

Abstract
Endocannabinoids can act as retrograde messengers that allow postsynaptic cells to regulate the strength of their synaptic inputs. In the cerebellum, Purkinje cells (PCs) release endocannabinoids through two mechanisms. Synaptic activation evokes local endocannabinoid release that relies on a pathway that involves the metabotropic glutamate receptor mGluR1 and phospholipase-C (PLC). In contrast, depolarization evokes endocannabinoid release from the entire dendritic arbor. This leads to depolarization-induced suppression of inhibitory (DSI) and excitatory (DSE) synapses by a mechanism that does not involve mGluR1 or PLC. This latter mechanism of endocannabinoid release has only been observed under artificial conditions that transiently elevate postsynaptic calcium to >5 μm. Here, we tested the possibility that this mechanism could lead to retrograde inhibition in response to more realistic calcium signals. At both climbing fiber and inhibitory synapses onto PCs, we found that prolonging the elevation of calcium significantly lowered the peak calcium required to evoke PLC-independent endocannabinoid release. This suggests that the mechanism of endocannabinoid release involved in DSI and DSE is likely to evoke endocannabinoid release in response to physiologically relevant levels of calcium. When dendritic calcium was elevated to 0.4–1 μm for 15 s or more, endocannabinoid release from PCs selectively suppressed inhibitory synapses. This suggests that inhibitory synapses are more sensitive to prolonged calcium increases. Thus, in contrast to localized retrograde inhibition evoked by synaptic activation, modest but sustained calcium elevation could globally suppress inhibitory synapses onto PCs.
Keywords: cerebellum, synaptic plasticity, endocannabinoids, calcium, Purkinje cell, two-photon imaging, retrograde inhibition
Introduction
Endocannabinoids can function as retrograde messengers that enable postsynaptic neurons to regulate the strength of their synaptic inputs. Many types of neurons release endocannabinoids from their dendrites that then bind to presynaptic CB1 cannabinoid receptors, leading to a reduction in the probability of neurotransmitter release (Freund et al., 2003; Piomelli, 2003). Two primary means of endocannabinoid release have been identified. Synaptically evoked endocannabinoid release usually requires a small elevation of calcium combined with activation of a Gq-coupled metabotropic receptor (Brenowitz and Regehr, 2005; Maejima et al., 2005). This activates phospholipase-C (PLC), producing diacylglycerol (DAG), which is converted to the endocannabinoid 2-arachidonylglycerol (2-AG) by the enzyme DAG lipase (Stella et al., 1997). Endocannabinoids are also liberated after postsynaptic depolarization, which leads to depolarization-evoked suppression of inhibitory (DSI) or excitatory (DSE) synapses (Kreitzer and Regehr, 2001a; Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001). In cerebellar Purkinje cells (PCs), this latter mechanism is independent of PLC (Safo and Regehr, 2005), and, for brief elevations of intracellular calcium, peak levels of 10–15 μm are required to produce half-maximal DSI and DSE (Brenowitz and Regehr, 2003; Maejima et al., 2005). Such calcium levels may occur only during periods when excitatory inputs are highly active or under pathological conditions. Therefore, it is unclear under what physiological conditions DSI or DSE might occur.
The possibility that depolarization triggers the release of endocannabinoids from Purkinje cells under physiological conditions is of interest because it would suggest a new functional role of endocannabinoids. In contrast to the highly localized modulation that can be achieved with synaptically evoked endocannabinoid release (Brown et al., 2003), the mechanisms underlying DSI and DSE could lead to more global modulation of synaptic inputs distributed throughout the dendrites of a PC. Because all of the synaptic inputs onto Purkinje cells contain CB1 receptors (CB1Rs) (Kreitzer and Regehr, 2001a,b), heightened activity could lead to the regulation of synapses from climbing fibers (CFs), granule cell parallel fibers (PFs), and inhibitory interneurons. Thus, depolarization-induced endocannabinoid release could allow Purkinje cells to control the manner in which they integrate their synaptic inputs. This may enable Purkinje cells to globally regulate synapses on rapid timescales. Although these possibilities are intriguing, the high calcium requirement for DSE and DSI suggests that such synaptic regulation may not occur under physiological conditions.
Here we test the possibility that prolonging the elevation of calcium lowers the peak levels of calcium required to evoke endocannabinoid release, and we examine whether PLC-independent mechanisms are likely to contribute to synaptic regulation under physiological conditions. We find that prolonging the duration of the calcium increase for 15 s or longer lowers the calcium needed for retrograde inhibition of both climbing fiber and inhibitory synapses. Measurements of calcium levels with two-photon imaging techniques indicate that, when peak calcium levels within spines and dendrites are ∼1 μm, endocannabinoid release selectively suppresses inhibitory synapses. This suggests that, for prolonged elevations of calcium, inhibitory synapses are more sensitive to retrograde suppression by endocannabinoids than are excitatory synapses. Moreover, the reduced calcium requirement indicates that physiological conditions that elevate dendritic calcium by several hundred nanomolar for many seconds can evoke endocannabinoid release and provides a means of globally suppressing retrograde inhibition onto Purkinje cells.
Materials and Methods
Electrophysiology.
Parasagittal slices (250–300 μm thick) were cut from the cerebellar vermis of Sprague Dawley rats. Dissections were performed in an ice-cold sucrose solution containing the following (in mm): 75 NaCl, 26 NaHCO3, 75 sucrose, 25 glucose, 2.5 KCl, 1.25 NaH2PO4, 7 MgCl2, and 0.5 CaCl2. Slices were incubated for 30 min at 32°C in sucrose solution, transferred to saline solution (in mm: 125 NaCl, 26 NaCO3, 1.25 NaH2PO4, 2.5 KCl, 1 MgCl2, 2 CaCl2, and 25 glucose), and after 30 min allowed to cool to room temperature. All solutions were bubbled with 95% O2/5% CO2.
To measure the calcium dependence of CF DSE, Purkinje cells from postnatal day 10 (P10) to P11 animals were voltage clamped at −60 mV using glass electrodes (1.3–1.7 MΩ) filled with an intracellular solution containing the following (in mm): 100 CsMeSO4, 35 CsCl, 15 HEPES, 2 HEDTA, 0.2 EGTA, 1 MgCl2, 15 tetraethylammonium-Cl, 2 MgATP, 0.3 NaGTP, 10 Tris-phoshocreatine, 2 QX-314 [2(triethylamino)-N-(2,6-dimethylphenyl) acetamine], and 0.5 fura-FF, adjusted to pH 7.30 with CsOH. Series resistance was compensated by 70–80% during recordings. Data were filtered at 3 kHz and sampled at 10–20 kHz. Access resistance and holding current were monitored, and experiments were rejected if these parameters changed significantly during recording. Climbing fibers were stimulated with a glass electrode filled with saline and placed in the granular layer. CF EPSCs were evoked with 200 μs pulses delivered to an isolated stimulus unit and were recorded in the presence of 30 μm picrotoxin to block GABAA receptors and 0.8–1 μm 2,3-dihydroxy-6-nitro-7-sulfonyl-benzo[f]quinoxaline (NBQX) to decrease CF EPSC amplitudes by >75% and reduce voltage-clamp errors (Foster et al., 2002). All experiments were performed at 34–36°C.
Calcium imaging during voltage-clamp protocols was performed with a Cooke (Romulus, MI) Sensicam QE CCD camera mounted on an Olympus Optical (Tokyo, Japan) BX51 upright microscope equipped with a 60×, 0.9 numerical aperture (NA) objective. A rapid wavelength switching monochromator (T.I.L.L. Polychrome IV; T.I.L.L. Photonics, Grafelfing, Germany) was used for fluorescence excitation in conjunction with a 455 nm dichroic mirror and a 455 long-pass filter for collecting fluorescence emission. Fura-FF fluorescence ratios were calculated as r = λ1/λ2, with λ1 = 383 nm and λ2 = 354 nm. Fluorescence ratios were converted to calcium concentration using the relationship Cai = KD × β × (R − Rmin)/(Rmax − R). Because λ2 is the isosbestic point of fura-FF, β = 1. The KD of fura-FF was measured by dissolving it in a buffer containing 150 mm CsMeSO4, 30 mm HEPES, and Ca that was buffered to known levels using 10 mm EGTA, to which varying amounts of CaCl2 were added (Tsien and Pozzan, 1989). Measurements were made using the experimental imaging system with a cuvette mounted in a temperature-controlled chamber at 34–36°C. The KD of fura-FF for Ca was determined to be 3.5 μm at 35°C compared with 7.7 μm at 24°C. Taking into account the differences in the KD at 24°C and 35°C, our estimate of the calcium dependence of DSE at CF synapses is similar to that of Maejima et al. (2005). The Rmin of fura-FF in our internal recording solution was measured at 34–36°C in the same cuvette used for determining the KD by adding 2.4 mm EGTA to our internal recording solution. The Rmax of fura-FF in our internal recording solution was measured in situ at 34–36°C by washing in the calcium ionophore ionomycin (20 μm) while imaging calcium in the dendrites of Purkinje cells. For CCD imaging of Purkinje cell dendritic calcium, 20 ms exposures were taken every 200 ms. Fluorescence excitation was restricted to a small region including the Purkinje dendrites and an adjacent cell-free region used for background correction. Calcium measurements were made from proximal dendrites of Purkinje cells corresponding to the location of CF synapses.
For experiments in which both synaptic stimulation and spontaneous activity were recorded in Purkinje cells from P15–P17 animals using a potassium-based intracellular solution that consisted of the following (in mm): 114 KMeSO4, 6 NaCl, 10 HEPES, 0.5 EGTA, 2 MgSO4, 0.16 CaCl2, 4 MgATP, 0.4 NaGTP, and 14 Tris-phosphocreatine. In some experiments (see Fig. 3G), P29–P31 animals were used. PF EPSCs were evoked with a stimulus electrode in the molecular layer in the presence of 10 μm picrotoxin to block GABAA receptors and 2 μm CGP55845 [(2S)-3-{[(15)-1-(3,4-dichlorophenyl)ethyl]amino-2-hydroxypropyl)(phenyl-methyl)phosphinic acid] to block GABAB receptors. Spontaneous (sIPSCs) and evoked (eIPSCs) IPSCs were recorded in the presence of 5–10 μm NBQX to block excitatory synapses and 2 μm CGP55845. Inhibitory responses were evoked by placing the stimulus electrode in the distal two-thirds of the molecular layer to preferentially activate stellate cells. Synaptic responses were evoked at 0.2 Hz in voltage clamp before and after spontaneous PC firing in current clamp. Synaptic stimulation at 0.2 Hz was continued during spontaneous firing to maintain steady-state levels of short-term plasticity that caused a small increase in response amplitudes of ∼10% as a result of facilitation during 0.2 Hz stimulation. The recording mode of the amplifier was switched between voltage clamp and current clamp using an external signal that was controlled by the data acquisition software.
Figure 3.
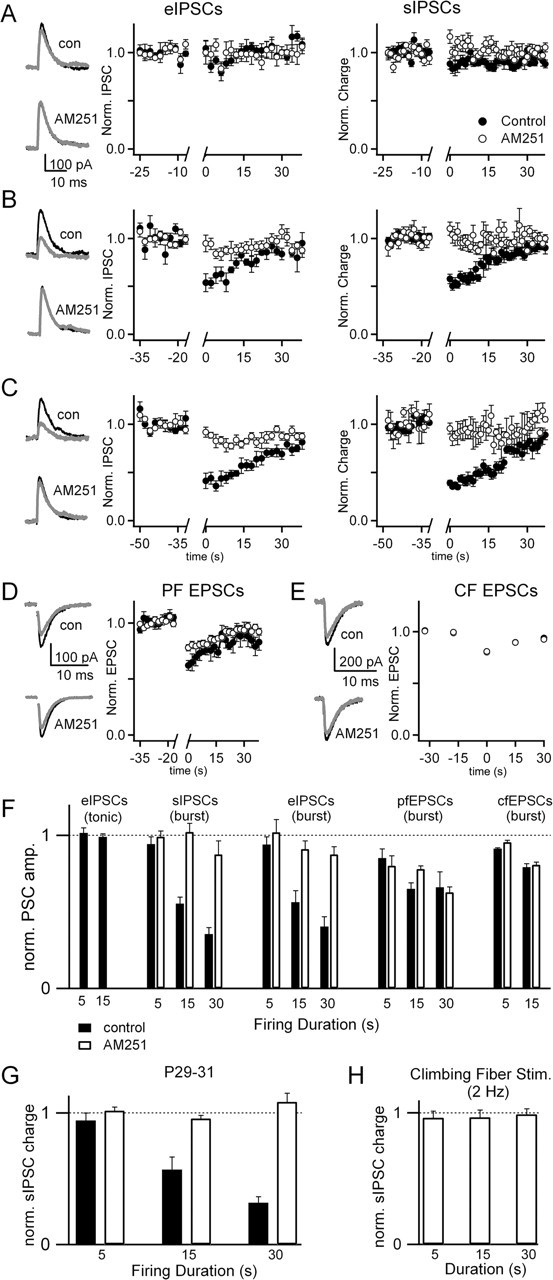
The effects of Purkinje cell activity on synaptic inputs from inhibitory interneurons, parallel fibers, and climbing fibers. Experiments were performed as in Figure 2. Evoked and sIPSCs were measured in the presence of NBQX, and the effects of burst mode firing for 5 s (A), 15 s (B), and 30 s (C) were assessed. Representative traces are shown for eIPSCs 2–6 s before (black) and 2–6 s after (gray) burst firing in control conditions and in the presence of AM251 (A–C, left). Summaries of the eIPSCs (A–C, middle) and sIPSCs (A–C, right) are shown for control conditions (filled circles) and in the presence of AM251 (open circles). Similar experiments with 15 s bursts were performed for PF EPSCs (D) and CF EPSCs (E), with picrotoxin in the bath to block IPSCs. F, The effects of tonic and burst firing are shown for the various types of synapses for control conditions and in the presence of AM251. The number of experiments performed under each condition ranged from 5 to 14. Data were analyzed by calculating the ratio of the average of three responses after burst firing to the average of three responses preceding the burst. G, Effects of burst firing on sIPSCs in P29–P31 animals was measured under control conditions and in the presence of AM251 (control, n = 4; AM251, n = 4). H, The effects of 2 Hz climbing fiber stimulation for durations of 5, 15, and 30 s on sIPSCs was measured (n = 10) in P15–P17 animals.
Two-photon imaging.
We used a custom two-photon laser-scanning microscope with a 60×, 0.9 NA objective and a titanium/sapphire laser (Mira; Coherent, Santa Clara, CA) tuned to 810 nm for excitation. Purkinje cells were loaded with Alexa 594 (20 μm) for visualization and fluo-5F (250 or 500 μm) for measurement of intracellular calcium. Fluorescence signals were collected in the epifluorescence and transfluorescence pathways (using a 1.4 NA oil-immersion condenser). Green and red fluorescence were separated using a 565 nm dichroic and filtered using BG22 glass and a 607/45 bandpass filter, respectively. Green and red fluorescence were collected using R3896 and H7422 photomultipliers (Hamamatsu, Hamamatsu City, Japan), respectively. Imaging and physiology were controlled with custom software written in Matlab (MathWorks, Natick, MA), which was kindly provided by Bernardo Sabatini. Line scans were performed at 500 Hz over regions that included a Purkinje cell dendrite and a spine, for a duration of 20 s. Trials in which photobleaching exceeded 5% were excluded from the analysis. Fluorescence signals were converted to calcium concentrations using values of Rmin and Rmax (Grynkiewicz et al., 1985) measured in intracellular solutions containing 0 mm Ca/3 mm EGTA and 3 mm Ca, respectively, using the experimental imaging system. The KD of fluo-5F measured in the potassium-based internal solution at 35°C using calibration solutions containing EGTA and varying amounts of CaCl2 (Tsien and Pozzan, 1989) was determined to be 550 nm.
AM251 [N-1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-1-piperidinyl-1H-pyrazole-3-carboxamide], NBQX, picrotoxin, carboxypeptidase A (CPA), cyclopropabchromen-1a-carboxylate ethyl ester (CPCCOEt), and CGP55845 were from Tocris Cookson (Ellisville, MO). Fura-FF, fluo-5F, Alexa 594, Cs-BAPTA, and ionomycin were from Invitrogen (Carlsbad, CA). U73122 (1-[6[[(17β)-3-methoxyestra-1,3,5(10)-trien-17-yl]amino]hexyl]-1H-pyrrole-2,5-dione) was from Biomol (Plymouth Meeting, PA). Tetrahydrolipstatin [THL (Orlistat)] was provided by Roche Pharmaceuticals (Nutley, NJ). All other chemicals were purchased from Sigma (St. Louis, MO).
Results
Prolonged Ca elevation reduces peak Ca required for DSE
We determined whether prolonging the duration of dendritic calcium levels lowered the peak calcium levels required to evoke endocannabinoid release and DSE. Purkinje cells from young animals were voltage clamped, and either a single voltage step was used to briefly elevate calcium or a series of voltage steps was used to evoke a prolonged increase in dendritic calcium. The resulting calcium signals were measured as described previously, and DSE at the CF was determined. Experiments were performed at 34–36°C. As shown in a representative experiment, depolarizations of 100, 200, and 2000 ms produced peak dendritic calcium levels of 1.9, 7.0, and 46 μm, respectively (Fig. 1A, left), and reduced synaptic strength to 87, 53, and 11% of control (Fig. 1A, right). In this experiment half, maximal DSE occurred at 8.9 μm calcium (Fig. 1C, circles). In the same cell, calcium was also increased by a series of depolarizations (nine depolarizations to 0 mV for 75 ms). Although peak calcium was only elevated to 1.8 μm (Fig. 1B, left), the EPSC was reduced to 55% of control (Fig. 1B, right). Much lower peak calcium levels are required to evoke DSE after a series of depolarizations than after a single step. This is shown for a representative experiment (Fig. 1C) and for a summary of nine experiments (Fig. 1F). The half-maximal calcium values for DSE after either a single voltage step or a series of voltage steps was determined to be 6.6 and 2.6 μm, respectively (Fig. 1F, solid lines).
Figure 1.

Prolonged calcium elevations lower the peak calcium required for endocannabinoid-dependent retrograde inhibition. Purkinje cells were voltage clamped with a Cs-based internal containing the calcium indicator fura-FF (500 μm). Cells were held at −60 mV and depolarized to 0 mV, the resulting calcium transient was measured, and the effect on the CF response was determined. A representative experiment (A–C) shows calcium transients arising from single depolarizations of 100, 200, and 2000 ms (A, left) and the corresponding CF EPSCs measured 13 s before and 2 s after these depolarizations (A, right). The calcium transient evoked in the same cell by a series of 75 ms depolarizations every 1.5 s (B, left) is also shown along with the corresponding CF EPSCs (B, right). C, A summary of the peak DSE as a function of the peak calcium levels produced by single depolarizations (open circles) is well approximated by the Hill equation DSE = (1 + (Ca0.5/Capost)m)−1, with Ca0.5 = 8.9 μm and m = 1.25. DSE arising from a series of brief (75 ms) depolarizations (open squares) does not lie on the curve. D, E, The calcium signal evoked by a series of brief depolarizations and the corresponding CF EPSCs 2 s before and 2 s after are shown for an experiment in which 2 μm AM251 was in the bath (D) and 20 mm BAPTA was included in the pipette (E). F, Summary of experiments in which the peak DSE is plotted as a function of the peak calcium for control conditions (open circles; n = 5) and for a series of brief depolarizations in control conditions (open squares; n = 9) in the presence of AM251 (triangle; n = 4) and with BAPTA in the pipette (inverted triangle; n = 4). The Hill equation was used to fit data from experiments with a single depolarization (thick line; Ca0.5 = 6.6 ± 0.4 μm and m = 1.7 ± 0.2) and with a series of 75 ms depolarizations (thin line; Ca0.5 = 2.6 ± 0.3 μm and m = 1.7 ± 0.3).
DSE after a single voltage step is known to reflect calcium-dependent endocannabinoid release that leads to activation of presynaptic CB1 receptors. We examined whether DSE evoked by a series of voltage steps shared these properties. DSE after a series of voltage steps was prevented by including either the CB1 receptor antagonist AM251 in the bath (Fig. 1D,F) or BAPTA in the recording pipette (Fig. 1E,F). Thus, as for single voltage steps, DSE evoked by a prolonged calcium increase after a series of voltage steps reflects calcium-dependent endocannabinoid release and activation of CB1 receptors.
These findings demonstrate that prolonging the duration of elevated calcium levels lowers the amount of calcium required to induce DSE at CF synapses. This suggests that modest increases in calcium could produce DSE under physiological conditions, provided calcium levels are elevated for many seconds.
It is of interest whether prolonging the duration of calcium elevation also lowers the peak calcium required to suppress PF synapses and inhibitory synapses. Previously, using voltage clamp and calcium measurements, we found that, at 24°C, the suppression of inhibitory synapses, CF synapses, and PF synapses all had a similar dependence on peak calcium levels for transient increases in calcium. We found that it was not practical to use this approach to study DSE of PF synapses and DSI at 35°C. Voltage control sufficient to achieve graded increases in dendritic calcium by brief voltage steps during recordings made at 35°C required that we record from Purkinje cells with small dendritic arbors in P10–P11 animals. In such young animals, CF synapses are readily studied, but inhibitory and PF synaptic responses are small and unstable and therefore unsuitable for studies of DSE and DSI at 35°C. A more general issue is that calcium entry cannot be controlled sufficiently to allow the study of DSE or DSI of any synapse in Purkinje cells from animals older than P12.
Examining retrograde signaling evoked by spontaneous Purkinje cell firing
The limitations of voltage clamp prompted us to use another approach that allowed us to study the effects of prolonged increases in dendritic calcium on retrograde inhibition. We elevated dendritic calcium levels by allowing Purkinje cells to fire spontaneously in current-clamp mode with a potassium-based internal solution. This allowed us to study retrograde inhibition of PF, CF, and inhibitory synapses onto Purkinje cells from P15–P17 animals.
Purkinje cells are known to fire spontaneously in either tonic mode (Fig. 2A, left) or in burst mode (Fig. 2A, right). In tonic mode, cells fire at approximately constant frequencies of 10–130 Hz (Womack and Khodakhah, 2004). In our experiments, Purkinje cells fired tonically at 55 ± 7 Hz (n = 19). In burst mode, calcium spikes occur in the dendrites and sodium spikes arise in the soma in bursts (Womack and Khodakhah, 2002; Swensen and Bean, 2003; Womack and Khodakhah, 2004). In these experiments, synaptic responses were measured in voltage clamp before and after periods of spontaneous PC firing in current clamp (Fig. 2B). During current-clamp recordings, small constant current injections (0–500 pA) caused the cell to fire in either tonic or burst mode, and the duration of spontaneous firing was controlled by the duration of the current injection in current clamp. Burst firing provided a convenient method that enabled us to examine the role of prolonged but moderate elevation of dendritic calcium on synaptic inputs to PCs that occurs during burst firing and under appropriate conditions could also be driven by synaptic excitation. Studies in brain slices indicate that the majority of Purkinje cells fire spontaneously in burst mode (Womack and Khodakhah, 2002), but it is not known whether this behavior occurs in vivo. Spontaneous firing by PCs in burst or tonic mode for periods of 5–30 s was used to study retrograde inhibition at PF, CF, and inhibitory synapses (see Figs. 3, 4) and was combined with two-photon imaging to determine the calcium dependence of retrograde inhibition (see Figs. 5–7).
Figure 2.

Assessing the effects of tonic firing and burst firing on inhibitory synaptic inputs to Purkinje cells. A, Examples of the typical firing patterns in tonic mode and burst mode are shown. B, Cells were voltage clamped with a potassium-based internal solution at −60 mV and then switched to current clamp in which the cell was allowed to fire in either tonic mode or burst mode, and then cells were returned to voltage clamp. While in voltage clamp, synaptic inputs were activated and the resulting synaptic currents were measured.
Figure 5.
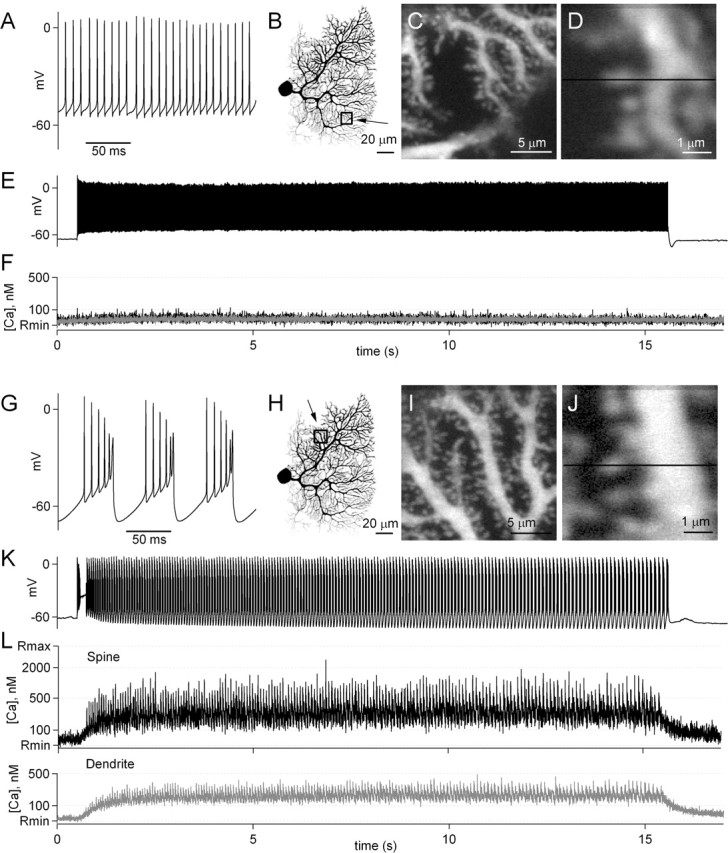
Dendritic calcium increases in Purkinje cells during firing in tonic and burst mode. A representative experiment is shown in which calcium levels within dendrites and spines were measured during Purkinje cell firing. Whole-cell current-clamp recordings were made from Purkinje cells with an electrode containing the calcium indicator fluo-5F, and calcium transients were measured as the Purkinje cell fired in tonic mode (A–F) and burst mode (G–L). As shown in an expanded view, this Purkinje cell fired at ∼100 Hz in tonic mode (A). B, A two-photon image has been inverted to show the morphology of the cell, and a box shows the region displayed on an expanded scale in C. An additional expanded view (D) shows the path of a line scan used to measure calcium levels within a spine and a dendrite. E, Spontaneous firing of the Purkinje cell was suppressed by small hyperpolarizing currents, and then a small depolarization caused the cell to tonically fire for 15 s. F, The calcium transients measured during tonic firing are shown for the spine and dendrite shown in D. Another trial is shown in which slightly larger currents were injected into the Purkinje cell, which caused the cell to fire in burst mode (G–L). Calcium was measured in the dendritic region shown in H–J. During 15 s of burst firing (K), calcium transients were measured in the spine and dendrite (L) in the region indicated by the line in J.
The effect of spontaneous Purkinje cell firing on retrograde suppression of synapses by endocannabinoids is shown in Figure 3. For inhibitory synapses, NBQX was included in the bath and a stimulus electrode was placed in the molecular layer to evoke IPSCs (Fig. 3A–C, left). sIPSCs (Fig. 3A–C, right) were also measured by integrating total charge over 1 s intervals. Experiments were performed both in control conditions and with AM251 in the bath. A transition to burst firing for 5 s (Fig. 3A) had no effect on eIPSCs or sIPSCs. However, 15 s of burst firing suppressed eIPSCs by 46 ± 9% (Fig. 3B), and this suppression recovered by 50% in 12–14 s. Traces for a representative cell (Fig. 3B, left) are shown before (black trace) and after (gray trace) burst firing. Bath application of the CB1 receptor antagonist AM251 prevented the suppression of eIPSCs. Similarly, suppression of sIPSCs was 47 ± 3%, recovered with a similar time course as that of eIPSCs, and was also blocked by AM251 (Fig. 3B, right). When the duration of burst firing was extended to 30 s (Fig. 3C), the extent of suppression of eIPSCs and sIPSCs was increased to 59 ± 8 and 61 ± 3%, and recovery was prolonged. In no cases did tonic firing of 5–15 s cause suppression of inhibitory synapses (Fig. 3F). These experiments demonstrate that burst firing but not tonic firing for 15–30 s evokes endocannabinoid release from Purkinje cells, activating CB1 receptors present on inhibitory nerve terminals and suppressing inhibitory synaptic strength.
After bursts of 15 s, PF EPSCs were suppressed by 33 ± 6%, but AM251 did not significantly reduce this effect (Fig. 3D) (p = 0.25, unpaired t test). In addition, CF EPSCs showed a small suppression of 20 ± 2% after 15 s bursts, but this was not affected by AM251 (Fig. 3E) (p = 0.84). Data are summarized in Figure 3F for each synapse. The results indicate that burst firing for 15–30 s causes endocannabinoid-mediated suppression of synaptic strength at inhibitory but not excitatory synapses onto Purkinje cells.
To determine whether endocannabinoid-mediated suppression of inhibitory synaptic inputs to Purkinje cells persists in older animals, experiments were performed on P29–P31 animals (Fig. 3G). After bursts of 5, 15, and 30 s, evoked IPSC amplitudes were suppressed by 6 ± 6, 43 ± 10, and 68 ± 4%, respectively. These values were not significantly different from results in P15–P17 animals (p > 0.4, t test).
We also tested whether physiological patterns of CF activity are capable of evoking endocannabinoid release from Purkinje cells. In vivo, climbing fibers fire at 1–2 Hz (Gilbert and Thach, 1977; Mano et al., 1986). Climbing fiber activation is known to evoke complex spikes that elevate postsynaptic calcium throughout Purkinje cell dendrites. In P11–P14 animals, CF activity causes an endocannabinoid-dependent reduction of sIPSC frequency (Duguid and Smart, 2004). Therefore, we tested the effect of 2 Hz CF stimulation for 5, 15, and 30 s on sIPSCs in P15–P17 animals (Fig. 3H). Under our experimental conditions, physiological patterns of CF activity do not evoke endocannabinoid-mediated suppression of inhibitory synapses.
Pharmacological properties of burst-evoked suppression
We clarified the mechanism of burst-evoked suppression at inhibitory synapses by testing its pharmacological sensitivity. Previous studies have shown that trains of parallel fiber stimulation evoke endocannabinoid release from Purkinje cells and retrograde inhibition of PF synapses (Brown et al., 2003). Such synaptically evoked suppression of excitation (SSE) requires elevation of intracellular calcium and activation of postsynaptic metabotropic glutamate receptor 1 (mGluR1) that leads to production of 2-AG. SSE is prevented by blocking activity of mGluR1, PLC, or DAG lipase. Depolarization-evoked suppression of excitation and inhibition relies on calcium elevation but none of the components of the mGluR1 cascade. To investigate possible mechanisms underlying burst-evoked endocannabinoid release from Purkinje cells, we examined the role of mGluR1, PLC, and DAG lipase on suppression of eIPSCs by 15 s of bursting by Purkinje cells (Fig. 4). Blocking mGluR1 with CPCCOEt, inhibiting PLC with U73122, or loading Purkinje cells with the DAG lipase inhibitor THL had no effect on burst-mediated suppression of eIPSCs (p = 0.56, 0.38, and 0.37 relative to control, t test). It was not possible in current-clamp experiments to demonstrate the effect of blocking postsynaptic calcium elevation with BAPTA on burst-evoked suppression because BAPTA prevents activation of calcium-activated conductances and profoundly alters the firing behavior of PCs. These results indicate that burst-evoked endocannabinoid release by Purkinje cells occurs by a mechanism that is similar to that of DSE/DSI and distinct from SSE.
Figure 4.

Pharmacological studies indicate that burst-evoked suppression of inhibitory synapses is similar to DSI and differs from synaptically evoked suppression of synaptic strength. The suppression of eIPSCs by 15 s in burst mode was examined as in Figures 2 and 3. A, Traces from representative experiments are shown for the eIPSCs measured before (black) and after (gray) burst firing in control conditions and after bath application of the mGluR1 antagonist CPCCOEt (100 μm), the PLC antagonist U73122 (5 μm, bath applied for 1 h), the DAG lipase inhibitor THL (2 μm) loaded in the recording pipette, and CPA, the inhibitor of endoplasmic reticulum calcium ATPase (30 μm, bath applied for 1 h). B, Results are summarized with numbers above the bars indicating the number of experiments performed for each condition.
Previous studies have shown in the hippocampus that intracellular calcium stores contribute to endocannabinoid release during DSI (Isokawa and Alger, 2006). To test for a role of internal calcium stores in burst-evoked endocannabinoid release from Purkinje cells, we applied the endoplasmic reticulum Ca-ATPase inhibitor CPA that depletes calcium stores. CPA had no effect on the suppression of eIPSCs after 15 s of burst firing (p = 0.33 relative to control, t test) (Fig. 4B), suggesting that release of calcium from internal stores does not contribute to endocannabinoid release evoked by burst firing in Purkinje cells.
Calcium signals during spontaneous firing
The properties of retrograde inhibition after burst firing raise a number of questions regarding calcium signaling during bursts. First, what are the dendritic calcium signals during prolonged bursts and do they provide insight into the observation that 15 s bursts produce retrograde inhibition but 5 s bursts do not? One possible explanation is that prolonged elevations of calcium lower the calcium levels needed to evoke endocannabinoid release, as demonstrated at CF synapses using voltage-clamp protocols (Fig. 1). Another possibility is that peak calcium levels during a 15 s burst are much higher than for a 5 s burst, and this underlies the greater efficacy of the longer burst. Second, can differential calcium signaling within dendrites account for the differential retrograde suppression of inhibitory and excitatory synapses? CF synapses are located on large proximal dendrites of Purkinje cells, and PF synapses are located on spines of fine distal dendrites. Stellate cell synapses are broadly distributed throughout PC dendrites. The greater sensitivity of inhibitory synapses to endocannabinoid release by burst firing could arise from larger calcium signals in the vicinity of inhibitory synapse than for CF or PF synapses.
To distinguish among these possibilities, we measured calcium during spontaneous firing in Purkinje cell dendrites and spines with high spatial and temporal resolution using two-photon laser scanning microscopy. Purkinje cells were loaded with the red fluorophore Alexa 594 to visualize dendrites and spines and the calcium indicator fluo-5F (250–500 μm) to measure calcium transients during spontaneous Purkinje cell firing. A representative experiment in which calcium was measured in the dendrite and spine during tonic and burst firing is illustrated in Figure 5.
During tonic firing (Fig. 5A), calcium was imaged in a region of a Purkinje cell dendrite illustrated in Figure 5B and at higher magnification in Figure 5C. A line scan was performed across a dendrite and spine as indicated by the line in Figure 5D. During a 15 s period of tonic firing (Fig. 5E), calcium showed no changes in either the dendrite or spine (Fig. 5F). This observation is consistent with the absence of sodium channels in Purkinje cell dendrites (Stuart and Hausser, 1994) and with our observation that tonic firing had no effect on eIPSC amplitudes. In the same cell, calcium was imaged during burst firing in a different dendritic region (Fig. 5G–J). After the onset of a current injection into the Purkinje cell soma by the recording pipette, the cell fired several spikes and then began bursting after a delay of ∼200 ms (Fig. 5K). During burst firing, calcium rose in the dendrite (Fig. 5L, gray trace) and spine (Fig. 5L, black trace) to a plateau of ∼200 nm from which small spikes were observed. The level of the plateau was the same in the dendrite and spine. In addition, individual calcium spikes were observed that reached peak levels of ∼300 nm in the dendrite and 500–2000 nm in the spine. After termination of the burst, calcium decayed with a time constant of ∼1 s.
To quantify calcium levels and determine the timing of calcium entry, burst-evoked fluorescence signals were averaged and then converted to calcium concentrations (Fig. 6). This approach is shown for a period of bursting corresponding to a period from 3 to 5 s after the onset of burst firing (Fig. 6A). The derivative of the dendritic calcium transient was used to determine the timing of the onset of each calcium transient (Fig. 6B, filled circles). These times were used to align the membrane potential traces (Fig. 6C, top) and the calcium transients measured in the spine (Fig. 6C, middle, gray traces) and the dendrite (Fig. 6C, bottom, gray traces). Calcium transients ceased abruptly as the burst firing stopped. The average of the calcium signals measured in dendrites and spines (Fig. 6C, black traces, D) were used to calculate calcium concentrations during burst firing (Fig. 6E).
Figure 6.

Timing and quantification of calcium signals during burst mode. Whole-cell recordings were made from Purkinje cells with an internal solution containing the green calcium indicator fluo-5F (500 μm) and the red indicator Alexa 594 (20 μm). Burst mode firing (A) evoked changes in calcium that resulted in a change in the ratio of green and red fluorescence (B). The first derivative of the fluorescence transients was used to determine the timing of the calcium transients, which are indicated by dots above the trace in B. The peaks determined from the dendritic calcium spikes were used to align the firing of the Purkinje cells (C, top), the spine calcium (C, middle), and the dendritic calcium (C, bottom). The average ΔG/R values (black) were determined from the individual traces (gray). The average ΔG/R values were replotted for the spine (black) and the dendrite (gray) (D) and were used to determine the calcium levels in the spine and dendrite (E).
We used this approach to compare dendrite and spine calcium signals during the interval from 3 to 5 s and from 13 to 15 s in different regions of the Purkinje cell dendritic arbor (Fig. 7). Three representative regions are illustrated from one Purkinje cell that range from a large proximal dendrite (Fig. 7B,C) to an intermediate dendrite (Fig. 7D,E) to a fine distal dendrite (Fig. 7F,G). The dendrite (Fig. 7B,D,F, top) and region of the line scan are shown (Fig. 7B,D,F, bottom). Calcium transients averaged over the interval between 3–5 and 13–15 s after the onset of burst firing are shown (Fig. 7C,E,G, left and right, respectively). In a large proximal dendrite, basal calcium rose slowly from resting levels during the onset of burst firing and reached a final plateau of ∼300 nm, with peak levels reaching ∼600 nm. In intermediate and distal dendrites, basal calcium quickly rose to a plateau of ∼300 nm within 1–2 s and remained at this level throughout the 15 s duration of burst firing. Peak calcium transients rose only slightly during burst firing, reaching ∼600 nm between 3 and 5 s of bursting and 800 nm after 13–15 s of bursting. A summary of data from 93 regions in 16 Purkinje cells (Fig. 7H, top row) shows that, on average, basal calcium in dendrites remained at 200–300 nm throughout 15 s bursts. Peak dendritic calcium levels reached 400–600 nm in dendrites. In spines, basal calcium was not significantly different from that of the neighboring dendrite, but peak levels were higher, reaching almost 900 nm at the end of the 15 s bursts. These experiments establish that elevation of dendritic calcium for 15 s to levels of <1 μm is sufficient to evoke endocannabinoid release from Purkinje cell dendrites. These experiments suggest that it is the prolonged elevation of calcium, rather than the minor differences in peak calcium levels at 5 and 15 s, that accounts for the retrograde inhibition for 15 s and not 5 s bursts.
Figure 7.
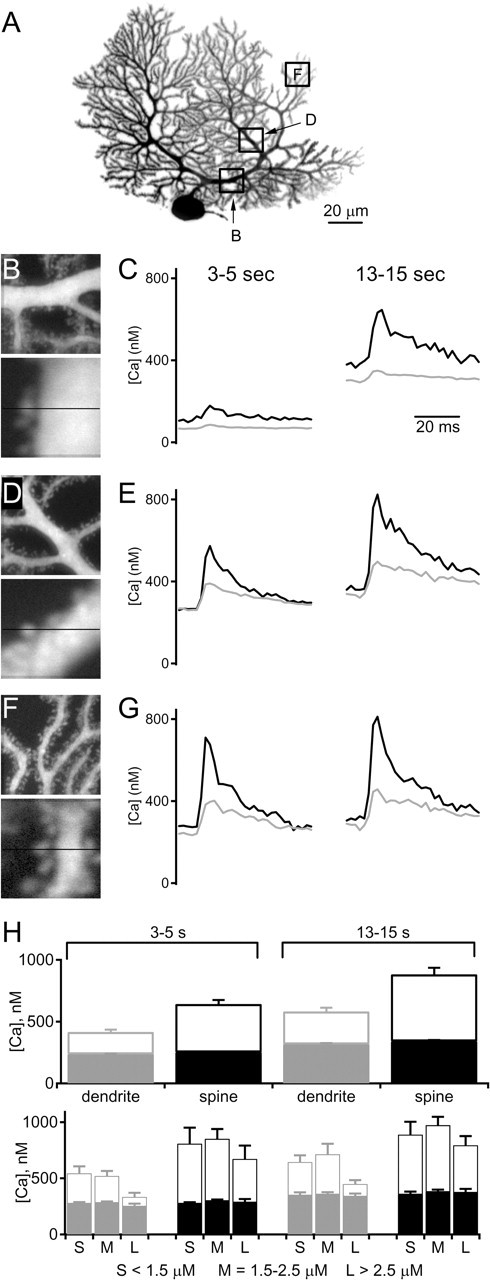
Calcium transients during Purkinje cell burst firing. A–G, Example of an experiment in which dendritic and spine calcium were determined in different parts of a cell during burst firing as in Figure 6. A, Image of a Purkinje cell showing representative regions in which calcium levels were measured. These regions are shown in an expanded view (B, D, F, top) and in an additional expanded view indicating the path of the line scan used to measure spine and dendritic calcium levels (B, D, F, bottom). Average calcium levels in spines (black) and dendrites (gray) for the period 3–5 s (C, E, G, left) and 13–15 s (C, E, G, right) from the onset of burst firing. H, Summary of the calcium transients measured in spines and dendrites during burst firing (top; n = 16 neurons, 93 regions). Filled bars indicate average baseline calcium, and open bars indicate peak calcium levels during bursts. In the bottom graph, data were divided into three groups consisting of dendrites <1.5, 1.5–2.5, and >2.5 μm in diameter
One possible explanation for our observation that endocannabinoid release selectively suppresses inhibitory synapses during burst firing is that burst-evoked dendritic calcium levels near inhibitory synapses are higher than those near excitatory synapses. We examined this issue by quantifying the calcium levels for dendrites of different diameters. We compared calcium levels in dendrites divided into groups consisting of thin dendrites <1.5 μm in diameter, intermediate dendrites between 1.5 and 2.5 μm, and large dendrites with diameters >2.5 μm (Fig. 7H, bottom row). These groups correspond to proximal, intermediate, and distal dendrites. Overall, calcium levels in dendrites and spines during bursts were similar in different regions of Purkinje cell dendrites. The levels of calcium were slightly higher in small dendrites, which correspond to the regions receiving PF synapses (which are not suppressed by endocannabinoids during bursts). The intermediate and large dendrites and their spines, which correspond to regions receiving inhibitory and CF synapses, had calcium increases that were slightly smaller in amplitude. There was no indication that dendritic regions receiving inhibitory synapses had larger calcium signals that could account for the preferential endocannabinoid release at inhibitory synapses.
Discussion
Here we show that, in contrast to the exceptionally high calcium levels required to evoke endocannabinoid release when calcium is increased transiently, modest but sustained elevations of dendritic calcium evoke endocannabinoid release from Purkinje cells. This suggests that Purkinje cells can globally regulate synapses via the PLC-independent release of endocannabinoids under physiological conditions. The observation that this mechanism of endocannabinoid release can selectively regulate inhibitory synapses suggests that sustained elevations of dendritic calcium could evoke endocannabinoid release that reduces feedforward inhibition at interneuron to PC synapses.
Calcium-dependent, PLC-independent endocannabinoid release
The endocannabinoid-mediated inhibition after prolonged elevation of calcium likely occurs by the same mechanism as DSE and DSI. The blockade of this synaptic suppression by CB1R antagonists, along with the presynaptic localization of CB1Rs on CF, PF, and interneuron boutons indicate that it reflects the retrograde action of endocannabinoids on synaptic inputs to PCs. The requirement for an increase in calcium was established by voltage-clamp studies of CF synapses, in which it was found that preventing a rise in dendritic calcium by including BAPTA in the recording electrode eliminated retrograde inhibition after a prolonged elevation of calcium. Finally, similar to DSE and DSI, retrograde inhibition after prolonged elevation of calcium was found to be independent of mGluR1s, PLC, and DAG lipase. These features distinguish this form of endocannabinoid-mediated synaptic suppression from that arising when modest calcium increases are combined with activation of Gq-coupled metabotropic receptors to activate a PLC-dependent pathway that produces the endocannabinoid 2-AG (Chevaleyre and Castillo, 2003; Maejima et al., 2005; Safo and Regehr, 2005).
There are several possible explanations for the observation that sustained calcium elevations lower the peak calcium increases needed to evoke endocannabinoid release. The simplest possibility is that endocannabinoids are slowly released during sustained increases in calcium, but, because endocannabinoid uptake is slow, there is an accumulation of endocannabinoids and synaptic suppression slowly builds. This is difficult to quantitatively evaluate because of numerous nonlinearities that can influence endocannabinoid release, uptake, and their effect on synaptic transmission. Another possibility is that a rate-limiting step in the pathway of calcium-evoked endocannabinoid release has slow kinetics and high calcium affinity and can be efficiently activated by prolonged elevation of calcium in the submicromolar range. Alternatively, the intracellular pathways involved in endocannabinoid production could have a low initial calcium affinity but may be subject to modulation by a high-affinity calcium sensor. This raises the possibility that endocannabinoid release could be regulated in an activity-dependent manner that resembles posttetanic potentiation, in which neurotransmitter release is enhanced when calcium in presynaptic terminals is elevated to low levels for prolonged periods (Zucker and Regehr, 2002). However, it is not currently possible to distinguish among these possibilities because little is known about the PLC-independent signaling pathway that links postsynaptic calcium elevation to endocannabinoid release.
The ability of sustained levels of intracellular calcium to lower the calcium requirement for endocannabinoid release suggests that the PLC-independent pathway is likely to be involved in synaptic regulation under physiological conditions in other types of neurons as well. For inhibitory synapses onto hippocampal pyramidal cells, when postsynaptic calcium is elevated for durations of ∼10 s, 4 μm calcium is required for half-maximal synaptic suppression (Wang and Zucker, 2001). This is a relatively high level of calcium that could be difficult to achieve under physiological conditions. However, if a sustained elevation of calcium also promotes endocannabinoid release at lower levels of dendritic calcium in hippocampal cells, as we observe here for Purkinje cells, it is likely that prolonged action potential trains could promote the release of endocannabinoids. In hippocampal cells and cortical pyramidal cells, trains of sodium action potentials evoke endocannabinoid release and retrograde inhibition, but these experiments have not been performed under conditions in which the PLC pathway is blocked and are often performed in the presence of bath-applied agonists of Gq-coupled receptors (Pitler and Alger, 1992; Morishita and Alger, 2001; Hampson et al., 2003; Bacci et al., 2004; Fortin et al., 2004). Thus, it remains uncertain whether PLC-independent pathways of endocannabinoid release contribute to the retrograde suppression after trains of spikes. Moreover, suppression after more sustained firing that leads to a prolonged elevation of calcium has not been assessed.
Synapse selectivity
Although PF, CF, and inhibitory synapses all express CB1Rs presynaptically and can be suppressed by postsynaptic depolarization, endocannabinoid release evoked by prolonged elevation of calcium to levels of 0.4–1 μm for 15 s only suppressed inhibitory synapses. Voltage-clamp and current-clamp studies are consistent with endocannabinoid-mediated suppression at CF synapses requiring higher calcium levels than inhibitory synapses. Current-clamp experiments established that endocannabinoid-dependent suppression occurs at inhibitory synapses when calcium levels are <1 μm and suggest that higher levels are required for CF and PF synapses. Voltage-clamp studies indicated that suppression of CF synapses requires postsynaptic calcium to be increased to 2 μm for 15 s. Unfortunately, differences in the properties of calcium signals in voltage-clamp and current-clamp experiments could complicate the interpretation of these experiments, and it was not possible to determine the calcium dependence of retrograde suppression for PF and inhibitory synapses at 35°C. At 24°C after brief calcium increases, DSE at CF and PF synapses and DSI all required similar calcium levels of ∼15 μm to evoke half-maximal synaptic suppression, but small elevations of calcium suppressed inhibitory synapses to a greater extent than CF and PF synapses. A difference in the calcium sensitivity of retrograde inhibition at different synapses could arise from differences in proteins that mediate endocannabinoid release, uptake mechanisms, or the sensitivity of presynaptic terminals to endocannabinoids. In the hippocampus, inhibitory synapses are more sensitive to endocannabinoids than excitatory synapses (Ohno-Shosaku et al., 2002), perhaps as a result of higher levels of CB1Rs at inhibitory boutons. Such differential endocannabinoid sensitivity could contribute to the selective suppression of inhibitory synapses we observe, but it is not known whether CB1Rs are expressed differentially at inhibitory and excitatory nerve terminals in the cerebellum. Another possibility is that dendritic regions near inhibitory synapses are exposed to larger calcium transients. This is unlikely because we find that calcium levels during bursts are slightly higher in dendritic regions receiving parallel fiber inputs than in those regions receiving the majority of inhibitory synapses. It remains possible, however, that differences in either local calcium buffering or in the proximity of proteins involved in endocannabinoid synthesis to calcium channels could result in differences in local calcium signals at inhibitory and excitatory synapses.
The observation that prolonged burst firing caused a suppression of PF and CF synapses that was not sensitive to the CB1 receptor antagonist AM251 indicates that additional mechanisms contribute to suppression of synapses after prolonged elevation of calcium. Additional studies will be required to determine whether this reflects a presynaptic change mediated by a retrograde messenger, a postsynaptic change in AMPA receptors, or shunting by calcium-activated conductances in the postsynaptic cell.
Implications for cerebellar function
Our results suggest that PLC-independent endocannabinoid release has the potential to globally regulate synapses under physiological conditions provided calcium is elevated to modest levels for prolonged periods. One way of elevating calcium is through spontaneous Purkinje cell firing. The ability of Purkinje cell spiking to evoke endocannabinoid release and regulate inhibitory synapses depends on the firing pattern. The lack of endocannabinoid-mediated modulation in tonic mode is consistent with known properties of Purkinje cells. Unlike many other types of neurons, Purkinje cells do not express sodium channels in their dendrites (Stuart and Hausser, 1994). Therefore, sodium spikes in Purkinje cells do not backpropagate into their dendrites. Moreover, potassium channels reduce the spread of depolarization within Purkinje cell dendrites to prevent dendritic calcium increases during tonic firing (Swensen and Bean, 2003; Womack and Khodakhah, 2003).
In contrast, burst firing elevated dendritic calcium and led to endocannabinoid release that suppressed inhibitory synapses. During burst firing, resurgent sodium current provides depolarizing drive that activates calcium channels in Purkinje cell dendrites (Raman et al., 1997). As the dendrite depolarizes, calcium levels increase and strongly activate calcium-activated potassium channels that hyperpolarize the dendrite, contributing to the termination of each cycle of the burst (Womack et al., 2004). Consistent with this mechanism, we observed that calcium levels rapidly reached an elevated steady-state plateau during bursts, from which small spikes arise in phase with the termination of each cycle of the burst. The timing of the calcium spike relative to the hyperpolarization of the cell after an individual cycle of a burst is consistent with calcium terminating the burst by activating calcium-activated potassium channels.
Although in brain slices Purkinje cells fire in tonic and burst mode, the firing pattern of Purkinje cells observed in vivo is more complicated because it is strongly affected by synaptic activity (Jaeger and Bower, 1994; Womack and Khodakhah, 2002). Moreover, the dendritic calcium signals that occur in vivo are not known. Our results indicate that calcium elevations evoked by ongoing complex spikes alone are not sufficient to cause endocannabinoid release. This is likely because rates of CF activity in vivo (<2 Hz) are much lower that rates of calcium spikes during bursts in vitro (10–20 Hz). Therefore, synaptic activation combined with activation of intrinsic conductances that lead to a more sustained elevation of calcium is likely to be required for endocannabinoid release through the mechanism we describe.
The high density of voltage-gated calcium channels in PC dendrites that underlie calcium signals during bursts is likely to elevate dendritic calcium during heightened periods of activity in vivo. Our results suggest that, if calcium increases are sustained, they will lead to the widespread release of endocannabinoids from Purkinje cell dendrites. For high levels of calcium, CF, PF, and inhibitory synapses will all be suppressed. For calcium elevations comparable with what we see in burst mode, endocannabinoids will selectively reduce the effect of inhibitory inputs. By activating both Purkinje cells and stellate cells, PF activity causes both direct excitation and feedforward inhibition of Purkinje cells. As a result, preferential suppression of stellate cell inputs to Purkinje cells by endocannabinoids will reduce this feedforward inhibition, thereby increasing the amplitude and duration of PF excitation as well as increasing spike output of Purkinje cells. This could facilitate the induction of long-term plasticity of excitatory synapses (Chevaleyre and Castillo, 2004).
Footnotes
*S.D.B. and A.R.B. contributed equally to this work.
This work was supported by National Institutes of Health Grants 5R01NS044396-04 (W.G.R.) and 5F32NS46842-02 (S.D.B.). We thank the members of the Regehr laboratory for critical reading of this manuscript and Bernardo Sabatini for assistance with two-photon microscopy.
References
- Bacci A, Huguenard JR, Prince DA (2004). Long-lasting self-inhibition of neocortical interneurons mediated by endocannabinoids. Nature 431:312–316. [DOI] [PubMed] [Google Scholar]
- Brenowitz SD, Regehr WG (2003). Calcium dependence of retrograde inhibition by endocannabinoids at synapses onto Purkinje cells. J Neurosci 23:6373–6384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenowitz SD, Regehr WG (2005). Associative short-term synaptic plasticity mediated by endocannabinoids. Neuron 45:419–431. [DOI] [PubMed] [Google Scholar]
- Brown SP, Brenowitz SD, Regehr WG (2003). Brief presynaptic bursts evoke synapse-specific retrograde inhibition mediated by endogenous cannabinoids. Nat Neurosci 6:1048–1057. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE (2003). Heterosynaptic LTD of hippocampal GABAergic synapses: a novel role of endocannabinoids in regulating excitability. Neuron 38:461–472. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE (2004). Endocannabinoid-mediated metaplasticity in the hippocampus. Neuron 43:871–881. [DOI] [PubMed] [Google Scholar]
- Duguid IC, Smart TG (2004). Retrograde activation of presynaptic NMDA receptors enhances GABA release at cerebellar interneuron-Purkinje cell synapses. Nat Neurosci 7:525–533. [DOI] [PubMed] [Google Scholar]
- Fortin DA, Trettel J, Levine ES (2004). Brief trains of action potentials enhance pyramidal neuron excitability via endocannabinoid-mediated suppression of inhibition. J Neurophysiol 92:2105–2112. [DOI] [PubMed] [Google Scholar]
- Foster KA, Kreitzer AC, Regehr WG (2002). Interaction of postsynaptic receptor saturation with presynaptic mechanisms produces a reliable synapse. Neuron 36:1115–1126. [DOI] [PubMed] [Google Scholar]
- Freund TF, Katona I, Piomelli D (2003). Role of endogenous cannabinoids in synaptic signaling. Physiol Rev 83:1017–1066. [DOI] [PubMed] [Google Scholar]
- Gilbert PF, Thach WT (1977). Purkinje cell activity during motor learning. Brain Res 128:309–328. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY (1985). A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem 260:3440–3450. [PubMed] [Google Scholar]
- Hampson RE, Zhuang SY, Weiner JL, Deadwyler SA (2003). Functional significance of cannabinoid-mediated, depolarization-induced suppression of inhibition (DSI) in the hippocampus. J Neurophysiol 90:55–64. [DOI] [PubMed] [Google Scholar]
- Isokawa M, Alger BE (2006). Ryanodine receptor regulates endogenous cannabinoid mobilization in the hippocampus. J Neurophysiol 95:3001–3011. [DOI] [PubMed] [Google Scholar]
- Jaeger D, Bower JM (1994). Prolonged responses in rat cerebellar Purkinje cells following activation of the granule cell layer: an intracellular in vitro and in vivo investigation. Exp Brain Res 100:200–214. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG (2001a). Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron 29:717–727. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG (2001b). Cerebellar depolarization-induced suppression of inhibition is mediated by endogenous cannabinoids. In: J Neurosci 21: RC174(1-5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maejima T, Oka S, Hashimotodani Y, Ohno-Shosaku T, Aiba A, Wu D, Waku K, Sugiura T, Kano M (2005). Synaptically driven endocannabinoid release requires Ca2+-assisted metabotropic glutamate receptor subtype 1 to phospholipase Cβ4 signaling cascade in the cerebellum. J Neurosci 25:6826–6835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mano N, Kanazawa I, Yamamoto K (1986). Complex-spike activity of cerebellar Purkinje cells related to wrist tracking movement in monkey. J Neurophysiol 56:137–158. [DOI] [PubMed] [Google Scholar]
- Morishita W, Alger BE (2001). Direct depolarization and antidromic action potentials transiently suppress dendritic IPSPs in hippocampal CA1 pyramidal cells. J Neurophysiol 85:480–484. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Maejima T, Kano M (2001). Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron 29:729–738. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Tsubokawa H, Mizushima I, Yoneda N, Zimmer A, Kano M (2002). Presynaptic cannabinoid sensitivity is a major determinant of depolarization-induced retrograde suppression at hippocampal synapses. J Neurosci 22:3864–3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piomelli D (2003). The molecular logic of endocannabinoid signalling. Nat Rev Neurosci 4:873–884. [DOI] [PubMed] [Google Scholar]
- Pitler TA, Alger BE (1992). Postsynaptic spike firing reduces synaptic GABAA responses in hippocampal pyramidal cells. J Neurosci 12:4122–4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Sprunger LK, Meisler MH, Bean BP (1997). Altered subthreshold sodium currents and disrupted firing patterns in Purkinje neurons of Scn8a mutant mice. Neuron 19:881–891. [DOI] [PubMed] [Google Scholar]
- Safo PK, Regehr WG (2005). Endocannabinoids control the induction of cerebellar LTD. Neuron 48:647–659. [DOI] [PubMed] [Google Scholar]
- Stella N, Schweitzer P, Piomelli D (1997). A second endogenous cannabinoid that modulates long-term potentiation. Nature 388:773–778. [DOI] [PubMed] [Google Scholar]
- Stuart G, Hausser M (1994). Initiation and spread of sodium action potentials in cerebellar Purkinje cells. Neuron 13:703–712. [DOI] [PubMed] [Google Scholar]
- Swensen AM, Bean BP (2003). Ionic mechanisms of burst firing in dissociated Purkinje neurons. J Neurosci 23:9650–9663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien R, Pozzan T (1989). Measurement of cytosolic free Ca2+ with quin2. Methods Enzymol 172:230–262. [DOI] [PubMed] [Google Scholar]
- Wang J, Zucker RS (2001). Photolysis-induced suppression of inhibition in rat hippocampal CA1 pyramidal neurons. J Physiol (Lond) 533:757–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA (2001). Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature 410:588–592. [DOI] [PubMed] [Google Scholar]
- Womack M, Khodakhah K (2002). Active contribution of dendrites to the tonic and trimodal patterns of activity in cerebellar Purkinje neurons. J Neurosci 22:10603–10612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womack MD, Khodakhah K (2003). Somatic and dendritic small-conductance calcium-activated potassium channels regulate the output of cerebellar Purkinje neurons. J Neurosci 23:2600–2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womack MD, Khodakhah K (2004). Dendritic control of spontaneous bursting in cerebellar Purkinje cells. J Neurosci 24:3511–3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womack MD, Chevez C, Khodakhah K (2004). Calcium-activated potassium channels are selectively coupled to P/Q-type calcium channels in cerebellar Purkinje neurons. J Neurosci 24:8818–8822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG (2002). Short-term synaptic plasticity. Annu Rev Physiol 64:355–405. [DOI] [PubMed] [Google Scholar]