

Abstract
Optimal management of neuropathic pain is a major clinical challenge. We investigated the involvement of c-Jun N-terminal kinase (JNK) in neuropathic pain produced by spinal nerve ligation (SNL) (L5). SNL induced a slow (>3 d) and persistent (>21 d) activation of JNK, in particular JNK1, in GFAP-expressing astrocytes in the spinal cord. In contrast, p38 mitogen-activated protein kinase activation was found in spinal microglia after SNL, which had fallen to near basal level by 21 d. Intrathecal infusion of a JNK peptide inhibitor, D-JNKI-1, did not affect normal pain responses but potently prevented and reversed SNL-induced mechanical allodynia, a major symptom of neuropathic pain. Intrathecal D-JNKI-1 also suppressed SNL-induced phosphorylation of the JNK substrate, c-Jun, in spinal astrocytes. However, SNL-induced upregulation of GFAP was not attenuated by spinal D-JNKI-1 infusion. Furthermore, SNL induced a rapid (<12 h) but transient activation of JNK in the L5 (injured) but not L4 (intact) DRG. JNK activation in the DRG was mainly found in small-sized C-fiber neurons. Infusion of D-JNKI-1 into the L5 DRG prevented but did not reverse SNL-induced mechanical allodynia. Finally, intrathecal administration of an astroglial toxin, l-α-aminoadipate, reversed mechanical allodynia. Our data suggest that JNK activation in the DRG and spinal cord play distinct roles in regulating the development and maintenance of neuropathic pain, respectively, and that spinal astrocytes contribute importantly to the persistence of mechanical allodynia. Targeting the JNK pathway in spinal astroglia may present a new and efficient way to treat neuropathic pain symptoms.
Keywords: glia, neuropathy, nerve injury, dorsal root ganglion, MAP kinase, spinal cord
Introduction
Damage to peripheral nerve very often results in neuropathic pain. Current treatments for this pain are only partially effective, and additional development is hindered by our incomplete knowledge of how neuropathic pain is induced and maintained. Increasing evidence shows that mitogen-activated protein kinases (MAPKs) play important roles in the induction and maintenance of chronic pain (for review, see Ji and Woolf, 2001; Ji and Strichartz, 2004; Obata and Noguchi, 2004). The MAPK family has three major members, extracellular signal-regulated kinase (ERK), p38, and c-Jun N-terminal kinase (JNK), representing three different signal transduction pathways. Compared with well documented roles of p38 and ERK in neuropathic pain, the distinct role of JNK in dorsal root ganglion (DRG) and spinal cord has not been investigated.
JNK contributes importantly to stress-induced apoptosis in various cell types (Xia et al., 1995; Borsello et al., 2003). For example, cerebral ischemia activates JNK, leading to excitotoxic neuronal death (Borsello et al., 2003). Recently, a cell-penetrating and proteinase-resistant peptide inhibitor of JNK (D-JNKI-1), derived from the JNK binding domain of JNK-interacting protein-1, was developed to competitively inhibit JNK binding to its substrates. D-JNKI-1 is an extremely effective neuroprotectant in vitro against excitotoxicity of cortical neurons and in vivo against two models of focal cerebral ischemia (Borsello et al., 2003; Borsello and Bonny, 2004), as well as in other neurodegenerative paradigms (Wang et al., 2003; Tezel et al., 2004).
Studies on pain research focus primarily on neuronal cells; less is known about the importance of glial cells. However, accumulating evidence indicates that spinal glia contribute to the pathogenesis of pain (DeLeo and Yezierski, 2001; Watkins et al., 2001; Tsuda et al., 2005). Both microglia and astrocytes are activated in the spinal cord after nerve injury (Jin et al., 2003; Raghavendra et al., 2003). Glial cells not only express various receptors for neurotransmitters but, on activation, also produce multiple inflammatory mediators [e.g., interleukin-1β (IL-1β), IL-6, tumor necrosis factor-α (TNF-α), and prostaglandin E2 (PGE2)] that are implicated in pain sensitization (Watkins et al., 2001; Watkins and Maier, 2003; DeLeo et al., 2004; Ji and Strichartz, 2004). Increasing evidence suggests that spinal microglia are essential to the initial development of neuropathic pain (Abbadie et al., 2003; Jin et al., 2003; Raghavendra et al., 2003; Tsuda et al., 2003, 2005; Coull et al., 2005; Tanga et al., 2005). Although astroglia are persistently activated in the spinal cord after nerve injury (Honore et al., 2000), the participation of this glial subtype in pain sensitization remains essentially unknown.
Here, we demonstrate another powerful but different role of JNK for controlling neuropathic pain. We report that JNK is activated in both DRG and spinal cord in a neuropathic pain condition produced by ligation of the L5 spinal nerve. Whereas transient activation of JNK in DRG neurons is involved in the induction of neuropathic pain, persistent activation of JNK in spinal astrocytes appears critical for the maintenance of neuropathic pain. In particular, D-JNKI-1 can effectively block mechanical allodynia, a characteristic behavioral response for neuropathic pain. Furthermore, an astroglial toxin administered in spinal fluid potently attenuates established neuropathic pain.
Materials and Methods
Animals and surgery.
Male adult Sprague Dawley rats (220–260 g) were used under Harvard Medical School Animal Care institutional guidelines. To produce a spinal nerve ligation (SNL), under pentobarbital anesthesia (40–50 mg/kg, i.p.), the L5 transverse process was removed to expose the L4 and L5 spinal nerves. The L5 spinal nerve was then isolated and tightly ligated with 6-0 silk thread (Kim and Chung, 1992).
Drug administration.
The peptide JNK inhibitor was synthesized by linking the 10 amino acid human immunodeficiency virus Tat (48–57) transporter sequence to a 20 amino acid JNK-binding motif (JBD20) of JNK-interacting protein-1/islet-brain 1. The peptide was also synthesized as protease-resistant D-form to expand its half-life in vivo (Borsello et al., 2003). A peptide with Tat sequence but lacking JBD20 was used as the control. The peptide inhibitor is highly selective and does not affect the activity of 40 other protein kinases (Borsello et al., 2003). The small chemical JNK inhibitor SP600125 (anthra[1,9-cd]pyrazol-6(2H)-one) and the astroglial toxin l-α-aminoadipate (l-α-AA) were purchased from Calbiochem (La Jolla, CA) and Sigma (St. Louis, MO), respectively.
For sustained drug infusion, laminectomy was performed at the spinal level, 2 cm below the lumbar enlargement, and a polyethylene (PE5) catheter was implanted into the intrathecal space of the spinal cord, ending at spinal L4–L5 level. The catheter was connected to an osmotic pump (Alzet, Cupertino, CA) for drug infusion. The drug-filled pumps were soaked in saline for 3 h before implantation and placed subcutaneously on the back of rats (Ji et al., 2002a,b). For drug infusion into the DRG, a PE5 catheter was inserted into the L5 spinal nerve and threaded toward to the L5 DRG either 30 min before or 10 d after the ligation of the same spinal nerve (L5). For single intrathecal injection, spinal cord puncture was made under brief sevofluorane anesthesia with a 27 gauge needle between the L5 and L6 level to deliver the reagents (20 μl) to the CSF (Zhuang et al., 2005). Immediately after the needle entry into subarachnoid space (change in resistance), a brisk tail flick could be observed.
Western blots.
As described previously (Zhuang et al., 2005), animals were rapidly killed, and the L5 spinal segments (dorsal part) were quickly removed and homogenized in a SDS sample buffer containing a mixture of proteinase and phosphatase inhibitors (Sigma). Protein samples (30 μg) were separated on SDS-PAGE gel and transferred to polyvinylidene difluoride blots. The blots were blocked with 5% milk and incubated overnight at 4°C with antibody against phosphorylated JNK (pJNK) or JNK (anti-rabbit, 1:1000; Cell Signaling Technology, Beverly, MA). These blots were further incubated with HRP-conjugated secondary antibody, developed in ECL solution, and exposed onto Hyperfilm (Amersham Biosciences, Arlington Heights, IL).
Immunohistochemistry.
Animals were terminally anesthetized with isoflurane and perfused through the ascending aorta with saline followed by 4% paraformaldehyde/1.5% picric acid. After the perfusion, the spinal cord segments (L5) and the DRGs (L4, L5) were removed and postfixed in the same fixative overnight. Spinal sections (transverse, free-floating, 30 μm) and DRG sections (15 μm) were cut in a cryostat and processed for immunofluorescence (Ji et al., 1995; Jin et al., 2003). All of the sections were blocked with 2% goat serum in 0.3% Triton X-100 for 1 h at room temperature (RT) and incubated overnight at 4°C with anti-pJNK antibody (anti-rabbit, 1:1000; Cell Signaling Technology). The sections were then incubated for 1 h at RT with cyanine 3-conjugated secondary antibody (1:400; Jackson ImmunoResearch, West Grove, PA). Other primary antibodies used in this study were polyclonal antibodies for JNK1 (rabbit, 1:2000; Santa Cruz Biotechnology, Santa Cruz, CA), pERK, p-p38, p-c-Jun (rabbit, 1:500, phosphorylated forms; Cell Signaling Technology), and monoclonal antibodies for neuronal-specific nuclear protein (NeuN) (mouse, 1:5000; Chemicon, Temecula, CA), glial fibrillary acidic protein (GFAP) (mouse, 1:5000; Chemicon), and neurofilament 200 kDa (NF-200) (mouse, 1:5000; Sigma). For double immunofluorescence, sections were incubated with a mixture of two primary antibodies (monoclonal and polyclonal), followed by a mixture of the two respective secondary antibodies. The specificity of the staining was tested by omission of primary antibodies or absorption with peptide antigens. Stained sections were examined with a Nikon (Tokyo, Japan(fluorescence microscope, and images were captured with a CCD Spot camera.
Behavioral analysis.
Animals were habituated to the testing environment daily for 3–7 d before baseline testing. For testing mechanical sensitivity, animals were put under inverted plastic boxes (11 × 13 × 24 cm) on an elevated mesh floor and allowed 30 min for habituation, before the threshold testing. Mechanical allodynia was tested using von Frey hairs in a blinded manner. The paw was pressed with one of a series of von Frey hairs with logarithmically incrementing stiffness (0.6, 1, 1.4, 2, 4, 6, 8, 10, 15, and 26 g) (Stoelting, Kiel, WI), presented perpendicular to the plantar surface (5–6 s for each hair). The 50% withdrawal threshold was determined using Dixon's up–down method (Chaplan et al., 1994).
Quantification and statistics.
To quantify positive cell profiles in the spinal cord, five to eight sections from the L5 spinal cord segments were randomly selected. An image in a square (450 × 338 μm) on the medial two-thirds of the superficial dorsal horn (laminas I–III), as described previously by Molander et al. (1984), was captured under 20× objective, and all of the positively stained cells in the area were counted by an investigator who did not know the treatment. Those cells with distinct contrast to the background were scored as positive. Background levels were obtained from sections incubated without primary antibody. Roughly, the densities of these positive cells should exhibit a signal/background ratio >2. This counting does not determine the total numbers of cells (Coggeshall and Lekan, 1996). Instead, it provides a comparison to evaluate differences between control and treated animals. To quantify pJNK expression in the DRG, the percentages of pJNK-positive neurons in the L5 and L4 DRG from four nonadjacent sections were determined as described previously (Ji et al., 1996). Data were presented as mean ± SEM. Differences between groups were compared using Student's t test or ANOVA, followed by Fisher's PLSD test. The criterion for statistical significance was p < 0.05.
Results
Activation of JNK in the spinal astrocytes after nerve injury
SNL produced rapidly appearing (<1 d) and persistent (>3 weeks) neuropathic pain (Kim and Chung, 1992). SNL also induced a marked activation of JNK in the spinal cord. Many more pJNK (active form of JNK)-immunoreactive (IR) cells were found in the ipsilateral than the contralateral spinal cord, predominantly in the superficial dorsal horn (laminas I–III) on day 10 (Fig. 1a–d) and day 21 (Fig. 1g,h). Double immunofluorescence indicated that pJNK was completely colocalized with GFAP, a marker for astrocytes, in the spinal cord at different times (days 3–21) of neuropathic pain development (Fig. 1e,f, i).
Figure 1.
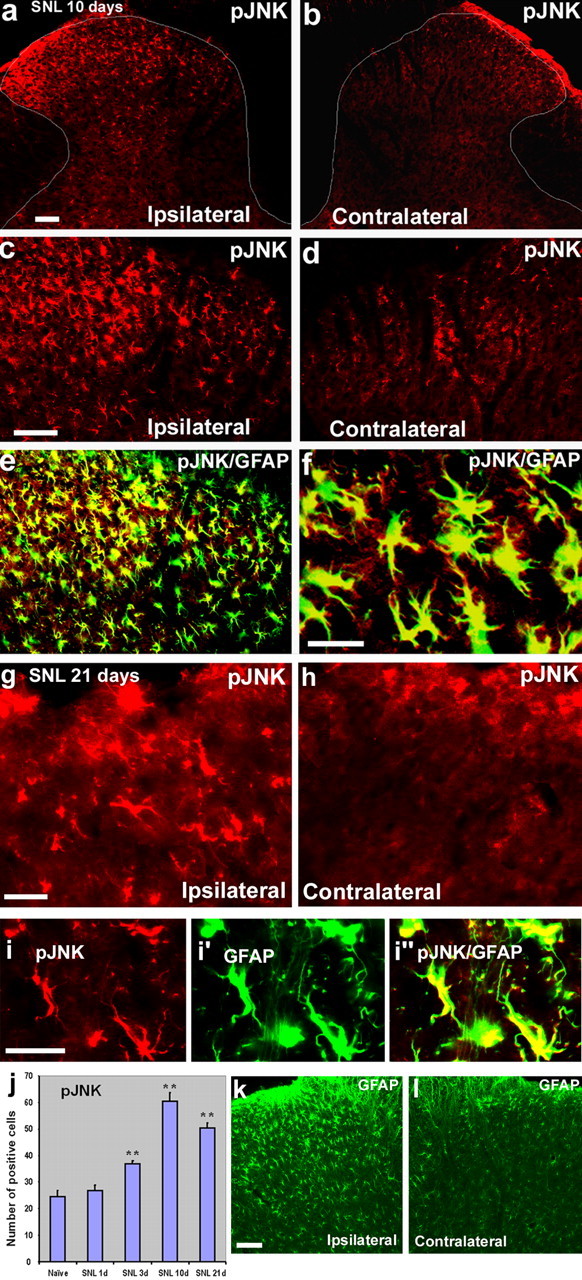
a–l, SNL induces persistent JNK activation in spinal astroglia. a, b, Immunohistochemistry reveals increases in pJNK levels in the ipsilateral spinal dorsal horn (L5) 10 d after SNL. White lines indicate the border of the dorsal horn gray matter. Scale bar, 50 μm. c, d, High-magnification images from a and b, respectively, showing pJNK staining in the medial superficial dorsal horn. Scale bar, 50 μm. e, Double immunofluorescence shows that pJNK (red) is completely colocalized with astroglia marker GFAP (green) in the medial superficial dorsal horn. Two single-stained images were merged. e has the same magnification as c. f, High-magnification image from e demonstrating the colocalization of pJNK and GFAP. Scale bar, 25 μm. g, h, SNL also induces JNK activation in the dorsal horn after 3 weeks. Scale bar, 25 μm. i, Colocalization of pJNK (i) and GFAP (i′) in the superficial dorsal horn 21 d after SNL. i″ is the merge of i and i′. Scale bar, 25 μm. j, Time course of pJNK induction in the L5 spinal cord after SNL, as indicated by the number of pJNK-IR cells in the superficial dorsal horn (laminas I–III). **p < 0.01 by ANOVA compared with control (n = 3 and 4). k, l, SNL induces persistent activation of astrocytes in the dorsal horn after 3 weeks, as indicated by GFAP upregulation. Scale bar, 50 μm.
To determine the percentage of cells that showed pJNK/GFAP double labeling, we quantified the number of pJNK and GFAP single-stained cells, as well as pJNK/GFAP double-stained cells in the medial laminas I–III (see Materials and Methods) in six spinal sections from three different rats perfused 10 d after SNL. Approximately 99% of pJNK-IR cells (358 of 363) were also GFAP-IR, suggesting that pJNK is almost exclusively expressed in astrocytes. However, only 33% of GFAP-IR cells (358 of 1080) were pJNK-IR, indicating that JNK is only activated in a fraction of spinal astrocytes.
A time course study showed that pJNK elevation was not evident at day 1, when symptoms of neuropathic pain began to develop (Fig. 1j). The rise in pJNK levels was significant on day 3, reached a peak on day 10, and remained at high levels on day 21 after SNL (Fig. 1j). Consistently, SNL induced a persistent upregulation of GFAP in the dorsal horn. GFAP expression remained at high level on day 21 (Fig. 1k,l).
Activation of JNK1 and c-Jun in spinal astrocytes after nerve injury
Western blot analysis confirmed an increase in spinal pJNK after SNL. Although both JNK1 (46 kDa) and JNK2 (54 kDa) were constitutively expressed in the spinal cord, only JNK1 was phosphorylated in the spinal cord (Fig. 2a). There was no significant change in pJNK expression after sham surgery (Fig. 2a). Double immunofluorescence showed that JNK1 was colocalized with GFAP but not with NeuN (neuronal marker) or with OX-42 (an antibody for the microglial marker CD11b) in the spinal cord after SNL (Fig. 2b–e). Therefore, JNK1 appears to be the isoform that is phosphorylated, exclusively in spinal astrocytes.
Figure 2.

a–e, JNK1 is activated in spinal astrocytes. a, Western blot analysis reveals an increase in the level of pJNK1 (46 kDa) in the L5 spinal cord 10 d after L5 SNL. b, Immunohistochemistry shows JNK1 expression in the medial superficial spinal cord 10 d after SNL. Scale bar, 20 μm. c–e, High-magnification images reveal double staining between JNK1 (red) and NeuN (c), OX-42 (d), or GFAP (e). Two single-stained images were merged. Scale bar, 10 μm. Note that the JNK1 is only colocalized with GFAP.
The transcription factor c-Jun is the best known substrate of JNK. JNK is known to activate p-c-Jun. SNL also induced an upregulation of p-c-Jun in the ipsilateral spinal cord (Fig. 3). Interestingly, p-c-Jun was also localized in GFAP-expressing astrocytes in the dorsal horn, with predominant localization in the nucleus (Fig. 3).
Figure 3.

a–d, SNL induces phosphorylation of c-Jun in spinal astroglia. a, b, Immunohistochemistry shows increases in p-c-Jun levels in the ipsilateral spinal dorsal horn (L5) 10 d after SNL. White lines indicate the border of the dorsal horn gray matter. Scale bar, 100 μm. c, Double immunofluorescence shows that p-c-Jun (red) is completely colocalized with astroglia marker GFAP (green) in the medial superficial dorsal horn. Scale bar, 50 μm. d, High-magnification image demonstrating the colocalization of pJNK and GFAP. Two single-stained images were merged. Note that p-c-Jun is expressed in the nucleus of GFAP-labeled cells. Scale bar, 10 μm.
Activation of p38 MAPK in spinal microglia after nerve injury
To determine whether different MAPKs show different activation patterns after nerve injury, we also examined the activation of p38 MAPK in the spinal cord after SNL. As shown previously (Jin et al., 2003), SNL strongly activated p38 in OX-42-expressing spinal microglia on day 3 (Fig. 4a–c). The number of p-p38-IR cells remained elevated in the ipsilateral spinal cord at 21 d after SNL but had markedly declined from the peak, and the p-p38 at this time was primarily localized in the nucleus of microglia (Fig. 4d–f). In parallel, microglial activation was maximal on day 3 but had fallen by day 21, although not to basal levels, as indicated by OX-42 staining (Fig. 4g–j).
Figure 4.
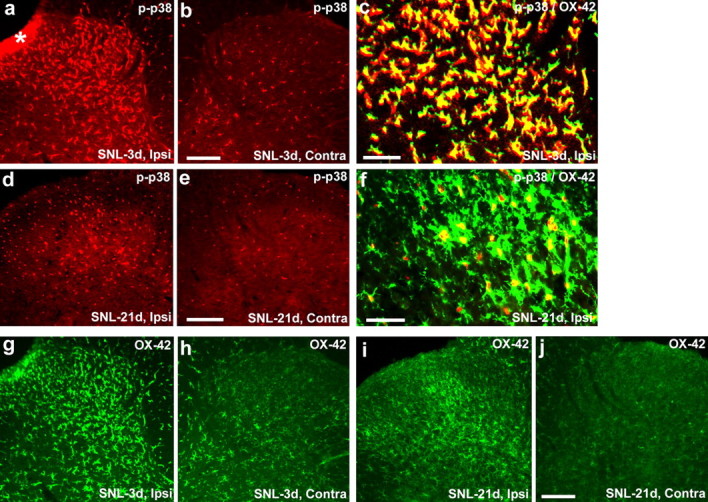
a–j, SNL activates p38 MAPK in spinal microglia. a–f, p-p38 immunofluorescence in the dorsal horn of the ipsilateral (Ipsi; a, c, d, f) and contralateral (Contra; b, e) side at 3 d (a–c) and 21 d (d–f) after SNL. c and d are double staining for p-p38 (red) and OX-42 (green). Note that p-p38 is expressed in OX-42-IR microglia at 3 and 21 d. Two single-stained images were merged. Scale bars: b, e, 100 μm; c, f, 50 μm. The asterisk indicates an artifact. g–j, OX-42 immunofluorescence in the dorsal horn of the ipsilateral (g, i) and contralateral (h, j) side at 3 d (g, h) and 21 d (i, j) after SNL. Scale bar, 100 μm.
Prevention and reversal of neuropathic pain by intrathecal infusion of JNK inhibitors
SNL-induced neuropathic pain is characterized by mechanical allodynia (pain-like response to preinjury innocuous mechanical stimulation) (Kim and Chung, 1992). To determine whether spinal JNK activation has a role in the development of mechanical allodynia, we infused the peptide inhibitor D-JNKI-1 via an osmotic pump for 2 weeks, starting 3 d before SNL. Although intrathecal infusion of D-JNKI-1 to spinal fluid did not change basal mechanical threshold tested before nerve injury, it prevented the development of mechanical allodynia for >10 d (Fig. 5). However, neuropathic pain was not permanently prevented, because allodynia developed again once the 2 week infusion ended (Fig. 5).
Figure 5.

Intrathecal perfusion of D-JNKI-1 prevents the development of neuropathic pain. The peptide JNK inhibitor D-JNKI-1 (50 μm) was infused into intrathecal space via an osmotic pump (0.5 μl/h for 2 weeks), starting 3 d before SNL. Note that the basal mechanical sensitivity is not changed after D-JNKI-1 infusion. The inhibitor almost completely blocks allodynia for >10 d, but neuropathic pain returns after infusion stops. *p < 0.05, **p < 0.01 by t test compared with corresponding vehicle controls (saline); n = 6.
To investigate whether JNK inhibition would reverse established neuropathic pain, a treatment mode more relevant to clinical situation, we infused the peptide inhibitor intrathecally via an osmotic pump 10 d after establishment of SNL-induced allodynia. This treatment (3 d infusion) effectively reversed SNL-induced allodynia for >4 d (Fig. 6a).
Figure 6.

a–d, Intrathecal administration of JNK inhibitors reverses neuropathic pain. a, Reversal of SNL-induced mechanical allodynia by intrathecal infusion of D-JNKI-1 (50 μm) via an osmotic pump (0.5 μl/h for 3 d) starting on post-SNL day 10. *p < 0.05 by t test compared with corresponding saline controls; n = 5. b, Reversal of SNL-induced mechanical allodynia by a bolus intrathecal injection of D-JNKI-1 (4 nmol) and SP600125 (50 nmol) at post-SNL day 10. *p < 0.05, **p < 0.01 by ANOVA compared with corresponding preinjection baseline; n = 6. c, Effects of D-JNKI-1 (0.2 and 4 nmol) and its control peptide (4 nmol), SP600125 (SP; 50 and 150 nmol) and its vehicle (20% DMSO), and saline, via intrathecal bolus injection, on mechanical allodynia on post-SNL day 10. **p < 0.01 compared with the vehicle (Veh); ##p < 0.01, compared with the control peptide; n = 6. The mechanical allodynia was tested at 3 h after all of the injections. d, Dose-dependent effects of D-JNKI-1 and SP600125, intrathecally injected on day 10, on reversing mechanical allodynia. Mechanical allodynia was tested 3 h after the injections. The ED50 for D-JNKI-1 and SP600125 is 0.3 and 14.9 nmol, respectively (n = 6).
To test the effects of a single bolus injection of JNK inhibitors on SNL-induced allodynia, we injected D-JNKI-1 and the small-molecule JNK inhibitor SP600125 intrathecally on post-SNL day 10. Both inhibitors could reverse mechanical allodynia for 6–12 h (Fig. 6b) in a dose-dependent manner (Fig. 6c). A control peptide (with Tat sequence but lacking the JBD20 sequence) had no effect (Fig. 6c). We also found that D-JNKI-1 was ∼50 times more potent than SP600125 in this reversal (Fig. 6c), the ED50 values being 0.3 and 14.9 nmol, respectively (Fig. 6d).
Regulation of c-Jun phosphorylation but not GFAP expression in astrocytes by JNK
To explore the mechanisms by which JNK activation would mediate neuropathic pain, we examined the effect of D-JNKI-1 on SNL-induced phosphorylation of c-Jun. Intrathecal infusion of D-JNKI-1, at a concentration that fully reversed allodynia (Fig. 6a), significantly suppressed SNL-induced c-Jun phosphorylation in spinal astrocytes (Fig. 7a). This suppression was only partial because of the fact that there were more p-c-Jun-IR cells than pJNK-IR cells in the spinal cord after SNL (Figs. 1j, 7a). Therefore, c-Jun phosphorylation in some cells may not be caused by pJNK.
Figure 7.

a, b, Effects of intrathecal D-JNKI-1 on SNL-induced c-Jun phosphorylation and GFAP expression in astrocytes. The numbers of p-c-Jun-IR (a) and GFAP-IR (b) cells in the medial two-thirds of superficial dorsal horn (laminas I–III) at 10 d after SNL were quantified. D-JNKI-1 was intrathecally infused via an osmotic pump (0.5 μl/h, 50 μm) at 7 d after SNL, and animals were perfused 3 d after the infusion. **p < 0.01, compared with controls; +p < 0.05, compared with SNL, ANOVA; n = 4.
SNL also induced a doubling of GFAP-expressing astrocytes in the spinal cord on day 10 (Fig. 7b). Astrocytes in cords from uninjured rats were in the resting state, exhibiting long and thin processes. After nerve injury, spinal astrocytes became reactive, demonstrating larger cell bodies and thicker processes. Although pJNK was colocalized with GFAP (Fig. 1), D-JNKI-1, at a concentration sufficient to reverse allodynia, did not change either the number of GFAP-expressing astrocytes (Fig. 7b) or the reactive morphology of astrocytes (data not shown).
Uptake of D-JNKI-1 by DRG cells after intrathecal injection
Although the intrathecal route is traditionally believed to target spinal cells, intrathecal injection of MAPK inhibitors has also been shown to affect DRG cells (Ji et al., 2002b; Obata et al., 2004a). To examine whether a larger peptide inhibitor can diffuse to and be taken up by DRG cells, we intrathecally injected a cumarin-labeled Tat peptide (4 nmol). Many fluorescence-labeled neurons and satellite cells were found in the DRG 3 h after the injection (Fig. 8), indicating that the peptide inhibitor is taken up by DRG cells after intrathecal injection.
Figure 8.
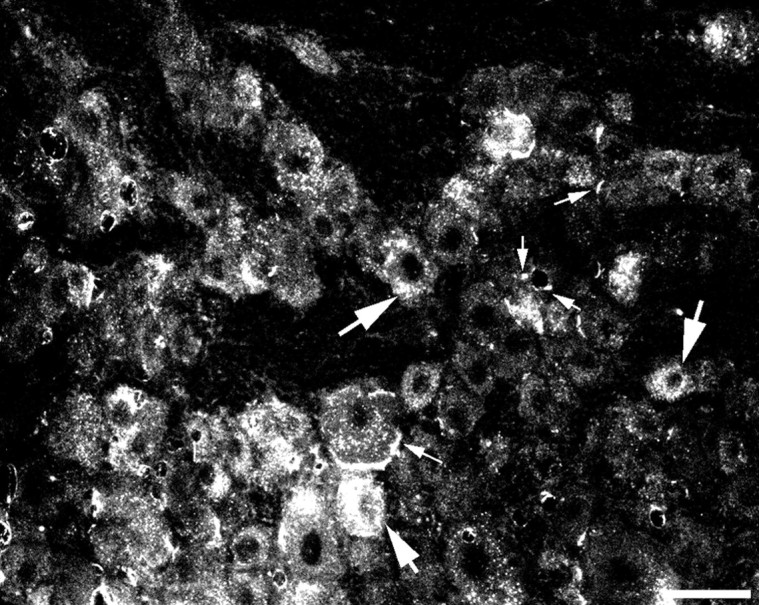
Uptake of the Tat peptide by DRG cells after intrathecal injection. Many fluorescence-labeled neurons and satellite cells are present in the DRG 3 h after intrathecal injection of a cumarin-labeled Tat peptide (4 nmol). Large and small arrows indicate neurons and satellite cells, respectively. Scale bar, 50 μm.
JNK activation in the injured small-sized neurons of the DRG
Because intrathecal D-JNKI-1 could also act on DRG cells, we further examined JNK activation in the DRG after nerve ligation. A previous study reported increased pJNK expression in DRG neurons 1 week after nerve injury (Obata et al., 2004a). However, we found a much earlier induction of pJNK in the DRG after SNL. In contrast to the kinetics in the spinal cord (Fig. 1j), the peak of pJNK induction in sensory neurons of the injured L5 DRG was at 12 h. At this time point, 35% of DRG neurons showed pJNK expression, whereas only 5% of neurons were positive in naive rats (Fig. 9a–c). There was only a modest increase of pJNK in the L5 DRG at 6 h after nerve injury (data not shown). Double staining indicated that pJNK was rarely colocalized with NF-200, a marker for large myelinated neurons, indicating that pJNK is mainly induced in C-type neurons (with C-fibers) in the L5 DRG after SNL (Fig. 9d). However, JNK activation in the L5 DRG was significantly reduced 10 d after SNL (Fig. 9c,e). Indeed, there are not many pJNK-IR neurons in the L5 DRG even 1 week after SNL (Obata et al., 2004a).
Figure 9.
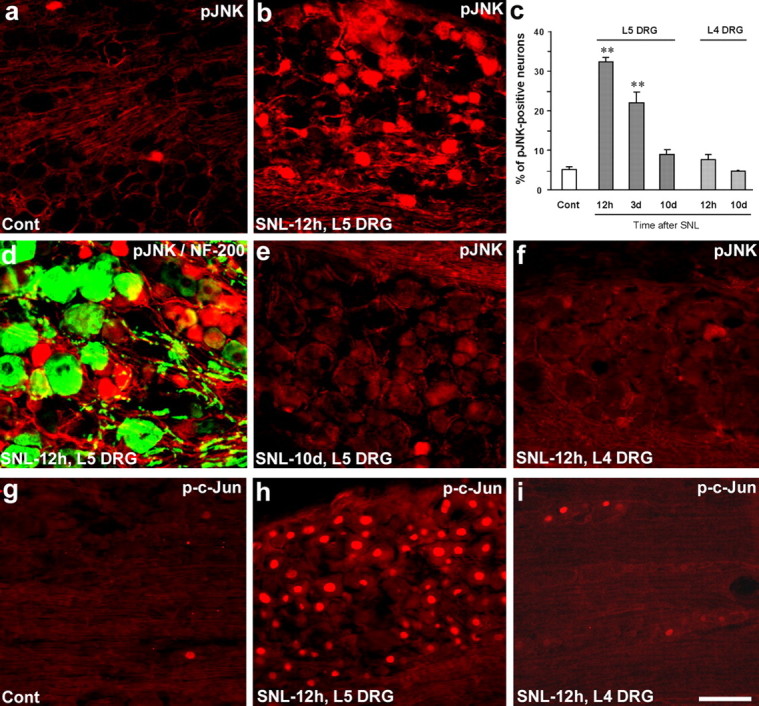
a–i, SNL induces rapid but transient JNK activation in the injured (L5) neurons of the DRG. a, b, SNL produces a rapid (at 12 h) pJNK increase in small-sized neurons of the L5 DRG. c, Percentage of pJNK-positive neuronal profiles in the L5 and L4 DRGs at different times after SNL. **p < 0.01 by ANOVA compared with control (naive); n = 4. d, Double immunofluorescence of pJNK (red) and NF-200 (green) in the L5 DRG. Note that pJNK is barely colocalized with NF-200. e, f, There is only low level of pJNK expression in the L5 DRG on day 10 (e) and in the L4 DRG on day 0.5 (f) after SNL. g–i, SNL induces p-c-Jun in the L5 DRG (h) but not in the L4 DRG (i). Scale bar, 50 μm.
Nerve injury has been known to produce changes not only in the injured neurons but also in the non-injured neurons that are adjacent to the injured neurons. For example, SNL (L5) induces p38 activation in the intact L4 DRG (Obata et al., 2004a,b). In contrast, pJNK was not induced in the L4 DRG (Fig. 9c,f), nor was pJNK induced in the L5 DRG of sham-operated rats (data not shown). These data suggest that axonal injury is required for JNK activation in sensory neurons.
SNL also induced very dramatic upregulation of p-c-Jun in the nucleus of the injured (L5) but not the intact (L4) DRG (Fig. 9g,h). However, the percentage of p-c-Jun-IR neurons was much higher than that of pJNK-IR neurons: >80% of neurons in the L5 DRG expressed p-c-Jun 12 h after SNL (Fig. 9h). This percentage could be even higher, because not every neuronal profile captures nucleus in 15-μm-thick DRG sections.
Prevention but not reversal of neuropathic pain by DRG perfusion of D-JNKI-1
To examine the role of JNK activation in the DRG for neuropathic pain, we directly infused D-JNKI-1 to the L5 DRG by inserting a PE5 catheter inside the L5 spinal nerve close to the DRG. Perfusion of the L5 DRG with D-JNKI-1 completely prevented the development of SNL-induced mechanical allodynia for more than 1 week (Fig. 10a). Mechanical allodynia matched that of untreated rats on post-SNL day 11 when no drug was left in the osmotic pump (Fig. 10a). In contrast to intrathecal infusion, perfusion of D-JNKI-1 into the L5 DRG 10 d after SNL did not reverse established allodynia (Fig. 10b). Together, our data suggest that JNK activation in small-sized (C-type) neurons of the L5 DRG is required for the induction but not the maintenance of mechanical allodynia after nerve injury.
Figure 10.
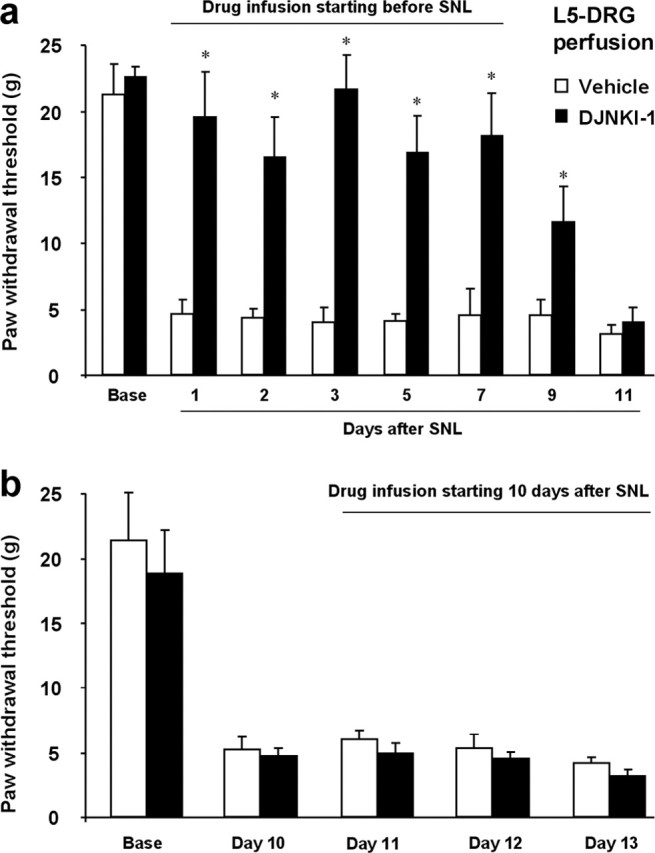
a, b, Perfusion of the L5 DRG with D-JNKI-1 prevents but does not reverse mechanical allodynia after SNL. The peptide JNK inhibitor D-JNKI-1 (50 μm) was infused into the L5 DRG via an osmotic pump (0.5 μl/h), starting 30 min before (a) or 10 d after (b) SNL. *p < 0.05 by t test compared with corresponding vehicle controls (saline); n = 5.
Reversal of neuropathic pain by an astroglial toxin
l-α-AA is a cytotoxin relatively specific for astrocytes. Ultrastructural evidence suggests that degeneration is confined to astrocytes after the injection of this toxin into the striatum (Huck et al., 1984; Khurgel et al., 1996; Rodriguez et al., 2004). To further examine whether spinal astrocytes are important for neuropathic pain, we administrated l-α-AA intrathecally to SNL rats on postoperative day 10. SNL-induced mechanical allodynia was suppressed by l-α-AA at 3, 6, 24, and 48 h after the injection, in a dose-dependent manner (Fig. 11a). The anti-allodynic effects of l-α-AA were reversible, recovering after 3 d (Fig. 11a). In agreement with previous studies (Khurgel et al., 1996; Rodriguez et al., 2004), l-α-AA (50 nmol) produced a marked reduction of GFAP-positive astrocytes in the dorsal horn (Fig. 11b,c). l-α-AA also decreased the expression of JNK1 (d, e), which was also expressed in spinal astrocytes. However, l-α-AA did not affect the number of NeuN-positive neurons in the spinal cord (Fig. 11d,e), in support of previous studies (Khurgel et al., 1996; Rodriguez et al., 2004), nor did l-α-AA alter the basal pain sensitivity in uninjured animals even at a high dose (150 nmol); the mechanical threshold was 15.7 ± 2.5, 16.0 ± 4.1, and 15.4 ± 2.8 g before and at 6 and 24 h after intrathecal l-α-AA injection (150 nmol), respectively (p > 0.05; n = 5).
Figure 11.
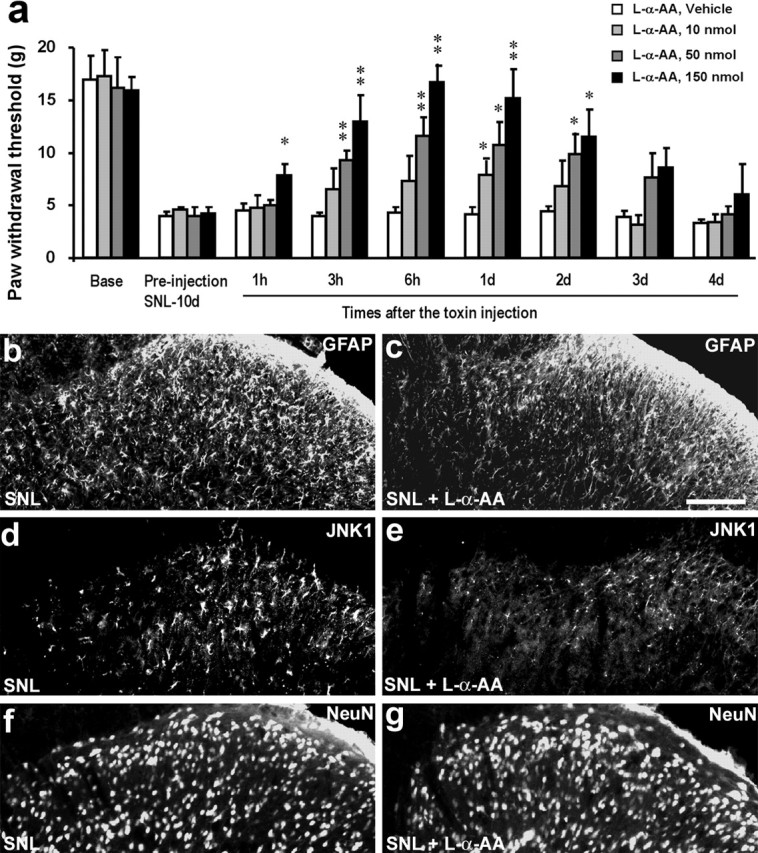
a–g, Astroglial toxin l-α-AA blocks neuropathic pain. a, l-α-AA (10, 50, and 150 nmol) was injected intrathecally at 10 d after SNL and reversed mechanical allodynia. *p < 0.05, **p < 0.01 by ANOVA compared with vehicle (saline) control; n = 6. b–g, Effects of l-α-AA (50 nmol) on GFAP (b, c), JNK1 (d, e), and NeuN (f, g) expressing cells in the superficial dorsal horn. Animals with SNL were perfused 24 h after the toxin injection for immunostaining. Scale bar, 50 μm.
Discussion
This study has revealed new mechanisms for neuropathic pain development and maintenance. We have several major findings in this study. First, we have shown that SNL rapidly (<12 h) but transiently (<10 d) activates JNK in small-sized (C-type) neurons of the injured (L5) DRG, which is important for the induction/development but not the maintenance of neuropathic pain. Second, we have demonstrated that SNL slowly (>3 d) but persistently (>21 d) activates JNK in spinal astrocytes, in which it is essential for the maintenance of neuropathic pain. The role of spinal astrocytes in maintaining neuropathic pain is further supported by our data showing reversal of mechanical allodynia after intrathecal infusion of an astroglial toxin. Third, we have shown that different MAPKs (p38 and JNK) are activated in different cell types (microglia and astrocytes), respectively, in the spinal cord after nerve injury.
JNK activation in the spinal cord and neuropathic pain
Our data show that JNK is slowly and persistently activated in spinal astroglia after SNL. JNK activation was also found in spinal astrocytes after amyotrophic lateral sclerosis (Migheli et al., 1997). Furthermore, JNK activation has been shown in spinal astrocytes after partial sciatic nerve lesion, but only one time point (21 d) was examined in this study (Ma and Quirion, 2002). It is important to investigate the cellular localization of pJNK at different times, because our previous studies have shown that ERK is activated in different cell types (microglia vs astrocytes) in the spinal cord at different times after nerve injury (Zhuang et al., 2005).
It is of particular interest that different MAPKs exhibit different activation patterns in the spinal cord after nerve injury. Whereas JNK is persistently activated in spinal astrocytes, p38 appears to be transiently activated in spinal microglia, with the peak activation in the first week (Table 1). Conversely, ERK activation is more dynamic; it is activated in spinal microglia in the first week after nerve injury, but only in spinal astrocytes at late times of nerve injury (Zhuang et al., 2005) (Table 1).
Table 1.
Summary of different activation patterns of three MAPK family members (ERK, p38, and JNK) in the DRG and spinal cord in normal and nerve injury (SNL) conditions
| Dorsal root ganglion |
Spinal cord dorsal horn (gray matter) |
|||||
|---|---|---|---|---|---|---|
| Basal expression | Injured (L5) | Noninjured (L4) | Basal expression | 3 d | 21 d | |
| pERK | Small neurons (+) | Large neurons (+) | No significant change | Not detectable | Microglia (+++) | Astrocytes (+++) |
| Satellite cells (+) | Satellite cells (+++) | |||||
| p-p38 | Small neurons (++) | Small neurons (+++) | Small neurons (+++) | Microglia (+) | Microglia (+++) | Microglia (++) |
| Satellite cells (+) | Large neurons (++) | Large neurons (+) | ||||
| pJNK | Small neurons (+) | Small neurons (+++) | No significant change | Astrocytes (+) | Astrocytes (++) | Astrocytes (+++) |
| Nerve fibers (+) | Nerve fibers (++) | |||||
| Satellite cells (+) | ||||||
Activation of MAPK is revealed by the expression of pMAPK. There is a low-level, basal expression of pMAPK in normal non-injured condition. +, ++, and +++ indicate low, medium, and high levels of expression of pMAPK, respectively. Small and large neurons of the DRG are described as NF-200-negative (C-fiber) and NF-200-positive (A-fiber) neurons, respectively. Switch of MAPKs to different cell types at late times has not been clearly studied in the DRG. pERK is also transiently induced in dorsal horn neurons in the first several hours of nerve injury. See the related references of Kim et al., 2002; Jin et al., 2003; Obata et al., 2003, 2004a,b; Schafers et al., 2003; Tsuda et al., 2004; Zhuang et al., 2005.
JNK has three isoforms: JNK1, JNK2, and JNK3. Whereas JNK3 is expressed in brain neurons, JNK1 is expressed in non-neuronal cells (e.g., immune cells) (Ip and Davis 1998; Borsello et al., 2003; Kuan et al., 2003). Our data have shown that JNK1 is the isoform that is activated in spinal astrocytes after SNL, because (1) only JNK1 was hyperphosphorylated in the spinal cord after SNL, and (2) JNK1 was expressed in spinal astrocytes in both injured and non-injured side of the spinal cord.
Our results demonstrate that intrathecal administration of JNK inhibitors, before or 10 d after nerve injury, prevented or reversed SNL-induced mechanical allodynia. The peptide inhibitor D-JNKI-1 was more potent than the small-molecule inhibitor SP600125, producing complete inhibition of SNL-induced allodynia at lower concentrations. Although it is generally believed that intrathecal drug infusion should target spinal cells, it is often neglected that the intrathecal route may also affect DRG cells. We found that D-JNKI-1 was taken up by many DRG cells after intrathecal injection. Therefore, the anti-allodynic effect of intrathecal D-JNKI-1 could be mediated by JNK activation at both DRG and spinal levels.
JNK activation in the DRG and neuropathic pain
We found that SNL activates JNK in the injured L5 DRG, in support of a previous study (Obata et al., 2004a). However, peak activation of JNK was missed in this previous study in which pJNK expression was examined 1 week after nerve injury. JNK activation in DRG neurons was more rapid than that of other MAPKs, becoming maximal within 12 h. As summarized in Table 1, the cell type expressing pJNK in the DRG is different from that expressing pERK and p-p38 after nerve injury. JNK was primarily activated in unmyelinated C-type neurons in the L5 DRG. Perfusion of the L5 DRG with D-JNKI-1 before nerve injury completely prevented SNL-induced mechanical allodynia, suggesting that JNK activation in C-type neurons in the L5 DRG is essential for the generation/development of mechanical allodynia. It remains to be investigated how JNK activation in small DRG neurons results in the generation of neuropathic pain. JNK may be involved in altered gene expression in the injured DRG neurons after nerve injury (Hokfelt et al., 1994; Ji and Strichartz, 2004). However, perfusion of D-JNKI-1 in the L5 DRG, starting 10 d after SNL, did not affect established allodynia. Consistently, JNK activation in the L5 DRG had significantly declined by day 10. Therefore, JNK activation in primary sensory neurons appears to be involved in the development but not the maintenance of neuropathic pain.
SNL also induced c-Jun phosphorylation in most neurons of the L5 DRG. Therefore, p-c-Jun can be used as a marker for the injured DRG neurons. However, pJNK is not a marker for overall axonal injury, because pJNK is only induced in 35% neurons of the L5 DRG with small size. Instead, pJNK is specifically related to C-fiber nociceptors.
Spinal astrocytes and neuropathic pain
Because (1) mechanical allodynia is reversed by intrathecal but not DRG (L5) perfusion of D-JNKI-1 and (2) JNK activation in the spinal cord is persistent, we conclude that JNK activation in the spinal cord contributes to the persistence of neuropathic pain. Our results also indicate an important role of spinal astrocytes in the maintenance of chronic mechanical allodynia. The importance of spinal astrocytes for persistent pain is further supported by our data showing that intrathecal injection of the astroglial toxin l-α-aminoadipate could effectively reverse mechanical allodynia. The effects of the toxin appear to be specific, because it only reduces the number of GFAP-expressing astrocytes, associated with a decrease in JNK1 expression, but not of NeuN-expressing neurons. Because basal pain sensitivity is not affected by this toxin, spinal astrocytes may not be important for normal pain perception. Interestingly, the effect of the toxin is reversible: neuropathic pain returns after 3 d. This reversal may be attributable to migration of astrocytes from other sites (Khurgel et al., 1996; Rodriguez et al., 2004).
Although increasing evidence suggests that spinal glia play an important role in pain control (Watkins et al., 2001; Ji and Strichartz, 2004; Tsuda et al., 2005), to our knowledge this is the first study that specifically addresses the role of spinal astrocytes in pain sensitization. The role of astrocytes in persistent pain is also supported by the following studies. First, spinal inhibition of ERK at a late time point, when pERK is expressed in spinal astrocytes, attenuates neuropathic pain (Zhuang et al., 2005). Second, implantation of neural stem cells into the injured spinal cord produces astrocytes and is followed by the development of mechanical allodynia (Hofstetter et al., 2005).
Although GFAP upregulation is one of the most distinct and persistent spinal changes in chronic pain conditions (Mantyh et al., 2002; DeLeo et al., 2004) and has been correlated with neuropathic pain (Garrison et al., 1994), we have shown that neuropathic pain can be blocked by D-JNKI-1 without accompanying reduction of GFAP expression. Our data suggest that spinal GFAP upregulation may be just associated with nerve injury but not a cause for neuropathic pain. Therefore, it could be misleading to use spinal GFAP change as an indicator of elevated pain states. It is the change of signaling molecules, not structural proteins, in astrocytes that is important for pain sensitization.
It remains to be investigated how JNK activation in spinal astroglia regulates pain hypersensitivity. Because D-JNKI-1 suppressed SNL-induced c-Jun phosphorylation in spinal astrocytes, JNK activation is likely to regulate gene transcription in spinal astrocytes via activation of the transcription factor c-Jun, as well as other transcription factors such as nuclear factor-κB and activating transcription factor-2. As a result, there is increased synthesis of neuroexcitatory and neurosensitizing substances (e.g., BDNF, IL-1β, TNF-α, PGE2, etc.) by spinal glia, leading to dorsal horn neuron sensitization (Samad et al., 2001; Watkins et al., 2001; DeLeo et al., 2004; Ji and Strichartz, 2004; Coull et al., 2005; Tsuda et al., 2005).
Concluding remarks
This study has identified the JNK cascade as a critical signaling pathway for neuropathic pain development and maintenance via distinct mechanisms in the DRG and spinal cord. In particular, we have shown that spinal astrocytes play an important role in maintaining persistent mechanical allodynia, although the underlying mechanisms remain to be investigated.
Many patients in the pain clinic suffer from neuropathic pain caused by injury to the peripheral nervous system or the CNS, and one of the most distinct symptoms is mechanical allodynia. At present, few drugs are effective in treating neuropathic pain, and their efficacy is only demonstrated in ≤30% of patients (Woolf and Mannion, 1999; Gardell et al., 2003). However, all current drugs were developed or considered to act against molecular targets in neurons. Targeting the JNK pathway in spinal glia and sensory neurons may offer a new option to treat intractable neuropathic pain.
Footnotes
*Z.-Y.Z. and Y.-R.W. contributed equally to this work.
This work was supported by National Institutes of Health Grants NS40698 and TW7180 (R.-R.J.), by Swiss National Science Foundation Grants 3232-066354 and 3200-066355, and by the Pierre Mercier Science Foundation (I.D.).
References
- Abbadie C, Lindia JA, Cumiskey AM, Peterson LB, Mudgett JS, Bayne EK, DeMartino JA, MacIntyre DE, Forrest MJ (2003). Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2. Proc Natl Acad Sci USA 100:7947–7952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borsello T, Bonny C (2004). Use of cell-permeable peptides to prevent neuronal degeneration. Trends Mol Med 10:239–244. [DOI] [PubMed] [Google Scholar]
- Borsello T, Clarke PG, Hirt L, Vercelli A, Repici M, Schorderet DF, Bogousslavsky J, Bonny C (2003). A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat Med 9:1180–1186. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL (1994). Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 53:55–63. [DOI] [PubMed] [Google Scholar]
- Coggeshall RE, Lekan HA (1996). Methods for determining numbers of cells and synapses: a case for more uniform standards of review. J Comp Neurol 364:6–15. [DOI] [PubMed] [Google Scholar]
- Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y (2005). BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 438:1017–1021. [DOI] [PubMed] [Google Scholar]
- DeLeo JA, Yezierski RP (2001). The role of neuroinflammation and neuroimmune activation in persistent pain. Pain 90:1–6. [DOI] [PubMed] [Google Scholar]
- DeLeo JA, Tanga FY, Tawfik VL (2004). Neuroimmune activation and neuroinflammation in chronic pain and opioid tolerance/hyperalgesia. The Neuroscientist 10:40–52. [DOI] [PubMed] [Google Scholar]
- Gardell LR, Wang R, Ehrenfels C, Ossipov MH, Rossomando AJ, Miller S, Buckley C, Cai AK, Tse A, Foley SF, Gong B, Walus L, Carmillo P, Worley D, Huang C, Engber T, Pepinsky B, Cate RL, Vanderah TW, Lai J, et al (2003). Multiple actions of systemic artemin in experimental neuropathy. Nat Med 9:1383–1389. [DOI] [PubMed] [Google Scholar]
- Garrison CJ, Dougherty PM, Carlton SM (1994). GFAP expression in lumbar spinal cord of naive and neuropathic rats treated with MK-801. Exp Neurol 129:237–243. [DOI] [PubMed] [Google Scholar]
- Hofstetter CP, Holmstrom NA, Lilja JA, Schweinhardt P, Hao J, Spenger C, Wiesenfeld-Hallin Z, Kurpad SN, Frisen J, Olson L (2005). Allodynia limits the usefulness of intraspinal neural stem cell grafts; directed differentiation improves outcome. Nat Neurosci 8:346–353. [DOI] [PubMed] [Google Scholar]
- Hokfelt T, Zhang X, Wiesenfeld-Hallin Z (1994). Messenger plasticity in primary sensory neurons following axotomy and its functional implications. Trends Neurosci 17:22–30. [DOI] [PubMed] [Google Scholar]
- Honore P, Rogers SD, Schwei MJ, Salak-Johnson JL, Luger NM, Sabino MC, Clohisy DR, Mantyh PW (2000). Murine models of inflammatory, neuropathic and cancer pain each generates a unique set of neurochemical changes in the spinal cord and sensory neurons. Neuroscience 98:585–598. [DOI] [PubMed] [Google Scholar]
- Huck S, Grass F, Hortnagl H (1984). The glutamate analogue α-aminoadipic acid is taken up by astrocytes before exerting its gliotoxic effect in vitro J Neurosci 4:2650–2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip YT, Davis RJ (1998). Signal transduction by the c-Jun N-terminal kinase (JNK): from inflammation to development. Curr Opin Cell Biol 10:205–219. [DOI] [PubMed] [Google Scholar]
- Ji RR, Strichartz G (2004). Cell signaling and the genesis of neuropathic pain. Sci STKE 2004:reE14. [DOI] [PubMed] [Google Scholar]
- Ji RR, Woolf CJ (2001). Neuronal plasticity and signal transduction in nociceptive neurons: implications for the initiation and maintenance of pathological pain. Neurobiol Dis 8:1–10. [DOI] [PubMed] [Google Scholar]
- Ji RR, Zhang Q, Law PY, Low HH, Elde R, Hokfelt T (1995). Expression of μ-, δ-, and κ-opioid receptor-like immunoreactivities in rat dorsal root ganglia after carrageenan-induced inflammation. J Neurosci 15:8156–8166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Zhang Q, Pettersson RF, Hokfelt T (1996). aFGF, bFGF and NGF differentially regulate neuropeptide expression in dorsal root ganglia after axotomy and induce autotomy. Regul Pept 66:179–189. [DOI] [PubMed] [Google Scholar]
- Ji RR, Befort K, Brenner GJ, Woolf CJ (2002a). ERK MAP kinase activation in superficial spinal cord neurons induces prodynorphin and NK-1 upregulation and contributes to persistent inflammatory pain hypersensitivity. J Neurosci 22:478–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Samad TA, Jin SX, Schmoll R, Woolf CJ (2002b). p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron 36:57–68. [DOI] [PubMed] [Google Scholar]
- Jin SX, Zhuang ZY, Woolf CJ, Ji RR (2003). p38 mitogen-activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. J Neurosci 23:4017–4022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurgel M, Koo AC, Ivy GO (1996). Selective ablation of astrocytes by intracerebral injections of alpha-aminoadipate. Glia 16:351–358. [DOI] [PubMed] [Google Scholar]
- Kim SH, Chung JM (1992). An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain 50:355–363. [DOI] [PubMed] [Google Scholar]
- Kim SY, Bae JC, Kim JY, Lee HL, Lee KM, Kim DS, Cho HJ (2002). Activation of p38 MAP kinase in the rat dorsal root ganglia and spinal cord following peripheral inflammation and nerve injury. NeuroReport 13:2483–2486. [DOI] [PubMed] [Google Scholar]
- Kuan CY, Whitmarsh AJ, Yang DD, Liao G, Schloemer AJ, Dong C, Bao J, Banasiak KJ, Haddad GG, Flavell RA, Davis RJ, Rakic P (2003). A critical role of neural-specific JNK3 for ischemic apoptosis. Proc Natl Acad Sci USA 100:15184–15189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Quirion R (2002). Partial sciatic nerve ligation induces increase in the phosphorylation of extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) in astrocytes in the lumbar spinal dorsal horn and the gracile nucleus. Pain 99:175–184. [DOI] [PubMed] [Google Scholar]
- Mantyh PW, Clohisy DR, Koltzenburg M, Hunt SP (2002). Molecular mechanisms of cancer pain. Nat Rev Cancer 2:201–209. [DOI] [PubMed] [Google Scholar]
- Migheli A, Piva R, Atzori C, Troost D, Schiffer D (1997). c-Jun, JNK/SAPK kinases and transcription factor NF-kappa B are selectively activated in astrocytes, but not motor neurons, in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 56:1314–1322. [DOI] [PubMed] [Google Scholar]
- Molander C, Xu Q, Grant G (1984). The cytoarchitectonic organization of the spinal cord in the rat. I. The lower thoracic and lumbosacral cord. J Comp Neurol 230:133–141. [DOI] [PubMed] [Google Scholar]
- Obata K, Noguchi K (2004). MAPK activation in nociceptive neurons and pain hypersensitivity. Life Sci 74:2643–2653. [DOI] [PubMed] [Google Scholar]
- Obata K, Yamanaka H, Dai Y, Tachibana T, Fukuoka T, Tokunaga A, Yoshikawa H, Noguchi K (2003). Differential activation of extracellular signal-regulated protein kinase in primary afferent neurons regulates brain-derived neurotrophic factor expression after peripheral inflammation and nerve injury. J Neurosci 23:4117–4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obata K, Yamanaka H, Kobayashi K, Dai Y, Mizushima T, Katsura H, Fukuoka T, Tokunaga A, Noguchi K (2004a). Role of mitogen-activated protein kinase activation in injured and intact primary afferent neurons for mechanical and heat hypersensitivity after spinal nerve ligation. J Neurosci 24:10211–10222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obata K, Yamanaka H, Dai Y, Mizushima T, Fukuoka T, Tokunaga A, Noguchi K (2004b). Differential activation of MAPK in injured and uninjured DRG neurons following chronic constriction injury of the sciatic nerve in rats. Eur J Neurosci 20:2881–2895. [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Tanga F, DeLeo JA (2003). Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. J Pharmacol Exp Ther 306:624–630. [DOI] [PubMed] [Google Scholar]
- Rodriguez MJ, Martinez-Sanchez M, Bernal F, Mahy N (2004). Heterogeneity between hippocampal and septal astroglia as a contributing factor to differential in vivo AMPA excitotoxicity. J Neurosci Res 77:344–353. [DOI] [PubMed] [Google Scholar]
- Samad TA, Moore KA, Sapirstein A, Billet S, Allchorne A, Poole S, Bonventre JV, Woolf CJ (2001). Interleukin-1beta-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature 410:471–475. [DOI] [PubMed] [Google Scholar]
- Schafers M, Svensson CI, Sommer C, Sorkin LS (2003). Tumor necrosis factor-α induces mechanical allodynia after spinal nerve ligation by activation of p38 MAPK in primary sensory neurons. J Neurosci 23:2517–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanga FY, Nutile-McMenemy N, DeLeo JA (2005). The CNS role of Toll-like receptor 4 in innate neuroimmunity and painful neuropathy. Proc Natl Acad Sci USA 102:5856–5861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tezel G, Yang X, Yang J, Wax MB (2004). Role of tumor necrosis factor receptor-1 in the death of retinal ganglion cells following optic nerve crush injury in mice. Brain Res 996:202–212. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Shigemoto-Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter MW, Inoue K (2003). P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature 424:778–783. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Mizokoshi A, Shigemoto-Mogami Y, Koizumi S, Inoue K (2004). Activation of p38 mitogen-activated protein kinase in spinal hyperactive microglia contributes to pain hypersensitivity following peripheral nerve injury. Glia 45:89–95. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Inoue K, Salter MW (2005). Neuropathic pain and spinal microglia: a big problem from molecules in “small” glia. Trends Neurosci 28:101–107. [DOI] [PubMed] [Google Scholar]
- Wang J, Van De Water TR, Bonny C, de Ribaupierre F, Puel JL, Zine A (2003). A peptide inhibitor of c-Jun N-terminal kinase protects against both aminoglycoside and acoustic trauma-induced auditory hair cell death and hearing loss. J Neurosci 23:8596–8607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins LR, Maier SF (2003). Glia: a novel drug discovery target for clinical pain. Nat Rev Drug Discov 2:973–985. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Milligan ED, Maier SF (2001). Glial activation: a driving force for pathological pain. Trends Neurosci 24:450–455. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Mannion RJ (1999). Neuropathic pain: aetiology, symptoms, mechanisms, and management. Lancet 353:1959–1964. [DOI] [PubMed] [Google Scholar]
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME (1995). Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 270:1326–1331. [DOI] [PubMed] [Google Scholar]
- Zhuang ZY, Gerner P, Woolf CJ, Ji RR (2005). ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. Pain 114:149–159. [DOI] [PubMed] [Google Scholar]