

Abstract
Neurotrophins have been shown to play a critical role in activity-dependent synaptic plasticity such as long-term potentiation (LTP) in the hippocampus. Although the role of brain-derived neurotrophic factor (BDNF) and its tyrosine kinase receptor [tyrosine receptor kinase B (TrkB)] is well documented, it still remains unresolved whether presynaptic or postsynaptic activation of TrkB is involved in the induction of LTP. To address this question, we locally and specifically interfered with a downstream target of the TrkB receptor, phospholipase Cγ (PLCγ). We prevented PLCγ signaling by overexpression of the PLCγ pleckstrin homology (PH) domain with a Sindbis virus vector. The isolated PH domain has an inhibitory effect and thereby blocks endogenous PLCγ signaling and consequently also IP3 production. Surprisingly, concurrent presynaptic and postsynaptic blockade of PLCγ signaling was required to reduce LTP to levels comparable with those in TrkB and BDNF knock-out mice. Blockade of presynaptic or postsynaptic signaling alone did not result in a significant reduction of LTP.
Keywords: synaptic plasticity, hippocampus, long-term potentiation, neurotrophins, neurotrophin receptors, intracellular signaling
Introduction
Brain-derived neurotrophic factor (BDNF), the major ligand of tyrosine receptor kinase B (TrkB), has been shown to be an important component controlling activity-dependent synaptic plasticity (Poo, 2001; Huang and Reichardt, 2003). Genetic approaches have shown that homozygous as well as heterozygous BDNF knock-out (KO) mice exhibit a marked reduction in long-term potentiation (LTP) (Korte et al., 1995; Patterson et al., 1996), which can be rescued by adenovirus-mediated local overexpression of BDNF (Korte et al., 1996) or by application of recombinant BDNF (Patterson et al., 1996). On the other hand, direct application of the recombinant protein to normal hippocampal slices enhances synaptic strength (Kang and Schuman, 1995) and the probability of LTP induction (Figurov et al., 1996). Moreover, the local application of BDNF together with a weak burst of presynaptic activity can induce LTP (Kovalchuk et al., 2002). Conversely, blockade of BDNF–TrkB interaction by TrkB-receptor bodies (Kang et al., 1997; Chen et al., 1999) or anti-BDNF antibodies (Chen et al., 1999) strongly reduces hippocampal LTP.
Experiments using conditional TrkB knock-out mice in which TrkB is removed postnatally from the forebrain have demonstrated that BDNF is mediating the effect on CA3–CA1 synaptic plasticity via activation of TrkB (Minichiello et al., 1999; Xu et al., 2000). Binding of BDNF leads to dimerization of TrkB receptors and to autophosphorylation of its tyrosine residues. This, on one hand, activates the Shc pathway and on the other hand leads to the association of phospholipase Cγ (PLCγ) with TrkB (Huang and Reichardt, 2003). We have shown previously that for hippocampal LTP, the PLCγ pathway plays a predominant role (Minichiello et al., 2002), whereas Shc pathway is of lesser if of any importance (Korte et al., 2000). After association with activated TrkB, PLCγ is phosphorylated and after translocation to the plasma membrane, cleaves phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2] into diacylglycerol (DAG) and inositol 1, 4, 5-triphosphate (IP3) (Huang and Reichardt, 2003). DAG stimulates protein kinase C (PKC), whereas IP3 releases Ca2+ from internal stores by binding to IP3 receptors (IP3Rs). The proper associations of activated PLCγ to the plasma membrane is mediated by the N-terminal pleckstrin homology (PH) domain of PLCγ (Falasca et al., 1998; Maffucci and Falasca, 2001). The PH domain conveys membrane association via binding to polyphosphoinositides PI(3,4,5)P3. We used this property and interfered with PLCγ signaling by overexpressing a green fluorescent protein (GFP)-tagged PLCγPH domain by a Sindbis (Sin) virus vector. The PLCγPH domain acts as a competitor of endogenous PLCγ and thereby blocks PLCγ signaling by a functional dominant-negative effect (Falasca et al., 1998). Thus, local injections of the Sindbis virus into the CA1 or the CA3 region allowed us to disrupt PLCγ signaling specifically on either presynaptic or postsynaptic side.
Materials and Methods
Acute hippocampal slice preparation.
Acute hippocampal slices were prepared from wild-type C57BL/6 mice (postnatal day 40–60) or from TrkBPLC/PLC mice following standard procedures. In brief, mice were anesthetized with diethyl ether and decapitated, and the brains were quickly dissected and placed into ice-cold constantly aerated (95% O2, 5% CO2) artificial CSF (in mm: 125 NaCl, 2.5 KCl, 1 MgCl2, 26 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, 25 glucose). Hippocampi were cut with a vibratome (350 μm; VT 1000S; Leica, Nussloch, Germany), and sections were kept submerged in a storage chamber in aerated ACSF. Before electrical recording or virus transduction, vibratome sections were left in the storage chamber for ∼1 h at room temperature so that neuronal activity could recover to baseline.
Preparation of dissociated hippocampal and cortical cell cultures.
Hippocampi or cortices were dissected from embryonic day 19 outbred Wistar rat embryos. Neurons were dissociated by incubation in trypsin-EDTA (20 min, 37°C; Sigma, Taufkirchen, Germany) and trituration in DMEM (Invitrogen, San Diego, CA)/10% horse serum (HS) (Sigma). Dissociated cells were plated on 13 mm polyornithin-coated glass coverslips in 24-well cell culture plates at a density of 60–70,000 cells/cm2 and kept at 37°C at 10% CO2. After 2–4 h, DMEM/10% HS was replaced with 500 μl per well of complete medium (Brewer and Cotman, 1989). Three days after plating, cytosine β-d-arabinofuranoside (1.5 μm; Sigma) was added to the medium to prevent propagation of glial cells. Cultures were used for experiments 7–14 d after preparation.
Construction of Sindbis viral vectors.
A replication-deficient Sindbis virus was produced by cotransfection of baby hamster kidney (BHK) cells by in vitro transcribed RNAs derived from a vector DH(26S)5′tRNA and the vector nsp2S carrying the transgene of interest (both vectors and nsp2S–EGFP were kindly provided by P. Osten, Max-Planck-Institute for Experimental Medicine, Heidelberg, Germany). The nsp2S variant of the Sindbis virus carries a mutation that minimizes the viral toxicity (Dryga et al., 1997). A PCR fragment of PLCγ–PHGFP (vector was kindly provided by M. Falasca, University College London, London, UK) was cloned into the nsp2S vector via SphI and XbaI sites. A β-galactosidase-containing Sindbis virus was produced using the Sindbis expression system (Invitrogen). Supernatants of cotransfected BHK cells were cleared by centrifugation and viral particles purified in a sucrose step gradient (20/55%). Inserted sequences were verified by sequencing and the expression of the correct protein in neurons by probing neuron lysates by Western blot using an anti-GFP antibody (Boehringer Mannheim, Mannheim, Germany).
Virus injection into acute hippocampal slices.
Hippocampal slices were transferred onto a Millicell filter membrane (Millipore, Schwalbach, Germany) in ACSF, forming a miniature interface system that prevented drying of the slice. Viral particles were pressure-injected (5–10 psi, 5–10 ms pulses) into the pyramidal cell layer of area CA1, CA3, or both by a Pneumatic PicoPump PV820 (World Precision Instruments, Sarasota, FL), connected to a patch pipette (tip diameter, 10 μm). The pipette was inserted into the slice under visual control through an inverted microscope. Injections were repeated at four to six locations to achieve efficient gene transfer into large parts of the CA1 or CA3 regions. The total time for injection was limited to <2 min to avoid slice deterioration. Injected slices were transferred to an oxygenated submersion chamber equipped with a heating wire and were stored at 37°C.
In all experiments, slices were injected with Sin–GFP at the same locations and treated equally, whereas Sin–PHGFP-injected served as positive control.
Extracellular recordings.
Starting 7–10 h after virus injection, slices were transferred to a recording chamber and kept submerged at 32°C while constantly being perfused (1 ml/min) with aerated ACSF. Schaffer collaterals in area CA3 were stimulated by monopolar tungsten electrodes, and field EPSPs (fEPSPs) were recorded by a glass electrode placed in the stratum radiatum of CA1. Electrodes were inserted in the region of GFP expression. To prove that the recording electrode was positioned within the transgene expression zone, optical glass fibers were used in most of the experiments. One end of the glass fiber was inserted into the recording electrode through a lateral opening in the electrode holder, while the other end was connected to a 15 V, 100 W halogen lamp. After insertion of the electrode into the slice, the illuminated electrode tip was visible, which was aligned with a fluorescence image of the GFP-expressing area. The tungsten stimulation electrode, which was visible by light microscopy, was positioned comparing the tip position with the fluorescence image. The stimulation electrode was placed close to the apical side of the CA3 pyramidal layer to ensure activation of axons from transduced neurons. In some experiments, a control pathway was used by inserting a second stimulus electrode in the CA3 region of the slice. By using a paired-pulse method, it was ensured that the test and control pathway were not overlapping. Synaptic field potentials were elicited with a frequency of 0.1 or 0.07 Hz and LTP induced by tetanic stimulation [three bursts of 30 stimuli at 100 Hz; interstimulus interval (ISI), 5 s; stimulus duration, 100 μs]. Data were collected with a LabView (National Instruments, Munich, Germany) based acquisition program and slopes of the fEPSPs were calculated.
Patch-clamp recordings.
After 1 week in culture, dissociated hippocampal neurons were transduced with Sin–GFP and incubated for 24 or 48 h. Neurons were perfused with external solution containing the following (in mm): 145 NaCl, 3 KCl, 10 HEPES, 3 CaCl2, 8 glucose, 2 MgCl2) at 0.5 ml/min. Patch pipettes, pulled with a Narishige (Tokyo, Japan) electrode puller (PP 830) to a resistance of 6–10 MΩ, were filled with internal solution containing the following (in mm): 136.5 K-gluconate, 17.5 KCl, 9 NaCl, 1 MgCl2, 10 HEPES, 0.2 EGTA, 2 Na-ATP, pH 7.2). Neurons were patched in whole-cell voltage-clamp mode (Axoclamp 2B amplifier; Molecular Devices, Sunnyvale, Ca). After a stable patch was established, the membrane potential was recorded in current-clamp mode. Only cells with resting membrane potentials below −50mV were included in the analysis. No holding current was applied. Data were sampled at 5 kHz, low-pass filtered at 2 kHz, and recorded with an acquisition program written in LabView. To generate current–voltage curves, steps of ∼0.02 nA (step duration, 100 ms) were applied. Data were analyzed and membrane resistance, time constant, and action potential size calculated with a LabView-based, customized analysis program.
IP3 measurement.
Hippocampal or cortical neurons were plated on 10 cm Falcon dishes at a density of 70,000 (hippocampal neurons) or 100,000 (cortical neurons) cells per cm2. After 5 d, neurons were transduced with Sindbis viral vectors yielding a transduction efficiency of ∼60–80% (as estimated by the observation of GFP fluorescence). After 6 and 24 h of transduction, one-half of the cultures were stimulated with 150 ng/ml BDNF for 10 min and the medium removed except for 500 μl. The reaction was stopped immediately by adding 500 μl of ice-cold trichloroacetic acid (15% v/v). IP3 levels of the ether-extracted supernatants were measured using the Biotrak (3H) IP3 assay system (TRK 1000; Amersham Biosciences, Braunschweig, Germany).
The experiment was performed five times using five different batches of neuronal cultures. Each experiment was accompanied by a control, which consisted of neurons transduced by Sin–GFP. For each experiment, we transduced one to three dishes per condition: (1) control neurons transduced with Sin–GFP not stimulated with BDNF, (2) control neurons transduced with Sin–GFP stimulated with BDNF, (3) Sin–PHGFP transduced neurons not stimulated, and (4) Sin–PHGFP transduced neurons stimulated with BDNF. We measured the IP3 release from each individual dish and compared basal IP3 levels (cells not stimulated with BDNF) with IP3 levels after BDNF stimulation. To compare different experiments, which show different levels of IP3 production, we needed to normalize levels of BDNF-induced IP3 production to the level of IP3 production of nonstimulated neurons (condition 2 is normalized to condition 1 and 4 to 3). Within a batch, this normalization was useful and possible, because all experimental parameters, such as quality and density of the neurons, transduction efficiency, and performance of the IP3 assay, were similar and thus the variance in IP3 levels small. For the final evaluation of the experiment, we compared the normalized and averaged values of five different sets of experiments and expressed the results as percentage from the basal (i.e., nonstimulated) IP3 levels.
Immunocytochemistry.
Hippocampal neurons were transduced with Sindbis viral vectors for 24 h, fixed with 4% paraformaldehyde (PFA) (20 min), and permeabilized with 0.2% Triton X-100/PBS. Background fluorescence was quenched by 0.1 m glycine/PBS, and unspecific binding sites were blocked by 20% normal goat serum (NGS; Sigma)/PBS. Cells were incubated overnight with the primary antibody [microtubule-associated protein 2 (MAP2), clone number AP-20; GFAP, clone number G-A-5; 1:200; Sigma; tau-1; Calbiochem, La Jolla, CA], washed, and incubated for 1 h with the secondary antibody (1:200; anti-mouse Alexa-594; Invitrogen, Karlsruhe, Germany). All steps were performed at room temperature; the incubation with the first antibody was performed at 4°C. The antibodies were diluted in 1% NGS PBS. Acute hippocampal slices were fixed for 30 min in 4% PFA and afterward incubated in sucrose. Cryostat sections (50 μm) were immunostained as above.
Pharmacology.
2-Aminoethyldiphenyl borate (2-APB) and U-73122 (PLC inhibitor) were obtained from Biotrend (Cologne, Germany), and U-73343 (inactive structural analog to U-73122) was obtained from Sigma.
Results
Sindbis viral vectors selectively transduce neurons
To make definitive statements about neuronal presynaptic or postsynaptic TrkB signaling, a selective neuronal transduction by the Sindbis viral constructs was of crucial importance. This was achieved by using the DH(26S)t' helper vector (Lustig et al., 1988). To test for neuronal specificity, dissociated hippocampal cultures were transduced by the Sindbis viral vector containing enhanced GFP (Sin–GFP). Figure 1A–F shows that transduction was very efficient and confined to neurons: only MAP2- (neuronal marker) but not GFAP- (glial marker) positive cells expressed GFP.
Figure 1.
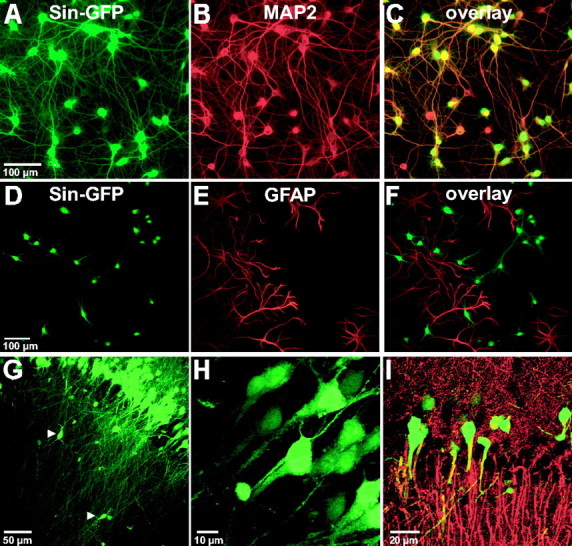
Sindbis viral vectors specifically target neurons in dissociated hippocampal cultures and acute slices. Dissociated hippocampal cultures (A–F) were transduced with Sin–GFP for 24 h. Only MAP2-positive cells (i.e., neurons) express GFP (C), whereas the glia-specific marker GFAP never labeled GFP-positive cells (F). The same specificity was seen in locally injected acute hippocampal slices (G–I) using the neuronal marker MAP2 (I) or by observing the very characteristic neuronal morphology of GFP-expressing cells (G, H). The arrowheads in G point to selected interneurons.
The same specificity was also observed in acute hippocampal slices. Neurons were very efficiently transduced by both Sin–GFP (Fig. 1G–I) and Sin–PHGFP (Fig. 2), as demonstrated by the coexpression of GFP and MAP2 (Fig. 1I) and the unambiguous morphology of GFP-positive cells, which were clearly identifiable as pyramidal neurons by their band-like arrangement (Fig. 1G) and their characteristic morphology (Fig. 1H). Long apical dendrites projecting far into stratum radiatum and shorter basal dendrites, extending into stratum oriens, were visible. Axons were labeled homogeneously and could be traced up to area CA1 from cells transduced in area CA3 (Fig. 3, schematic drawing in E and insets in F, G). Some small, dispersed, nonpyramidal neurons, most likely interneurons, were also transduced (Fig. 1G,H, arrows). We never observed transduced glial cells, corroborating the high specificity of the virus for neurons.
Figure 2.

The PLCγ1PH domain is associated to intracellular and extracellular membranes. A, B, Single confocal planes of dissociated hippocampal cells transduced by Sin–GFP (A) or Sin–PHGFP (B). The PHGFP domain shows a characteristic patchy distribution of a membrane associated protein, whereas Sin–GFP is evenly distributed in the cytoplasm and the nucleus. C, The PLCγ1PH domain is associated to intramembrane and plasma membrane. Two individual confocal planes of one hippocampal neuron expressing the GFP-tagged PLCPH domain are shown. In the middle plane, a clear accumulation in intracellular membranes is visible, whereas a plane through the bottom of the cell demonstrates the accumulation of the protein in the plasma membrane. D, The PLCγ1PH domain is expressed and transported into tips of axons (arrowhead) and dendrites (arrow) already 6 h after Sindbis viral-mediated transduction. In the top panel, the staining with the axonal marker tau-1 is shown, and in the bottom panel, the patchy GFP fluorescence of the GFP-tagged PH domain is shown. Scale bars, 10 μm.
Figure 3.
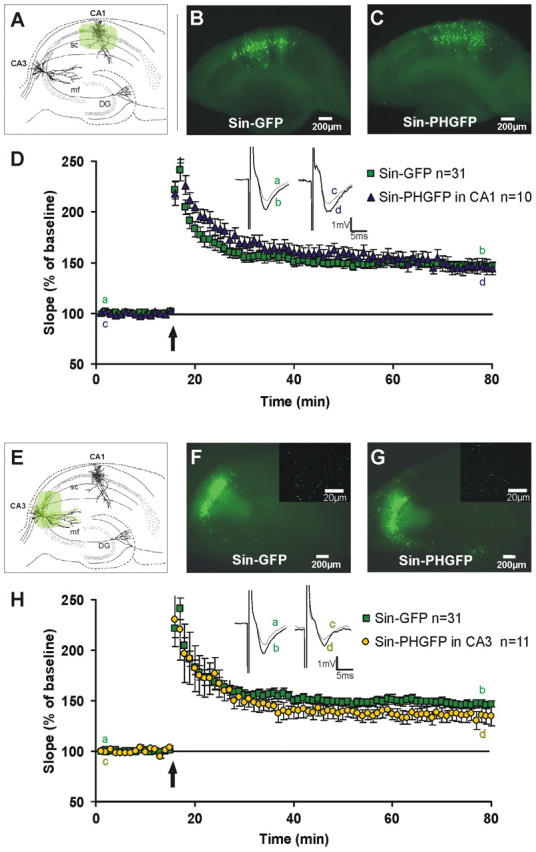
Local expression of the PLCγPH domain exclusively in area CA1 or exclusively CA3 does not decrease LTP significantly. Schematic and representative images of acute hippocampal slices injected in area CA1 (A–C) or CA3 (E–G) with either Sin–GFP (B, F) or Sin–PHGFP (C, G) are shown. No apparent difference was observable between extent and intensity of transgene expression in Sin–GFP- or Sin–PHGFP-transduced slices. After at least 6 h of incubation, Schaffer collaterals were stimulated and fEPSPs recorded in area CA1. EPSP slope size is shown before and after tetanic stimulation (3 × 30 pulses; 100 Hz; arrow) for Sin–PHGFP and Sin–GFP control slices in CA1 (D) and CA3 (H). There is no significant difference between Sin–GFP and Sin–PHGFP 55–60 min after tetanic stimulation (p > 0.1 for CA1 and CA3, t test). D, H, Insets show original sweeps from representative individual experiments. Letters correspond to the time point when traces were taken. F, G, Insets show axonal labeling of Sin–GFP and Sin–PHGFP, respectively. Error bars represent SEM. DG, Dentate gyrus; mf, mossy fibers; sc, Schaffer collaterals.
Confocal images of dissociated hippocampal neurons expressing the GFP (Fig. 2A) or GFP-tagged PLCγ1-PH domain (Fig. 2B–D, PHGFP) show that PHGFP is excluded from the nucleus and is localized, reminiscent of a membrane associated protein, in distinct spots along the plasma membrane and in the cytoplasm surrounding the nucleus.
Time course of virus-mediated transgene expression
The limited life time of acute hippocampal slices forced us to restrict the incubation time after viral injection to a minimum. In order to determine the time required for adequate transgene expression, acute hippocampal slices were injected with a Sindbis virus coding for β-galactosidase (Sin–βGal), because, especially at low levels of expression, β-Gal enzymatic activity is easier to detect than a weak GFP fluorescence. Sindbis-injected slices showed considerable β-Gal expression already 3 h after transduction, suggesting that transgene expression occurred at even earlier time points (data not shown). The number of cells expressing β-Gal increased rapidly, and after 6 h incubation, the majority of cells within the respective area were labeled (data not shown). Because the onset of visible expression of GFP or PHGFP is only slightly extended as a result of the need of the oxidation of GFP (time constant, ∼30 min), we chose to start the experiments after a minimal incubation time of 6 h for all our experiments. At this time, the GFP fluorescence was clearly visible in all experiments with acute slices. Moreover, at this time, we could detect the GFP or PHGFP fluorescence in the terminals of CA3 axons reaching the CA1 region (Figs. 2D, 3, insets), indicating that the time was sufficient for the delivery of the PH domain along the axons to the axon terminals.
To exclude that Sindbis virus cytotoxicity would influence our measurements, we analyzed membrane characteristics of Sin–GFP-transduced dissociated hippocampal pyramidal neurons by whole-cell recordings 24 and 48 h after transduction: resting membrane potential, action potential amplitude, time constant, and membrane resistance were similar to nontransduced controls at corresponding time points (supplemental Table 1, available at www.jneurosci.org as supplemental material). This made us confident that Sindbis virus transduction does not alter physiological properties of hippocampal pyramidal cells at least up to 48 h after transduction, a time point far beyond the duration of any of our experiments.
In addition, it is important to note that we could not detect any interference by possible cytotoxic viral effects in acute hippocampal slices extensively injected with Sin–GFP to naive control slices. The magnitude of LTP, which would be very sensitive to changes in the viability of neurons in the injected area, is neither altered by the virus expression nor by the mechanical manipulation attributable to the injection itself (control slices, noninjected, n = 8, 165.3 ± 5.2%; Sin–GFP-injected slices, n = 6, 158.1 ± 7.2; 50–55 min after LTP induction; p = 0.26, two-tailed t test).
PLCγPH domain overexpression leads to diminished PLCγ signaling and a reduction in BDNF induced IP3 production
Confocal images of dissociated hippocampal neurons expressing the GFP-tagged PLCγ PH domain show that PHGFP is excluded from the nucleus and is localized in distinct spots along the plasma membrane and in the cytoplasm surrounding the nucleus (Fig. 2). After extraction of soluble proteins by saponin, the fluorescent signal decreased but the typical patchy distribution remained, indicating a membrane association. To prove that the PLCγPH domain indeed interferes with endogenous PLCγ signaling and thus diminishes IP3 production (Falasca et al., 1998), we measured total IP3 levels in hippocampal and cortical neurons after stimulation with BDNF, comparing BDNF-induced IP3 production in Sin–PHGFP-transduced neurons with that in Sin–GFP-transduced control neurons. BDNF-induced IP3 production was normalized to basal IP3 levels in Sin–GFP- and Sin–PHGFP-transduced neurons, which were not stimulated with BDNF. Our data show that transduction with Sin–PHGFP blocks BDNF-induced IP3 production completely, whereas Sin–GFP-transduced neurons responded normally with IP3 production to the BDNF stimulus [Sin–PHGFP, 102 ± 8%; Sin–GFP, 179 ± 19% (mean ± SEM); p = 0.006]
LTP is not affected in selectively presynaptically or postsynaptically Sin–PHGFP-transduced slices
To interfere with PLCγ signaling exclusively in presynaptic or in postsynaptic neurons, Sin–PHGFP was injected into the CA3 or CA1 area of acute hippocampal slices. Control slices were injected with Sindbis vectors expressing only GFP. We injected large parts of area CA3, because the axons (Schaffer collaterals) do not project in a topographically defined order to the CA1 pyramidal cells (Ishizuka et al., 1990). Conversely, in area CA1, efficient transduction was necessary, because otherwise nontransduced neurons will contribute to the population response and “dilute” a potentially blocking effect of PLCγPH on LTP.
The virus injection and expression of a neutral transgene such as GFP did not alter basal synaptic transmission (EPSP slope and paired-pulse facilitation were normal; data not shown). Also, CA3–CA1 synaptic plasticity was not altered, as we demonstrated by measuring the magnitude of LTP in slices injected with Sin–GFP in area CA3 and CA1 very extensively, which is as high as in noninjected slices (see above).
Moreover, potentiation magnitudes after tetanic stimulation were the same in slices transduced with Sin–GFP in CA3 or CA1 alone, or in CA3 and CA1 simultaneously (Sin–GFP in CA3, 143.1 ± 6.7%, n = 13; Sin–GFP in CA1, 144.2 ± 3.9%, n = 10; Sin–GFP in CA3/CA1, 149.4 ± 5.5%, n = 8; LTP magnitude at 50–55 min after tetanic stimulation; p > 0.1 for all conditions, t test). Therefore, we grouped LTP data obtained from single (CA3 or CA1) and double (CA3 and CA1) Sin–GFP-transduced slices together as controls.
We then went on to the real experiment testing whether interference with PLCγ signaling in CA3 or CA1 would alter LTP. To this end, up to five injections were placed in area CA3 or CA1 to achieve the widest possible transgene expression in the respective area. After 6 h, large parts of the pyramidal cell layer were GFP positive, with a similar degree of expression efficiency and size of the labeled area in Sin–PHGFP- and Sin–GFP-transduced slices (Fig. 3B,C,F,G). Sin–GFP-injected slices showed a characteristically different distribution of GFP-expressing pyramidal cells in the CA1 and CA3 areas. Although most pyramidal neurons in the injected area were GFP positive, pyramidal cells in CA1 appeared more scattered (Fig. 3B,C) compared with the band-like appearance of labeled pyramidal neurons in CA3 (Fig. 3F,G).
In slices injected with Sin–PHGFP or Sin–GFP in postsynaptic area CA1, a solid LTP was inducible, which remained stable for at least 1 h after tetanic stimulation under both conditions. There was no significant difference between the magnitude of the potentiation of Sin–GFP control slices (Fig. 3D) (146.3 ± 3.8%; n = 31; 50–55 min after tetanus application) and Sin–PHGFP CA1 slices (Fig. 3D) (151.6 ± 7.5%; n = 10; p = 0.18, t test). Similarly, for slices transduced presynaptically in area CA3 with either Sin–PHGFP or Sin–GFP, there also was no statistically significant difference between LTP amplitudes (Fig. 3H) (146.3 ± 3.8% in control slices, n = 31; 137.4 ± 7.8% in Sin–PHGFP CA3 slices; 50–55 min after induction; n = 11; p = 0.12, t test), although a tendency toward a reduction of LTP was visible for Sin–PHGFP. Therefore, interference with PLCγ signaling in either hippocampal region (CA3 or CA1) alone did not reduce LTP in a significant manner.
LTP is reduced in presynaptically and postsynaptically Sin–PHGFP-injected slices
We therefore tested whether simultaneous injection of Sin–PHGFP into both CA3 and CA1 region might lead to a change in the magnitude of LTP. Again, despite virus injection into several sites within both areas, the slices retained a healthy appearance with well defined cell layers, unfrayed edges, and with normal basal synaptic transmission, measured by EPSP slope at different fiber volley sizes (data not shown) and paired-pulse facilitation (Sin–GFP-injected controls, 10 ms ISI, 170.4 ± 8.9; Sin–PHGFP, 167 ± 10.2, p > 0.1, t test; 20 ms ISI, 180.1 ± 9.3; Sin–PHGFP, 176 ± 12.1, p > 0.1, t test). In addition, a control pathway was recorded and did not show any decline in signal strength during the time course of the experiment (55–60 min after tetanus application in the test pathway; 96.3 ± 4.9; n = 3). There was no difference in transduction efficacy and area of transduction between Sin–PHGFP and Sin–GFP slices (Fig. 4B,C).
Figure 4.
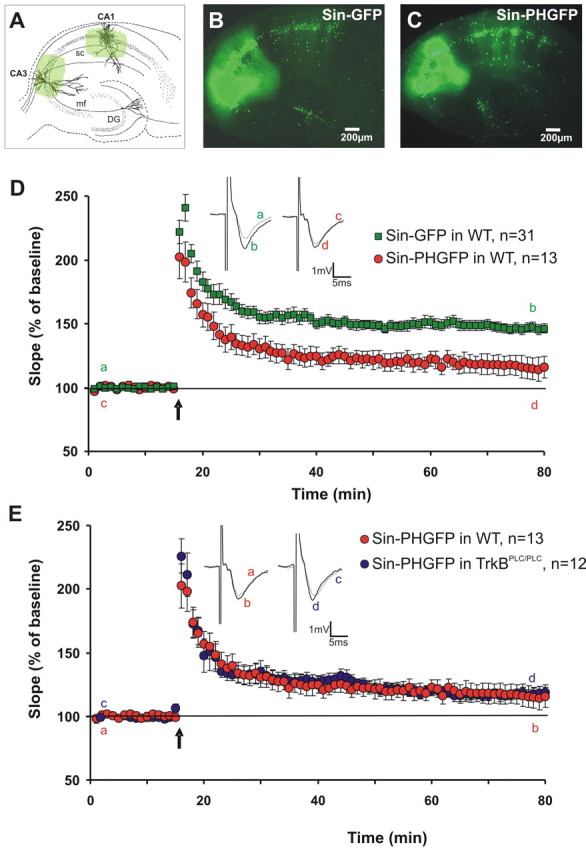
Expression of the PLCγ1PH domain in both the CA1 and CA3 region significantly decreases LTP. A–C, Schematic and representative images of acute hippocampal slices injected simultaneously in area CA3 and CA1 with either Sin–GFP (B) or Sin–PHGFP (C). After at least 6 h of incubation, synaptic field potentials in area CA1 were elicited through stimulation of Schaffer collaterals. D, E, Grouped recordings are shown before and after tetanic stimulation (3 × 30 pulses; 100 Hz; arrow) for double area injected Sin–PHGFP and Sin–GFP wild-type (WT) control slices and in TrkBPLC/PLC mice. The difference in LTP magnitude at 55–60 min after tetanic stimulation is significantly different from controls (p = 0.012, t test), whereas injection of Sin–PHGFP into TrkBPLC/PLC slice leads to the same low level of potentiation as Sin–PHGFP in wild-type slices (p = 0.72, t test). It is important to note that Sin–PHGFP injection into TrkBPLC/PLC slice did not further decrease the remaining low level of potentiation. D, E, Insets show original sweeps from representative individual experiments. Letters correspond to the time point when traces were taken. Error bars represent SEM. DG, Dentate gyrus; mf, mossy fibers; sc, Schaffer collaterals.
However, LTP recordings in presynaptically and postsynaptically Sin–PHGFP-injected slices now revealed a clear and statistically significant difference compared with LTP recordings in Sin–GFP-injected control slices (Fig. 4D) (Sin–GFP control, 146.3 ± 3.8%, n = 31; double-injected Sin–PHGFP slices, 115.6 ± 8.5%; 50–55 min after LTP induction; n = 13; p = 0.0014, t test). This reduction was visible directly after PTP (posttetanic potentiation), suggesting that PLCγ signaling is required already during the induction period of LTP. This reduction in LTP magnitude after presynaptic and postsynaptic blockade of PLCγ is comparable with that obtained in slices from BDNF and TrkB KO mice (Korte et al., 1995; Patterson et al., 1996; Minichiello et al., 1999; Xu et al., 2000). Our data, therefore, show that only concurrent presynaptic and postsynaptic blockade of PLCγ signaling interrupts hippocampal LTP.
Although the observed reduction in LTP clearly demonstrates the necessity of PLCγ signaling in the establishment of LTP, we determined the contribution of TrkB mediated PLCγ signaling. To distinguish between the involvement of the TrkB and other potential pathways signaling to PLCγ, we made use of mice carrying a homozygote mutation in the TrkB receptor: TrkBPLC/PLC mice (Minichiello et al., 2002). In these mice, the tyrosine 816 has been mutated into phenylalanine, thus disrupting very specific the activation of PLCγ after TrkB activation. In contrast, the Shc can still be activated by TrkB (Minichiello et al., 2002). These mice show a reduced LTP with ∼20% residual potentiation. We used these mice to test whether concurrent presynaptic and postsynaptic blockade of PLCγ by Sin–PHGFP would yield an additional decrease in LTP. If this were the case, other signaling mechanisms, independent of TrkB, might be responsible for the effect we observed. If, in contrast, no additional decrease were to occur, the effect would result from TrkB-mediated PLCγ signaling. This indeed was the case (Fig. 4E). Control slices from TrkBPLC/PLC mice injected with Sin–GFP showed a potentiation of 120 ± 4.8% at 50–55 min after tetanus (n = 5; data not shown), which again was undistinguishable from untransduced TrkBPLC/PLC slices (p > 0.1). Importantly, when we transduced slices of TrkBPLC/PLC mice with Sin–PHGFP, the residual LTP was undistinguishable (117.4 ± 5.1%; n = 12; p > 0.1) to both untransduced slices and GFP-transduced control slices of the same transgenic mice (Fig. 4E). This demonstrates that PLCγ signaling exclusively via the TrkB receptor is responsible for the diminished LTP.
Moreover, these experiments indicate the specificity of our approach using the PLCγPH domain. Because the TrkB receptor in the TrkBPLC/PLC slices is exclusively deficient in the signal transduction via PLCγ and not via any other TrkB-signaling pathway, one would expect an additional decrease in the magnitude of LTP after the transduction with the PLCγPH domain if the PH domain would also block other LTP-relevant pathways involved besides PLCγ. This is not the case, providing proof of an exclusive effect of the blockade of PLCγ on LTP.
Pharmacological blockade of IP3 receptors prevents induction of LTP
To use a virus-independent blockade of PLC signaling, we used pharmacological means to investigate the role of PLC signaling in CA3–CA1 synaptic plasticity. This approach is less specific, but we considered it to be important for obtaining independent pharmacological proof about PLC involvement in hippocampal CA3–CA1 synaptic plasticity and to further characterize downstream signaling. We first monitored LTP in slices, which were either treated with a selective membrane-permeable PLC inhibitor U-73122 (10 μm) or with the structural inactive analog U-73343 (10 μm) (Reyes-Harde and Stanton, 1998). The PLC inhibitor but not the inactive analog effectively blocked LTP (Fig. 5A). The difference between U-73343-treated and U-73122 slices was highly significant (control, U-73343, 155.8 ± 12.8%, 50–55 min after the tetanus application, n = 5; PLC inhibitor, U-73122, 121.1 ± 6.2%, n = 6; p = 0.013, t test). No effect on baseline synaptic transmission (fEPSP size or paired-pulse facilitation) was observed.
Figure 5.
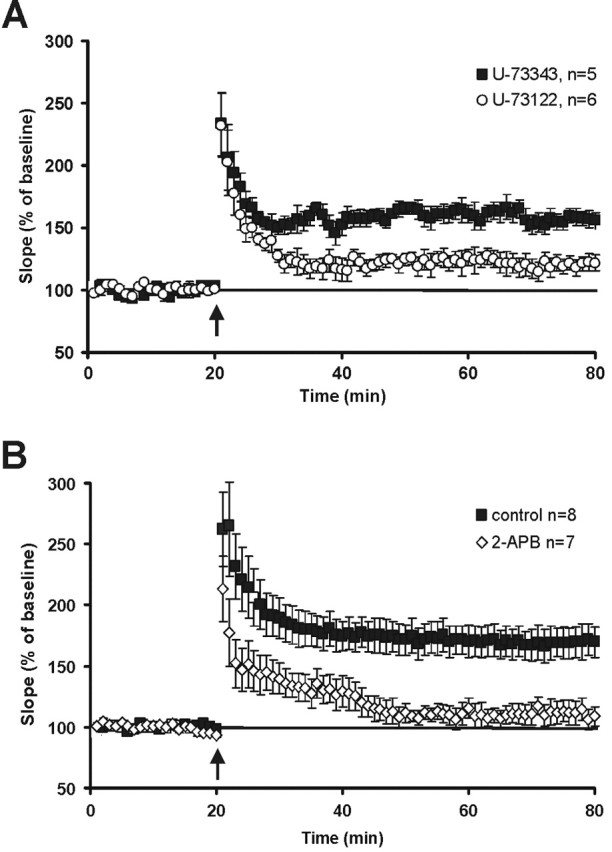
LTP is strongly reduced in hippocampal slices pretreated with PLC inhibitors or with the IP3 receptor blocker 2-APB. A, Slices treated with the selective membrane-permeable PLC inhibitor U-73122 (10 μm) show a significantly lower LTP (tetanic stimulation, 3 × 30, 100 Hz; arrow) than slices treated with the ineffective control substance U-73343 (10 μm). p = 0.003, t test. B, After tetanic stimulation (3 × 30 pulses; 100 Hz; arrow) of the Schaffer collaterals, control slices express pronounced potentiation, whereas slices incubated in 2-APB show no LTP (p = 0.0024, t test). Error bars represent SEM.
To further evaluate the downstream signaling components of the PLC pathway, we explored the involvement of IP3 receptor activation in synaptic enhancement. In this case, LTP was recorded in acute hippocampal slices that were incubated in 2-APB (75 μm), a membrane-permeable IP3 receptor antagonist that blocks IP3-mediated Ca2+ release from internal stores (Maruyama et al., 1997; Prakriya and Lewis, 2001). Slices were placed in 2-APB medium at least 20 min before tetanic stimulation and remained in 2-APB medium throughout the experiment. Incubation in 2-APB showed a strong effect on LTP (Fig. 5B) (control, 168.2 ± 6.1%, 50–55 min after the tetanus application, n = 8; 2-APB, 112.4 ± 13.2%, n = 7; p = 0.0054, t test). A clear disparity between the traces is apparent shortly after tetanic stimulation. Hence, the release of Ca2+ from intracellular stores seems to be necessary already during LTP induction.
Discussion
In the present paper, we interfered locally and specifically with PLCγ signaling downstream of the TrkB neurotrophin receptor. Previous studies have by and large interfered with the ligand BDNF. They were inconclusive as to whether BDNF acts in an anterograde, retrograde, or autocrine manner because they did not permit to distinguish between presynaptic and postsynaptic action of BDNF. In contrast, our new approach, which is based on interference on the receptor side, enabled us to investigate directly the relative contribution of presynaptic and postsynaptic TrkB signaling to CA3–CA1 synaptic plasticity. To our surprise, we found that signaling on either presynaptic or postsynaptic side is sufficient to support LTP, and only the interruption of the signaling cascade on both sides produced a marked reduction comparable with TrkB or BDNF KO mice.
In our experiments, PLCγ signaling was inhibited by virus-mediated overexpression of the GFP-tagged PH domain of PLCγ1. The use of Sindbis viruses enabled us to achieve a highly selective neuronal (Fig. 1) and at the same time, a rapid and efficient expression of the inhibitory PH domain or GFP as control (Figs. 3, 4). Moreover, importantly, local injections enabled us to confine the area of transgene expression to an exclusively presynaptic or postsynaptic locus (Fig. 3). Because this blockade of endogenous signaling is, in contrast to conventional gene deletion studies, only temporary, interference with the normal development can be excluded.
Our observations suggest that the effect of LTP reduction is fully accounted for by signaling via PLCγ, because the reduction in LTP is of a similar magnitude to that observed with genetic approaches, in which conditional TrkB KO mice (Minichiello et al., 1999; Xu et al., 2000) and BDNF KO mice (Korte et al., 1995; Patterson et al., 1996) were used. It is also unlikely that other IP3 generating pathways could have been affected, because the PH domain of PLCγ specifically binds PI(3,4,5)P3, whereas, for example, PLCδ binds with high affinity to PI(4,5)P2 (Lemmon et al., 1995) and its signaling should therefore not be affected. Furthermore, our results resembles the LTP reduction seen in mice that were mutated in the PLCγ recruitment site of TrkB (Minichiello et al., 2002), arguing further that BDNF mediated plasticity is completely carried by the PLCγ pathway. An additional strong argument for this assertion comes from experiments in which we show that overexpression of the PH domain in TrkBPLC/PLC mice yields no further reduction in LTP (Fig. 4E).
The fact that in the hippocampus BDNF/TrkB-mediated CA3–CA1 synaptic plasticity seems to be completely accomplished by the PLCγ pathway is reminiscent of observations at the neuromuscular junction, where NT3-mediated synaptic potentiation is solely dependent on TrkC activated PLCγ and IP3 production (Yang et al., 2001). Not in line to these and our data are observations in which IP3R type I KO mice show a deficient long-term depression in the cerebellum (Inoue et al., 1998) and normal LTP in the hippocampus (Fujii et al., 2000). This difference could be because of the different stimulus protocol (a weak tetanus was used), the targeting of only IP3R type I, and the use of rather young mice before weaning. The major difference is the IP3R was knocked out from early developmental stages, causing already a different development reflected by the perinatal death of most homozygous animals (causing the problem of a specific selection).
Because PLCγ activation results not only in the formation of IP3 but also of DAG, which in turn activates PKC, a contribution of the PKC pathway (Abeliovich et al., 1993) cannot be excluded. However, in this context, it should be noted that the involvement of PKC in LTP has recently been questioned, because the evidence for this is mainly based on the use of inhibitors with inconsistent specificities (Malenka and Nicoll, 1999) or PKC activation by phorbol esters, which seem to activate munc13 rather than PKC (Brose and Rosenmund, 2002).
Interestingly, LTP was only significantly reduced when the PH domain was expressed concomitantly in the CA3 and CA1 region, indicating that TrkB activation can act on both the presynaptic as well as postsynaptic side during LTP induction. This shows a redundancy of TrkB-mediated synaptic potentiation. This is in line with the following published results: BDNF and also other neurotrophins can be secreted presynaptically and postsynaptically in an activity-dependent manner (Gärtner et al., 2000; Hartmann et al., 2001; Kohara et al., 2001; Balkowiec and Katz, 2002; Gärtner and Staiger, 2002), and BDNF is required during the induction period of LTP (Kossel et al., 2001; Kovalchuk et al., 2002). BDNF can bind directly to available presynaptic or postsynaptic TrkB receptors (Drake et al., 1999), and it can thereby trigger presynaptic and postsynaptic PLCγ activation and Ca2+ release from endogenous Ca2+ stores (Li et al., 1998b; Canossa et al., 2001). Therefore, both sides of the synapse contain the molecular machinery for BDNF release as well as TrkB receptors, which subsequently could activate PLCγ. In fact, it has been shown that BDNF can modulate presynaptic and postsynaptic functions. Presynaptically, it was demonstrated that BDNF can increase the release of neurotransmitters via TrkB (Lessmann et al., 1994; Li et al., 1998a; Schinder et al., 2000). Postsynaptically, BDNF can modulate NMDA receptor function (Suen et al., 1997; Levine et al., 1998), Nav1.9 activation (Blum et al., 2002), and AMPA receptor expression (Lin et al., 1998; Narisawa-Saito et al., 1999).
Our experiments, in addition to demonstrating the involvement of PLCγ, show that the downstream IP3 receptor activation is of critical importance (Fig. 5B, 2-APB experiments). In line with these results are experiments that have used xestospongine, which blocks the function of IP3Rs at a different binding site than 2-APB, with similar effects on LTP induction (Raymond and Redman, 2002). This result is in accordance with experiments showing that interference with internal Ca2+ stores impairs LTP induction (Behnisch and Reymann, 1995; Harvey, 1995). Interestingly, also the presynaptic effect of increase in transmitter release mentioned previously is mediated by the internal calcium stores and IP3 (Li et al., 1998b; Kang and Schuman, 2000). All these observations are in line with our results, because they show that signaling molecules downstream of TrkB/PLCγ are important for LTP.
There are three previous studies that directly addressed the question of whether BDNF/TrkB acts presynaptically or postsynaptically in hippocampal LTP. Xu et al. (2000) tested whether completely ablating the TrkB receptors in CA1 and CA3 influenced LTP, and indeed it did. These authors also found that mice lacking TrkB in the CA1 region have an unchanged LTP, in comparison to the already reduced LTP in mice in which the TrkB receptor was ablated in CA1 and CA3. Overall, this is in line with our observation. From these data, they concluded that BDNF has an exclusively presynaptic role during LTP. Our data, although completely in line with their observation, show that things are more complicated as blocking TrkB only on the presynaptic side also shows no significant effect on LTP.
On the other hand, it was suggested that BDNF-mediated perforant path LTP is induced postsynaptically, because postsynaptic BDNF application on granule cells in the dentate gyrus together with increased presynaptic activity induced LTP (Kovalchuk et al., 2002). Apart from the fact that these authors have worked on a different synapse, their approach is fundamentally different from ours so that the results are difficult to compare. In fact, the authors themselves explicitly state that their experiments do not exclude an additional presynaptic mechanism.
Finally, Zakharenko et al. (2003) reported that in the CA3–CA1 pathway in hippocampal slices, BDNF is released acting only presynaptically and that there is no postsynaptic effect on synaptic plasticity. At first sight, their results seems contradictory to our findings. However, on closer inspection they are not. As far as the side of release is concerned, our study did not address this question. In terms of the side of action of BDNF, our data do not contradict theirs; we do, however, draw different conclusions. We think that our data interfering with TrkB signaling provide strong evidence that presynaptic and postsynaptic BDNF action can support hippocampal LTP, whereas their data mainly address the question of where BDNF is produced and released. In general, it seems to us that only experiments working with the receptor can provide conclusive answers with respect to the side of action of BDNF. Experiments interfering only with the ligand will in principle not be able to distinguish between an autocrine and a paracrine action. Our data are, therefore, in good agreement with previous observations. However, we extend the previous findings in an important manner in that we show that only the blockade on both sides of the synapse effectively reduces LTP.
In summary, our experiments show that presynaptic as well as postsynaptic TrkB signaling can support hippocampal synaptic plasticity. Our findings support the view that both are active during normal synaptic enhancement, and it is therefore tempting to speculate that this redundancy might constitute a safety mechanism to ensure that synaptic strengthening in the hippocampus is performed in a reliable and robust manner.
Footnotes
*A.G. and D.G. contributed equally to this work.
A. Gärtner's present address: Cavalieri Ottolenghi Scientific Institute, University of Turin, Regione Gonzole 10, 10043 Orbassano, Turin, Italy.
This work was supported by the Max-Planck Society as well as the Deutsche Forschungsgemeinschaft (Heisenberg-Stipend to M.K.) and the Volkswagen Foundation (M.K., T.B.). We thank Arthur Konnerth for valuable comments on a previous version of this manuscript and Pavel Osten and Marco Falasca for reagents.
References
- Abeliovich A, Chen C, Goda Y, Silva AJ, Stevens CF, Tonegawa S (1993). Modified hippocampal long-term potentiation in PKCgamma-mutant mice. Cell 75:1253–1262. [DOI] [PubMed] [Google Scholar]
- Balkowiec A, Katz DM (2002). Cellular mechanisms regulating activity-dependent release of native brain-derived neurotrophic factor from hippocampal neurons. J Neurosci 22:10399–10407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behnisch T, Reymann KG (1995). Thapsigargin blocks long-term potentiation induced by weak, but not strong tetanisation in rat hippocampal CA1 neurons. Neurosci Lett 192:185–188. [DOI] [PubMed] [Google Scholar]
- Blum R, Kafitz KW, Konnerth A (2002). Neurotrophin-evoked depolarization requires the sodium channel Na(V)1.9. Nature 419:687–693. [DOI] [PubMed] [Google Scholar]
- Brewer GJ, Cotman CW (1989). Survival and growth of hippocampal neurons in defined medium at low density: advantages of a sandwich culture technique or low oxygen. Brain Res 494:65–74. [DOI] [PubMed] [Google Scholar]
- Brose N, Rosenmund C (2002). Move over protein kinase C, you've got company: alternative cellular effectors of diacylglycerol and phorbol esters. J Cell Sci 115:4399–4411. [DOI] [PubMed] [Google Scholar]
- Canossa M, Gartner A, Campana G, Inagaki N, Thoenen H (2001). Regulated secretion of neurotrophins by metabotropic glutamate group I (mGluRI) and Trk receptor activation is mediated via phospholipase C signalling pathways. EMBO J 20:1640–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Kolbeck R, Barde YA, Bonhoeffer T, Kossel A (1999). Relative contribution of endogenous neurotrophins in hippocampal long-term potentiation. J Neurosci 19:7983–7990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake CT, Milner TA, Patterson SL (1999). Ultrastructural localization of full-length trkB immunoreactivity in rat hippocampus suggests multiple roles in modulating activity-dependent synaptic plasticity. J Neurosci 19:8009–8026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dryga SA, Dryga OA, Schlesinger S (1997). Identification of mutations in a Sindbis virus variant able to establish persistent infection in BHK cells: the importance of a mutation in the nsP2 gene. Virology 228:74–83. [DOI] [PubMed] [Google Scholar]
- Falasca M, Logan SK, Lehto VP, Baccante G, Lemmon MA, Schlessinger J (1998). Activation of phospholipase C gamma by PI 3-kinase-induced PH domain-mediated membrane targeting. EMBO J 17:414–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figurov A, Miller LDP, Olafsson P, Wang T, Lu B (1996). Neurotrophins regulate synaptic responses to tetanic stimulation and LTP in the hippocampus. Nature 381:706–709. [DOI] [PubMed] [Google Scholar]
- Fujii S, Matsumoto M, Igarashi K, Kato H, Mikoshiba K (2000). Synaptic plasticity in hippocampal CA1 neurons of mice lacking type 1 inositol-1,4,5-trisphosphate receptors. Learn Mem 7:312–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gärtner A, Staiger V (2002). Neurotrophin secretion from hippocampal neurons evoked by long-term-potentiation-inducing electrical stimulation patterns. Proc Natl Acad Sci USA 99:6386–6391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gärtner A, Shostak Y, Hackel N, Ethell IM, Thoenen H (2000). Ultrastructural identification of storage compartments and localization of activity-dependent secretion of neurotrophin 6 in hippocampal neurons. Mol Cell Neurosci 15:215–234. [DOI] [PubMed] [Google Scholar]
- Hartmann M, Heumann R, Lessmann V (2001). Synaptic secretion of BDNF after high-frequency stimulation of glutamatergic synapses. EMBO J 20:5887–5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey AL (1995). The pharmacology of galanthamine and its analogues. Pharmacol Ther 68:113–128. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF (2003). TRK receptors: roles in neuronal signal transduction. Annu Rev Biochem 72:609–642. [DOI] [PubMed] [Google Scholar]
- Inoue T, Kato K, Kohda K, Mikoshiba K (1998). Type 1 inositol 1,4,5-trisphosphate receptor is required for induction of long-term depression in cerebellar Purkinje neurons. J Neurosci 18:5366–5373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizuka N, Weber J, Amaral DG (1990). Organization of intrahippocampal projections originating from CA3 pyramidal cells in the rat. J Comp Neurol 295:580–623. [DOI] [PubMed] [Google Scholar]
- Kang H, Schuman EM (1995). Long-lasting neutrotrophin-induced enhancement of synaptic transmisson in the adult hippocampus. Science 267:1658–1662. [DOI] [PubMed] [Google Scholar]
- Kang H, Schuman EM (2000). Intracellular Ca(2+) signaling is required for neurotrophin-induced potentiation in the adult rat hippocampus. Neurosci Lett 282:141–144. [DOI] [PubMed] [Google Scholar]
- Kang H, Welcher AA, Shelton D, Schuman EM (1997). Neurotrophins and time: different roles for trkb signaling in hippocampal long-term potentiation. Neuron 19:653–664. [DOI] [PubMed] [Google Scholar]
- Kohara K, Kitamura A, Morishima M, Tsumoto T (2001). Activity-dependent transfer of brain-derived neurotrophic factor to postsynaptic neurons. Science 291:2419–2423. [DOI] [PubMed] [Google Scholar]
- Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T (1995). Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci USA 92:8856–8860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korte M, Griesbeck O, Gravel C, Carroll P, Staiger V, Thoenen H, Bonhoeffer T (1996). Virus-mediated gene transfer into hippocampal CA1 region restores long-term potentiation in brain-derived neurotrophic factor mutant mice. Proc Natl Acad Sci USA 93:12547–12552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korte M, Minichiello L, Klein R, Bonhoeffer T (2000). Shc-binding site in the TrkB receptor is not required for hippocampal long-term potentiation. Neuropharmacology 39:717–724. [DOI] [PubMed] [Google Scholar]
- Kossel AH, Cambridge SB, Wagner U, Bonhoeffer T (2001). A caged Ab reveals an immediate/instructive effect of BDNF during hippocampal synaptic potentiation. Proc Natl Acad Sci USA 98:14702–14707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalchuk Y, Hanse E, Kafitz KW, Konnerth A (2002). Postsynaptic induction of BDNF-mediated long-term potentiation. Science 295:1729–1734. [DOI] [PubMed] [Google Scholar]
- Lemmon MA, Ferguson KM, O'Brien R, Sigler PB, Schlessinger J (1995). Specific and high-affinity binding of inositol phosphates to an isolated pleckstrin homology domain. Proc Natl Acad Sci USA 92:10472–10476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessmann V, Gottmann K, Heumann R (1994). BDNF and NT-4/5 enhance glutamatergic synaptic transmission in cultured hippocampal neurones. NeuroReport 6:21–25. [DOI] [PubMed] [Google Scholar]
- Levine ES, Crozier RA, Black IB, Plummer MR (1998). Brain-derived neurotrophic factor modulates hippocampal synaptic transmission by increasing N-methyl-d-aspartic acid receptor activity. Proc Natl Acad Sci USA 95:10235–10239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YX, Xu Y, Ju D, Lester HA, Davidson N, Schuman EM (1998a). Expression of a dominant negative TrkB receptor, T1, reveals a requirement for presynaptic signaling in BDNF-induced synaptic potentiation in cultured hippocampal neurons. Proc Natl Acad Sci USA 95:10884–10889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YX, Zhang YO, Lester HA, Schuman EM, Davidson N (1998b). Enhancement of neurotransmitter release induced by brain-derived neurotrophic factor in cultured hippocampal neurons. J Neurosci 18:10231–10240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SY, Wu K, Levine ES, Mount HT, Suen PC, Black IB (1998). BDNF acutely increases tyrosine phosphorylation of the NMDA receptor subunit 2B in cortical and hippocampal postsynaptic densities. Mol Brain Res 55:20–27. [DOI] [PubMed] [Google Scholar]
- Lustig S, Jackson AC, Hahn CS, Griffin DE, Strauss EG, Strauss JH (1988). Molecular basis of Sindbis virus neurovirulence in mice. J Virol 62:2329–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffucci T, Falasca M (2001). Specificity in pleckstrin homology (PH) domain membrane targeting: a role for a phosphoinositide-protein co-operative mechanism. FEBS Lett 506:173–179. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA (1999). Long-term potentiation–a decade of progress? Science 285:1870–1874. [DOI] [PubMed] [Google Scholar]
- Maruyama T, Kanaji T, Nakade S, Kanno T, Mikoshiba K (1997). 2APB, 2-aminoethoxydiphenyl borate, a membrane-penetrable modulator of Ins(1,4,5)P3-induced Ca2+ release. J Biochem (Tokyo) 122:498–505. [DOI] [PubMed] [Google Scholar]
- Minichiello L, Korte M, Wolfer D, Kühn R, Unsicker K, Cestari V, Rossi-Arnaud C, Lipp HP, Bonhoeffer T, Klein R (1999). Essential role for TrkB receptors in hippocampus-mediated learning. Neuron 24:401–414. [DOI] [PubMed] [Google Scholar]
- Minichiello L, Calella A, Medina D, Bonhoeffer T, Klein R, Korte M (2002). Mechanism of TrkB-mediated hippocampal long-term potentiation. Neuron 36:121–137. [DOI] [PubMed] [Google Scholar]
- Narisawa-Saito M, Carnahan J, Araki K, Yamaguchi T, Nawa H (1999). Brain-derived neurotrophic factor regulates the expression of AMPA receptor proteins in neocortical neurons. Neuroscience 88:1009–1014. [DOI] [PubMed] [Google Scholar]
- Patterson SL, Abel T, Deuel TAS, Martin KC, Rose JC, Kandel ER (1996). Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron 16:1137–1145. [DOI] [PubMed] [Google Scholar]
- Poo MM (2001). Neurotrophins as synaptic modulators. Nat Rev Neurosci 2:24–32. [DOI] [PubMed] [Google Scholar]
- Prakriya M, Lewis RS (2001). Potentiation and inhibition of Ca(2+) release-activated Ca(2+) channels by 2-aminoethyldiphenyl borate (2-APB) occurs independently of IP(3) receptors. J Physiol (Lond) 536:3–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond CR, Redman SJ (2002). Different calcium sources are narrowly tuned to the induction of different forms of LTP. J Neurophysiol 88:249–255. [DOI] [PubMed] [Google Scholar]
- Reyes-Harde M, Stanton PK (1998). Postsynaptic phospholipase C activity is required for the induction of homosynaptic long-term depression in rat hippocampus. Neurosci Lett 252:155–158. [DOI] [PubMed] [Google Scholar]
- Schinder AF, Berninger B, Poo MM (2000). Postsynaptic target specificity of neurotrophin-induced presynaptic potentiation. Neuron 25:151–163. [DOI] [PubMed] [Google Scholar]
- Suen PC, Wu K, Levine ES, Mount HT, Xu JL, Lin SY, Black IB (1997). Brain-derived neurotrophic factor rapidly enhances phosphorylation of the postsynaptic N-methyl-d-aspartate receptor subunit 1. Proc Natl Acad Sci USA 94:8191–8195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Gottschalk W, Chow A, Wilson RI, Schnell E, Zang K, Wang D, Nicoll RA, Lu B, Reichardt LF (2000). The role of brain-derived neurotrophic factor receptors in the mature hippocampus: modulation of long-term potentiation through a presynaptic mechanism involving TrkB. J Neurosci 20:6888–6897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, He X, Feng L, Mizuno K, Liu XW, Russell J, Xiong WC, Lu B (2001). PI-3 kinase and IP3 are both necessary and sufficient to mediate NT3-induced synaptic potentiation. Nat Neurosci 4:19–28. [DOI] [PubMed] [Google Scholar]
- Zakharenko SS, Patterson SL, Dragatsis I, Zeitlin SO, Siegelbaum SA, Kandel ER, Morozov A (2003). Presynaptic BDNF required for a presynaptic but not postsynaptic component of LTP at hippocampal CA1-CA3 synapses. Neuron 39:975–990. [DOI] [PubMed] [Google Scholar]