

Abstract
Age-dependent changes in multiple Ca2+-related electrophysiological processes in the hippocampus appear to be consistent biomarkers of aging, and several also correlate with cognitive decline. These findings have led to the hypothesis that a common mechanism of Ca2+ dyshomeostasis underlies aspects of aging-dependent brain impairment. However, some key predictions of this view remain untested, including that multiple Ca2+-related biomarkers should emerge concurrently during aging and their onset should also precede/coincide with initial signs of cognitive decline. Moreover, blocking a putative common source of dysregulated Ca2+ should eliminate aging differences. Here, we tested these predictions using combined electrophysiological, imaging, and pharmacological approaches in CA1 neurons to determine the ages of onset (across 4-, 10-, 12-, 14-, and 23-month-old F344 rats) of several established biomarkers, including the increases in the slow afterhyperpolarization, spike accommodation, and [Ca2+]i rise during repetitive synaptic stimulation. In addition, we tested the hypothesis that altered Ca2+-induced Ca2+ release (CICR) from ryanodine receptors, which can be triggered by L-type Ca2+ channels, provides a common source of dysregulated Ca2+ in aging. Results showed that multiple aging biomarkers were first detectable at about the same age (12 months of age; approximately midlife), sufficiently early to influence initial cognitive decline. Furthermore, selectively blocking CICR with ryanodine slowed the Ca2+ rise during synaptic stimulation more in aged rat neurons and, notably, reduced or eliminated aging differences in the biomarkers. Thus, this study provides the first evidence that altered CICR plays a role in driving the early and simultaneous emergence in hippocampus of multiple Ca2+-related biomarkers of aging.
Keywords: aging, ryanodine, afterhyperpolarization, calcium imaging, calcium channels, endoplasmic reticulum
Introduction
It has long been recognized that aging-dependent changes occur in multiple brain electrophysiological processes and that some of these correlate closely with functional decline. Deficits in synaptic transmission and plasticity have been found in several brain regions and species (deToledo-Morrell et al., 1988; Landfield, 1988; Barnes, 1994; Bickford, 1995; Foster and Norris, 1997; Bach et al., 1999; Disterhoft et al., 2004). Furthermore, alterations in a number of Ca2+-dependent/mediated processes have been found to be consistent biomarkers of aging in hippocampal neurons, including those in the Ca2+-dependent slow afterhyperpolarization (sAHP), spike accommodation, the Ca2+ action potential, and whole-cell Ca2+ currents (Landfield and Pitler, 1984; Disterhoft et al., 1996; Norris et al., 1998; Thibault et al., 1998; Disterhoft et al., 2004; Tombaugh et al., 2005). The activity of L-type voltage-gated Ca2+ channels (Thibault and Landfield, 1996) and the rise of [Ca2+]i during postsynaptic action potential generation (Thibault et al., 2001; Hemond and Jaffe, 2005) are also increased in hippocampal neurons from aging animals.
These and other findings, from different neuronal cell types and technical approaches, have given rise to several versions of the general hypothesis that a common mechanism of Ca2+ dysregulation underlies many aspects of functional aging and, potentially, Alzheimer's disease (AD) (Landfield and Pitler, 1984; Gibson and Peterson, 1987; Landfield, 1987; Khachaturian, 1989; Disterhoft et al., 1996; Michaelis et al., 1996; Thibault et al., 1998; Verkhratsky and Toescu, 1998; Murchison et al., 2004; Toescu et al., 2004). However, several critical questions and predictions of this hypothesis remain untested. First, if the hypothesis is correct, the onset of Ca2+ dysregulation should precede or coincide with initial deficits in hippocampally dependent cognitive tasks. These cognitive deficits begin to appear in mammals as early in adulthood as midlife (Aitken and Meaney, 1989; Fischer et al., 1992; Kadar et al., 1994; Frick et al., 1995; Bach et al., 1999; Markowska, 1999; Knuttinen et al., 2001). Second, multiple Ca2+-related biomarkers of hippocampal aging should emerge approximately simultaneously, rather than asynchronously. Third, blocking a common source of dysregulated Ca2+ should eliminate aging differences in multiple Ca2+-mediated biomarkers.
An increase in Ca2+ influx through L-type voltage-gated Ca2+ channels (L-VGCCs) has been implicated previously as a possible Ca2+ source for several hippocampal electrophysiological and cognitive aging changes (Thibault et al., 1998; Disterhoft et al., 2004). However, there are other candidate sources of dysregulated Ca2+. In particular, Ca2+-induced Ca2+ release (CICR) from ryanodine receptors (RyRs) on the endoplasmic reticulum (ER), which can be triggered by Ca2+ influx via L-VGCCs (Chavis et al., 1996; Empson and Galione, 1997; Fagni et al., 2000; Sukhareva et al., 2002; Verkhratsky, 2005), may be altered in some models of aging or AD (Gibson et al., 1996; Verkhratsky and Toescu, 1998; Leissring et al., 2000; Clodfelter et al., 2002; Kumar and Foster, 2004). Therefore, it seems possible that amplified CICR also plays a role in generating Ca2+-related markers of aging. If so, high-dose ryanodine, which selectively locks RyR channels into a low-conductance state (Bezprozvanny et al., 1991; Coronado et al., 1994; Humerickhouse et al., 1994), should substantially reduce aging differences in these markers, by counteracting the larger CICR components in older age groups.
Here, we tested key questions and predictions of this hypothesis using combined electrophysiological and imaging analyses of the age of onset and of the ryanodine sensitivity of multiple biomarkers of hippocampal aging. The results lend strong new support to a unified Ca2+ dyshomeostasis hypothesis.
Materials and Methods
Slice preparations.
All experiments were conducted in compliance with the institutional guidelines of the Animal Care and Use Committee at the University of Kentucky. Male F344 rats were obtained from the National Institute on Aging aged rat colony in subsets, each containing rats for all age points. The subsets were staggered such that, when studied, animals in the different age groups averaged 4, 10, 12, 14, and 23 months of age. Animals were anesthetized in a CO2 chamber before rapid decapitation. Brains were rapidly removed and transverse hippocampal slices (350 μm) were cut with a vibratome (TPI, St. Louis, MO) into cold oxygenated artificial CSF (ACF) of the following composition (in mm): 128 NaCl, 1.25 KH2PO4, 10 glucose, 26 NaHCO3, 3 KCl, 0.1 CaCl2, and 2 MgCl2 (Thibault et al., 2001). Intact slices were placed in an interface-type chamber containing ACF with 2 mm CaCl2 (Ca-ACF) at 32°C and gassed with 95% O2–5% CO2 until used for recording. After at least 1 h of recovery, individual slices were then transferred for recording to a perfusion chamber (Warner Instruments, Hamden, CT) equipped with a bottom net for Ca-ACF perfusion beneath the slice. The oxygenated Ca-ACF, delivered at 1.5–2 ml/min, was maintained at 32 ± 1°C using an inline heater (TC2Bip; Cell Micro Controls, Norfolk, VA) positioned 1 cm before the chamber inlet.
Electrophysiology.
Recording data were acquired and analyzed using pCLAMP 8, a sharp-electrode amplifier (Axoclamp 2A), and a DigiData 1320 board for digitization (Molecular Devices, Union City, CA). Sharp intracellular electrodes were pulled from microhematocrit glass capillaries (Fisher Scientific, Pittsburgh, PA) on a Sutter Instruments (Novato, CA) pipette puller, and had tip resistances of 80–120 MΩ when filled with 2 m KmeSO4, 10 mm HEPES, and 10 mm bis-Fura-2, pH 7.4. After impalement and stabilization, neurons were held at −70 mV in current clamp with minimal injected current for ∼5 min to obtain input resistance measures and to allow indicator filling. All experiments were conducted in current-clamp mode with bridge balance compensation and capacitance neutralization. Voltage records were digitized at 2–20 kHz and low-pass filtered at 1 kHz.
The afterhyperpolarization (AHP) was triggered with the membrane held at −60 mV using a 100 ms current depolarization pulse delivered through the electrode at sufficient intensity to generate three Na+ action potentials. AHP amplitudes were measured at the negative voltage peak immediately after the depolarization pulse, reflecting the medium AHP (mAHP), which typically lasts several hundred milliseconds in hippocampal pyramidal neurons, and at 1 s after the end of the step, during the sAHP, which has durations in the 1–3 s range (Lancaster and Nicoll, 1987; Storm, 1990; Williamson and Alger, 1990; Sah and Faber, 2002; Stocker, 2004). Area and duration of the AHP were measured from the end of the depolarization step until return of the membrane voltage to baseline (−60 mV). AHPs were elicited every 30 s, and measures were averaged from 10–15 consecutive AHPs recorded in each cell.
Spike-frequency accommodation was also determined from −60 mV using current intensity just sufficient to generate three Na+ action potentials within the first 100 ms of an 800 ms depolarization pulse. Accommodation was measured as the number of action potentials during the entire current step.
Repetitive synaptic stimulation (RSS) for ratiometric Ca2+ imaging was performed using a twisted bipolar stimulation electrode (0.0045 inch coated stainless steel; A-M Systems, Everett, WA) positioned in the Schaffer-collaterals/commissural fibers of stratum radiatum ∼500 μm from the recorded neuron. For all synaptic stimulation, pulse duration was 100 μs and was delivered by a SD9K stimulator (Astro Med, Grass Instruments, Warwick, RI). Input–output (I/O) relationships were determined in every cell during baseline periods (0.2 Hz) before RSS. The RSS train was delivered at 7 Hz for 20 s. Stimulus intensity during RSS was set to deliver pulses at 50% above Na+ action potential threshold, to ensure action potential generation at 7 Hz throughout the 20 s train. Double spikes to a single pulse were rare and counted as single spikes.
Cell health and exclusion criteria (Tables 1, 2).
Table 1.
Basic neuronal and experimental properties: neuronal parameters for all recorded CA1 neurons
| Age |
|||||
|---|---|---|---|---|---|
| 4 months (n = 19) | 10 months (n = 16) | 12 months (n = 21) | 14 months (n = 15) | 23 months (n = 21) | |
| Input resistance (MΩ) | 55.76 ± 2.84 | 56.25 ± 3.66 | 52.55 ± 2.647 | 56.02 ± 3.42 | 50.87 ± 2.87 |
| Resting membrane potential (mV) | −62.0 ± 0.41 | −62.44 ± 0.61 | −62.71 ± 0.47 | −61.47 ± 0.60 | −62.67 ± 0.50 |
| Action potential amplitude (mV) | 77.37 ± 1.32 | 77.13 ± 1.22 | 76.91 ± 1.19 | 79.20 ± 1.50 | 78.0 ± 1.15 |
| Electrode resistance (MΩ) | 100.11 ± 7.09 | 106.13 ± 6.34 | 99.90 ± 5.81 | 100.93 ± 8.91 | 97.8 ± 3.97 |
| RSS hyperpolarization (mV) | −2.77 ± 0.40 | −3.21 ± 0.48 | −2.75 ± 0.28 | −2.23 ± 0.34 | −2.55 ± 0.26 |
Basic membrane and experimental variables for CA1 pyramidal neurons across all age groups. No significant aging effect was detected on any of these baseline variables. RSS hyperpolarization was measured at its peak level during RSS from a holding potential of −70 mV. All values are means ± SEM.
Table 2.
Basic neuronal and experimental properties: imaging parameters for neurons both recorded and imaged
| Age |
|||||
|---|---|---|---|---|---|
| 4 months (n = 11) | 10 months (n = 10) | 12 months (n = 11) | 14 months (n = 11) | 23 months (n = 10) | |
| Resting calcium (nm) | 101.50 ± 9.05 | 95.77 ± 6.50 | 93.68 ± 10.54 | 89.25 ± 6.18 | 100.71 ± 6.54 |
| Cell depth (μm) | 70.91 ± 4.95 | 71.00 ± 4.46 | 69.55 ± 5.62 | 65.46 ± 5.54 | 61.00 ± 8.88 |
| Action potential number during 20 s, 7 Hz RSS | 139 ± 0.89 | 139 ± 0.57 | 139 ± 0.34 | 137 ± 0.89 | 139 ± 0.65 |
Basic membrane and experimental variables for CA1 pyramidal neurons across all age groups. No significant aging effect was detected on any of these baseline variables. Cell depth is the depth of imaged cells below the surface of the slice. Action potential numbers reflect spikes generated during the 20 s, 7 Hz RSS train. All values are means ± SEM.
For imaging studies, cells with resting [Ca2+]i >200 nm were excluded from analysis. For recording analyses, neurons with input resistance <35 MΩ, action potential amplitude <70 mV (measured from −60 mV), or holding current more than −200 pA at −70 mV were excluded from the study. Overall yield of recorded neurons that met all criteria (including those not imaged because of depth) was approximately two cells per daily animal preparation. The yield of neurons both imaged and recorded per animal was somewhat less (∼1.5), and neither yield differed across aging. As previously reported (Thibault et al., 2001), gray values taken at rest for either the 357 or the 380 nm wavelength (see below) did not differ significantly with age, nor did cell depth measures (Table 2), indicating that age differences in Ca2+ indicator loading or tissue opacity did not contribute significantly to the quantitative measures of [Ca2+]i reported here.
Fluorometric [Ca2+]i measurements.
Individual neurons loaded with the ratiometric Ca2+ indicator bis-Fura-2 (10 mm) were imaged on the stage of a Nikon (Tokyo, Japan) E600 microscope equipped with a 40× water immersion objective and a CCD camera (Roper Scientific, Princeton Instruments, Trenton, NJ). The fluorophore was excited using a wavelength switcher (Sutter Lambda DG-4) and software control (Axon Imaging Workbench, version 2.2.1.54; Molecular Devices). The 510 nm wavelength was monitored during both 357 and 380 nm wavelength excitation through a dichroic mirror centered at 430 nm. To increase the speed of image acquisition during concomitant electrophysiology (Jaffe and Brown, 1994) while maintaining adequate spatial resolution, we imaged neurons using the isosbestic method (Grynkiewicz et al., 1985) and limited our images to defined regions of interest around the neuronal soma (Magee and Johnston, 1997). Frequency of image acquisition was synchronized to the 7 Hz frequency of synaptic stimulation. Individual images from neurons were sequentially captured at 7 Hz for the 380 nm wavelengths, and a 357 nm wavelength was captured every 5 s. To obtain ratio images at 7 Hz, the linear relationship between each subsequent 357 nm image was determined and used to extrapolate 357 nm values for every captured 380 nm image. As expected, the 357 nm images did not significantly increase during stimulation, because at the isosbestic point, fluorescence emission is [Ca2+]-insensitive. The final ratio was obtained by dividing the 380 nm images with the extrapolated 357 nm images. All fluorescence intensities were background subtracted from an area near to the indicator-filled neuron that showed little fluorescence scatter. Acquisition of images was begun before synaptic stimulation and was maintained past the end of RSS train to obtain data on resting levels and decay kinetics of [Ca2+]i (see Figs. 3, 4). Rise and decay time constants (τ) of somatic [Ca2+]i were determined using a standard exponential fit with a simplex method in Clampfit (Molecular Devices). All fit values showed correlation coefficients >0.93. During RSS, [Ca2+]i rose rapidly, reaching peak values within a few seconds, and then stabilized at that level for the remainder of the train. Area under the curve (AUC) of the Ca2+ response was calculated from the integral of the area of the response during the first 10 s of the 20 s RSS, using SigmaPlot (version 9.0) and a user-defined transform.
Figure 3.
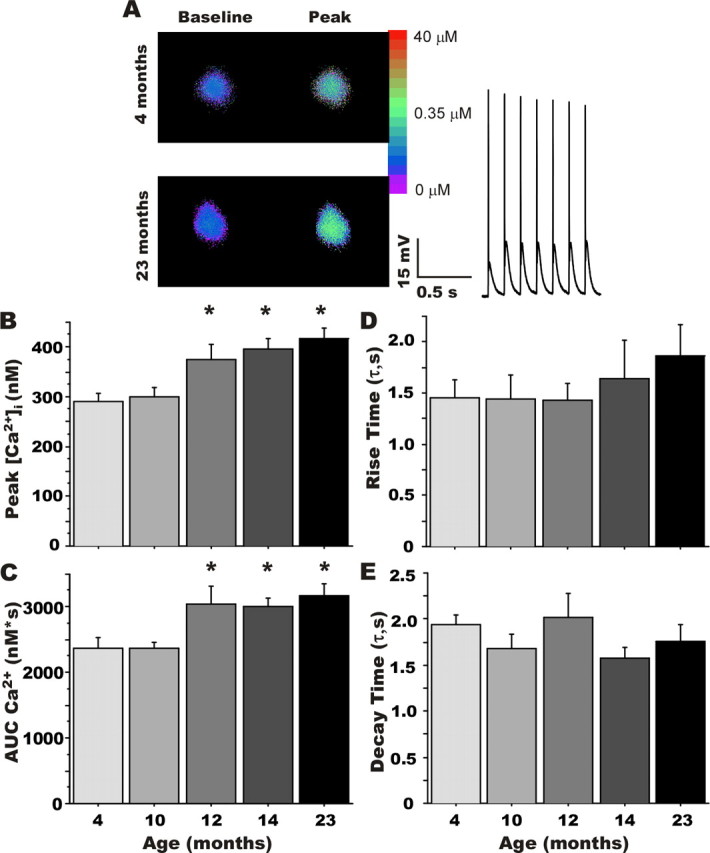
Rapid ratiometric Ca2+ imaging. A, Representative pseudocolor ratiometric images of bis-Fura-2-filled CA1 pyramidal neurons from a young-adult (4 months; top) and an aged (23 months; bottom) animal. Somatic regions are displayed at rest (left) and during repetitive synaptic stimulation (peak; right). The inset shows the first second of the postsynaptic response during 7 Hz RSS above action potential threshold in a young-adult neuron. Group comparisons revealed a significant effect of aging on peak [Ca2+]i and the area under the curve (AUC) of the somatic Ca2+ response (B, C), but no significant differences in the rising and decaying phases of the [Ca2+]i response (D, E, respectively). *Different from the 4-month-old group at p < 0.05. Means ± SEM are shown.
Figure 4.
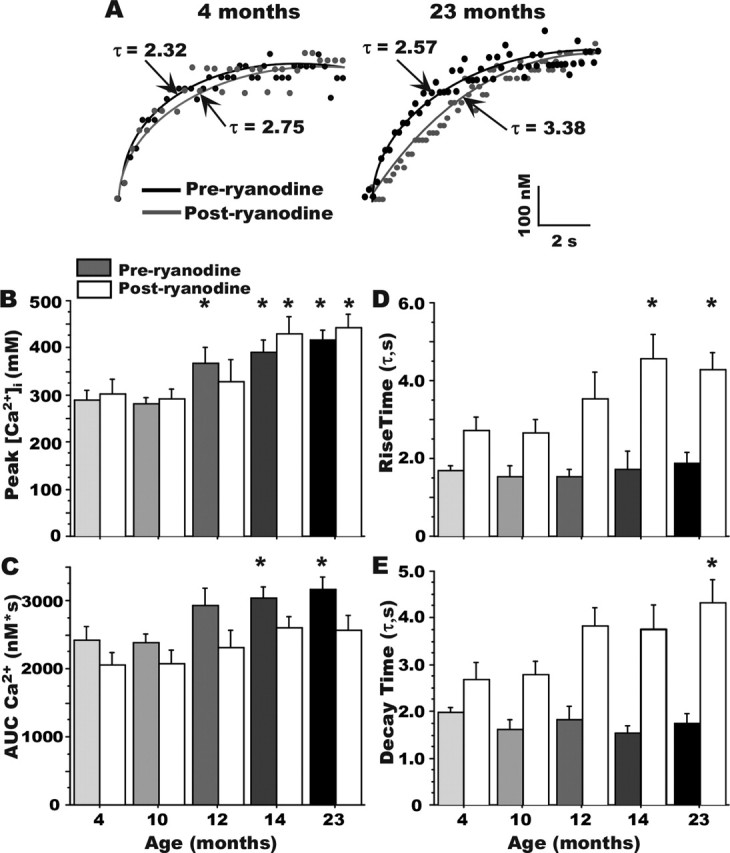
Effect of ryanodine on kinetics of the [Ca2+]i response. A, Examples of the effects of ryanodine in slowing the rising phase of somatic Ca2+ at the onset of RSS in a 4-month-old (left) and 23-month-old (right) animal. B, Ryanodine did not significantly affect peak Ca2+ levels measured during RSS. Grayscale bars, Preryanodine. White bars, Postryanodine. Aging effect persists after ryanodine (asterisks). C, Ryanodine eliminated the effect of aging on measures of the AUC. Although the rising (D) and decaying (E) time constants of Ca2+ did not differ with aging, ryanodine treatment significantly lengthened both time constants. All aging comparisons are made within a treatment condition (i.e., preryanodine only or postryanodine only). Note that peak Ca2+ levels are increased with aging in cells before the ryanodine treatment (asterisks above grayscale bars) and also after ryanodine treatment (asterisks above white bars). *Different from the 4-month-old group appropriate for preryanodine and postryanodine comparisons at p < 0.05. Means ± SEM are shown.
Ratio values were calibrated to generate estimates of free [Ca2+]i using a standardized series of droplets containing 50 μm bis-Fura 2 and nine increasing Ca2+ concentrations (0–39 μm free Ca2+, 1 mm Mg2+; Molecular Probes, Eugene OR). The standard curve obtained from this in vitro calibration was fitted to yield several parameters: Rmin, the minimum ratio in the absence of Ca2+; Rmax, the maximum ratio at saturating Ca2+ concentrations; Kd, the dissociation constant for Ca2+ binding to the indicator; and a constant (β) equal to the ratio of the 380 nm image in zero Ca2+ to the 380 nm image in 39 μm Ca2+. The extracted values were 0.6 for Rmin, 2.38 for Rmax, and 2.86 (in micromolar concentration) for Kdβ. Image ratios were converted to free Ca2+ concentration ([Ca2+]i) using the following equation: [Ca2+]i = Kdβ(R − Rmin)/(Rmax − R), in which R is the 357/380 nm fluorescence emission ratio of the imaged cell (Grynkiewicz et al., 1985).
Drug application.
Ryanodine (Alomone Labs, Jerusalem, Israel) is a plant alkaloid with high specificity and potency at RyRs, and at >10 μm selectively blocks the receptors, eliminating CICR without influencing Ca2+ influx from other sources (Friel and Tsien, 1992; Alford et al., 1993; Belousov et al., 1995; Garaschuk et al., 1997). Ryanodine was applied using whole-bath perfusion and a syringe pump (model 341B; Sage Instruments, Orion Research, Cambridge, MA). The ryanodine stock solution (20 mm in distilled water) was introduced into the flow of the oxygenated Ca-ACF at a rate 1000 times slower than the main perfusate, resulting in a final ryanodine concentration estimated at 20 μm. The drug was allowed to infuse into the recording chamber for 15 min. To ensure that neither the initial RSS, the inter-RSS delay, nor the vehicle, accounted for the effects of ryanodine, control experiments with vehicle (distilled water) were conducted using the same delivery method and duration, and all markers (AHP measures, Ca2+ concentration during rest and during RSS, input resistance, and spike height) were measured again ∼15 min after the first RSS. In 16 cells (3–4 per age group), no statistical effects or trends of initial RSS, vehicle, or time were detected on any measures in any age group or in all cells combined.
Statistical analyses.
Variables were analyzed for main effects of aging using one-way ANOVA across groups, and Fisher's protected least significant difference post hoc comparisons against the youngest group were used to determine at which age point an effect first appeared (Statview; SAS Institute, Cary, NC). Values of p < 0.05 were considered significant. The main effect of aging, both before and after drug application, on the different Ca2+-dependent markers was also determined in a subset of cells that received ryanodine (see Figs. 2, 4). Repeated-measure two-way ANOVAs comparing the same cells treated before and after ryanodine application also were used to determine whether the effects of ryanodine differed as a function of age.
Figure 2.
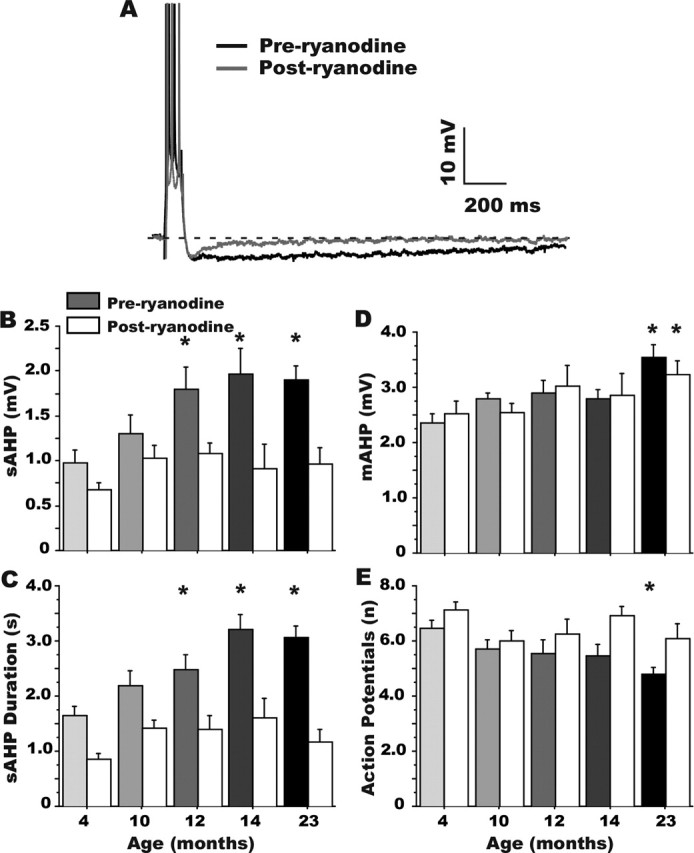
Ryanodine eliminates the aging effect on the sAHP. A, Representative examples of the effect of 20 μm ryanodine on the AHP recorded from a 23-month-old rat CA1 neuron. Only a subgroup of the cells presented in Figure 1 were treated with ryanodine, and thus the aging effect was also analyzed separately for that subgroup, both before (grayscale bars) and after (white bars) ryanodine treatment (B–E). Note that ryanodine reduced measures of sAHP amplitude (A, B), sAHP duration (C), and accommodation (E) more in aged animals and eliminated the aging effect in each measure. However, the mAHP (D) was unaffected by ryanodine. *Different at p < 0.05 from the 4-month-old group appropriate for preryanodine and postryanodine comparisons. Means ± SEM are shown.
Results
As shown in Tables 1 and 2, basic experimental and neuronal parameters reflecting preparation health and comparability did not differ significantly with age. The data were obtained from nine animals per age group, except for the 10 month group (n = 10 animals), yielding electrophysiological measures on a total of 92 neurons from 46 animals. Concomitant imaging data were obtained in a subset of 53 cells from 37 animals (8 animals for the 4 and 23 month groups and 7 animals each for all other age groups). Statistical outcomes were highly similar whether cells or animals (with cell values averaged) were used as the sampling population (Thibault and Landfield, 1996; Thibault et al., 2001).
Age course of changes in AHP properties
Figure 1A illustrates the AHP triggered in CA1 pyramidal neurons from a young-adult (4 months) and an aged (23 months) animal after generation of three action potentials. As seen previously (see Introduction), the sAHP (amplitude measured 1 s after the end of the depolarization) was increased substantially as a function of aging (F(4,89) = 6.634; p < 0.0001) (Fig. 1B). This aging effect was first detectable statistically in cells recorded from 12-month-old animals and was maintained at age points older than 12 months. Measures of sAHP duration also revealed a significant increase with aging (F(4,89) = 9.417; p < 0.0001) (Fig. 1C) that paralleled the onset of changes in sAHP amplitude. The maximum duration was observed in animals at 23 months and was significantly different from duration at 12 months (p < 0.01). The mAHP measured at the deepest point of deflection after the end of the depolarization showed similar age-dependent changes (F(4,89) = 6.701; p < 0.0001) (Fig. 1D), beginning at 12 months of age. In addition, the maximum mAHP amplitude was observed in 23-month-old animals, which also differed significantly from 14-month-old animals (p < 0.01).
Figure 1.
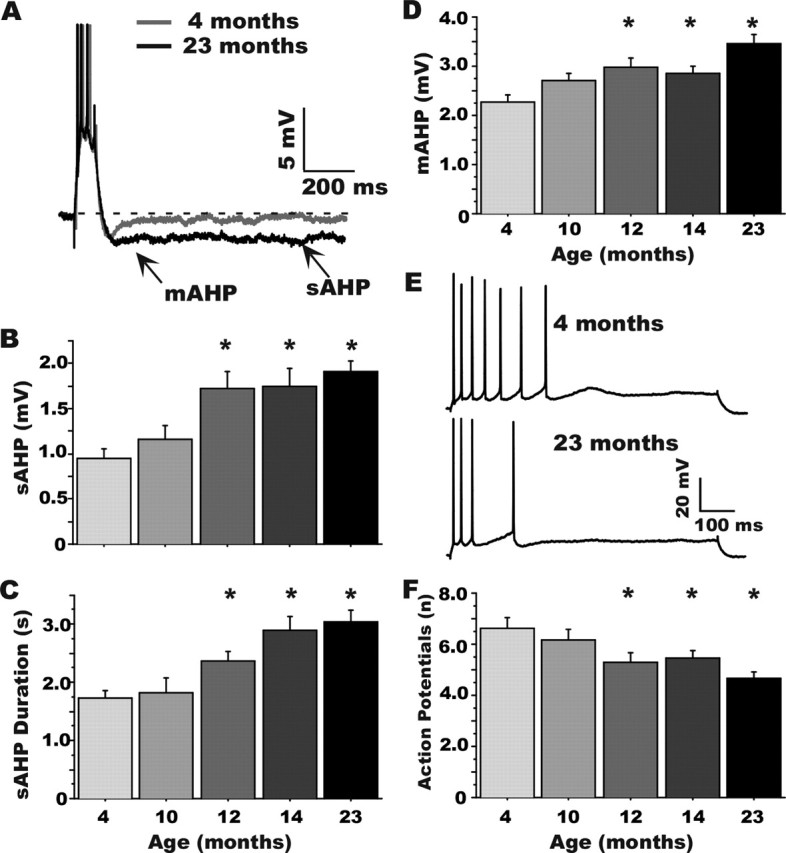
Age course of alterations in Ca2+-dependent AHPs in CA1 neurons during aging. A, Representative examples of AHPs recorded in CA1 neurons from a young-adult (4 months) and an aged (23 months) animal illustrating where measurements were taken (arrows). B–D, Quantitative group changes in sAHP amplitude and duration and mAHP amplitude, respectively. E, Representative examples of spike frequency accommodation in a 4-month-old (top) and a 23-month-old (bottom) animal. F, Group results on measures of spike-frequency accommodation (number of action potentials generated during an 800 ms depolarization). *Significantly different from the 4-month-old group at p < 0.05. Means ± SEM are shown. Note that the earliest detectable changes occurred for all variables at 12 months of age.
Age course of changes in spike-frequency accommodation
In a subset of neurons (n = 72) from all age groups, we also measured spike-frequency accommodation. During a sustained intracellular depolarization pulse, neurons initially respond with high-frequency firing, followed by a decreasing action potential firing rate (spike-frequency accommodation) as the sAHP develops and membrane conductance increases. As previously reported (Moyer et al., 1992; Disterhoft et al., 1996), an age-dependent increase in accommodation (decreased number of action potentials) during the depolarization was observed (Fig. 1E) (F(4,68) = 4.734; p < 0.01). The greatest accommodation was detected in the 23-month-old group, and, as with sAHP measures, significant differences were observed in animals as early as 12 months (Fig. 1F).
Ca2+-induced Ca2+ release contribution to aging changes in the AHP
To determine the degree of CICR contribution to the development of larger AHPs with aging, we assessed the effects of high ryanodine concentration (20 μm) in a subset of the cells shown in Figure 1.Figure 2A illustrates the ryanodine effect in a neuron of an aged animal (23 months) and highlights the specificity of ryanodine for the sAHP relative to the mAHP. Ryanodine significantly reduced the amplitude of the sAHP (white bars; F(1,97) = 24.65; p < 0.0001) (Fig. 2B). Post hoc analyses on measures of both absolute decrease and percent decrease induced by ryanodine, revealed a substantially greater effect of ryanodine on the sAHP at 12 versus 4 months of age, which was maintained until 23 months of age (Fig. 2B). Consequently, CICR appeared to account for essentially the full aging-related increase in the sAHP as ryanodine eliminated the aging differences in the sAHP amplitude (before ryanodine, aging effect: F(4,45) = 2.76; p < 0.05, see asterisks; postryanodine aging effect: F(4,45) = 0.93; p = 0.46) (Fig. 2B). Quantitatively similar effects of ryanodine were observed on measures of AHP duration (Fig. 2C). However, ryanodine did not significantly affect the mAHP (white bars; F(1,97) = 0.065; p = 0.80) (Fig. 2D). Before ryanodine application, the mAHP was increased significantly with aging (F(4,45) = 2.82; p < 0.05), and after ryanodine, this aging effect was still present (F(4,45) = 3.25; p < 0.05), indicating little or no effect of ryanodine on the mAHP. As with the sAHP, however, ryanodine also significantly reduced spike frequency accommodation in aged neurons, increasing the number of action potentials fired during the step depolarization (F(1,74) = 12.32; p < 0.001) (Fig. 2E) and eliminating the effect of aging. Before ryanodine application, accommodation was increased with aging (F(4,34) = 3.16; p < 0.05), whereas after ryanodine, accommodation was no longer increased with aging (F(4,34) = 0.92; p = 0.46).
Ratiometric imaging of the stimulation-induced rise in [Ca2+]i in CA1 neurons
[Ca2+]i measures were obtained before, during, and after an RSS train of pulses delivered at 7 Hz with pulse intensity set 50% above action potential threshold. This protocol largely avoided spike failure, ensuring comparable levels of postsynaptic action potential generation throughout the train (Table 2), and mimicked physiologically relevant (theta) frequencies of synaptic activation. This RSS protocol causes significant Ca2+ influx into the dendrites and somata of pyramidal neurons (Thibault et al., 2001), because repetitive spike generation triggers a large postsynaptic [Ca2+]i response (Jaffe et al., 1992; Regehr and Tank, 1992; Brown and Jaffe, 1994; Helmchen et al., 1996; Jacobs and Meyer, 1997; Thibault et al., 2001).
Figure 3A illustrates [Ca2+]i responses obtained at rest and during peak [Ca2+]i induced by RSS (inset; first 1 s) in representative CA1 neurons from a 4-month-old (top) and a 23-month-old (bottom) animal. As previously reported (Thibault et al., 2001; Xiong et al., 2002), resting [Ca2+]i did not differ significantly with aging (Table 2). During RSS, however, [Ca2+]i increased steadily in the somata of neurons in all age groups, reaching maximal values within 5 s, and maintaining equilibrium at approximately that level for the remainder of the RSS. In response to comparable degrees of postsynaptic spike generation, peak [Ca2+]i was increased significantly as a function of aging (F(4,48) = 7.017; p < 0.001) (Fig. 3B). [Ca2+]i remained significantly elevated in aged animals throughout the train and analysis of AUC of the Ca2+ response during RSS also revealed a significant increase above the 4 month group, beginning at 12 months of age (F(4,48) = 3.64; p < 0.05) (Fig. 3C). Time constants for the rising (Fig. 3D) and decaying (Fig. 3E) phases of somatic [Ca2+]i at the beginning and the end of RSS, respectively, as determined by single exponential fit parameters, did not differ significantly with age.
CICR contribution to aging changes in [Ca2+]i transients
Ryanodine application exerted no detectable effects on baseline parameters of input resistance, action potential threshold or amplitude, or EPSP size. During RSS, however, ryanodine dramatically slowed the rise time, thereby reducing AUC, of the Ca2+ response, particularly in aged rat neurons (Fig. 4A). The peak [Ca2+]i eventually attained during RSS continued to show an aging effect after as well as before ryanodine (Fig. 4B, asterisks above white and grayscale bars). However, the aging-specific action of ryanodine in slowing rise time reduced the overall AUC of the Ca2+ response substantially more in aged animal neurons (F(1,57) = 13.4; p < 0.01) eliminating the age effect on AUC (Fig. 4C). Thus, ryanodine significantly increased both the rise and decay time constants of somatic [Ca2+]i (F(1,57) = 53.5; p < 0.0001; and F(1,57) = 70.4; p < 0.0001, respectively) (Fig. 4D,E), again, substantially more in aged than young rat neurons (Fig. 4A,D). In addition, the ryanodine-sensitive component of the [Ca2+]i response (the difference between preryanodine and postryanodine AUC, or Δ) analyzed on a per-second basis during RSS, confirmed that the primary contribution of enhanced CICR in aging rat neurons occurred relatively rapidly (1–5 s of RSS), and diminished thereafter during the RSS train (data not shown) (main effect of aging on the action of ryanodine on Δ AUC: F(4,24) = 4.417; p < 0.01).
Discussion
Concurrent emergence of ryanodine-sensitive aging biomarkers
Although there is wide recognition of the value of examining intermediate age points in biological aging research (Miller and Nadon, 2000; Coleman et al., 2004), the age of first appearance has not been determined with high resolution for most brain biomarkers. Nevertheless, age-of-onset data can provide important insights into the sequence and potential mechanistic interactions of multiple age-dependent processes. For example, hippocampal-dependent learning impairment, among the few biomarkers for which onset has been assessed (see Introduction), appears to emerge around midlife (∼12 months of age in F344 rats) or earlier in several mammalian species. Presumably, therefore, the onset of physiological biomarkers that underlie this functional impairment must precede or coincide with the onset of cognitive decline.
Here, we tested with relatively high resolution (∼2 month intervals) the earliest ages at which established electrophysiological and imaging Ca2+-mediated biomarkers of aging were detectable statistically. The results provide the first evidence that, in F344 rats, multiple Ca2+-mediated markers of brain aging emerge concurrently, as predicted by the hypothesis of an underlying common mechanism, and emerge by 12 months of age, as predicted by their putative roles in the pathogenesis of cognitive decline. Moreover, these biomarkers exhibited aging-specific enhanced sensitivity to ryanodine, also beginning at ∼12 months of age (Figs. 2, 4), resulting in the effective elimination by ryanodine of most aging differences in Ca2+-related processes. Together, therefore, these findings provide substantial new evidence that aging-dependent Ca2+ dyshomeostasis in hippocampal CA1 neurons depends on a common mechanism, partly comprising altered CICR from RyRs, that emerges early enough in adulthood to play a role in initiating cognitive decline.
Our findings on altered CICR in brain aging also are consistent with work in other models indicating that overfilling of the ER with Ca2+ may be present in some Alzheimer's disease models (Gibson et al., 1996; Leissring et al., 2000; Paschen and Frandsen, 2001), and that larger ER Ca2+ pools develop in older and/or more vulnerable neurons (Verkhratsky and Toescu, 1998; Clodfelter et al., 2002). Recent reports also suggest that alterations in CICR may contribute to aging differences in synaptic plasticity, potentially through effects on the sAHP (Kumar and Foster, 2004).
CICR interactions with L-type channels in Ca2+ dysregulation
As noted, altered activity of L-type voltage-gated Ca2+ channels also has been implicated as a candidate mechanism in aging-dependent Ca2+ dyshomeostasis. L-VGCC agonists and antagonists strongly modulate the sAHP, spike accommodation, Ca2+ action potential, and the synaptically activated rise in [Ca2+]i, as well as aging-dependent cognitive function (Disterhoft et al., 1996, 2004; Norris et al., 1998; Thibault et al., 1998, 2001). Furthermore, available L-VGCCs increase in CA1 neurons with aging (Thibault and Landfield, 1996; Veng et al., 2003). Recent data obtained in a separate age course study also showed that increased L-VGCC activity first appeared at about the same age as these Ca2+-mediated biomarkers and the enhanced ryanodine sensitivity (O. Thibault, personal communication).
However, evidence of a role for L-VGCCs in Ca2+ dysregulation is not necessarily inconsistent with the findings here on CICR. Clearly, Ca2+ influx via VGCCs can trigger substantial CICR and, as in muscle, CICR from RyRs may be in series with L-VGCCs in at least some types of neurons (Chavis et al., 1996; Empson and Galione, 1997; Borde et al., 2000; Fagni et al., 2000; Sukhareva et al., 2002; Verkhratsky, 2005). Therefore, elevated L-VGCC activity and amplified CICR could reflect different stages of a single pathway that acts to dysregulate Ca2+ in aging (Fig. 5, model). If so, an important question still to be resolved is whether increased L-VGCC activity is a sufficient trigger for amplifying CICR or whether CICR is altered independently.
Figure 5.

Schematic model of proposed interactions of L-VGCC and altered CICR in driving Ca2+-related processes in the hippocampus of young (A) and aged (B) animals. The increased L-VGCC activity and enhanced CICR are in series, amplifying the impact of Ca2+ influx on multiple functions. The thickness of arrows schematically represents the relative weight of contribution to [Ca2+]i levels and responses. Despite similar spike-mediated depolarization, with aging, the contribution is increased at several stages. The dashed arrows indicate a possible direct parallel contribution of L-VGCCs to [Ca2+]i.
Of course, L-VGCC activity may not be the only source of elevated Ca2+ influx. However, hippocampal NMDA receptor activity, a major source of postsynaptic Ca2+ influx during synaptic activation (Alford et al., 1993; Emptage et al., 1999; Kovalchuk et al., 2000; Sabatini et al., 2002), appears to decrease rather than increase with aging (Barnes et al., 1997; Magnusson, 1998; Potier et al., 2000; Clayton et al., 2002; Rosenzweig and Barnes, 2003). Nonetheless, other sources may be altered with aging. In addition, several studies have raised the possibility that other ion conductances also contribute to these aging biomarkers (Power et al., 2002; Wu et al., 2004; Tombaugh et al., 2005). Clearly, therefore, additional studies will be needed to determine more precisely the degree to which alterations in the L-VGCC–CICR pathway account for Ca2+-related biomarkers of aging.
Potential consequences of amplified CICR in vivo
Ca2+-related processes were studied here primarily as outcome biomarkers and, consequently, RSS stimulus pulse intensity was maintained at high levels to ensure consistent spike generation (Table 2). However, it should be noted that, under more physiological conditions, the larger Ca2+ transients in aged animal neurons might well induce greater spike failure. For example, EPSP amplitude during RSS is decreased by aging-dependent Ca2+ elevation (Thibault et al., 2001), which would likely translate into reduced spike generation in vivo. Furthermore, the enhanced sAHP seen with aging might well also increase spike failure via accommodation (Moyer et al., 1996; Thompson et al., 1996; Disterhoft et al., 2004; Tombaugh et al., 2005). Although clear aging differences in pyramidal neuron firing frequency have not yet been found in behaving rats, the effects of a larger sAHP or reduced facilitation of the EPSP may be more likely to appear during bursts or to be reflected in interspike intervals and network dynamics rather than in total spikes over a lengthy period (Rosenzweig and Barnes, 2003). Similarly, greater hyperpolarization during repetitive synaptic activation of spikes was not seen here in aging animal neurons (Table 1), in contrast to the larger AHP after spikes induced by intracellular stimulation (Fig. 1A). However, several factors could account for this. For one, multiple conductances contribute to synaptically induced hyperpolarization, which may mask the enhanced sAHP component (Storm, 1990; Lancaster et al., 2001; Wu et al., 2004). Nevertheless, an enhanced sAHP, even if masked, might still inhibit synaptic throughput and spike generation, depending on cellular regions of localization of AHP and excitatory conductances (Jaffe et al., 1992; Lancaster et al., 2001; Sah and Faber, 2002; Johnston et al., 2003). However, it is clear that additional studies are needed on the nature of in vivo effects of Ca2+-mediated biomarkers of aging.
Kinetics of the CICR effect
In agreement with a previous study (Thibault et al., 2001), higher peak Ca2+ concentrations and an increased AUC of the Ca2+ response were present in neurons from aged animals during suprathreshold RSS (Figs. 3, 4), whereas no differences in resting [Ca2+]i were found. In addition, greater temporal sampling resolution in the present study revealed that peak [Ca2+]i levels were generally attained within 5 s of RSS at 7 Hz (Fig. 4A), and there were no age differences in the rise or decay time constants (Fig. 3). Ryanodine slowed the Ca2+ rise and decay time constants (Fig. 4D,E), and did so more in aged neurons (Fig. 4A,D), although it did not alter peak Ca2+ concentrations eventually attained (Fig. 4A,B). As a consequence of slowed rise time, ryanodine also reduced the AUC more in aged rat neurons and eliminated the aging difference in the AUC biomarker during RSS (Fig. 4C). Together, these results are consistent with the interpretation that RyRs provide a large and early booster source of Ca2+ to the soma during the first few seconds of repetitive action potential activity, particularly in CA1 neurons from aged rats. The observation that [Ca2+]i reached the same peak after ryanodine, albeit more slowly in older animal neurons, could reflect slowed clearance mechanisms and altered kinetics (Murchison et al., 2004; Toescu and Verkhratsky, 2004; Toescu et al., 2004). In addition, the slower initial rise of the component remaining after ryanodine in aged rat neurons could result from decreased influx/release from ligand-gated channels or other sources. Conceivably, therefore, increased L-type channel activity and CICR could be compensatory responses for decreases in other Ca2+ sources or signaling pathways.
An integrative model of hippocampal Ca2+ dyshomeostasis
Together with previous work, the present studies suggest a new integrative, albeit simplified, model of Ca2+ dysregulation in hippocampal neurons during aging (Fig. 5). In this model, increased activity in the L-VGCC-RyR axis begins around midlife, initiating a cascade of Ca2+ dysregulation and altered physiology that impedes synaptic throughput and spike generation and, in turn, impairs cognitive function. Moreover, the dysregulated Ca2+ may well also increase activation of Ca2+ pathways involving calcineurin, calpains, and gene regulation, thereby disrupting cell signaling and heightening neuronal vulnerability. Thus, if supported by additional studies, this model may suggest new therapeutic strategies for intervening in aging-dependent cognitive impairment, as well as provide a mechanistic framework for the growing evidence that some potential therapeutic interventions are more effective if begun soon after midlife (Zandi et al., 2002).
Footnotes
This work was supported by National Institute on Aging Grants AG04542 and AG10836.
References
- Aitken DH, Meaney MJ (1989). Temporally graded, age-related impairments in spatial memory in the rat. Neurobiol Aging 10:273–276. [DOI] [PubMed] [Google Scholar]
- Alford S, Frenguelli BG, Schofield JG, Collingridge GL (1993). Characterization of Ca2+ signals induced in hippocampal CA1 neurones by the synaptic activation of NMDA receptors. J Physiol (Lond) 469:693–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach ME, Barad M, Son H, Zhuo M, Lu YF, Shih R, Mansuy I, Hawkins RD, Kandel ER (1999). Age-related defects in spatial memory are correlated with defects in the late phase of hippocampal long-term potentiation in vitro and are attenuated by drugs that enhance the cAMP signaling pathway. Proc Natl Acad Sci USA 96:5280–5285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes CA (1994). Normal aging: regionally specific changes in hippocampal synaptic transmission. Trends Neurosci 17:13–18. [DOI] [PubMed] [Google Scholar]
- Barnes CA, Rao G, Shen J (1997). Age-related decrease in the N-methyl-d-aspartateR-mediated excitatory postsynaptic potential in hippocampal region CA1. Neurobiol Aging 18:445–452. [DOI] [PubMed] [Google Scholar]
- Belousov AB, Godfraind JM, Krnjevic K (1995). Internal Ca2+ stores involved in anoxic responses of rat hippocampal neurons. J Physiol (Lond) 486:547–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezprozvanny I, Watras J, Ehrlich BE (1991). Bell-shaped calcium-response curves of Ins(1,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature 351:751–754. [DOI] [PubMed] [Google Scholar]
- Bickford P (1995). Aging and motor learning: a possible role for norepinephrine in cerebellar plasticity. Rev Neurosci 6:35–46. [DOI] [PubMed] [Google Scholar]
- Borde M, Bonansco C, de Sevilla F, Le Ray D, Buno W (2000). Voltage-clamp analysis of the potentiation of the slow Ca2+-activated K+ current in hippocampal pyramidal neurons. Hippocampus 10:198–206. [DOI] [PubMed] [Google Scholar]
- Brown TH, Jaffe DB (1994). Calcium imaging in hippocampal neurons using confocal microscopy. Ann NY Acad Sci 747:313–324. [DOI] [PubMed] [Google Scholar]
- Chavis P, Fagni L, Lansman JB, Bockaert J (1996). Functional coupling between ryanodine receptors and L-type calcium channels in neurons. Nature 382:719–722. [DOI] [PubMed] [Google Scholar]
- Clayton DA, Mesches MH, Alvarez E, Bickford PC, Browning MD (2002). A hippocampal NR2B deficit can mimic age-related changes in long-term potentiation and spatial learning in the Fischer 344 rat. J Neurosci 22:3628–3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clodfelter GV, Porter NM, Landfield PW, Thibault O (2002). Sustained Ca2+-induced Ca2+-release underlies the post-glutamate lethal Ca2+ plateau in older cultured hippocampal neurons. Eur J Pharmacol 447:189–200. [DOI] [PubMed] [Google Scholar]
- Coleman P, Finch C, Joseph J (2004). The need for multiple time points in aging studies. Neurobiol Aging 25:3–4. [DOI] [PubMed] [Google Scholar]
- Coronado R, Morrissette J, Sukhareva M, Vaughan DM (1994). Structure and function of ryanodine receptors. Am J Physiol 266:C1485–C1504. [DOI] [PubMed] [Google Scholar]
- deToledo-Morrell L, Geinisman Y, Morrell F (1988). Age-dependent alterations in hippocampal synaptic plasticity: relation to memory disorders. Neurobiol Aging 9:581–590. [DOI] [PubMed] [Google Scholar]
- Disterhoft JF, Thompson LT, Moyer JR Jr, Mogul DJ (1996). Calcium-dependent afterhyperpolarization and learning in young and aging hippocampus. Life Sci 59:413–420. [DOI] [PubMed] [Google Scholar]
- Disterhoft JF, Wu WW, Ohno M (2004). Biophysical alterations of hippocampal pyramidal neurons in learning, ageing and Alzheimer's disease. Ageing Res Rev 3:383–406. [DOI] [PubMed] [Google Scholar]
- Empson RM, Galione A (1997). Cyclic ADP-ribose enhances coupling between voltage-gated Ca2+ entry and intracellular Ca2+ release. J Biol Chem 272:20967–20970. [DOI] [PubMed] [Google Scholar]
- Emptage N, Bliss TV, Fine A (1999). Single synaptic events evoke NMDA receptor-mediated release of calcium from internal stores in hippocampal dendritic spines. Neuron 22:115–124. [DOI] [PubMed] [Google Scholar]
- Fagni L, Chavis P, Ango F, Bockaert J (2000). Complex interactions between mGluRs, intracellular Ca2+ stores and ion channels in neurons. Trends Neurosci 23:80–88. [DOI] [PubMed] [Google Scholar]
- Fischer W, Chen KS, Gage FH, Bjorklund A (1992). Progressive decline in spatial learning and integrity of forebrain cholinergic neurons in rats during aging. Neurobiol Aging 13:9–23. [DOI] [PubMed] [Google Scholar]
- Foster TC, Norris CM (1997). Age-associated changes in Ca2+-dependent processes: relation to hippocampal synaptic plasticity. Hippocampus 7:602–612. [DOI] [PubMed] [Google Scholar]
- Frick KM, Baxter MG, Markowska AL, Olton DS, Price DL (1995). Age-related spatial reference and working memory deficits assessed in the water maze. Neurobiol Aging 16:149–160. [DOI] [PubMed] [Google Scholar]
- Friel DD, Tsien RW (1992). A caffeine- and ryanodine-sensitive Ca2+ store in bullfrog sympathetic neurones modulates effects of Ca2+ entry on [Ca2+]i J Physiol (Lond) 450:217–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garaschuk O, Yaari Y, Konnerth A (1997). Release and sequestration of calcium by ryanodine-sensitive stores in rat hippocampal neurones. J Physiol (Lond) 502:13–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson G, Martins R, Blass J, Gandy S (1996). Altered oxidation and signal transduction systems in fibroblasts from Alzheimer patients. Life Sci 59:477–489. [DOI] [PubMed] [Google Scholar]
- Gibson GE, Peterson C (1987). Calcium and the aging nervous system. Neurobiol Aging 8:329–343. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY (1985). A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem 260:3440–3450. [PubMed] [Google Scholar]
- Helmchen F, Imoto K, Sakmann B (1996). Ca2+ buffering and action potential-evoked Ca2+ signaling in dendrites of pyramidal neurons. Biophys J 70:1069–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemond P, Jaffe DB (2005). Caloric restriction prevents aging-associated changes in spike-mediated Ca2+ accumulation and the slow afterhyperpolarization in hippocampal CA1 pyramidal neurons. Neuroscience 135:413–420. [DOI] [PubMed] [Google Scholar]
- Humerickhouse RA, Bidasee KR, Gerzon K, Emmick JT, Kwon S, Sutko JL, Ruest L, Besch HR Jr (1994). High affinity C10-Oeq ester derivatives of ryanodine. Activator-selective agonists of the sarcoplasmic reticulum calcium release channel. J Biol Chem 269:30243–30253. [PubMed] [Google Scholar]
- Jacobs JM, Meyer T (1997). Control of action potential-induced Ca2+ signaling in the soma of hippocampal neurons by Ca2+ release from intracellular stores. J Neurosci 17:4129–4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe DB, Brown TH (1994). Confocal imaging of dendritic Ca2+ transients in hippocampal brain slices during simultaneous current- and voltage-clamp recording. Microsc Res Tech 29:279–289. [DOI] [PubMed] [Google Scholar]
- Jaffe DB, Johnston D, Lasser-Ross N, Lisman JE, Miyakawa H, Ross WN (1992). The spread of Na+ spikes determines the pattern of dendritic Ca2+ entry into hippocampal neurons. Nature 357:244–246. [DOI] [PubMed] [Google Scholar]
- Johnston D, Christie BR, Frick A, Gray R, Hoffman DA, Schexnayder LK, Watanabe S, Yuan LL (2003). Active dendrites, potassium channels and synaptic plasticity. Philos Trans R Soc Lond B Biol Sci 358:667–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadar T, Arbel I, Silbermann M, Levy A (1994). Morphological hippocampal changes during normal aging and their relation to cognitive deterioration. J Neural Transm Suppl 44:133–143. [DOI] [PubMed] [Google Scholar]
- Khachaturian ZS (1989). Calcium, membranes, aging, and Alzheimer's disease. Introduction and overview. Ann NY Acad Sci 568:1–4. [DOI] [PubMed] [Google Scholar]
- Knuttinen MG, Gamelli AE, Weiss C, Power JM, Disterhoft JF (2001). Age-related effects on eyeblink conditioning in the F344 × BN F1 hybrid rat. Neurobiol Aging 22:1–8. [DOI] [PubMed] [Google Scholar]
- Kovalchuk Y, Eilers J, Lisman J, Konnerth A (2000). NMDA receptor-mediated subthreshold Ca2+ signals in spines of hippocampal neurons. J Neurosci 20:1791–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Foster TC (2004). Enhanced long-term potentiation during aging is masked by processes involving intracellular calcium stores. J Neurophysiol 91:2437–2444. [DOI] [PubMed] [Google Scholar]
- Lancaster B, Nicoll RA (1987). Properties of two calcium-activated hyperpolarizations in rat hippocampal neurones. J Physiol (Lond) 389:187–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster B, Hu H, Ramakers GM, Storm JF (2001). Interaction between synaptic excitation and slow afterhyperpolarization current in rat hippocampal pyramidal cells. J Physiol (Lond) 536:809–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landfield PW (1987). “Increased calcium-current” hypothesis of brain aging. Neurobiol Aging 8:346–347. [DOI] [PubMed] [Google Scholar]
- Landfield PW (1988). Hippocampal neurobiological mechanisms of age-related memory dysfunction. Neurobiol Aging 9:571–579. [DOI] [PubMed] [Google Scholar]
- Landfield PW, Pitler TA (1984). Prolonged Ca2+-dependent afterhyperpolarizations in hippocampal neurons of aged rats. Science 226:1089–1092. [DOI] [PubMed] [Google Scholar]
- Leissring MA, Akbari Y, Fanger CM, Cahalan MD, Mattson MP, LaFerla FM (2000). Capacitative calcium entry deficits and elevated luminal calcium content in mutant presenilin-1 knockin mice. J Cell Biol 149:793–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC, Johnston D (1997). A synaptically controlled, associative signal for Hebbian plasticity in hippocampal neurons. Science 275:209–213. [DOI] [PubMed] [Google Scholar]
- Magnusson KR (1998). The aging of the NMDA receptor complex. Front Biosci 3:e70–e80. [DOI] [PubMed] [Google Scholar]
- Markowska AL (1999). Sex dimorphisms in the rate of age-related decline in spatial memory: relevance to alterations in the estrous cycle. J Neurosci 19:8122–8133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelis ML, Bigelow DJ, Schoneich C, Williams TD, Ramonda L, Yin D, Huhmer AF, Yao Y, Gao J, Squier TC (1996). Decreased plasma membrane calcium transport activity in aging brain. Life Sci 59:405–412. [DOI] [PubMed] [Google Scholar]
- Miller RA, Nadon NL (2000). Principles of animal use for gerontological research. J Gerontol A Biol Sci Med Sci 55:B117–B123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer JR Jr, Thompson LT, Black JP, Disterhoft JF (1992). Nimodipine increases excitability of rabbit CA1 pyramidal neurons in an age- and concentration-dependent manner. J Neurophysiol 68:2100–2109. [DOI] [PubMed] [Google Scholar]
- Moyer JR Jr, Thompson LT, Disterhoft JF (1996). Trace eyeblink conditioning increases CA1 excitability in a transient and learning-specific manner. J Neurosci 16:5536–5546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murchison D, Zawieja DC, Griffith WH (2004). Reduced mitochondrial buffering of voltage-gated calcium influx in aged rat basal forebrain neurons. Cell Calcium 36:61–75. [DOI] [PubMed] [Google Scholar]
- Norris CM, Halpain S, Foster TC (1998). Reversal of age-related alterations in synaptic plasticity by blockade of L-type Ca2+ channels. J Neurosci 18:3171–3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschen W, Frandsen A (2001). Endoplasmic reticulum dysfunction—a common denominator for cell injury in acute and degenerative diseases of the brain? J Neurochem 79:719–725. [DOI] [PubMed] [Google Scholar]
- Potier B, Poindessous-Jazat F, Dutar P, Billard JM (2000). NMDA receptor activation in the aged rat hippocampus. Exp Gerontol 35:1185–1199. [DOI] [PubMed] [Google Scholar]
- Power JM, Wu WW, Sametsky E, Oh MM, Disterhoft JF (2002). Age-related enhancement of the slow outward calcium-activated potassium current in hippocampal CA1 pyramidal neurons in vitro. J Neurosci 22:7234–7243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regehr WG, Tank DW (1992). Calcium concentration dynamics produced by synaptic activation of CA1 hippocampal pyramidal cells. J Neurosci 12:4202–4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig ES, Barnes CA (2003). Impact of aging on hippocampal function: plasticity, network dynamics, and cognition. Prog Neurobiol 69:143–179. [DOI] [PubMed] [Google Scholar]
- Sabatini BL, Oertner TG, Svoboda K (2002). The life cycle of Ca2+ ions in dendritic spines. Neuron 33:439–452. [DOI] [PubMed] [Google Scholar]
- Sah P, Faber ES (2002). Channels underlying neuronal calcium-activated potassium currents. Prog Neurobiol 66:345–353. [DOI] [PubMed] [Google Scholar]
- Stocker M (2004). Ca2+-activated K+ channels: molecular determinants and function of the SK family. Nat Rev Neurosci 5:758–770. [DOI] [PubMed] [Google Scholar]
- Storm JF (1990). Potassium currents in hippocampal pyramidal cells. Prog Brain Res 83:161–187. [DOI] [PubMed] [Google Scholar]
- Sukhareva M, Smith SV, Maric D, Barker JL (2002). Functional properties of ryanodine receptors in hippocampal neurons change during early differentiation in culture. J Neurophysiol 88:1077–1087. [DOI] [PubMed] [Google Scholar]
- Thibault O, Landfield PW (1996). Increase in single L-type calcium channels in hippocampal neurons during aging. Science 272:1017–1020. [DOI] [PubMed] [Google Scholar]
- Thibault O, Porter NM, Chen KC, Blalock EM, Kaminker PG, Clodfelter GV, Brewer LD, Landfield PW (1998). Calcium dysregulation in neuronal aging and Alzheimer's disease: history and new directions. Cell Calcium 24:417–433. [DOI] [PubMed] [Google Scholar]
- Thibault O, Hadley R, Landfield PW (2001). Elevated postsynaptic [Ca2+]i and L-type calcium channel activity in aged hippocampal neurons: relationship to impaired synaptic plasticity. J Neurosci 21:9744–9756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson LT, Moyer JR Jr, Disterhoft JF (1996). Transient changes in excitability of rabbit CA3 neurons with a time course appropriate to support memory consolidation. J Neurophysiol 76:1836–1849. [DOI] [PubMed] [Google Scholar]
- Toescu EC, Verkhratsky A (2004). Ca2+ and mitochondria as substrates for deficits in synaptic plasticity in normal brain ageing. J Cell Mol Med 8:181–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toescu EC, Verkhratsky A, Landfield PW (2004). Ca2+ regulation and gene expression in normal brain aging. Trends Neurosci 27:614–620. [DOI] [PubMed] [Google Scholar]
- Tombaugh GC, Rowe WB, Rose GM (2005). The slow afterhyperpolarization in hippocampal CA1 neurons covaries with spatial learning ability in aged Fisher 344 rats. J Neurosci 25:2609–2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veng LM, Mesches MH, Browning MD (2003). Age-related working memory impairment is correlated with increases in the L-type calcium channel protein alpha1D (Cav1.3) in area CA1 of the hippocampus and both are ameliorated by chronic nimodipine treatment. Brain Res Mol Brain Res 110:193–202. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A (2005). Physiology and pathophysiology of the calcium store in the endoplasmic reticulum of neurons. Physiol Rev 85:201–279. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Toescu EC (1998). Calcium and neuronal ageing. Trends Neurosci 21:2–7. [DOI] [PubMed] [Google Scholar]
- Williamson A, Alger BE (1990). Characterization of an early afterhyperpolarization after a brief train of action potentials in rat hippocampal neurons in vitro. J Neurophysiol 63:72–81. [DOI] [PubMed] [Google Scholar]
- Wu WW, Chan CS, Disterhoft JF (2004). Slow afterhyperpolarization governs the development of NMDA receptor-dependent afterdepolarization in CA1 pyramidal neurons during synaptic stimulation. J Neurophysiol 92:2346–2356. [DOI] [PubMed] [Google Scholar]
- Xiong J, Verkhratsky A, Toescu EC (2002). Changes in mitochondrial status associated with altered Ca2+ homeostasis in aged cerebellar granule neurons in brain slices. J Neurosci 22:10761–10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandi PP, Breitner JC, Anthony JC (2002). Is pharmacological prevention of Alzheimer's a realistic goal? Expert Opin Pharmacother 3:365–380. [DOI] [PubMed] [Google Scholar]