

Abstract
The microtubule binding protein tau is implicated in neurodegenerative tauopathies, including frontotemporal dementia (FTD) with Parkinsonism caused by diverse mutations in the tau gene. Hyperphosphorylation of tau is considered crucial in the age-related formation of neurofibrillary tangles (NFTs) correlating well with neurotoxicity and cognitive defects. Transgenic mice expressing FTD mutant tau-P301L recapitulate the human pathology with progressive neuronal impairment and accumulation of NFT. Here, we studied tau-P301L mice for parameters of learning and memory at a young age, before hyperphosphorylation and tauopathy were apparent. Unexpectedly, in young tau-P301L mice, increased long-term potentiation in the dentate gyrus was observed in parallel with improved cognitive performance in object recognition tests. Neither tau phosphorylation, neurogenesis, nor other morphological parameters that were analyzed could account for these cognitive changes. The data demonstrate that learning and memory processes in the hippocampus of young tau-P301L mice are not impaired and actually improved in the absence of marked phosphorylation of human tau. We conclude that protein tau plays an important beneficial role in normal neuronal processes of hippocampal memory, and conversely, that not tau mutations per se, but the ensuing hyperphosphorylation must be critical for cognitive decline in tauopathies.
Keywords: LTP, neurodegeneration, memory, dendate gyrus, neurogenesis, Golgi impregnation
Introduction
Protein tau is a microtubule-associated protein involved in the assembly and stabilization of microtubuli (MTs). The affinity of tau for MT is actively regulated by phosphorylation and by changes in the ratio of its isoforms, containing either three or four MT binding domains (tau-3R, tau-4R) (Andorfer and Davies, 2000; Buée et al., 2000; Goedert and Jakes, 2005). In an increasing number of neurodegenerative disorders, including Alzheimer's disease (AD), intracellular accumulation of hyperphosphorylated tau as neurofibrillary tangles (NFTs) parallel memory disturbances (Tolnay and Probst, 1999). In a diverse group of inherited tauopathies known as frontotemporal dementia with Parkinsonism (FTDP-17), mutations in the tau gene located on chromosome 17 are the underlying genetic causes. AD and FTDP-17 patients share the diagnostic accumulations of hyperphosphorylated tau as NFTs (Goedert and Jakes, 2005).
FTDP-17 mouse models show that increased phosphorylation of tau results in severe tauopathy, often with premature death (for review, see Lee et al., 2005). Furthermore, FTDP-17 modeling transgenic mice have memory impairment (Tatebayashi et al., 2002; Arendash et al., 2004; Pennanen et al., 2004) and severe hyperphosphorylation of tau at later ages (Chen, 2005; Sun et al., 2005; Terwel et al., 2005).
In most studies, memory was tested when phosphorylation of tau was evident. In the present study, we questioned whether mutant tau per se alters memory and learning (i.e., in the absence of increased phosphorylation and before NFT formation). We analyzed transgenic mice expressing tau-P301L in the human tau-2N/4R isoform in an otherwise normal genetic background (Terwel et al., 2005). Hippocampal learning and memory was studied at a young age, when phosphorylation of tau is normal or even somewhat lower than in wild-type (WT) mice (Terwel et al., 2005). Moreover, the behavioral tests were performed before the onset of any motor defects, as tested by rotarod performance that showed motor impairment in older tau transgenic mice (Lee et al., 2005; Terwel et al., 2005). Long-term potentiation (LTP) in both the CA1 and dentate gyrus (DG) was measured in parallel with a hippocampus-dependent memory task that imposes a low level of stress [i.e., the object recognition task (ORT)]. The CA1 is early and severely affected in AD, whereas the DG is severely affected in FTDP-17 patients (van Swieten et al., 1999; Kobayashi et al., 2003; Bronner et al., 2005). Adult neurogenesis was studied as a possible factor contributing to tau-related changes in hippocampal memory function (Gould et al., 1999; van Praag et al., 1999; Shors et al., 2002; Jin et al., 2004; Shors, 2004), particularly because disruption or overexpression of human protein tau affects the cell cycle, neuronal maturation, and axonal elongation (Takei et al., 2000; Dawson et al., 2001; Zhao et al., 2003; Andorfer et al., 2005). A second factor possibly contributing to hippocampal memory function (i.e., the morphology CA1 pyramidal and DG granular cells) was also studied in detail. However, the surprising outcome of improved learning and memory in young tau-P301L mice was not correlated with increased neurogenesis nor with changes in hippocampal morphology.
Materials and Methods
Transgenic mice
Unless indicated otherwise, 8- to 10-week-old male tau-P301L transgenic mice in the FVB/N genetic background (Terwel et al., 2005) were compared with age- and sex-matched FVB/N nontransgenic mice. In the transgenic mice, the longest human tau isoform bearing the P301L mutation (tau-4R/2N-P301L) is expressed under control of the mouse thy1 gene promoter, resulting specifically in neuronal expression only, which begins in the second postnatal week. Several independent founder lines were obtained and phenotyped initially, demonstrating similar genotypic and phenotypic characteristics. We selected one strain for additional experiments based on a very similar expression of the tau-P301L transgene as the previously characterized tau-4R strain (Spittaels et al., 1999, 2000), and both strains were characterized in extensive detail (Terwel et al., 2005). Homozygous tau-P301L mice obtained by inbreeding are normally fertile and transmit the transgene in a stable manner to their offspring.
Behavioral testing
Mice were housed in groups from three to seven animals per cage and placed in the behavioral testing room at least 30 min before the experiments commenced. Two age groups of mice, 5 and 9 weeks of age, were subjected to the following tests, performed in chronological order, as described below.
First, motor ability was tested by rotarod on a revolving horizontal rod of 3.2 cm in diameter (Med Associates, Georgia, VT). After one (5-week-old mice) or two (9-week-old mice) training sessions of 5 min at 16 rotations per minute (rpm), the mice were finally tested on the rod, accelerating from 4 to 40 rpm over a 3 min period. The time that mice remained on the rod was recorded automatically.
Exploratory and motor capacities were subsequently analyzed in an open field setting. Each mouse was placed for 5 min on an elevated, white opaque Plexiglas surface of 52 × 52 cm without bordering walls, dimly lit from underneath with fluorescent tubes. The travel path was recorded by a video camera linked to a computer and analyzed using dedicated software (Ethovision; Noldus, Wageningen, The Netherlands). The center of the open field was defined as an inner square of 40 × 40 cm, and the time spent in the center was calculated.
Learning and memory characteristics were analyzed by means of the ORT (Dewachter et al., 2002). Individual mice were habituated on day 1 for two sessions of 5 min in a Plexiglas box (52 × 52 × 40 cm), with a white opaque floor and black walls softly illuminated from underneath. On day 2, in the acquisition phase, mice were presented for 10 min with two identical objects placed in two adjacent quadrants of the box. The time the animal spent exploring an object, with its snout directed toward the object within ± 2 cm, was recorded manually. The travel paths were recorded by computerized video imaging and analyzed using dedicated software, as above. A 10 min retention trial was given after a delay of either 1 or 3.5 h. During the retention trial, the mice were confronted with one familiar and one novel object. We used two sets of identical objects. Combinations of objects and their positions in the box (left, right) were randomized to avoid preferences not based on novelty. However, after analysis, neither WT nor P301L mice showed a preference for the different objects or for the left or right side of the box. After each trial, objects were cleaned to eliminate odor cues. The level of discrimination (d2) was calculated by the formula d2 = (b − a)/(b + a), where a and b are the exploration times spent on the old and novel objects, respectively. Furthermore, exploration time during acquisition and retention and the absolute discrimination index, d1 (= b − a), were analyzed (data not shown).
Electrophysiology
Slice preparation and recording conditions.
After decapitation between 9:00 and 10:00 A.M., brains were removed immediately and kept at 4°C in artificial CSF (ACSF) containing the following (in mm): 120 NaCl, 3.5 KCl, 5.0 MgSO4, 1.25 NaH2PO4, 0.2 CaCl2, 10 d-glucose, and 25 NaHCO3, gassed with 95% O2 and 5% CO2. The right hemisphere was immersion fixed for histology, and the left hemisphere was used for field potential recordings. For Schaffer collateral recordings, 400 μm transversal hippocampal sections were prepared using a tissue chopper; for perforant path recordings, 400 μm horizontal forebrain sections were obtained using a vibroslicer (Leica VT 1000S; Leica Microsystems, Nussloch, Germany). After 1 h of incubation at room temperature (RT) in oxygenated recording ACSF (in mm: 120 NaCl, 3.5 KCl, 1.3 MgSO4, 1.25 NaH2PO4, 1.25 CaCl2, 10 d-glucose, 25 NaHCO3), slices were transferred to a recording chamber and perfused with oxygenated ACSF at 31.5°C.
Bipolar, stainless steel, isolated (except for the tip) stimulating electrodes were placed in the Schaffer collaterals or in the perforant path to record field EPSPs (fEPSPs) using a glass microelectrode (2–5 MΩ filled with ACSF) placed in the stratum radiatum of CA1 or the middle third of the molecular layer of the DG, respectively. To evoke robust LTP in the DG, GABA-mediated activity was blocked by adding 10 μm bicuculline methiodide (Sigma, St. Louis, MO) to the ACSF (Alfarez et al., 2003).
Stimulation protocol
Before baseline recording commenced, the maximal fEPSP amplitude and slope were determined by gradually increasing the stimulus intensity (60 s interstimulus interval) until the response saturated. The relationship between the stimulus intensity and the evoked response was fit to a Bolzmann equation: R(i)=Rmax/[1 + exp(i − ih)/(S)], in which R(i) is the response at stimulus intensity (i), Rmax is the maximal response, ih is the intensity at which the half-maximal response is observed, and slope factor S is the index that is proportional to the slope of the stimulus–response curve. The half-maximal stimulation intensity was used to record baseline responses for at least 20 min. Recordings in which the baseline was not stable or the maximal fEPSP amplitude was <1.5 mV were rejected.
After baseline recordings, paired-pulse responses with interstimulus intervals of 50, 100, 200, and 500 ms were tested in the DG to ensure the medial perforant path was stimulated. Recordings that showed paired pulse facilitation were discarded. If all of the above criteria were met, LTP was evoked in both the CA1 and the DG using a theta burst protocol consisting of two trains of four pulses at 100 Hz intermitted by 200 ms. This procedure was repeated five times with an interval of 30 s. After theta burst stimulation, LTP was recorded for 60 min.
Brain histology
The histological experiments described below were performed on the same animals, with the other hemisphere used for electrophysiological recordings, except for the histological Golgi staining, which was performed on a separate set of mice of the same age. After decapitation, the right cerebrum was immersion fixed in 4% paraformaldehyde in 0.1 m PBS, pH 7.4, for 7 d. The hemisphere was washed in PBS, equilibrated in 30% sucrose overnight, and frozen on dry ice, after which 30 μm coronal sections were cut using a sliding microtome. The left cerebrum was used for field potential recordings as described above.
Total volume and cellular density of the DG and other hippocampal subregions were determined by a stereological approach as described previously (Heine et al., 2004a). In short, every 10th section was hematoxylin stained and used to determine the total volume of the granular cell layer (GCL), the hilus, and the total CA1, calculated according to Cavalieri's direct estimator. On each section, areas of interest were photographed using a standard magnification, and surface areas were measured on a Macintosh computer using the public domain program Object-Image (an extended version of NIH Image, developed at the National Institutes of Health and at the University of Amsterdam; available at http://simon.bio.uva.nl). Total volume was calculated using the formula V = ΣAxT, where ΣA is the sum of area measurements, and T is the intersection distance (300 μm).
The same sections were used to determine the three-dimensional numerical density of neurons (NV, neurons per cubic millimeter) in the GCL, using the optical dissector method. Counting frames were placed randomly over the GCL. Individual neurons were visualized (Axiophot microscope, 100×; numerical aperture, 1.30 oil objective; Zeiss, Thornwood, NY) and counted, according to the dissector approach, if their nuclear profile was present and if they were positioned within the counting frame or intersected by its inclusion edges (i.e., the top and the right edges). Per hippocampus, at least 100 dissectors were used to calculate the numerical density (NV) of neurons from NV = ΣQ/ΣVDIS, where ΣQ is the sum of the neurons counted in all dissectors, and ΣVDIS is the sum of the dissector volumes. One dissector volume is 7.5 × 104 μm3, as calculated from VDIS = aDISxh, where aDIS is the area of the square counting frame, measuring 50 μm on a side, and h is the dissector height of 30 μm.
Golgi staining
Mice were decapitated, and their brains were removed and placed in Golgi-Cox solution (1.04% potassium dichromate, 1.04% mercury chloride, 0.83% potassium chromate, dissolved in double distilled water). After rinsing in water for 5 min and dehydration in 70% EtOH (O/N), 96% EtOH (O/N), 100% EtOH (8 h), and 1:2 EtOH/ether (O/N), brains were saturated by consecutive overnight incubations in 3, 6, and 12% celloidine. Celloidine was cleared with chloroform before 200 μm coronal sections were cut. Staining was developed by a 5 min rinse in water, 30 min in 16% ammonia, a 2 min rinse in water, 7 min in 1% sodium thiosulphate, and a 10 min rinse in water followed by dehydration for 5 min in 70% in EtOH, 5 min in 96% EtOH, 5 min in butanol, and 5 min in Histo-clear (Biozym, Landgraaf, The Netherlands). The sections were mounted in Histomount (National Diagnostics, Atlanta, GA) under glass coverslips.
In the thick impregnated sections, dentate granule cells were selected from the middle third of the inner pyramidal blade at approximately bregma −2.54 mm. Pyramidal cells were selected from the same level of the CA1 area opposite to the middle third of the inner pyramidal blade of the DG. If neurons were completely stained and horizontally orientated within the section, 99 Z-stacks of 1 μm were recorded with the program Image-Pro Plus, version 5.1.1.38 (Media Cybernetics, Silver Spring, MD), using an Axioplan 2 (Zeiss) microscope, equipped with an Evolution QEi FAST (monochrome, 12 bit) camera (Media Cybernetics) at a 40× magnification (Plan-Apochromat). The drawing tool NeuroDraw (Image-Pro Plus; developed by K. de Vos, J. van Heerikhuize, and C. W. Pool, Netherlands Institute for Brain Research, Amsterdam, The Netherlands) was used to determine total dendritic length per neuron, number of dendrites per cell, cell area, length per dendrite, number of dendritic ramifications, number of terminal segments, mean terminal segment length, and the mean intersegment length (Ramakers et al., 1998).
Immunohistochemistry
Immunohistochemistry (IHC) for phosphorylation-independent human tau with monoclonal antibody (mAb) HT7 (1:10,000; Innogenetics, Gent, Belgium) and for the specific phospho-epitope Ser(P)396/Ser(P)404(AD2, 1:1000; A. Delacourte), Thr(P)231 (AT180, 1:250; Innogenetics), and Ser(P)202/Thr(P)205 (AT8, 1:1000; Innogenetics) was as described previously (Terwel et al., 2005). In short, mice were anesthetized with pentobarbital (120 mg/kg, i.p.) and fixed by transcardiac perfusion with 4% paraformaldehyde in PBS (2 ml/min for 10 min). The brain was removed, postfixed overnight in 4% paraformaldehyde, and stored in PBS, 0.1% sodium azide, at 4°C. Sagittal sections (40 μm) were cut, rinsed in PBS, and treated with 1.5% H2O2 in 50% methanol to inactivate endogenous peroxidases. Nonspecific binding of antibodies was blocked by treatment with 10% fetal calf serum and 3% bovine serum albumin in PBS (blocking buffer). The sections were incubated at room temperature overnight with primary antibodies in blocking buffer and 0.1% Triton X-100. The sections were then incubated for 1 h with goat anti-mouse or anti-rabbit IgG-horseradish peroxidase, diluted 1:500 in blocking buffer and 0.1% Triton X-100. Next, the sections were washed with PBS and 50 mm Tris-HCl, pH 7.6, for 5 min and developed with 3,3′-diaminobenzidine (DAB), 0.3% H2O2 in 50 mm Tris-HCl, pH 7.6, for 3–5 min.
Western blotting
Western blotting was performed as described previously (Terwel et al., 2005) on extracts of isolated hippocampi of three individual mice per genotype and age group. The total amount of tau was detected using mAb Tau-5 (1:1000; PharMingen, San Diego, CA), and selected phosphoepitopes were detected with mAbs AT8 (1:200), AD180 (1:200), and AD2 (1:5000). Tau-4R isoforms were specifically detected with the rabbit polyclonal antibody directed against the second microtubule binding domain (Takuma et al., 2003).
Analysis of neurogenesis
Because hippocampal LTP and learning and memory have been related to parallel changes in neurogenesis, we analyzed three different neurogenesis markers: (1) doublecortin, as a cumulative marker for young migratory neurons (3 d to 3 weeks of age) (Couillard-Despres et al., 2005); (2) the birth date marker bromodeoxyuridine (BrdU); and (3) Ki-67, an endogenous DNA binding protein present only in proliferating cells.
BrdU immunohistochemistry.
To study the effects of mutated tau on adult generated cell survival, 1-month-old mice were injected in the morning with BrdU (5 mg/ml dissolved in 0.007 m NaOH/0.9% NaCl, i.p.) at a dose of 50 mg/kg, repeated for seven consecutive days. The mice were killed 4 weeks after the first injection. After blocking endogenous peroxidase with 1% H2O2 for 20 min and denaturing DNA in hot formamide and 2 N HCl, free-floating sections were incubated in the primary antibody mouse α-BrdU (1: 3000; Roche, Woerden, The Netherlands), diluted in phosphate buffer/0.1% bovine serum albumin/0.3% Triton X-100/1% goat serum for 1 h at room temperature and then overnight at 4°C. The rest of the procedure was identical to that described previously (Heine et al., 2004b).
Doublecortin immunohistochemistry.
Sections were rinsed in 50 mm Tris buffered saline (TBS), pH 7.6, three times for 5 min. After blocking endogenous peroxidase activity by 2% H2O2, sections were incubated for 1 h at room temperature and overnight at 4°C with the primary goat anti-doublecortin antiserum (C-18; Santa Cruz Biotechnology, Santa Cruz, CA) diluted 1:600 in 0.25% gelatin/0.5% Triton X-100 in TBS. The secondary antibody (donkey anti-goat Ig; 1:500) was applied for 2 h at room temperature. Subsequently, the reaction was amplified for 2 h with the Elite Vectastain avidin-biotin complex (ABC kit; Vector Laboratories, Burlingame, CA). The signal was further amplified with biotinylated tyramide (1:500) and 0.01% peroxide for 30 min followed by 45 min incubation with ABC. DAB was used to visualize antibody labeling.
Ki-67 immunohistochemistry.
The Ki-67 antigen is a 345–395 kDa nonhistone protein complex present in proliferating cells during G1, S, G2, and M but not the G0 phase of the cell cycle (Gerdes et al., 1984; Endl and Gerdes, 2000; Duchrow et al., 2001). Ki-67 is widely used in tumor biology and considered a good proliferation marker. Moreover, the numbers of cells visualized with BrdU labeling after a short survival time are highly comparable with the numbers obtained with Ki-67 staining (Kee et al., 2002).
Microwave antigen retrieval was applied before antibody incubation. Sections were mounted on Menzel Superfrost Plus glass sections. After air drying, sections were placed in 400 ml of 0.01 m citrate buffer, pH 6.0, and heated in a household microwave device starting at 800 W and gradually decreasing to 100 W once boiling commenced. Sections were allowed to cool down to RT. The following procedure was identical to that used for doublecortin immunohistochemistry; however, instead of 0.05 m TBS, 0.1 m TBS was used. The primary antibody solution was 1:1500 (NCL-Ki67p; Novocastra, New Castle, UK). The secondary antibody solution was 1:200 (biotinylated anti-rabbit; Amersham Biosciences, Piscataway, NJ).
Statistical analysis
Except for the Golgi data, which were analyzed using a nonparametric Mann–Whitney U test, differences between strains were tested using a two-way ANOVA.
Results
Tau expression and phosphorylation
Previously, we analyzed the phosphorylation status of protein tau in whole brain and in spinal cord and its progression with age. We observed that in young tau-P301L mice, mutant tau was less phosphorylated than wild-type tau-4R in the brains of age-matched tau-4R mice, which differ only in the mutation (Spittaels et al., 1999; Terwel et al., 2005).
Here, we compared specifically the phosphorylation status of tau in the hippocampal formation in both the transgenic strains, and relative to nontransgenic mice, all at 2 months of age. First, IHC with mAb HT7 specific for human tau demonstrated expression in all hippocampal subareas in tau-P301L mice (Fig. 1). Some differences in localization (i.e., tau-P301L more in somatodendritic compartments than tau-4R) were evident in the CA1, dentate gyrus, and perforant pathway (Fig. 1). Second, IHC with mAbs AT8, AT180, and AD2, directed against specified phosphoepitopes, revealed that in the brains of tau-P301L mice, less phosphorylated epitopes were present than in tau-4R mice, and even hardly more than in nontransgenic mice (Fig. 1).
Figure 1.
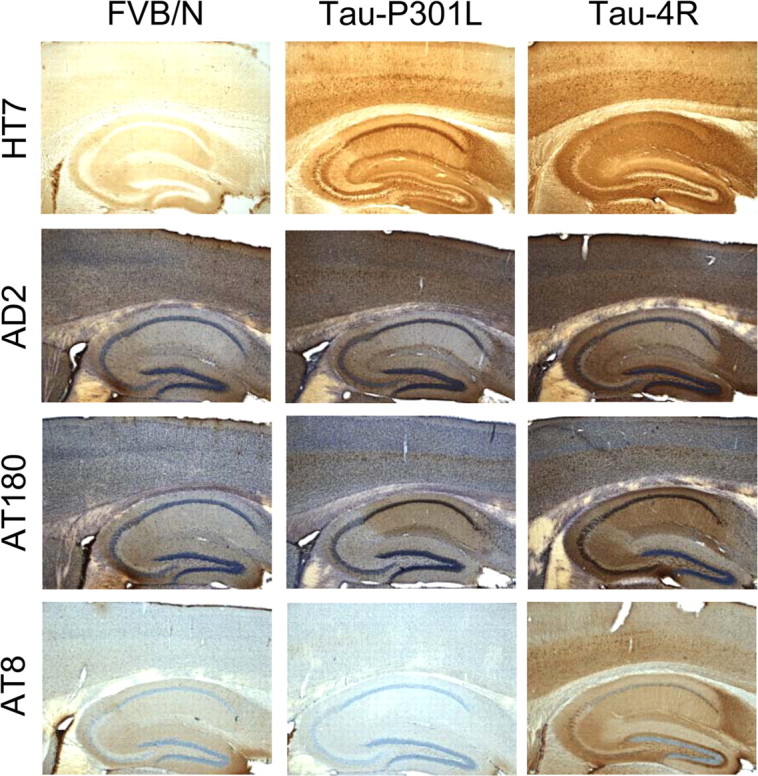
Immunohistochemistry for human tau and phosphorylated tau in the hippocampus of young nontransgenic and of tau-4R and tau-P301L transgenic mice. Human protein tau was detected with mAb HT7 in the hippocampal formation of tau-P301L and tau-4R transgenic mice (8 weeks of age) but not in nontransgenic mice (FVB/N). IHC with the phosphorylation-specific antibodies AD2, AT8, and AT180 demonstrated less phosphorylation in tau-P301L mice. Except for IHC with HT7, all sections were counterstained with hematoxylin (original magnification, 5×).
Because IHC is not well suited for quantitative estimations, we quantified by Western blotting the IHC results, using the same phosphoepitope-specific antibodies AT8, AT180, and AD2 as well as the pan-tau antibody Tau-5. Particularly, the AT8 epitope was less abundant in hippocampal extracts from tau-P301L mice, but also the other phosphoepitopes were less prominent (Fig. 2). The lesser phosphorylation of transgenic tau-P301L was also inferred from its higher electrophoretic mobility in SDS-PAGE (Fig. 2), as observed previously (Terwel et al., 2005).
Figure 2.

Western blotting for human tau and phosphorylated tau in the hippocampus of young nontransgenic and tau-4R and tau-P301L transgenic mice. Protein extracts from the hippocampus of nontransgenic mice (FVB) and tau-P301L and tau-4R transgenic mice (n = 3, each) were analyzed by Western blotting with mAb Tau5 to detect total tau (i.e., mouse tau and transgenic human tau). Western blotting with phosphorylation-dependent antibodies AT8, AT180, and AD2 demonstrated that transgenic tau-P301L was less phosphorylated than wild-type mouse tau and transgenic tau-4R. Only tau-4R isoforms were present as demonstrated with antibody 2R (bottom panel) directed against the second microtubule binding domain in protein tau-4R (Takuma et al., 2003). The different tau-4R isoforms are indicated. Protein loaded was threefold higher for the nontransgenic than for the transgenic mice to compensate for the overexpression relative to endogenous mouse tau for different levels of the epitopes and for differences in titer and avidity of the respective antibodies. Samples loaded for tau-P301L and tau-4R transgenic mice were equivalent to 2.8 μg of protein for mAbs Tau-5 and AD2, 9 μg for mAb AT180, 18 μg for mAb AT8, and 5.6 μg for 2R antibody.
Protein tau-isoforms with three or four microtubule binding domains were differentiated in Western blotting with an antiserum that specifically recognizes the second microtubule binding domain present only in tau-4R and not in tau-3R (Takuma et al., 2003). The data unequivocally demonstrate that the tau isoforms expressed in the brain of young adult mice examined here was tau-4R (Fig. 2, bottom), in accordance with the reported complete postnatal switch in expression from tau-3R to tau-4R in the mouse brain (Andorfer et al., 2000; Takuma et al., 2003).
The current observation of lesser phosphorylation of tau-P301L relative to tau-4R completely conforms to our previous findings in brain and spinal cord of older tau-P301L transgenic mice (Terwel et al., 2005). Although in young mice, tau-P301L is relatively less phosphorylated at the AT8 and AD2 epitopes, no immunoreactivity at all was detected with mAb AT100, which defines a typical pathological epitope (Terwel et al., 2005). Moreover, the electrophoretic mobility of tau-P301L was not markedly slow, which further underlines the minimal degree of posttranslational phosphorylation.
We therefore conclude that in young tau-P301L mice, the phosphorylation of protein tau in the hippocampus is, if anything, lower than in age-matched nontransgenic mice.
Electrophysiology
To characterize typical hippocampal properties, we recorded field potentials and induced LTP in brain sections, both in the DG and in CA1. By fitting the input–output curves to a Bolzmann equation, the three major determinants for basal transmission were calculated (i.e., maximal amplitude or slope of the fEPSP, half-maximal stimulation intensity, and slope factor) (Table 1). None of these parameters were significantly different in brain sections of young tau-P301L mice compared with age-matched nontransgenic mice, regardless of whether recording was in the CA1 or DG.
Table 1.
Basal neural transmission in tau-P301L transgenic and nontransgenic mice
| WT CA1 (n = 12) | P301L CA1 (n = 7) | WT DG (n = 7) | P301L DG (n = 5) | |
|---|---|---|---|---|
| Slope | ||||
| Rmax (mV/ms) | −2.3 ± 0.6 | −2.2 ± 0.4 | −1.7 ± 0.7 | −1.8 ± 0.7 |
| Ih (arbitrary units) | 196 ± 4 | 200 ± 6 | 197 ± 9 | 206 ± 8 |
| Slope factor | −6.0 ± 0.7 | −9.8 ± 1.8 | −4.1 ± 1.7 | −7.0 ± 3.2 |
| Amplitude | ||||
| Rmax (mV) | −3.0 ± 0.1 | −2.4 ± 0.2 | −1.8 ± 0.4 | −2.2 ± 0.3 |
| Ih (arbitrary units) | 196 ± 3 | 188 ± 4 | 193 ± 10 | 199 ± 6 |
| Slope factor | −8.8 ± 0.7 | −7.1 ± 1.3 | −4.3 ± 2.9 | −6.2 ± 1.3 |
Basal neural transmission is unchanged in tau-P301L transgenic mice, whether recorded in the CA1 or DG. Values are mean ± SEM. Rmax, Maximal response; Ih, half-maximal stimulation intensity.
LTP in the CA1 area was unaffected (Fig. 3A), but LTP in the DG was significantly increased in brain sections from tau-P301L mice, both when analyzed over the complete 1 h recording period (p = 0.031; F = 6.28; n = 5) and over the last 5 min of the session (p = 0.036; F = 5.84; n = 5) (Fig. 3B).
Figure 3.
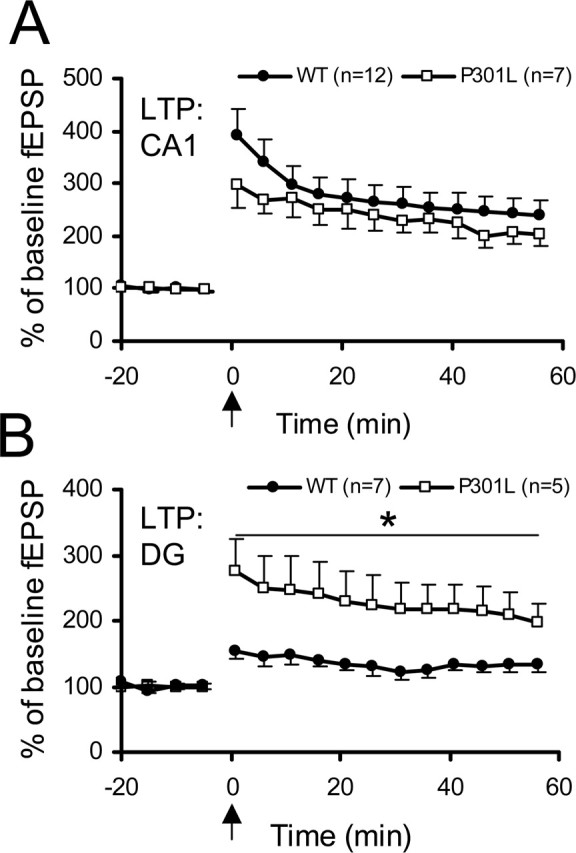
Long-term potentiation in CA1 and DG of young mice at 9 weeks of age. A, LTP is similar in CA1 of nontransgenic and tau-P301L transgenic mice. B, LTP in the DG is significantly increased in tau-P301L transgenic mice compared with nontransgenic mice (WT) over the whole 60 min period (*p = 0.031) as well as over the last 5 min (right panel; *p = 0.036). Although potentiation was low in the WT, it was still significantly increased between 55 and 60 min compared with the pretetanus situation (p = 0.027). The level of 100% was defined as the average of the slope of 20 fEPSP recordings before the induction of LTP by theta burst stimulation (arrow). Error bars represent SEM.
Because facilitated LTP in the DG is potentially indicative of improved cognition, the mice were tested for motor and cognitive parameters.
Behavioral testing
Before the object recognition test, the basal motor and behavioral parameters were tested by rotarod and open field tests.
Tau-P301L mice fell off the rotating rod significantly earlier than nontransgenic mice (182 ± 39 vs 268 ± 12 s; mean ± SEM; p = 0.039; F = 4.86; n = 10 and 11, respectively), indicating some motor impairment already at the young age of 9 weeks.
Locomotor activity in an open field revealed that tau-P301L mice traveled overall significantly more distance than nontransgenic mice (i.e., 2030 ± 292 vs 1223 ± 136 cm; p = 0.006; F = 9.74; n = 11 and 10, respectively). Both genotypes exhibited equal relative times spent in the center of the open field (17.2 ± 4.9 vs 417.9 ± 3.0%), demonstrating no difference in anxiety or exploratory behavior.
We then substantiated whether the electrophysiological change (i.e., increased LTP in the DG was paralleled by concurrent alteration of hippocampal memory function) by subjecting the mice to the ORT. Tau-P301L mice performed similar to nontransgenic mice in the ORT with a 1 h delay. High d2 values indicated that both groups had a good recall of the familiar object (0.49 ± 0.05 vs 0.45 ± 0.11, respectively, for 11 wild-type vs 10 transgenic mice). At the 3.5 h interval, however, the nontransgenic mice showed low d2 values, indicating they had low recall of the familiar object in contrast to tau-P301L mice that performed similar at the 3.5 h as at the 1 h interval (Fig. 4), demonstrating that they still recognized the familiar object. Because this difference was highly significant (p = 0.046; F = 4.65), we conclude that young tau-P301L mice display improved memory in this task compared with age-matched nontransgenic mice.
Figure 4.

Novel object recognition in young mice at 9 weeks of age. Young age-matched nontransgenic and tau-P301L transgenic mice were assessed in the novel object recognition task with 1 h and 3.5 h delay intervals after acquisition. Both genotypes showed a similar preference for the novel object at 1 h, whereas at 3.5 h, the preference of the tau-P301L transgenic mice for the novel object remained high, as opposed to the decline in nontransgenic (WT) mice (*p = 0.02) This demonstration of improved memory was also observed in younger tau-P301L mice (at 5 weeks of age) (for details, see Results, Behavioral testing). Error bars represent SEM.
Because the rotarod test pointed to motor problems, we additionally tested even younger tau-P301L mice in ORT (i.e., at the age of 5 weeks). At this age, neither a motor impairment was evident in the rotarod test nor increased travel distance in the open field (results not shown). Nevertheless, also at 5 weeks of age, the tau-P301L mice performed significantly better in ORT at the 3.5 h interval than age-matched nontransgenic mice (0.42 ± 0.07 vs 0.05 ± 0.09; p = 0.011; F = 8.00; n = 12 and 10, respectively).
Hippocampal morphology
To address possible morphological correlates of the increased LTP in the DG and the improved cognition, we investigated structural changes in several hippocampal subareas of tau-P301L mice in direct comparison with nontransgenic mice of the same age, same gender, and same genetic background. No major differences were observed in the volume of the hippocampal subregions or in cellular density (Fig. 5).
Figure 5.

Volume of the hippocampal formation. Total volume of three hippocampal subareas per hemisphere was not different in tau-P301L transgenic mice relative to age-matched nontransgenic mice (WT). Cell density in the DG granular layer was similar in age-matched tau-P301L and nontransgenic mice (WT) (see Results for details and discussion of other parameters). Error bars represent SEM.
Then, we analyzed the morphology of CA1 and DG at the individual, single-cell level by Golgi impregnation. Various parameters of the dendritic tree were measured (i.e., cell area, dendritic length, number of dendritic ramifications, number of terminal segments, mean terminal segment length, and mean intersegment length). Two parameters eventually relevant for the problem at hand (e.g., total dendritic length and total number of dendrites) were not different in either brain region of tau-P301L and nontransgenic mice (Fig. 6). In contrast, in CA1, the area of individual soma was significantly smaller in tau-P301L mice (160 ± 6 μm2; n = 41) than in nontransgenic mice (180 ± 6 μm2; n = 44; p < 0.01). The length of the terminal branch of the apical dendrites was increased from 46 ± 2 μm in nontransgenic mice (n = 44) to 52 ± 2 μm in tau-P301L mice (n = 41; p = 0.037). In the DG, the intersegment length was increased from 22.2 ± 1.7 μm in nontransgenic mice (n = 91) to 28.8 ± 2.2 μm in tau-P301L mice (n = 98; p = 0.018). All other parameters did not differ between the two genotypes.
Figure 6.

Dendritic morphology in young tau-P301L transgenic mice. Examples of Golgi–Cox impregnated cells in DG and CA1 are shown. Quantitative analysis of Golgi–Cox impregnated cells in CA1 revealed no major dendritic changes in different cellular parameters in the tau-P301L transgenic mice relative to age-matched nontransgenic mice (WT) (see Results for details). Error bars represent SEM.
Neurogenesis
Stereological measurements were performed using three independent markers for different stages of the neurogenesis process: doublecortin (DCX) as marker for young migrating neurons, Ki-67 as marker for cycling cells, and BrdU as a “cell-age” marker, analyzed here 4 weeks after injection of BrdU (Fig. 7). Neither of these markers revealed a major difference in either the number of newborn, proliferating, or surviving young neurons in tau-P301L mice compared with nontransgenic mice (Table 2).
Figure 7.
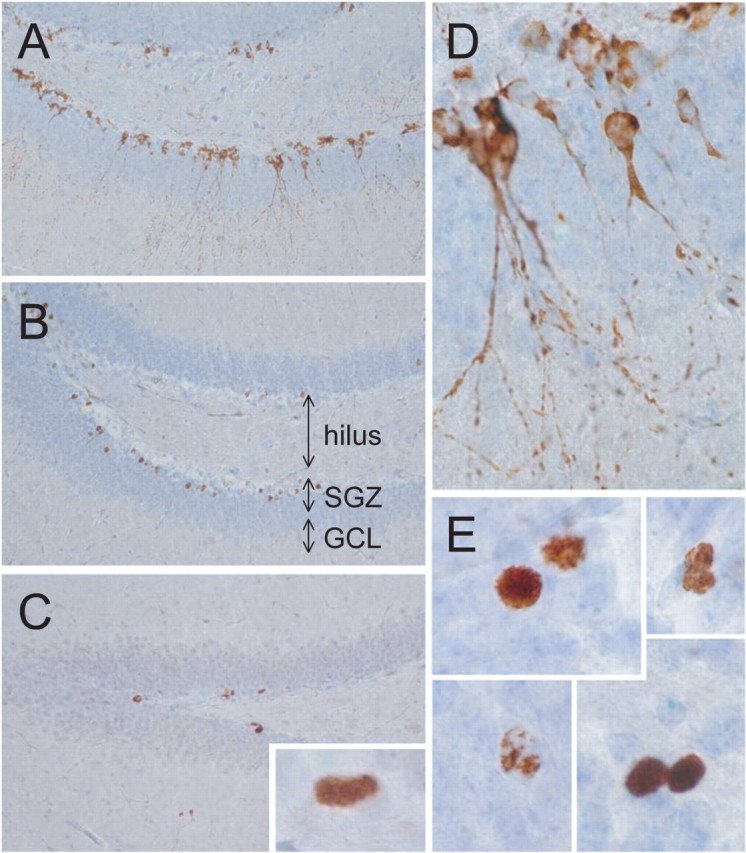
Newborn cells and survival in young tau-P301L transgenic mice. Different markers were examined to define putative changes in cell genesis and/or turnover. A, D, Immunohistochemistry for doublecortin marks young neurons. B, E, BrdU was analyzed 4 weeks after injection as a measure of cell age (see Results for details). C, Immunohistochemistry for the Ki-67 antigen as a marker of proliferating cells. A–C show an overview of the dentate gyrus, and representative individual cells or groups of cells are illustrated in D and E. Sections were counterstained with hematoxylin.
Table 2.
Cell birth and survival is unchanged in tau-P301L transgenic mice
| Hilus (WT) | Hilus (P301L) | SGZ (WT) | SGZ (P301L) | GCL (WT) | GCL (P301L) | |
|---|---|---|---|---|---|---|
| Ki-67 (10 vs 5) | 129 ± 21 | 108 ± 25 | 665 ± 57 | 760 ± 70 | 55 ± 12 | 94 ± 19 |
| BrdU (10 vs 7) | 248 ± 30 | 215 ± 46 | 2684 ± 134 | 2573 ± 183 | 184 ± 23 | 163 ± 25 |
| DCX (8 vs 3) | 24 ± 6 | 42 ± 23 | 7424 ± 754 | 7740 ± 840 | 256 ± 33 | 313 ± 12 |
The markers used are Ki-67, which labels proliferating cells; BrdU, a marker for cell survival (4 weeks); and DCX, which labels young migrating neurons. The numbers indicate total cells per hemisphere ±SEM. The number of animals per group is in parentheses (WT vs P301L). SGZ, Subgranular zone.
The analysis therefore excludes that changes in basal neurogenesis contributed to the demonstrated functional alterations in the hippocampus of young tau-P301L mice.
Discussion
The current study demonstrates for the first time that neuronal expression of a mutant protein tau does not impair hippocampal functions per se. On the contrary, the remarkable and unexpected increase in LTP in the DG of young tau-P301L mice was paralleled by a significant improvement in cognitive performance in the object recognition task. At this young age, no tau-pathology was evident, and the phosphorylation of protein tau was normal or, if anything, less than in age-matched nontransgenic mice. Finally, no major morphological or neurogenic abnormalities were detected.
Our previous biochemical and pathological analysis of the tau-P301L mice already established the progressive and age-related nature of the phosphorylation of protein tau, leading to conformational changes and extensive neurofibrillary pathology (Terwel et al., 2005). The progressive morbidity with age is accompanied by progressive motor defects and increasing hyperphosphorylation. In the early stages of life, phosphorylation of protein tau is low without any signs of axonal dilations or inclusions (Terwel et al., 2005; our study).
The combined data lend strong support to the hypothesis that not mutant tau itself but the progressive hyperphosphorylation with age is critical in FTDP-17 tauopathies, inflicting or at least signaling the onset of memory impairment and neurodegeneration. Moreover, the current data point to an important physiological function of protein tau in contributing to memory performance in the hippocampus.
Tau transgenic mice, including those expressing tau-P301L, have shown cognitive impairment (Arendash et al., 2004; Pennanen et al., 2004; Santa Cruz et al., 2005). Most of these transgenic mice were tested, however, when tau hyperphosphorylation and pathology was already evident. Most recently, suppression of tau-P301L expression in an inducible transgenic model ameliorated memory functions without reversing the tauopathy (Santa Cruz et al., 2005). All of these data are in line with our previous and current studies, whereas the combined results demonstrate that hyperphosphorylated tau, and not NFT, must be the cause of the observed memory defects. This conclusion is based on observations that hyperphosphorylation of tau invariably precedes the formation of any tau aggregates in all model systems studied (see Introduction). In an analogy with the evolution of concepts in the “refined” amyloid cascade hypothesis proposing soluble oligomers rather than insoluble polymers of amyloid peptides to be the actual culprits in AD (Selkoe, 2005), we underwrite the hypothesis that NFTs are not detrimental for neuronal functions (Andorfer et al., 2005) but the soluble isoforms of abnormally phosphorylated tau (Terwel et al., 2005; our study).
Motor impairment has been observed in tau transgenic mice, progressing with age (Spittaels et al., 1999, 2000; Lewis et al., 2000; Arendash et al., 2004; Ikeda et al., 2005; Terwel et al., 2005). To exclude possible confounding parameters, we performed rotarod and open field experiments before cognitive testing. The higher locomotor activity of tau-P301L mice at age 9 weeks in the open field corroborates the increased explorative activity in other tau transgenic mice (Tanemura et al., 2002; Pennanen et al., 2004). Hippocampal alterations are known to affect explorative activity (Decker et al., 1995; Harley and Martin, 1999; Crusio, 2001). Using a demanding test protocol in the rotarod paradigm [i.e., involving less training sessions than in a previous study (Terwel et al., 2005)], we demonstrate that 5-week-old tau-P301L mice do not suffer any motor deficit. Importantly, the cognitive improvement was not influenced to any degree by hyperactivity or motor problems, because tau-P301L mice at 5 weeks of age displayed a similar improvement in memory performance in the object recognition paradigm, without showing any alterations in motor or locomotor faculties. Therefore, the improved cognitive performance in the ORT task is not related to or caused by motor disabilities or problems.
The alternative test for cognitive performance (i.e., the water-maze task) proved not suitable for tau-P301L mice, because this test depends heavily on swimming ability, and performance is negatively influenced by motor defects. Moreover, the FVB mouse strain carries the retinal degeneration gene that can hamper visual tasks, particularly at older age (Pugh et al., 2004). In contrast, the ORT task depends on short-range visual, as well as tactile stimuli, is less stressing than the water maze task and requires limited motor skills. Interestingly, a similar improvement in learning performance of tau-P301L mice is observed in a water maze task in an independent strain of tau-P301L mice with a different genetic background (A. Blokland and H. J. Schröder, personal communication).
Moreover, hippocampal LTP in CA1 has been demonstrated to correlate well with memory functions measured by ORT (Miller et al., 2002; Wang et al., 2004). The suggestion that ORT would not depend on hippocampal functions (Mumby, 2001) was mostly based on tests with short delay protocols in which animals were tested immediately (minutes) after training. Recent studies demonstrate that the delay length determines hippocampal involvement (Hammond et al., 2004), and this is crucial for object recognition at a longer delay (hours), similar to what we studied here. However, we do acknowledge that associated structures like perirhinal and postrhinal cortex may influence ORT performance (Bussey et al., 1999; Kesner et al., 2001), and alterations in these brain regions could contribute to the changes observed in the young tau-P301L mice.
The current data illustrate the clear correlation between improved memory performance and increased LTP in the DG but not CA1 hippocampal areas. To define possible causes of these interesting regional effects, we analyzed several cellular and structural correlates. Adult neurogenesis, uniquely occurring in the DG, has been implicated in memory function (Gould et al., 1999; van Praag et al., 1999; Shors et al., 2001, 2002). Despite improvements in cognition and LTP in the DG, no changes in either proliferation or survival of the newborn cells or in the extent of neurogenesis were observed in the tau-P301L mice. The tau-P301L mice thereby represent an example of improved cognition not accompanied or paralleled by alterations in neurogenesis, proving that the mutant tau protein affects hippocampal functions through other mechanisms.
Because only relative minor changes were observed in dendritic properties and in other morphological parameters of hippocampal neurons, and only in DG, we conclude that subtle intracellular effects must underlie the changes in LTP and cognition. One possibility is a differential stabilization of MTs by tau that could contribute importantly to vesicular transport (Zhang et al., 2005) and in turn to synaptic plasticity and memory. The trafficking of NMDA and AMPA receptors, critically implicated in LTP and memory formation (Davis et al., 1992; Malinow and Malenka, 2002; Bast et al., 2005), depends heavily on stable MT-mediated transport (Setou et al., 2000, 2002; Yuen et al., 2005). Possibly, MT stability is improved in the tau-P301L mice, despite a possible reduced binding of mutant tau (Hong et al., 1998; Perez et al., 2000; Zhang et al., 2004), which could, however, be balanced by the higher concentration of tau. Moreover, the mutant tau-P301L is embedded in the tau-4R isoform, which binds more avidly to MT than the tau-3R isoform.
Whether the alterations are the result of expression of mutant tau or overexpression of tau-4R is unresolved. Although tau-2N/4R mice are available that overexpress the same isoform as the tau-P301L mice, evidently without the mutation, the same experiments cannot be performed in these mice, because they suffer a severe axonopathy (Spittaels et al., 1999; Terwel et al., 2005). In comparison, an interesting analogy is observed in transgenic mice that overexpress p25, because these, also unexpectedly, showed improved hippocampus-dependent memory functions (Angelo et al., 2003). The calpain-truncated cdk5 activator subunit p25 is associated with pathological hyperphosphorylation of tau and was expected to induce neurodegeneration (for review, see Tsai et al., 2004). Increased tau phosphorylation was not observed in young p25 transgenic mice, but tau expression was increased (Angelo et al., 2003). This is consistent with the hypothesis that increased concentration of protein tau, and not the mutation per se, underlies the improved cognitive performance in young tau-P301L mice.
Although the present transgenic mice replicate many features of the corresponding human tauopathy, including adult onset, progressive neurodegeneration, accumulation of abnormal tau-aggregates, and premature death, we are not aware of any reports of similar improvements in memory performance in young adult human subjects bearing tau-P301L or other FTDP-17 mutations. In part, this could also be a result of the clinical heterogeneity of FTDP-17 patients (Ingram and Spillantini, 2002). Alternatively, improved hippocampal functioning may be unique to the present type of transgenic model that is essentially different from the human situation in that it expresses only the 2N/4R splice variant of tau.
In summary, we describe an unexpected parallel improvement in hippocampal LTP and memory in young tau-P301L mice before the onset of hyperphosphorylation or aggregation of protein tau. We thereby demonstrate that not the tau-P301L mutation per se is critical for cognitive decline but conclude that excess phosphorylation of tau, triggered by the mutation is pathogenic. Moreover, the results highlight an important novel role for protein tau in hippocampal memory function.
Footnotes
This work was supported by the Internationale Stichting Alzheimer Onderzoek, the Instituut voor Wetenschappelijk en Technologisch onderzoek, the Fonds voor Wetenschappelijk Onderzoek–Vlaanderen, the K. U. Leuven Special Research Fund, and the K. U. Leuven–Research and Development. We thank Dr. M. Oitzl (Leiden/Amsterdam Center for Drug Research, Leiden University, Amsterdam, The Netherlands), J. van Heerikhuize (Netherlands Institute for Brain Research, Amsterdam, The Netherlands), S. Maslam (Swammerdam Institute for Life Sciences–Center for Neuroscience, University of Amsterdam, Amsterdam, The Netherlands), and H. Devijver (Laboratorium Experimentele Genetica en Transgenese–Experimental Genetics Group, Leuven, Belgium) for expert advice and technical support and Dr. A. Campbell (Swammerdam Institute for Life Sciences–Center for Neuroscience) for correcting English writing. K.B. received a predoctoral training grant from the European Robotics Research Network (European Economic Community–Marie Curie Training Site; QLK6-CT-2000-60042).
References
- Alfarez DN, Joels M, Krugers HJ (2003). Chronic unpredictable stress impairs long-term potentiation in rat hippocampal CA1 area and dentate gyrus in vitro. Eur J Neurosci 17:1928–1934. [DOI] [PubMed] [Google Scholar]
- Andorfer C, Davies P (2000). PKA phosphorylation on tau: developmental studies in the mouse. Dev Neurosci 4:303–309. [DOI] [PubMed] [Google Scholar]
- Andorfer C, Acker CM, Kress Y, Hof PR, Duff K, Davies P (2005). Cell-cycle reentry and cell death in transgenic mice expressing wild-type human tau isoforms. J Neurosci 25:5446–5454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelo M, Plattner F, Irvine EE, Giese KP (2003). Improved reversal learning and altered fear conditioning in transgenic mice with regionally restricted p25 expression. Eur J Neurosci 18:423–431. [DOI] [PubMed] [Google Scholar]
- Arendash GW, Lewis J, Leighty RE, McGowan E, Cracchiolo JR, Hutton M, Garcia MF (2004). Multi-metric behavioral comparison of APPsw and P301L models for Alzheimer's disease: linkage of poorer cognitive performance to tau pathology in forebrain. Brain Res 1012:29–41. [DOI] [PubMed] [Google Scholar]
- Bast T, da Silva BM, Morris RG (2005). Distinct contributions of hippocampal NMDA and AMPA receptors to encoding and retrieval of one-trial place memory. J Neurosci 25:5845–5856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronner IF, Ter Meulen BC, Azmani A, Severijnen LA, Willemsen R, Kamphorst W, Ravid R, Heutink P, van Swieten JC (2005). Hereditary Pick's disease with the G272V tau mutation shows predominant three-repeat tau pathology. Brain 128:2645–2653. [DOI] [PubMed] [Google Scholar]
- Buée L, Bussiere T, Buée-Scherrer V, Delacourte A, Hof PR (2000). Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev 33:95–130. [DOI] [PubMed] [Google Scholar]
- Bussey TJ, Muir JL, Aggleton JP (1999). Functionally dissociating aspects of event memory: the effects of combined perirhinal and postrhinal cortex lesions on object and place memory in the rat. J Neurosci 19:495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YG (2005). Specific tau phosphorylation sites in hippocampus correlate with impairment of step-down inhibitory avoidance task in rats. Behav Brain Res 158:277–284. [DOI] [PubMed] [Google Scholar]
- Couillard-Despres S, Winner B, Schaubeck S, Aigner R, Vroemen M, Weidner N, Bogdahn U, Winkler J, Kuhn HG, Aigner L (2005). Doublecortin expression levels in adult brain reflect neurogenesis. Eur J Neurosci 21:1–14. [DOI] [PubMed] [Google Scholar]
- Crusio WE (2001). Genetic dissection of mouse exploratory behaviour. Behav Brain Res 125:127–132. [DOI] [PubMed] [Google Scholar]
- Davis S, Butcher SP, Morris RG (1992). The NMDA receptor antagonist d-2-amino-5-phosphonopentanoate (d-AP-5) impairs spatial learning and LTP in vivo at intracerebral concentrations comparable to those that block LTP in vitro J Neurosci 12:21–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson HN, Ferreira A, Eyster MV, Ghoshal N, Binder LI, Vitek MP (2001). Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J Cell Sci 114:1179–1187. [DOI] [PubMed] [Google Scholar]
- Decker MW, Curzon P, Brioni JD (1995). Influence of separate and combined septal and amygdala lesions on memory, acoustic startle, anxiety, and locomotor activity in rats. Neurobiol Learn Mem 64:156–168. [DOI] [PubMed] [Google Scholar]
- Dewachter I, Reverse D, Caluwaerts N, Ris L, Kuiperi C, Van Den Haute C, Spittaels K, Umans L, Serneels L, Thiry E, Moechars D, Mercken M, Godaux E, Van Leuven F (2002). Neuronal deficiency of presenilin 1 inhibits amyloid plaque formation and corrects hippocampal long-term potentiation but not a cognitive defect of amyloid precursor protein [V717I] transgenic mice. J Neurosci 22:3445–3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchrow M, Schmidt MH, Zingler M, Anemuller S, Bruch HP, Broll R (2001). Suppression of cell division by pKi-67 antisense-RNA and recombinant protein. Cell Physiol Biochem 11:331–338. [DOI] [PubMed] [Google Scholar]
- Endl E, Gerdes J (2000). The Ki-67 protein: fascinating forms and an unknown function. Exp Cell Res 257:231–237. [DOI] [PubMed] [Google Scholar]
- Gerdes J, Lemke H, Baisch H, Wacker HH, Schwab U, Stein H 4 (1984). Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J Immunol 133:1710–1715. [PubMed] [Google Scholar]
- Goedert M, Jakes R (2005). Mutations causing neurodegenerative tauopathies. Biochim Biophys Acta 1739:240–250. [DOI] [PubMed] [Google Scholar]
- Gould E, Beylin A, Tanapat P, Reeves A, Shors TJ (1999). Learning enhances adult neurogenesis in the hippocampal formation. Nat Neurosci 2:260–265. [DOI] [PubMed] [Google Scholar]
- Hammond RS, Tull LE, Stackman RW (2004). On the delay-dependent involvement of the hippocampus in object recognition memory. Neurobiol Learn Mem 82:26–34. [DOI] [PubMed] [Google Scholar]
- Harley CW, Martin GM (1999). Open field motor patterns and object marking, but not object sniffing, are altered by ibotenate lesions of the hippocampus. Neurobiol Learn Mem 72:202–214. [DOI] [PubMed] [Google Scholar]
- Heine VM, Maslam S, Joels M, Lucassen PJ (2004a). Prominent decline of newborn cell proliferation, differentiation, and apoptosis in the aging dentate gyrus, in absence of an age-related hypothalamus-pituitary-adrenal axis activation. Neurobiol Aging 25:361–375. [DOI] [PubMed] [Google Scholar]
- Heine VM, Maslam S, Zareno J, Joels M, Lucassen PJ 1 (2004b). Suppressed proliferation and apoptotic changes in the rat dentate gyrus after acute and chronic stress are reversible. Eur J Neurosci 19:131–144. [DOI] [PubMed] [Google Scholar]
- Hong M, Zhukareva V, Vogelsberg-Ragaglia V, Wszolek Z, Reed L, Miller BI, Geschwind DH, Bird TD, McKeel D, Goate A, Morris JC, Wilhelmsen KC, Schellenberg GD, Trojanowski JQ, Lee VM (1998). Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science 282:1914–1917. [DOI] [PubMed] [Google Scholar]
- Ikeda M, Shoji M, Kawarai T, Kawarabayashi T, Matsubara E, Murakami T, Sasaki A, Tomidokoro Y, Ikarashi Y, Kuribara H, Ishiguro K, Hasegawa M, Yen SH, Chishti MA, Harigaya Y, Abe K, Okamoto K, St George-Hyslop P, Westaway D (2005). Accumulation of filamentous tau in the cerebral cortex of human tau R406W transgenic mice. Am J Pathol 166:521–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingram EM, Spillantini MG (2002). Tau gene mutations: dissecting the pathogenesis of FTDP-17. Trends Mol Med 8:555–562. [DOI] [PubMed] [Google Scholar]
- Jin K, Peel AL, Mao XO, Xie L, Cottrell BA, Henshall DC, Greenberg DA (2004). Increased hippocampal neurogenesis in Alzheimer's disease. Proc Natl Acad Sci USA 101:343–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kee N, Sivalingam S, Boonstra R, Wojtowicz JM (2002). The utility of Ki-67 and BrdU as proliferative markers of adult neurogenesis. J Neurosci Methods 115:97–105. [DOI] [PubMed] [Google Scholar]
- Kesner RP, Ravindranathan A, Jackson P, Giles R, Chiba AA (2001). A neural circuit analysis of visual recognition memory: role of perirhinal, medial, and lateral entorhinal cortex. Learn Mem 8:87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Kidani T, Ujike H, Hayashi M, Ishihara T, Miyazu K, Kuroda S, Koshino Y (2003). Another phenotype of frontotemporal dementia and parkinsonism linked to chromosome-17 (FTDP-17) with a missense mutation of S305N closely resembling Pick's disease. J Neurol 250:990–992. [DOI] [PubMed] [Google Scholar]
- Lee VM, Kenyon TK, Trojanowski JQ (2005). Transgenic animal models of tauopathies. Biochim Biophys Acta 1739:251–259. [DOI] [PubMed] [Google Scholar]
- Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Paul Murphy M, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M (2000). Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet 25:402–405. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC (2002). AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci 25:103–126. [DOI] [PubMed] [Google Scholar]
- Miller S, Yasuda M, Coats JK, Jones Y, Martone ME, Mayford M (2002). Disruption of dendritic translation of CaMKIIalpha impairs stabilization of synaptic plasticity and memory consolidation. Neuron 36:507–519. [DOI] [PubMed] [Google Scholar]
- Mumby DG (2001). Perspectives on object-recognition memory following hippocampal damage: lessons from studies in rats. Behav Brain Res 127:159–181. [DOI] [PubMed] [Google Scholar]
- Pennanen L, Welzl H, D'Adamo P, Nitsch RM, Gotz J (2004). Accelerated extinction of conditioned taste aversion in P301L tau transgenic mice. Neurobiol Dis 15:500–509. [DOI] [PubMed] [Google Scholar]
- Perez M, Lim F, Arrasate M, Avila J (2000). The FTDP-17-linked mutation R406W abolishes the interaction of phosphorylated tau with microtubules. J Neurochem 74:2583–2589. [DOI] [PubMed] [Google Scholar]
- Pugh PL, Ahmed SF, Smith MI, Upton N, Hunter AJ (2004). A behavioral characterization of the FVB/N mouse strain. Behav Brain Res 155:283–289. [DOI] [PubMed] [Google Scholar]
- Ramakers GJ, Winter J, Hoogland TM, Lequin MB, van Hulten P, van Pelt J, Pool CW (1998). Depolarization stimulates lamellipodia formation and axonal but not dendritic branching in cultured rat cerebral cortex neurons. Brain Res Dev Brain Res 108:205–216. [DOI] [PubMed] [Google Scholar]
- Santa Cruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH (2005). Tau suppression in a neurodegenerative mouse model improves memory function. Science 309:476–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ (2005). Defining molecular targets to prevent Alzheimer disease. Arch Neurol 62:192–195. [DOI] [PubMed] [Google Scholar]
- Setou M, Nakagawa T, Seog DH, Hirokawa N (2000). Kinesin superfamily motor protein KIF17 and mLin-10 in NMDA receptor-containing vesicle transport. Science 288:1796–1802. [DOI] [PubMed] [Google Scholar]
- Setou M, Seog DH, Tanaka Y, Kanai Y, Takei Y, Kawagishi M, Hirokawa N (2002). Glutamate-receptor-interacting protein GRIP1 directly steers kinesin to dendrites. Nature 417:83–87. [DOI] [PubMed] [Google Scholar]
- Shors TJ (2004). Memory traces of trace memories: neurogenesis, synaptogenesis and awareness. Trends Neurosci 27:250–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shors TJ, Miesegaes G, Beylin A, Zhao M, Rydel T, Gould E (2001). Neurogenesis in the adult is involved in the formation of trace memories. Nature 410:372–376. [DOI] [PubMed] [Google Scholar]
- Shors TJ, Townsend DA, Zhao M, Kozorovitskiy Y, Gould E (2002). Neurogenesis may relate to some but not all types of hippocampal-dependent learning. Hippocampus 12:578–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spittaels K, Van den Haute C, Van Dorpe J, Bruynseels K, Vandezande K, Laenen I, Geerts H, Mercken M, Sciot R, Van Lommel A, Loos R, Van Leuven F (1999). Prominent axonopathy in the brain and spinal cord of transgenic mice overexpressing four-repeat human tau protein. Am J Pathol 155:2153–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spittaels K, Van den Haute C, Van Dorpe J, Geerts H, Mercken M, Bruynseels K, Lasrado R, Vandezande K, Laenen I, Boon T, Van Lint J, Vandenheede J, Moechars D, Loos R, Van Leuven F (2000). Glycogen synthase kinase-3beta phosphorylates protein tau and rescues the axonopathy in the central nervous system of human four-repeat tau transgenic mice. J Biol Chem 275:41340–41349. [DOI] [PubMed] [Google Scholar]
- Sun L, Wang X, Liu S, Wang Q, Wang J, Bennecib M, Gong CX, Sengupta A, Grundke-Iqbal I, Iqbal K (2005). Bilateral injection of isoproterenol into hippocampus induces Alzheimer-like hyperphosphorylation of tau and spatial memory deficit in rat. FEBS Lett 579:251–258. [DOI] [PubMed] [Google Scholar]
- Takei Y, Teng J, Harada A, Hirokawa N (2000). Defects in axonal elongation and neuronal migration in mice with disrupted tau and map1b genes. J Cell Biol 150:989–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takuma H, Arawaka S, Mori H (2003). Isoforms changes of tau protein during development in various species. Brain Res Dev Brain Res 142:121–127. [DOI] [PubMed] [Google Scholar]
- Tanemura K, Murayama M, Akagi T, Hashikawa T, Tominaga T, Ichikawa M, Yamaguchi H, Takashima A (2002). Neurodegeneration with tau accumulation in a transgenic mouse expressing V337M human tau. J Neurosci 22:133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatebayashi Y, Miyasaka T, Chui DH, Akagi T, Mishima K, Iwasaki K, Fujiwara M, Tanemura K, Murayama M, Ishiguro K, Planel E, Sato S, Hashikawa T, Takashima A (2002). Tau filament formation and associative memory deficit in aged mice expressing mutant (R406W) human tau. Proc Natl Acad Sci USA 99:13896–13901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwel D, Lasrado R, Snauwaert J, Vandeweert E, Van Haesendonck C, Borghgraef P, Van Leuven F (2005). Changed conformation of mutant Tau-P301L underlies the moribund tauopathy, absent in progressive, non-lethal axonopathy of Tau-4R/2N transgenic mice. J Biol Chem 280:3963–3973. [DOI] [PubMed] [Google Scholar]
- Tolnay M, Probst A (1999). Tau protein pathology in Alzheimer's disease and related disorders. Neuropathol Appl Neurobiol 25:171–187. [DOI] [PubMed] [Google Scholar]
- Tsai LH, Lee MS, Cruz J (2004). Cdk5, a therapeutic target for Alzheimer's disease? Biochim Biophys Acta 1697:137–142. [DOI] [PubMed] [Google Scholar]
- van Praag H, Christie BR, Sejnowski TJ, Gage FH (1999). Running enhances neurogenesis, learning, and long-term potentiation in mice. Proc Natl Acad Sci USA 96:13427–13431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Swieten JC, Stevens M, Rosso SM, Rizzu P, Joosse M, de Koning I, Kamphorst W, Ravid R, Spillantini MG, Niermeijer MG, Heutink P (1999). Phenotypic variation in hereditary frontotemporal dementia with tau mutations. Ann Neurol 46:617–626. [DOI] [PubMed] [Google Scholar]
- Wang H, Ferguson GD, Pineda VV, Cundiff PE, Storm DR (2004). Overexpression of type-1 adenylyl cyclase in mouse forebrain enhances recognition memory and LTP. Nat Neurosci 7:635–642. [DOI] [PubMed] [Google Scholar]
- Yuen EY, Jiang Q, Feng J, Yan Z (2005). Microtubule regulation of N-methyl-d-aspartate receptor channels in neurons. J Biol Chem 280:29420–29427. [DOI] [PubMed] [Google Scholar]
- Zhang B, Higuchi M, Yoshiyama Y, Ishihara T, Forman MS, Martinez D, Joyce S, Trojanowski JQ, Lee VM (2004). Retarded axonal transport of R406W mutant tau in transgenic mice with a neurodegenerative tauopathy. J Neurosci 24:4657–4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Maiti A, Shively S, Lakhani F, McDonald-Jones G, Bruce J, Lee EB, Xie SX, Joyce S, Li C, Toleikis PM, Lee VM, Trojanowski JQ (2005). Microtubule-binding drugs offset tau sequestration by stabilizing microtubules and reversing fast axonal transport deficits in a tauopathy model. Proc Natl Acad Sci USA 102:227–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Ho L, Suh J, Qin W, Pyo H, Pompl P, Ksiezak-Reding H, Pasinetti GM (2003). A role of P301L tau mutant in anti-apoptotic gene expression, cell cycle and apoptosis. Mol Cell Neurosci 24:367–379. [DOI] [PubMed] [Google Scholar]