SM, Klutch R, Pfeiffer P, Bouvier S, Odesa S, Wu TC, Voskuhl RR (2002). Treatment of multiple sclerosis with the pregnancy hormone estriol. Ann Neurol 52:421–428. [DOI] [PubMed] [Google Scholar]
- Sierra A, Azcoitia I, Garcia-Segura L (2003). Endogenous estrogen formation is neuroprotective in model of cerebellar ataxia. Endocrine 21:43–51. [DOI] [PubMed] [Google Scholar]
- Soldan SS, Retuerto AI, Sicotte NL, Voskuhl RR (2003). Immune modulation in multiple sclerosis patients treated with the pregnancy hormone estriol. J Immunol 171:6267–6274. [DOI] [PubMed] [Google Scholar]
- Sribnick EA, Wingrave JM, Matzelle DD, Ray SK, Banik NL (2003). Estrogen as a neuroprotective agent in the treatment of spinal cord injury. Ann NY Acad Sci 993:125–133. discussion 159–160. [DOI] [PubMed] [Google Scholar]
- Sribnick EA, Ray SK, Nowak MW, Li L, Banik NL (2004). 17beta-estradiol attenuates glutamate-induced apoptosis and preserves electrophysiologic function in primary cortical neurons. J Neurosci Res 76:688–696. [DOI] [PubMed] [Google Scholar]
- Sribnick EA, Wingrave JM, Matzelle DD, Wilford GG, Ray SK, Banik NL (2005). Estrogen attenuated markers of inflammation and decreased lesion volume in acute spinal cord injury in rats. J Neurosci Res 82:283–293. [DOI] [PubMed] [Google Scholar]
- Subramanian S, Matejuk A, Zamora A, Vandenbark AA, Offner H (2003). Oral feeding with ethinyl estradiol suppresses and treats experimental autoimmune encephalomyelitis in SJL mice and inhibits the recruitment of inflammatory cells into the central nervous system. J Immunol 170:1548–1555. [DOI] [PubMed] [Google Scholar]
- Suen WE, Bergman CM, Hjelmstrom P, Ruddle NH (1997). A critical role for lymphotoxin in experimental allergic encephalomyelitis. J Exp Med 186:1233–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sur P, Sribnick EA, Wingrave JM, Nowak MW, Ray SK, Banik NL (2003). Estrogen attenuates oxidative stress-induced apoptosis in C6 glial cells. Brain Res 971:178–188. [DOI] [PubMed] [Google Scholar]
- Takao T, Flint N, Lee L, Ying X, Merrill J, Chandross KJ (2004). 17beta-estradiol protects oligodendrocytes from cytotoxicity induced cell death. J Neurochem 89:660–673. [DOI] [PubMed] [Google Scholar]
- Tchorzewski H, Krasomski G, Biesiada L, Glowacka E, Banasik M, Lewkowicz P (2000). IL-12, IL-6 and IFN-gamma production by lymphocytes of pregnant women with rheumatoid arthritis remission during pregnancy. Mediators Inflamm 9:289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiberio M, Chard DT, Altmann DR, Davies G, Griffin CM, Rashid W, Sastre-Garriga J, Thompson AJ, Miller DH (2005). Gray and white matter volume changes in early RRMS: a 2-year longitudinal study. Neurology 64:1001–1007. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mörk S, Bö L (1998). Axonal transection in the lesions of multiple sclerosis [see comments]. N Engl J Med 338:278–285. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Bö L, Mörk S, Chang A (1999). Pathogenesis of tissue injury in MS lesions. J Neuroimmunol 98:49–56. [DOI] [PubMed] [Google Scholar]
- van Groen T, Kadish I (2005). Transgenic AD model mice, effects of potential anti-AD treatments on inflammation and pathology. Brain Res Brain Res Rev 48:370–378. [DOI] [PubMed] [Google Scholar]
- Vegeto E, Bonincontro C, Pollio G, Sala A, Viappiani S, Nardi F, Brusadelli A, Viviani B, Ciana P, Maggi A (2001). Estrogen prevents the lipopolysaccharide-induced inflammatory response in microglia. J Neurosci 21:1809–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veliskova J, Velisek L, Galanopoulou AS, Sperber EF (2000). Neuroprotective effects of estrogens on hippocampal cells in adult female rats after status epilepticus. Epilepsia 41:Suppl 6, S30–S35. [DOI] [PubMed] [Google Scholar]
- Vercellino M, Plano F, Votta B, Mutani R, Giordana MT, Cavalla P (2005). Grey matter pathology in multiple sclerosis. J Neuropathol Exp Neurol 64:1101–1107. [DOI] [PubMed] [Google Scholar]
- Verghese J, Kuslansky G, Katz MJ, Sliwinski M, Crystal HA, Buschke H, Lipton RB (2000). Cognitive performance in surgically menopausal women on estrogen. Neurology 55:872–874. [DOI] [PubMed] [Google Scholar]
- Viscoli CM, Brass LM, Kernan WN, Sarrel PM, Suissa S, Horwitz RI (2005). Estrogen therapy and risk of cognitive decline: results from the Women’s Estrogen for Stroke Trial (WEST). Am J Obstet Gynecol 192:387–393. [DOI] [PubMed] [Google Scholar]
- Voskuhl R (2003). Sex hormomes and other pregnancy-related factors with therapeutic potential in multiple sclerosis. In: Multiple sclerosis therapeutics (Cohen D, Rudick R, eds) Ed 2 pp. 535–549. London: Martin Dunitz.
- Weiss DJ, Gurpide E (1988). Non-genomic effects of estrogens and antiestrogens. J Steroid Biochem 31:671–676. [DOI] [PubMed] [Google Scholar]
- Whitacre CC, Reingold SC, O’Looney PA (1999). A gender gap in autoimmunity. Science 283:1277–1278. [DOI] [PubMed] [Google Scholar]
- Wise PM, Dubal DB, Wilson ME, Rau SW, Bottner M (2001). Minireview: neuroprotective effects of estrogen-new insights into mechanisms of action. Endocrinology 142:969–973. [DOI] [PubMed] [Google Scholar]
- Wujek JR, Bjartmar C, Richer E, Ransohoff RM, Yu M, Tuohy VK, Trapp BD (2002). Axon loss in the spinal cord determines permanent neurological disability in an animal model of multiple sclerosis. J Neuropathol Exp Neurol 61:23–32. [DOI] [PubMed] [Google Scholar]
- Xiao BG, Liu X, Link H (2004). Antigen-specific T cell functions are suppressed over the estrogen-dendritic cell-indoleamine 2,3-dioxygenase axis. Steroids 69:653–659. [DOI] [PubMed] [Google Scholar]
- Zang YC, Halder JB, Hong J, Rivera VM, Zhang JZ (2002). Regulatory effects of estriol on T cell migration and cytokine profile: inhibition of transcription factor NF-kappa B. J Neuroimmunol 124:106–114. [DOI] [PubMed] [Google Scholar]