

Abstract
Somatosensory information, conveyed through the gracilis nucleus (GN), is regulated by descending corticofugal (CF) glutamatergic fibers. In addition, the GN receives cholinergic inputs with still unclear source and functional significance. Using both the in vitro slice and intracellular recording with sharp and patch electrodes and in vivo extracellular single-unit recordings, we analyzed the effects of activation of cholinergic receptors on synaptic, intrinsic, and functional properties of rat GN neurons. The cholinergic agonist carbamilcholine-chloride [carbachol (CCh); 1–10 μm] in vitro (1) induced presynaptic inhibition of EPSPs evoked by both dorsal column and CF stimulation, (2) increased postsynaptic excitability, and (3) amplified the spike output of GN neurons. The inhibition by atropine (1 μm) and pirenzepine (10 μm) of all presynaptic and postsynaptic effects of CCh suggests actions through muscarinic M1 receptors. The above effects were insensitive to nicotinic antagonists. We searched the anatomical origin of the cholinergic projection to the GN throughout the hindbrain and forebrain, and we found that the cholinergic fibers originated mainly in the pontine reticular nucleus (PRN). Electrical stimulation of the PRN amplified sensory responses in the GN in vivo, an effect prevented by topical application of atropine. Our results demonstrate for the first time that cholinergic agonists induce both presynaptic and postsynaptic effects on GN neurons and suggest an important regulatory action of inputs from cholinergic neuronal groups in the pontine reticular formation in the functional control of somatosensory information flow in the GN.
Keywords: gracile nuclei, cuneate nuclei, glutamatergic transmission, presynaptic inhibition, M1 mAChR, somatosensory information, in vitro, in vivo
Introduction
The gracilis nucleus (GN) of the dorsal column (DC) nuclei receives somatosensory information from the hindlimbs through afferents running in the dorsal columns (Nyberg and Blomqvist, 1982; Rustioni and Weinberg, 1989; DeBiasi et al., 1994). DC fibers are glutamatergic and contact both projecting neurons and inhibitory interneurons (Rustioni and Weinberg, 1989; Broman, 1994; DeBiasi et al., 1994). Stimulation of the peripheral receptive field or electrical stimulation of the DC evokes EPSPs and IPSPs in dorsal column nucleus neurons in vivo (Andersen et al., 1964; Canedo et al., 1998). In addition, dorsal column nuclei receive corticofugal (CF) descending glutamatergic fibers mainly from the sensorimotor cortex (Jabbur and Towe, 1961; Valverde, 1966). Intracellular recordings in an in vitro preparation have shown that the stimulation of CF evokes EPSPs with both NMDA and non-NMDA components, whereas the DC input elicits EPSPs via activation of non-NMDA receptors (Nuñez and Buño, 1999, 2001).
Dorsal column nuclei show acetylcholinesterase activity and acetylcholinesterase-positive fibers (Simon et al., 1981; Avendaño and Dykes, 1996), and choline acetyltransferase (ChAT)-positive terminals have been reported in the neuropil (Henderson and Sherriff, 1991). However, the source and functional significance of the cholinergic input to the dorsal column nuclei remains unclear. Cholinergic interaction with glutamatergic transmission (Fernández de Sevilla et al., 2002; Fernández de Sevilla and Buño, 2003) (for review, see Aigner, 1995) is of key importance in the control of normal and pathological behaviors because it can influence forms of synaptic plasticity that underlie learning and memory (Madison et al., 1991; Hasselmo, 1999), and lesions of the cholinergic system may produce cognitive deficits (Hasselmo, 1999). Cholinergic participation in somatosensory control is less understood, although acetylcholine (ACh) amplifies responses at thalamocortical connections. This effect is reduced by disruption of cortical cholinergic activity and blocked by application of atropine (Rasmusson and Dykes 1988; Baskerville et al., 1997; Maalouf et al., 1998) (for review, see Shulz et al., 2003), suggesting actions via muscarinic acetylcholine receptors (mAChRs). Cholinergic inputs could also play an important role in the modulation of somatosensory transmission through the GN, in which forms of short-term plasticity have been described (Nuñez and Buño, 2001).
The aim of the present study was to both characterize the action of the cholinergic agonist carbamylcholine-chloride [carbachol (CCh)] on the intrinsic and synaptic properties of rat GN neurons using an in vitro slice preparation and determine the physiological role of the cholinergic activity by analyzing in vivo the effects of electrical stimulation of descending cholinergic inputs on somatosensory signals in the GN. We report that CCh reduces synaptic responses and increases GN neuronal excitability and spike responses in vitro via activation of postsynaptic and presynaptic M1 mAChR, respectively. These actions modify the integrative properties of GN neurons facilitating transmission of sustained sensory inputs. In addition, electrical stimulation of cholinergic inputs amplifies somatosensory responses in vivo via activation of muscarinic receptors. The possible origin of cholinergic projections to the GN was mainly from the pontine reticular formation investigated using double labeling with the neuronal retrograde tracer cholera-toxin β subunit (CTb) and ChAT.
Materials and Methods
In vitro experiments
Young Wistar rats (12–14 d old) were anesthetized with ether and decapitated, and the brain was quickly removed and submerged in cold (∼4°C) artificial CSF (ACSF) maintained at pH 7.3 by bubbling with carbogen (95% O2–5% CO2). The ACSF composition was as follows (in mm): 124 NaCl, 2.69 KCl, 1.25 KH2PO4, 2 MgSO4, 26 NaHCO3, 2 CaCl2, and glucose. In the Mg2+-free solution, MgSO4 was equimolarly replaced with CaCl2.
Sagittal slices (400 μm), cut with a Vibratome (Pelco, St. Louis, MO), were incubated for at least 1 h in carbogen-bubbled ACSF at room temperature (20–22°C). Individual slices were transferred to an immersion recording chamber (2 ml) placed either on the stage of an inverted microscope (Diaphot-TMD; Nikon, Tokyo, Japan) or an upright microscope (BX50WI; Olympus Optical, Tokyo, Japan) equipped with infrared and differential interference contrast image devices and with a 40× water-immersion objective. Slices were superfused at a rate of 1 ml/min with gassed ACSF at room temperature, and total renewal of the bath solution was in ∼3 min.
Intracellular recordings with sharp electrodes.
The GN neurons were impaled under visual guidance with 90–120 MΩ potassium acetate (3 m) filled micropipettes (pulled with a P87 puller; Sutter Instruments, Novato, CA). The microelectrode tip was positioned according to the atlas of Paxinos and Watson (1986). Signals were amplified with an Axoclamp 2B (Molecular Devices, Palo Alto, CA), and the traditional bridge current-clamp method was used. Cells were rejected when, in control ACSF, the membrane potential (Vm) fell below −50 mV during the experiment.
Patch-clamp whole-cell recordings.
Patch-clamp recordings in the whole-cell configuration were performed using 3–6 MΩ fire-polished pipettes (the same pipette puller was used). The internal patch pipette solution contained the following (in mm): 100.0 potassium gluconate; 5.0 EGTA, 10.0 HEPES, 32.5 KCl, 1.0 MgCl2, 4.0 Na2-ATP, and 0.4 Na2-GTP at pH 7.3 (adjusted with KOH). Pipettes were connected to an Axoclamp 2B amplifier (Molecular Devices) or a PC-ONE amplifier (Dagan, Minneapolis, MN), and cells were recorded in the continuous single-electrode current or voltage-clamp mode. Under voltage clamp, cells were held at a Vm of −60 mV unless otherwise specified. The pipette was positioned in the GN, and neurons were viewed with the Olympus Optical microscope. Fast and slow capacitances were neutralized, and series resistance was compensated (∼80%). Patch recordings were rejected when the access resistance (7–15 MΩ) increased >20% during the experiment. Leak and the linear capacitance current components were subtracted with the Clampex program (Molecular Devices) method. Current pulses, voltage commands, acquisition, and display of the recorded signals were generated with the Clampex program in both sharp and patch electrode recordings. No significant differences in the effects of CCh were found between sharp and parch electrode current-clamp recordings, indicating that the intracellular dialysis that occurs with patch electrodes was not affecting the intracellular signal transduction mechanisms involved. Once this was established, patch electrodes were used because they enabled voltage-clamp recordings and prolonged current-clamp recordings with favorable signal-to-noise ratios.
Stimulation.
A schematic diagram of the localization of the dorsal column nuclei within the slice and of the position of the stimulating and recording electrodes is shown in Figure 1A (left). Electrical stimulation of the DC and CF was through bipolar nichrome electrodes (60 μm diameter) insulated except at the tips and connected to a Grass Instruments (Quincy, MA) stimulator triggered by the Clampex program. Single pairs or barrages of electrical stimuli (0.3 ms, 0.2–1.0 V) applied at the DC or CF (see Fig. 1A, left) induced DC-EPSPs and CF-EPSPs or DC-EPSCs and CF-EPSCs in GN neurons, respectively.
Figure 1.
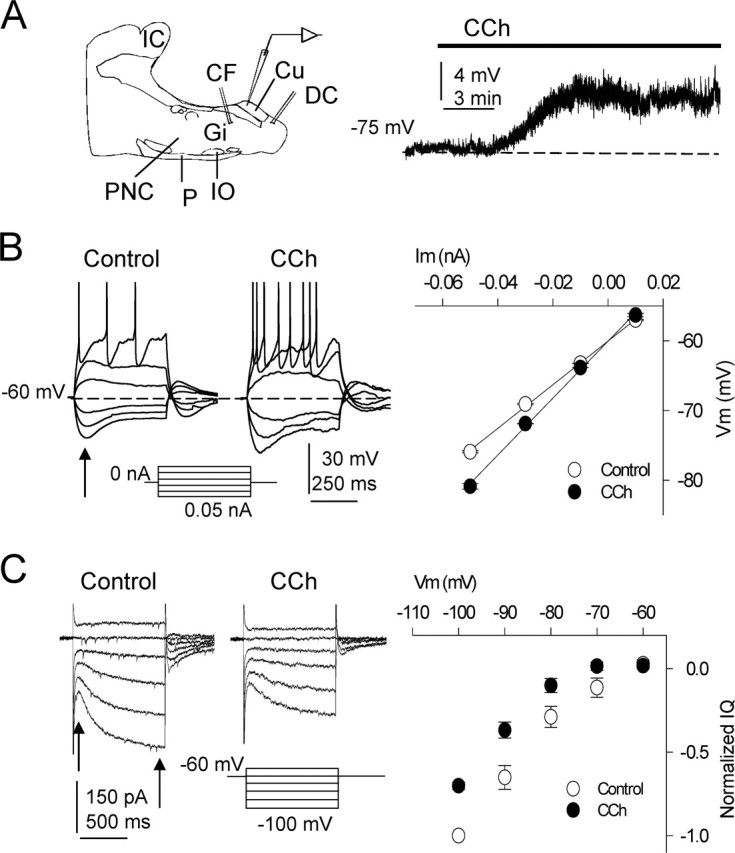
Modifications of intrinsic properties induced by CCh. A, Left, Diagram of recording and stimulating electrode locations in the slice. CF, Corticofugal fibers; Cu, cuneate nucleus; CF, cuneate fasciculus; DC, dorsal column fibers; Gi, gigantocellular reticular nucleus; IC, inferior colliculus; IO, inferior olive; P, pyramidal tract; PnC, caudal pontine reticular nucleus. A, Right, Superfusion with CCh (10 μm) depolarized DC neurons. B, Left, Current-clamp responses evoked by hyperpolarizing and depolarizing current pulses in control ACSF and during CCh in a representative experiment. The cell was returned to the resting potential by injecting a sustained current during the CCh-induced depolarization. Note the larger hyperpolarizing responses, the increased excitability, and the lower current spike threshold with CCh. B, Right, Average I–V relationships (n = 9) calculated with hyperpolarizing and depolarizing current pulses in control ACSF (open circles) and with 10 μm CCh (filled circles). The values were measured at the peak hyperpolarization (arrow in B, left) and were well fitted to a linear regression showing the steeper slope with CCh, indicating an increased membrane resistance (n = 9; p < 0.01). C, Left, Representative voltage-clamp recordings evoked by hyperpolarizing and depolarizing voltage commands in control ACSF and during 10 μm CCh. Hyperpolarization activated a slowly increasing non-inactivating inward current (i.e., Q-current) that was markedly inhibited by CCh. C, Right, Average I–V relationship in control ACSF (open circles) and with CCh (filled circles) showing the CCh-induced inhibition of the Q-current (n = 9; p < 0.01). Values were the difference between currents at onset and end of voltage commands (arrows) and normalized to the largest control current (n = 20; p < 0.01). The resting Vm is indicated in each current-clamp record, as in the following figures.
Pharmacology and data analysis.
CCh (1–10 μm), atropine (1 μm), a concentration that does not interfere with nicotinic receptors (Alkondon et al., 2000), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) (20 μm), tetrodotoxin (TTX) (1 μm), mecamylamine (MMA) (50 μm), and methylycaconitine (MLA) (0.1 μm) were added to the ACSF and superfused. Recordings were performed under 50 μm picrotoxin to block GABAA inhibitory postsynaptic responses. All chemicals were from Sigma-Aldrich Quimica (Madrid, Spain).
Data were low-pass filtered at 3 kHz and sampled at rates of 10 kHz through a Digidata 1200B interface (Molecular Devices) connected to a Pentium-based computer. Data was stored on the hard disk of the computer with the Clampex program that also generated stimulus timing signals, transmembrane current pulses, and voltage commands. The mean input resistance (Rin) was either estimated from voltage responses evoked by a 0.2 nA hyperpolarizing pulse applied at the resting Vm or from linear fits of current–voltage (I–V) relationships obtained with hyperpolarizing pulses. Analysis of TTX-insensitive miniature EPSCs (mEPSCs) was performed using MiniAnalysis (version 5.2.1; Synaptosoft, Decatur, GA) with minimal acceptable amplitude of mEPSCs at 6 pA. Values are expressed as the mean ± SE, and data were compared using Student’s pair or unpaired t test as appropriate.
In vivo experiments
Data were obtained from 19 urethane-anesthetized (1.6 g/kg, i.p.) young adult Wistar rats of either sex, weighing 180–250 g. Animals were placed in a stereotaxic device. The body temperature was maintained at 37°C, and the end-tidal CO2 concentration was controlled. Experiments were performed in accordance with the European Communities Council Directive (86/609/EEC).
Tungsten microelectrodes (5 MΩ) were used to obtain single-unit recordings in the GN [anterior (A), −13.6 to −14.6; lateral (L), 0.2–1.0 from the bregma; height (H), 0.0–0.5 mm from the surface of the nucleus (according to the atlas of Paxinos and Watson, 1986)]. The recording electrode was introduced at a 60° angle to the surface of the nucleus after opening the cisterna magna. The dura mater was removed, and the nucleus surface was covered with mineral oil to prevent drying. Single-unit recordings were filtered (0.3–3 kHz) and amplified via an AC P15 preamplifier (Grass Instruments). Spike amplitude and shape were continuously monitored on-line in an analog oscilloscope. Continuously recorded data were sampled at 8 kHz and fed into a Macintosh computer (Apple Computers, Cupertino, CA) for off-line analysis with Spike 2 software (Cambridge Electronic Design, Cambridge, UK). Recording were performed in control ACSF and during of topic application of atropine sulfate (a muscarinic receptor antagonist; 0.5 mm; 1 μl) on the gracilis nucleus. They were applied with a Hamilton microsyringe placed on the gracilis nucleus, tight to the recording place. Chemicals were purchased from Sigma-Aldrich Quimica.
Tactile stimulation was performed by an electronically gated solenoid with a probe of 1 mm in diameter that induced <0.5 mm skin deflection. The control tactile stimulation consisted in pulses lasting 20 ms and delivered at 0.5 Hz, which were directed to a small area of the hindlimb (receptive field).
Electrical stimulation at the caudal pontine reticular nuclei (PRN) (A, −10 mm; L, 1.0 mm; H, 9 mm) was performed by means of bipolar electrodes, using rectangular pulses (0.3 ms duration, 10–100 μA) at 0.5 Hz or with trains of pulses at 50 Hz during 500 ms.
Summed peristimulus time histograms (PSTHs) (2 ms bins) were calculated using Spike 2 software. The mean tactile response was measured from the PSTH as the number of spikes evoked at 10–50 ms after stimulus onset divided by the number of stimuli. To measure the time course of tactile response, the mean response to five tactile stimuli were calculated before and after reticular formation stimulation. All data are shown as mean ± SE. Data were compared using Student’s paired test (*p < 0.05; **p < 0.01; ***p < 0.001).
Cholinergic cell groups projecting to the GN: double-labeling experiments.
Under general anesthesia (a mixture of 50% ketamine, 40% atropine, and 10% valium, i.p.), rats were injected with ∼200 nl of CTb (List Biologic, Campbell, CA) in the GN with a 500 nl blunt-point Hamilton syringe. Under microscopic control, the cisterna magna was opened and the dura reflected to introduce the syringe, attached to a micromanipulator, in the gracile nucleus. To control the volume of product injected, CTb was delivered in four successive injections (each ∼50 nl) separated several minutes to minimize the washout attributable to the flow of CSF. Animals were housed and cared for according to the European Community Council Directive (86/609/EEC) and allowed to survive 7 d. Animals were then perfused transcardially with a mixture of 3% paraformaldehyde, 15% picric acid, and 0.1% glutaraldehyde in 0.1 m buffer phosphate, followed by increasing concentrations of phosphate-buffered sucrose. Brains were postfixed overnight in the same fixative and cryoprotected by storing in 30% sucrose 5 d to be frozen sectioned at 40 μm. Sections were collected in three consecutive series to be processed for CTb revealed with Nissl staining and double-labeling procedure. Series processed for CTb were revealed with IntenSE BL silver enhancement kit (Amersham Biosciences, Piscataway, NJ) after washing in citrate-acetate buffer, pH 5.5; sections were fixed in 5% sodium tiosulphate and stored in 0.1 m phosphate buffer before mounting. Series devoted to double labeling were first processed for CTb and then incubated with 1:100 goat anti-ChAT antiserum (AB144P; Chemicon, Temecula, CA) in a solution containing 20% normal rabbit serum, 5% bovine serum albumin, and 0.5% Triton X-100 in PBS for 36 h; then, sections were incubated in 1:40 rabbit anti-goat IgG and 1:600 goat peroxidase-antiperoxidase in the previous solution for 1.5 h each and revealed with 0.05% 3–3′ DAB and 0.003% H2O2. Nissl series was used for delimiting structures. Microscopic analysis of sections was under bright-field and/or dark-field illumination at 40× (Axioskop; Zeiss, Jena, Germany). Only brain sections from animals in which the injections were restricted to the GN were considered (n = 7).
Results
In vitro experiments
The intracellular current-clamp recordings with sharp electrodes included in this study were from 73 GN neurons that showed a resting Vm of −72.7 ± 1.0 mV, an Rin of 48.6 ± 4.3 MΩ, and fired overshooting action potentials with mean peak amplitude of 75.5 ± 1.2 mV. In addition, whole-cell patch-clamp recordings in either current-clamp or voltage-clamp modes of 78 neurons showing Vm of −61 ± 2.0 mV and Rin of 291.1 ± 18.4 MΩ were also included.
CCh modified excitability and intrinsic membrane properties
In current-clamp sharp electrode recordings, CCh depolarized most GN neurons (38 of 52, or 73%) in a dose-dependent manner, as occurs in other systems (Benardo and Prince, 1982; McCormick, 1992; Nuñez et al., 1997; Yajeya et al., 1999: Borde et al., 2000). At CCh concentrations between 1 and 10 μm, the Vm depolarized between 2 and 10 mV, and the average depolarization induced by 10 μm CCh was 6.2 ± 0.7 mV (Fig. 1A, right). In those cells depolarized by CCh (n = 38), the Rin was also increased from 48.6 ± 4.3 to 56.8 ± 5.1 MΩ (16.9% increase; n = 38; p = 0.025) when using sharp electrodes and from 312.8 ± 10.3 to 408.4 ± 12.1 MΩ (a 30.5% increase; n = 20; p = 0.008) when using patch electrodes. Therefore, the I–V relationships, calculated with data obtained in response to hyperpolarizing pulses under current clamp with patch electrodes, displayed steeper slopes under CCh than in control conditions (n = 20 same cells) (Fig. 1B). The I–V relationships were measured from peak voltage values under current clamp at the onset of the hyperpolarization before the initiation of depolarizing sags (Fig. 1B). The linear slopes of the I–V relationships measured in both the control and CCh-treated cells are consistent with CCh acting on voltage-insensitive conductances. Under current clamp, most cells (20 of 31, or 64%) showed a prominent depolarizing sag during hyperpolarizing pulses (i.e., also termed delayed inward rectification) (Fig. 1B, left) that is caused by the activation of the Na+- and K+-mediated Q-current that tends to drive the Vm to more depolarized values (Nuñez and Buño, 1999). Similar I–V relationships were also calculated with values obtained at the end of the hyperpolarizing pulses. These I–V relationships departed from linearity and suggested the activation of a depolarizing conductance during the hyperpolarization (data not shown).
Under voltage clamp, hyperpolarizing pulses from −60 to −100 mV activated the Q-current, as revealed by the gradually activating inward current that was markedly inhibited by CCh (from 100.2 ± 16.6 to 75.3 ± 15.5 pA under 10 μm CCh; a 25% reduction; n = 9; p = 0.003) (Fig. 1C). Superfusion with TTX (1 μm; n = 4) failed to modify the above described effects of CCh (data not shown). Some neurons (n = 10) that were not depolarized by CCh increased the input resistance 5–10%, whereas a few neurons (n = 4) were not affected by CCh; these four cells were not analyzed further. Although CCh always reduced the Q-current under voltage clamp, the depolarizing sag mediated by the Q-current activation could increase in amplitude with the CCh challenge (Fig. 1B), an effect that was probably caused by the marked membrane conductance drop that paralleled the effects of CCh.
The responses to depolarizing current pulses revealed that CCh reduced the current threshold for spike generation from 0.04 ± 0.03 to 0.02 ± 0.01 nA (a 50% reduction; p = 0.02; n = 15) and increased the number of spikes (from 3.1 ± 0.3 to 6.5 ± 0.6; a 109.6% increase; p < 0.001; n = 15) evoked by identical 0.05 nA depolarizing current pulses (Fig. 1B, left). All neurons showed an increased Rin under CCh. Therefore, the significant increase in spike output is caused by the rise in the input resistance. This effect occurred without evident modifications of the delay to the first action potential evoked by depolarizing pulses, which is mediated by the transient K+-mediated A-current (Nuñez and Buño, 1999). The effects of CCh on the membrane resistance and excitability were similar in neurons that showed or lacked the Q-current.
CCh inhibited both DC and CF inputs
We analyzed under current and voltage clamp the effects of CCh on DC-EPSPs and -EPSCs that display a non-NMDA but lack the NMDA component (Nuñez and Buño, 2001). To minimize changes caused by modifications in the driving force, measurements of EPSP amplitudes were at the initial resting Vm, which was set by injecting a hyperpolarizing current if the cell was depolarized by CCh. As occurs in other systems (Hounsgaard, 1978; Valentino and Dingledine, 1981; Fernández de Sevilla et al., 2002), bath-applied CCh (10 μm) decreased the DC-EPSP peak amplitude (27 of 38 neurons, or 71%) from 4.1 ± 0.45 to 2.2 ± 0.37 mV (a 46.3% decrease; p = 0.002) (Fig. 2A, left traces). In voltage-clamp conditions, the DC-EPSC was also reduced by 10 μm CCh (38.5% reduction; p < 0.01; n = 10) (Fig. 2A, middle traces). Synaptic responses were not modified by the CCh challenge in the rest of the neurons (n = 11).
Figure 2.
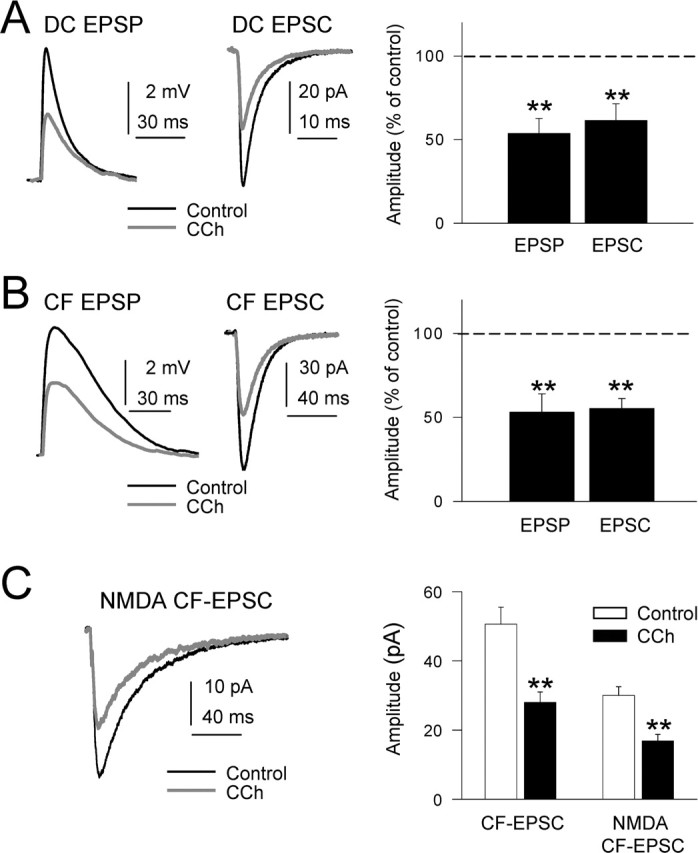
Inhibition of DC-EPSPs and -EPSCs by CCh. A, Left, Superimposed DC-EPSPs recorded in current-clamp conditions in control ACSF (black trace) and with 10 μm CCh (gray trace). During CCh, the Vm was adjusted at the resting value (−65 mV) by continuous current injection. A, Middle, Same as A, left, but EPSCs recorded in voltage-clamp conditions at −65 mV holding potential. A, Right, Summary data showing the inhibition of DC-EPSP and -EPSC by CCh (n = 27; p < 0.01 in both cases). B, Left and right, Same as A, left and right, but for CF-EPSPs and -EPSCs. C, Left, Superimposed isolated NMDA EPSCs (Mg2+-free ACSF plus 20 μm CNQX; black trace) and when CCh was added (gray trace). C, Right, Summary data showing the inhibition of CF-EPSP and isolated NMDA CF-EPSC by CCh (n = 7; p < 0.01).
We had shown previously that CF-EPSC displayed both non-NMDA and NMDA components (Nuñez and Buño, 2001). We analyzed the effects of CCh on CF-EPSCs under voltage-clamp in control ACSF and in Mg2+-free ACSF with 20 μm CNQX. At a Vm of −60 mV in control ACSF, only the non-NMDA component of CF-EPSC was recorded because of the voltage-dependent block by extracellular Mg2+ of NMDA receptor channels. The CCh (10 μm) challenge reduced the mean non-NMDA CF-EPSC peak amplitude from 50.6 ± 4.9 pA in the controls to 28.0 ± 3.0 pA under CCh (a 44.4% reduction; n = 10; p = 0.008) as well as the CF-EPSP peak amplitude (Fig. 2B). In Mg2+-free ACSF plus CNQX, the block by extracellular Mg+2 of NMDA receptor channels is absent and the non-NMDA EPSC component is blocked; thus, an isolated NMDA CF-EPSC component is recorded. In these conditions, the mean amplitude of the isolated NMDA CF-EPSC was reduced from 30.0 ± 2.5 to 16.8 ± 1.9 pA (a 44.1% reduction; n = 7; p = 0.003) (Fig. 2C). These results together suggest that the effects could be mediated by a presynaptic action of CCh because it inhibits to a similar degree the DC-EPSC and both the NMDA and non-NMDA components of CF-EPSPs. However, these results do not completely confirm presynaptically mediated actions.
Finally, we analyzed the dose–response relationship for the cholinergic inhibition of the EPSPs and the increase in the Rin. EPSPs peak amplitude and Rin at different concentrations of CCh (0.5, 1, 5, 10, 20, and 50 μm) were normalized to the control values in each cell, and the normalized values of different cells were averaged and plotted as a function of drug concentration. The plot was well fitted (r2 = 0.99 in all cases) to the formalism y = y0+ a/1 + (x/c)b, derived from the Hill equation, where c is the IC50. The estimated IC50 values were 1.5, 1.2, and 3.3 μm for the CF-EPSP, DC-EPSP, and Rin, respectively (data not shown).
The EPSP amplitude reduction may be underestimated because of the important membrane conductance drop induced by CCh. Therefore, we plotted the amplitude of the EPSPs as a function of Rin, which revealed a negative correlation (r2 = 0.95), suggesting a possible underestimation of the action of CCh on EPSP amplitude.
CCh inhibits DC and CF inputs presynaptically
Paired-pulse modulation is a form of short-term presynaptic plasticity characterized by a change in the peak amplitude of the second EPSP (R2) when it is elicited shortly after (<100 ms) a preceding (R1) stimulation (Kamiya and Zucker, 1994; Fernández de Sevilla and Buño, 2002; Martin and Buño, 2003) (for review, see Thomson, 2000). Changes in short-term plasticity during paired-pulse stimulation have been recognized as an indication of a presynaptically mediated effect. Therefore, we analyzed the actions of CCh on paired-pulse responses (50–80 ms interval) of both DC- and CF-EPSPs and EPSCs under current and voltage clamp, respectively.
Paired-pulse plasticity of DC-EPSPs was typified by a reduced R2 EPSP relative to R1 or paired-pulse depression (PPD). Superfusion with CCh (10 μm) reduced the PPD and, on average, converted it to paired-pulse facilitation (PPF). Indeed, the mean R1 peak amplitude was 4.5 ± 0.5 mV in control conditions and was reduced by CCh to 3.2 ± 0.1 mV. However, CCh did not modify the amplitude of R2, which was 3.7 ± 0.2 mV in control solution and 3.64 ± 0.3 mV under CCh (data not shown). Therefore, CCh increased the paired-pulse ratio (defined as R2/R1) from 0.83 ± 0.05 in control conditions to 1.12 ± 0.04 under CCh (a 35% increase; p = 0.001; n = 10), a change from PPD to PPF, suggesting a presynaptically mediated effect.
In contrast, the short-term plasticity of CF-EPSPs was characterized by a PPF. The mean control CF-EPSP peak amplitude was 5.5 ± 0.5 mV and CCh inhibited R1 to 3.6 ± 0.4 mV, whereas the mean peak amplitude of R2 remained unaltered (6.0 ± 0.5 mV in control and 6.1 ± 0.4 mV under CCh). Therefore, CCh reduced the R1 more than R2, thus increasing the PPF and the paired-pulse ratio from 1.09 ± 0.07 in control conditions to 1.69 ± 0.1 under CCh (a 55% increase; p = 0.001; n = 8; data not shown). Although in control conditions the R1 and R2 differences were not statistically significant under current clamp, they were significant during the CCh challenge and in both control and under CCh in voltage-clamp conditions. These differences could be caused by the conductance increase and the depolarization during R1 that could shunt and reduce the driving force of R2.
Similar results were obtained under voltage clamp in which the PPD that characterized DC-EPSC in control solution, with a mean paired-pulse ratio of 0.81 ± 0.07, was converted to a PPF by CCh, with a paired-pulse ratio of 1.3 ± 0.09 (a 60% increase; p = 0.005; n = 10) (Fig. 3A,B). In addition, the PPF displayed by CF-EPSC in control conditions, with a paired-pulse ratio of 1.15 ± 0.08, was increased by CCh to 1.54 ± 0.06 (a 34% increase; p = 0.006; n = 10) (Fig. 3A,B).
Figure 3.

CCh inhibits CF- and DC-EPSCs via a presynaptic mechanism. A, Top, DC-EPSCs evoked by paired-pulse stimulation (80 ms interval) in control ACSF (black trace), during CCh (gray trace), and superimposed and scaled to the first EPSC (R1). Note that, in control ACSF, R1 was larger than the second response (R2), indicating PPD. CCh reduced R1 more than R2, changing the PPD to PPF. A, Bottom, Same as A, top, but for CF-EPSC. Note that, in control ACSF, R1 was smaller than the second response R2, indicating PPF, and that CCh increased PPF. B, Summary data showing the paired-pulse ratio (R2/R1) in control ACSF (open bars) and with CCh (filled bars) for both DC- and CF-EPSCs (n = 10; p < 0.01 in both cases). C, Top, Representative recordings of mEPSCs in control (with superfusion of 0.5 μm TTX) and when 10 μm CCh was added. Note the decreased mEPSCs frequency induced by CCh. C, Bottom, Summary data (n = 8) showing mean mEPSCs frequency and amplitude in control ACSF plus TTX (open bars) and when CCh was added (filled bars). D, Top, Cumulative probability plots of mean inter-mEPSC interval in control ACSF plus TTX (filled circles) and when CCh was added (open circles). D, Bottom, Same as D, top, but for mean mEPSCs amplitude. Note that CCh increased the mean inter-mEPSC interval (p < 0.05; Kolmogorov–Smirnov test) without changing the mean mEPSCs amplitude (same cells as in C).
A modification of the mEPSCs frequency is another universally recognized sign of presynaptically mediated effects. Therefore, we measured the changes in mEPSC frequency in the presence of TTX (1 μm). The CCh (10 μm) challenge decreased the mEPSC frequency from 0.91 ± 0.11 Hz in control to 0.44 ± 0.02 Hz under CCh (a 51.6% decrease; n = 8; p = 0.011; Kolmogorov–Smirnov test), a modification that was not paralleled by amplitude changes (18.1 ± 3.3 pA in control and 17.4 ± 2.3 pA under CCh; p < 0.05) (Fig. 3C,D).
The above results together are consistent with CCh acting presynaptically via activation of cholinergic receptors to inhibit transmitter release by reducing release probability at synaptic terminals of both cortical and dorsal column fibers.
M1 mAChRs mediate the presynaptic and postsynaptic effects of CCh
Preincubation with the wide-spectrum mAChR antagonist atropine (1 μm) prevented all of the effects of 10 μm CCh. The increase in Rin and the excitability, the slow depolarization, and the presynaptic inhibition of DC and CF inputs were all blocked by atropine, suggesting that CCh acted both presynaptically and postsynaptically via activation of muscarinic receptors (Fig. 4A,B,D).
Figure 4.

Both presynaptic and postsynaptic effects of CCh are exclusively via activation of M1 muscarinic receptors. A, Top, Representative current-clamp responses evoked by depolarizing and hyperpolarizing current pulses in control ACSF and with 10 μm CCh obtained 10 min after the onset of superfusion with atropine (1 μm). Note the absence of the cholinergic effects. A, Bottom, Same as A, top, but under MMA (50 μm) and MLA (0.1 μm); note the unchanged CCh effects with nicotinic antagonists. B, Summary data showing the mean input resistance (in megaohms) in control (open bars) and during CCh (filled bars) with control ACSF (n = 10; p < 0.01), atropine (n = 10; p > 0.05), pirenzepine (n = 6; p > 0.05), methoctramine (n = 6; p < 0.01), and MMA plus MLA (n = 9; p < 0.01). C, Top, Superimposed DC-EPSPs in control (black trace) and during CCh (gray trace) when superfusing pirenzepine, methoctramine, and MMA plus MLA. C, Bottom, Same as C, top, but for CF-EPSC. Note that only pirenzepine blocks the presynaptic inhibition of EPSCs. D, Summary data showing mean DC-EPSP and CF-EPSP amplitudes (open and filled bars, respectively) during 10 μm CCh (represented as a percentage of control amplitudes) in control ACSF (n = 10; p < 0.01), atropine (n = 10; p > 0.05), pirenzepine (n = 6; p > 0.05), methoctramine (n = 6; p < 0.01), and MMA plus MLA (n = 9; p < 0.01).
It has been suggested that atropine can also inhibit nicotinic receptors when applied at high concentrations (Alkondon et al., 2000). To test whether nicotinic receptors were involved, we recorded GN neurons during superfusion with MMA (50 μm), the α3β4 nicotinic acetylcholine receptor (nAChR) antagonist, and the α7 nAChR antagonist MLA (0.1 μm). In those conditions, CCh (10 μm) induced a slow depolarization that reached values of 5.6 ± 1.2 mV and increased both the Rin in voltage-clamp recordings (from 290.1 ± 18.8 MΩ in control to 381.1 ± 21.9 MΩ with CCh; n = 9; p < 0.001) and the excitability, indicating that the postsynaptic effects of CCh on GN neurons persisted after blocking nAChRs (Fig. 4A,B).
Under MMA and MLA, CCh also reduced DC- and CF-EPSP amplitudes (4.3 ± 0.5 mV in control to 2.1 ± 0.2 mV under CCh; n = 9; p = 0.015; and 3.1 ± 0.3 mV in control to 1.17 ± 0.1 mV under CCh; n = 9; p = 0.012, respectively) (Fig. 4C,D), and the paired-pulse ratio of both DC-EPSCs and CF-EPSCs were significantly increased from 0.8 ± 0.06 and 1.01 ± 0.04 to 1.21 ± 0.08 and 1.38 ± 0.07 by CCh, respectively (n = 9; p = 0.015 and p = 0.001, respectively; data not shown). These results indicate that nAChRs are not engaged in the presynaptic actions of CCh.
It has been reported that both M1 and M2 mAChRs are found in the pons medulla oblongata (Ge et al., 1995). Therefore, to determine the type of mAChR that mediated the cholinergic effects, we superfused CCh (10 μm) in the presence of the M1 mAChR antagonist pirenzepine and the M2 mAChR antagonist methoctramine, respectively. The effects of CCh on the Vm, excitability, and spike threshold were prevented when pirenzepine (10 μm) was added to the bath (Fig. 4B). In addition, the DC- and CF-EPSP amplitude reductions induced by CCh were prevented by pirenzepine (3.3 ± 1.0 to 3.9 ± 0.94 and 3.4 ± 1.17 to 3.2 ± 1.56 mV, respectively; p > 0.05 in both cases) (Fig. 4C,D). In contrast, the M2 mAChR antagonist methoctramine (10 μm) had no effect on the postsynaptic and presynaptic actions of CCh (Fig. 4C,D). The above results indicate that both presynaptic and postsynaptic actions of CCh are mediated by M1 mAChRs.
Effects of CCh on the spike output in response to synaptic input
To investigate the possible functional role of the cholinergic regulation of the integrative properties of GN neurons, we analyzed the effect of CCh on the GN neuron spike output in response to suprathreshold synaptic stimulation. EPSPs were elicited with repeated stimulus barrages (10 Hz, 1.9 s at 0.1/s) applied at the DC and CF fibers. In control conditions, DC stimulation evoked a transient response with an initial pair of spikes, followed by subthreshold EPSPs that decreased gradually in amplitude during the stimulation, a “phasic” spike response consistent with EPSP depression and little summation (Fig. 5A, top left). The 10 μm CCh challenge depolarized the GN neurons (6.5 ± 2.1 mV; n = 6) and increased both EPSP summation and the number of spikes evoked by the barrage, resulting in a sustained, more “tonic,” spike response (Fig. 5A, top right). On average, 3.1 ± 1.1 spikes per stimulus were evoked in control conditions, whereas CCh increased the number of spikes to 9.2 ± 0.4 (a 296.7% increase; p = 0.002; n = 6).
Figure 5.

Effects of CCh on spike responses evoked by DC and CF barrages. A, Representative responses, obtained from a single GN neuron, evoked by suprathreshold stimulus barrages (10 Hz, 1.9 s duration at a rate of 0.1/s) applied at the DC (DC stimulation) and CF (CF stimulation) fibers in controls solution (left) and during superfusion with 10 μm CCh (right). Note the depolarization, the increased summation, and the more sustained response evoked under CCh in both DC- and CF-evoked responses. B, Summary data (n = 6) showing the average number of spikes per stimulus in control ACSF and when 10 μm CCh was added both for DC and CF stimulations. Note the amplified responses evoked by both stimulations under CCh. C, Representative examples of subthreshold stimulus barrages applied at the DC and CF fibers (top and bottom, respectively) in control and CCh. D, Summary data (n = 5) showing the average summation index (Rlast/R1) in control and under CCh for DC- and CF-EPSPs.
The control response evoked by CF stimulation was characterized by initial subthreshold EPSPs that facilitated and summated to evoke an initial burst (two spikes), followed by three spikes separated by silent periods with EPSPs that failed to evoke spikes (Fig. 5B, bottom left). The CCh challenge reduced the amplitude of the initial EPSPs and increased the facilitation and summation of subsequent EPSPs, resulting in a more sustained response with additional spikes and very few failures of EPSPs (Fig. 5B, bottom right). The number of spikes per stimulus was 3.5 ± 0.7 in control condition and increased to 9.6 ± 0.5 with CCh (a 274.2% increase; p = 0.003; n = 6).
We also analyzed the effects of CCh on DC and CF subthreshold synaptic signals. We applied the same synaptic stimulation at lower intensities at DC and CF fibers, both in control conditions and during superfusion of 10 μm CCh (Fig. 5C). The profiles of the control DC and CF responses were different because the DC input showed marked depression with little summation (Fig. 5C, top, Control), whereas the CF input evoked an initial facilitation and summation followed by depression (Fig. 5C, bottom, Control). The CCh challenge reduced the peak amplitudes of both R1 DC-EPSP (45.6 ± 5.8%; p = 0.005; n = 6) and CF-EPSP (55.2 ± 4.7%; p = 0.004; n = 6). However, the profiles of the effects of CCh on the subsequent responses were markedly different for the DC and CF stimulations (Fig. 5C, CCh, top and bottom). The initial depression of DC-EPSPs in control conditions was changed to facilitation by CCh, whereas the final depression was not much affected (102.5 ± 10.1%; p = 0.08; n = 6). The initial facilitation of CF-EPSPs was increased by CCh, and the final depression was abolished. In addition, CCh increased the temporal summation of DC-EPSP (208.2 ± 15.2%; p = 0.0008; n = 6).
These results show that CCh is able to modify the time course of the responses evoked by stimulation barrages as a result of reducing the first EPSPs of the response but not the last one. Therefore, the temporal summation of the EPSPs estimated by a summation ratio (defined as Rlast/R1) was 0.5 ± 0.1 in control and 0.7 ± 0.3 during CCh (DC input, n = 5, p > 0.5, 0.7 ± 0.2 in control and 3.0 ± 0.4 during CCh; for CF input, n = 5, p < 0.001) (Fig. 5D). These results could be explained by the increase in the membrane time constant (τ) for the DC input (τ = Rm * Cm) and by the increase in both τ and NMDA receptor component in the CF input attributable to the depolarization induced by CCh.
The above results together indicate that activation of M1 mAChRs modifies both the transfer of somatosensory information and its descending control by favoring the conduction of sustained versus transient signals in the GN.
Anatomical identification of cells of origin of the cholinergic input
The anatomical material revealed that the CTb deposits were restricted to the GN, specifically in the GN in all animals selected (n = 7) (Fig. 6A). The distribution of neurons positive for ChAT immunoreactivity was very similar to that described by several authors using different techniques (Jacobowitz and Palkovits, 1974; Palkovits and Jacobowitz, 1974; Armstrong et al., 1983; Mesulam et al., 1983; Jones, 1990; Oh et al., 1992; Schäfer et al., 1998). The ChAT-labeled cells were in the basal prosencephalic structures, the brainstem motor nuclei, the pedunculopontine tegmental and laterodorsal nuclei, and scattered in the pontine and bulbar reticular formation. The CTb-positive neurons were not very numerous but were widely distributed and located in ipsilateral and/or contralateral structures along both forebrain and brainstem, in layer V of the contralateral cerebral cortex, ipsilateral zona incerta, contralateral red nucleus, ipsilateral and contralateral mesencephalic reticular formation, oral and caudal pontine reticular nuclei, magnocellular bulbar reticular formation, and vestibular nuclei (Fig. 6B,C). However, CTb-positive neurons could not be detected in well defined cholinergic structures such as the pedunculopontine or the laterodorsal tegmental nuclei or basal forebrain. Double-labeled neurons for both CTb and ChAT immunoreactivity were located both ipsilaterally and contralaterally in caudal structures of the brainstem mainly in the caudal pontine reticular formation (Fig. 6B,C).
Figure 6.
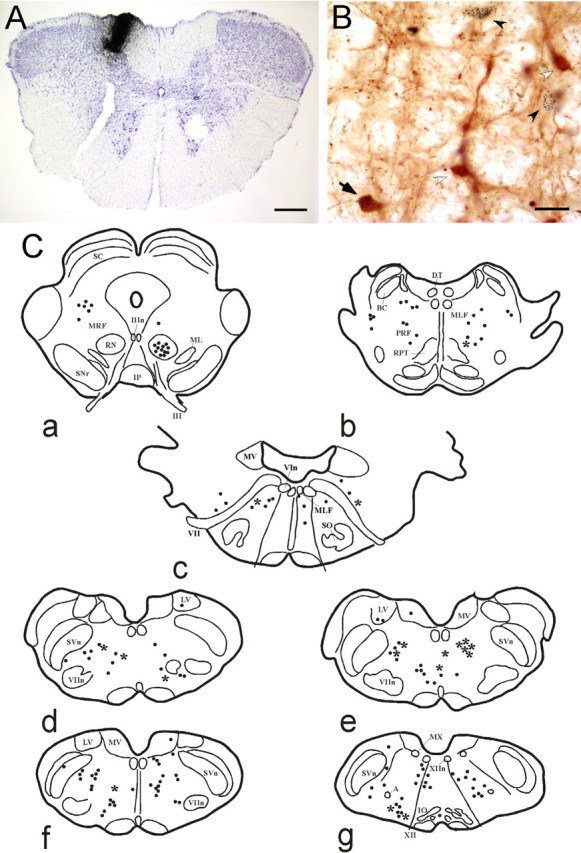
Origin of cholinergic afferents to the GN. A, Microphotograph of a medullary coronal section showing a CTb injection (i.e., CTb staining) in the gracile nucleus. Scale bar, 450 μm. B, Samples of ChAT (white arrowhead), CTb-positive (black arrowhead), and double-labeled (arrow) neurons in the gigantocellular bulbar reticular nucleus. Scale bar, 40 μm. C, Schematic drawings of coronal sections from rostral (a) to caudal (g) of the rat brain in one of our cases, showing the distribution of CTb-positive neurons (dots) and double-labeled neurons (asterisk). A, Ambiguous nucleus; BC, brachium conjunctivum; DT, dorsal tegmental nucleus of Gudden; IO, inferior olive; Ip, interpeduncular nucleus; LV, lateral vestibular nucleus; ML, medial lemniscus; MLF, medial longitudinal fascicle; MRF, mesencephalic reticular formation; MV, medial vestibular nucleus; MX, motor nucleus of the vagus nerve; PRF, pontine reticular formation; RN, red nucleus; RPT, pontine reticular nucleus; SC, superior colliculus; SNr, sustantia nigra pars reticulata; SO, superior olive; SVn, spinal trigeminal nucleus; III, oculomotor nerve; IIIn, nucleus of the oculomotor nerve; VIn, nucleus of the abducens nerve; VII, facial nerve; VIIn, nucleus of the facial nerve; XII, hypoglossal nerve; XIIn, nucleus of the hypoglossal nerve.
In vivo experiments
Functional role of ACh release in the GN
Thirty-five GN neurons were selected for analysis; they displayed a rapidly adapting “phasic” response to tactile stimulation delivered on the glabrous skin of the hindpaw. Neurons were recorded during the application of ACSF on the GN (control condition). This neuronal population comprised neurons with a nonrhythmic firing pattern and low spontaneous firing rate (<5 Hz). Previous works have identified these cells as projecting neurons (Panetsos et al., 1998; Nuñez et al., 2000).
The spontaneous firing rate of GN cells was 1.8 ± 0.11 spikes/s in control conditions (n = 35). Application of a brief barrage of stimulating pulses at the PRN (50 Hz, 500 ms duration) induced a large increase of the firing rate to 12.3 ± 0.25 spikes/s (p = 0.001; n = 27) (Fig. 7A, ACSF, black trace) that could last up to 3 min. Remained neurons were not affected by PRN stimulation (n = 8). This effect was abolished when atropine (0.5 mm; 10 μl; n = 8) was applied to the GN before PRN stimulation (Fig. 7A, Atropine, gray trace). Application of atropine did not modify the spontaneous activity in our sample. A closer analysis of the increase in the firing rate of GN neurons by PRN stimulation did not reveal synchronization between PRN stimuli and GN spikes, suggesting that stimulation was not “driving” GN neurons.
Figure 7.
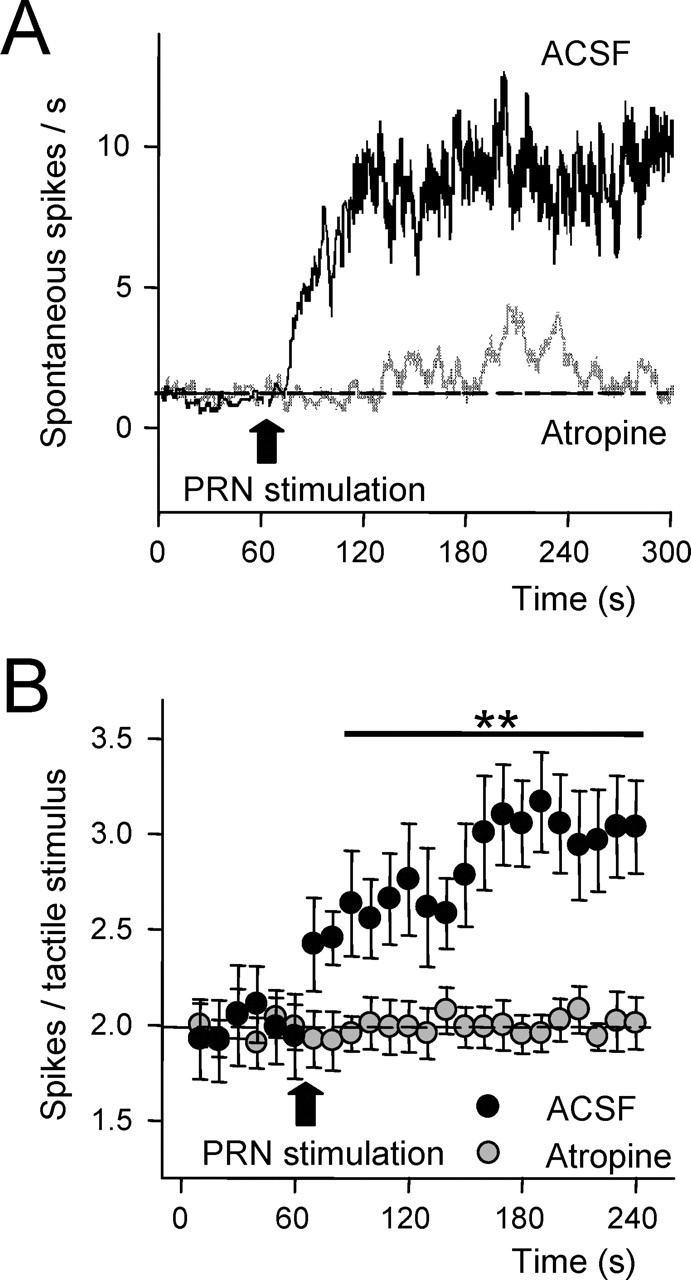
Cholinergic modulation of tactile responses in the gracilis nucleus. A, Plot of the spontaneous firing rate of a representative GN neuron in spontaneous conditions and after electrical stimulation of the PRN (50 Hz during 500 ms). Reticular formation stimulation increased the firing rate in control condition (after application of ACSF on the GN; n = 27). Atropine application on GN (0.5 mm; 1 μl; n = 8) abolished the increase of the firing rate evoked by PRN stimulation. B, Plot of the mean response (spikes per stimuli) evoked by 20 ms tactile stimuli delivered at the receptive field of GN neurons at 0.5 Hz (n = 27). Each value indicates the response average of five stimuli. Electrical stimulation of the PRN (50 Hz during 500 ms) increased tactile responses in control (ACSF). However, response facilitation was abolished by application of atropine on GN (n = 8; Atropine).
Tactile stimuli applied at the receptive field of GN neurons (n = 27) at 1/s evoked 2.0 ± 0.26 spikes per stimulus in control conditions. Stimulus at the PRN with a brief pulse barrage (50 Hz during 500 ms) increased tactile responses during at least 3 min in all neurons (3.1 ± 0.24 spikes per stimulus; p = 0.008) (Fig. 7B). This potentiation was abolished by previous application of atropine on the GN (n = 8) (Fig. 7B).
Discussion
Cholinergic receptors are widely distributed in the CNS in which at least five genetically distinct mAChRs (Levey et al., 1991) and 16 genetically different nAChRs (Lukas et al., 1999) are present. In our regions of interest in which presynaptic cholinergic and postsynaptic GN neurons are present, M1 and M2 mAChRs are found (Ge et al., 1995). Consistent with those reports, present results demonstrate that cholinergic agonists regulate neuronal activity in the GN as revealed in vitro by the effects of bath-applied CCh. The CCh challenge induced three major effects: (1) a presynaptic inhibition of CF- and DC-EPSPs, (2) an increase in postsynaptic excitability, and (3) an amplification of the spike responses evoked both by CF and DC stimulation. These effects were mediated by M1-type mAChRs. In addition, electrical stimulation in the PRN in which presynaptic cholinergic neurons are present induces an atropine-sensitive increase in spontaneous spike activity and amplifies somatosensory-evoked responses. These original findings suggest an important action of cholinergic inputs acting via M1 mAChRs in the regulation of somatosensory information flow at the level of the first synaptic relay station.
Modification of intrinsic membrane properties
Depolarizing muscarinic responses are caused by reductions of potassium conductances via a G-protein-coupled signaling mechanism in other systems (Benardo and Prince, 1982; McCormick, 1992; Borde et al., 1995, 2000; Nuñez et al., 1997; Yajeya et al., 1999). In GN neurons, the CCh-induced depolarization is inhibited by muscarinic antagonists atropine and pirenzepine, indicating that CCh acted via activation of M1 mAChRs. The CCh challenge was associated with an increased membrane resistance, probably mediated by the inhibition of several K+ channel types normally active at the resting Vm. Present results are consistent with CCh inhibiting both voltage-insensitive and voltage-gated conductances. This conclusion is consistent with the I–V relationships calculated early during voltage deflections evoked by hyperpolarizing pulses that were linear both in control conditions and under CCh, suggesting inhibition of a leak channel by CCh. However, late depolarizing sags that increased with hyperpolarization were present, and I–V relationships clearly departed from a linear model during the sags, consistent with the activation of voltage-gated conductances.
The characteristic time course and voltage dependence of the sag evoked by hyperpolarizing pulses (present results) and its block by low concentrations of Cs+ (Nuñez and Buño, 1999) are consistent with the activation of the Na+- and K+-mediated Q-current (also termed Ih) (Adams and Halliwell, 1982; Edman and Grampp, 1989; McCormick and Pape, 1990). The Q-current was markedly inhibited by CCh, as revealed under voltage clamp, a reduction that could also contribute to increased Rin (present results). These changes in intrinsic properties were paralleled by an increased excitability, as revealed by both a reduced current threshold to evoke spikes and the enhanced spike frequency evoked by identical depolarizing current pulses.
Regulation of synaptic responses
Immunohistochemical studies in the rat show that DC and CF inputs to dorsal column nuclei are glutamatergic (Rustioni and Weinberg, 1989; Broman, 1994; DeBiasi et al., 1994) and that dorsal column nuclei neurons express both AMPA and NMDA receptors (Watanabe et al., 1994; Kus et al., 1995; Popratiloff et al., 1997). Consistent with those findings, we had reported that responses of GN neurons evoked by both DC and CF inputs were glutamatergic and that non-NMDA receptors mediate DC-EPSPs, whereas CF-EPSPs also display an NMDA component (Nuñez and Buño, 1999, 2001).
Present results suggest that the CCh challenge diminished the peak amplitude of both DC- and CF-EPSPs most likely by reducing transmitter release through activation of presynaptic receptors, as suggested by the changes in paired-pulse responses and the decreased mEPSC frequency that parallel the EPSC inhibition (Kamiya and Zucker, 1994; Fernández de Sevilla et al., 2002; Fernández de Sevilla and Buño, 2003). The presynaptic effects of CCh were inhibited by low concentrations of atropine, indicating that CCh acted through the activation of presynaptic muscarinic receptors at both DC and CF terminals, as occurs in other systems (Hounsgaard, 1978; Qian and Saggau, 1997; Alkondon et al., 2000; Fernández de Sevilla et al., 2002). The effects of CCh on synaptic potentials were blocked by the M1 mAChR antagonist pirenzepine but were not affected by the M2 mAChR antagonist methoctramine, consistent with a presynaptic action via M1 mAChR activation. In addition, nAChR antagonists did not modify CCh effects, indicating that nicotinic receptors were not engaged in the effects of CCh.
In other systems, the presynaptic muscarinic inhibition is mediated through the block of N-type voltage-gated Ca2+ channels by mAChR that act via a G-protein-coupled signaling pathway (Qian and Saggau, 1997). The resulting reduced Ca2+ influx inhibits glutamate release by decreasing transmitter release probability (Valentino and Dingledine, 1981; Fernández de Sevilla et al., 2002). Therefore, it is likely that similar presynaptic mechanisms mediate the muscarinic inhibition in the GN.
Morphological study
ChAT-positive fibers and terminals have been reported in the dorsal column nuclei (Simon et al., 1981; Henderson and Sherriff, 1991; Avendaño and Dykes, 1996), suggesting an important role of the cholinergic input to the GN in sensorimotor modulation. Cholinergic projections from the laterodorsal and pedunculopontine tegmental nuclei to the pontine tegmentum have been reported in cats and rats (Mitani et al., 1988; Shiromani et al., 1988; Semba et al., 1990). However, the source of the cholinergic innervation described in the GN in the rat remained unclear. In the present results, the number of CTb-positive neurons found to project to the GN nuclei is low, probably because of the superficial location of the GN nuclei and the difficulty of avoiding the washout produced by the flow of CSF, which probably decreased the CTb concentration at the injection site. However, a consistent number of CTb-positive neurons were located in forebrain and brainstem. Interestingly, CTb-positive neurons and doubled-labeled cells could not be detected in the neuronal Ch5 and Ch6 cholinergic groups described by Mesulam et al. (1983). These results agree with the description that cholinergic neurons of the laterodorsal and pedunculopontine tegmental nuclei project to prosencephalic structures, whereas noncholinergic neurons project in a descending pattern (Satoh and Fibiger, 1986; Goldsmith and van der Kooy, 1988; Spann and Grofova, 1989; Semba et al., 1990). Our results indicate that the cholinergic projections to the GN arise at the pontine and bulbar reticular formation in which cholinergic cells have also been described (Armstrong et al., 1983; Cuello and Sofroniew, 1984). Therefore, present morphological results indicate that this cholinergic input may originate in brainstem reticular neurons and could underlie the changes in synaptic transmission and neuronal activity described here, as suggested by the effects of their stimulation in vivo.
Functional implications
Present results suggest that cholinergic activity controls the flow of somatosensory signals in the GN by presynaptically inhibiting both ascending DC and descending CF fibers and by postsynaptically regulating the output by increasing the excitability of GN neurons. Interestingly, activation of descending CF fibers also amplifies DC neuronal responses to DC stimulation in vitro (Nuñez and Buño, 2001), suggesting that cholinergic activity acting cooperatively with descending glutamatergic CF fibers may potentiate the flow of somatosensory information in the GN. Results obtained in anesthetized rats showed that electrical stimulation of the caudal pontine reticular formation increased the firing rate of GN cells and facilitated sensory responses through activation of cholinergic receptors. Previous recordings of neurons located in the pontine gigantocellular nucleus in behavioral rats showed that these neurons increased the firing rate during waking, waking with movements, and paradoxical sleep (Vertes, 1977). We found that some of these neurons are cholinergic and project to the GN. Thus, it is possible that this increment of cholinergic neuron activity may facilitate sensory transmission through the GN during waking and paradoxical sleep.
In addition, a diversity of firing modes and response types characterize in vivo recordings of dorsal column nuclei neurons (Andersen et al., 1964; Canedo et al., 1998; Malmierca and Nuñez, 1998; Panetsos et al., 1998; Nuñez et al., 2000). Most of these firing behaviors are absent in the deafferented slice preparation (Nuñez and Buño, 1999, 2001), although low-threshold membrane oscillations could be observed in cultures of dorsal column nuclei neurons (Reboreda et al., 2003). However, these discharge patterns can be reproduced in vitro under muscarinic activation, probably indicating that the natural functional repertoire of GN neurons is under the complex modulation by cholinergic input.
Humans participating in a task requiring alertness and attention display increased regional blood flow in the midbrain reticular formation (Kinomura et al., 1996), in which cholinergic neurons are located. We can speculate that the change in neuronal activity of cholinergic brainstem reticular formation neurons during these cognitive tasks may favor transmission of the relevant information at the first somatosensory relay station, the GN. This hypothesis may be corroborated by the existence of collateral ascending axons from sensory pathways that make synapses with neurons located in the brainstem reticular formation (Scheibel, 1980).
Footnotes
This work was supported by Spanish Ministerio de Ciencia y Tecnología Grant BFI2002-01107 (D.F-S., postdoctoral researcher) and Comunidad Autónoma de Madrid Grant GR/SAL/0877/2004.
References
- Adams PR, Halliwell JV (1982). A hyperpolarization-induced inward current in hippocampal pyramidal cells. J Physiol (Lond) 324:62p. [Google Scholar]
- Aigner TG (1995). Pharmacology of memory: cholinergic- glutamatergic interactions. Curr Opin Neurobiol 5:155–160. [DOI] [PubMed] [Google Scholar]
- Alkondon M, Pereira EF, Almeida LE, Randall WR, Albuquerque EX (2000). Nicotine at concentrations found in cigarette smokers activates and desensitizes nicotinic acetylcholine receptors in CA1 interneurons of rat hippocampus. Neuropharmacology 39:2726–2739. [DOI] [PubMed] [Google Scholar]
- Andersen P, Eccles JC, Oshima T, Schmidt RF (1964). Mechanisms of synaptic transmission in the cuneate nucleus. J Neurophysiol 27:1096–1116. [DOI] [PubMed] [Google Scholar]
- Armstrong DM, Saper CB, Levey AI, Wainer BH, Terry RD (1983). Distribution of cholinergic neurons in the rat brain: demonstrated by the immunocytochemical localization of choline acetyltransferase. J Comp Neurol 216:53–68. [DOI] [PubMed] [Google Scholar]
- Avendaño C, Dykes RW (1996). Evolution of morphological and histochemical changes in the adult cat cuneate nucleus following forelimb denervation. J Comp Neurol 370:479–490. [DOI] [PubMed] [Google Scholar]
- Baskerville KA, Schweitzer JB, Herron P (1997). Effects of cholinergic depletion on experience-dependent plasticity in the cortex of the rat. Neuroscience 80:1159–1169. [DOI] [PubMed] [Google Scholar]
- Benardo LS, Prince DA (1982). Ionic mechanisms of cholinergic excitation in mammalian hippocampal pyramidal cells. Brain Res 249:333–344. [DOI] [PubMed] [Google Scholar]
- Borde M, Cazalets JR, Buño JR (1995). Activity-dependent response depression in rat hippocampal CA1 pyramidal neurons in vitro. J Neurophysiol 74:1714–1729. [DOI] [PubMed] [Google Scholar]
- Borde M, Bonansco C, Fernández de Sevilla F, Le Ray D, Buño W (2000). Voltage-clamp analysis of the potentiation of the slow Ca2+-activated K+ current in hippocampal pyramidal neurons. Hippocampus 10:198–206. [DOI] [PubMed] [Google Scholar]
- Broman J (1994). Neurotransmitters in subcortical somatosensory pathways. Anat Embryol 189:181–214. [DOI] [PubMed] [Google Scholar]
- Canedo A, Martinez L, Mariño J (1998). Tonic and bursting activity in the cuneate nucleus of the chloralose-anesthetized cat. Neuroscience 84:603–617. [DOI] [PubMed] [Google Scholar]
- Cuello AC, Sofroniew MV (1984). The anatomy of the CNS cholinergic neurons. Trends Neurosci 7:74–78. [Google Scholar]
- DeBiasi S, Vitellaro-Zuccarello L, Bernardi P, Valtschanoff JG, Weinberg R (1994). Ultrastructural and immunocytochemical characterization of terminals of postsynaptic ascending dorsal column fibers in the rat cuneate nucleus. J Comp Neurol 353:109–118. [DOI] [PubMed] [Google Scholar]
- Edman A, Grampp W (1989). Ion permeation through hyperpolarization activated membrane channels (Q-channels) in the lobster stretch receptor neurone. Pflügers Arch 413:249–255. [DOI] [PubMed] [Google Scholar]
- Fernández de Sevilla D, Buño W (2003). Presynaptic inhibition of Schaffer collateral synapses by stimulation of hippocampal cholinergic afferent fibres. Eur J Neurosci 17:555–558. [DOI] [PubMed] [Google Scholar]
- Fernández de Sevilla D, Cabezas C, Ochima de Prada AN, Sánchez-Jimenez A, Buño W (2002). Selective muscarinic regulation of functional glutamatergic Schaffer collateral synapses in rat CA1 pyramidal neurons. J Physiol (Lond) 545:51–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge XQ, Hu B, Yao B, Xu PC, Bian CF (1995). Relationship between muscarinic receptor subtypes and cyclic nucleotides in pons-medulla oblongata. Zhongguo Yao Li Xue Bao 16:408–411. [PubMed] [Google Scholar]
- Goldsmith M, van der Kooy D (1988). Separate non-cholinergic descending projections and cholinergic ascending projections from the nucleus tegmenti pedunculopontinus. Brain Res 445:386–391. [DOI] [PubMed] [Google Scholar]
- Hasselmo ME (1999). Neuromodulation: acetylcholine and memory consolidation. Trends Cogn Sci 3:351–359. [DOI] [PubMed] [Google Scholar]
- Henderson Z, Sherriff FE (1991). Distribution of choline acetyltransferase immunoreactive axons and terminals in the rat and ferret brainstem. J Comp Neurol 314:147–163. [DOI] [PubMed] [Google Scholar]
- Hounsgaard J (1978). Presynaptic inhibitory action of acetylcholine in area CA1 of the hippocampus. Exp Neurol 62:787–797. [DOI] [PubMed] [Google Scholar]
- Jabbur SJ, Towe AL (1961). Cortical excitation of neurons in dorsal column nuclei of cat, including an analysis of pathways. J Neurophysiol 24:499–509. [DOI] [PubMed] [Google Scholar]
- Jacobowitz DM, Palkovits M (1974). Topographic atlas of catecholamine and acetylcholinesterase-containing neurons in the rat brain. I. Forebrain (telencephalon, diencephalon). J Comp Neurol 157:13–28. [DOI] [PubMed] [Google Scholar]
- Jones B (1990). Immunohistochemical study of choline acetyltransferase-immunoreactive processes and cells innervating the pontomedullary reticular formation in the rat. J Comp Neurol 295:485–514. [DOI] [PubMed] [Google Scholar]
- Kamiya H, Zucker RS (1994). Residual Ca2+ and short-term synaptic plasticity. Nature 371:603–606. [DOI] [PubMed] [Google Scholar]
- Kinomura S, Larsson J, Gulyás B, Roland PE (1996). Activation by attention of the human reticular formation and thalamic intralaminar nuclei. Science 271:512–515. [DOI] [PubMed] [Google Scholar]
- Kus L, Saxon D, Beitz AJ (1995). NMDA R1 mRNA distribution in motor and thalamic projecting sensory neurons in the rat spinal cord and brain stem. Neurosci Lett 25:201–204. [DOI] [PubMed] [Google Scholar]
- Levey AI, Kitt CA, Simonds WF, Price DL, Brann MR (1991). Identification and localization of muscarinic acetylcholine receptor proteins in brain with subtype-specific antibodies. J Neurosci 11:3218–3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukas RJ, Changeux JP, le Novère N, Albuquerque EX, Balfour DJK, Berg DK, Bertrand D, Chiappinelli VA, Clarke PBS, Collins AC, Dani JA, Grady SR, Kellar KJ, Lindstrom JM, Marks MJ, Quik M, Taylor PW, Wonnacott S (1999). International Union of Pharmacology. XX. Current status of the nomenclature for nicotinic acetylcholine receptors and their subunits. Pharmacol Rev 51:397–401. [PubMed] [Google Scholar]
- Maalouf M, Miasnikov AA, Dykes RW (1998). Blockade of cholinergic receptors in rat barrel cortex prevents long-term changes in the evoked potential during sensory preconditioning. J Neurophysiol 80:529–545. [DOI] [PubMed] [Google Scholar]
- Madison DV, Malenka RC, Nicoll RA (1991). Mechanisms underlying long-term potentiation of synaptic transmission. Annu Rev Neurosci 14:379–397. [DOI] [PubMed] [Google Scholar]
- Malmierca E, Nuñez A (1998). Corticofugal action on somatosensory response properties of rat nucleus gracilis cells. Brain Res 810:172–180. [DOI] [PubMed] [Google Scholar]
- Martin ED, Buño W (2003). Caffeine-mediated presynaptic long-term potentiation in hippocampal CA1 pyramidal neurons. J Neurophysiol 89:3029–3038. [DOI] [PubMed] [Google Scholar]
- McCormick DA (1992). Cellular mechanisms underlying cholinergic and noradrenergic modulation of neuronal firing mode in the cat and guinea pig dorsal lateral geniculate nucleus. J Neurosci 12:278–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA, Pape HC (1990). Properties of a hyperpolarization-activated cation current and its role in rhythmic oscillation in thalamic relay neurones. J Physiol (Lond) 431:291–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesulam M-M, Mufson EJ, Wainer BH, Levey AI (1983). Central cholinergic pathways in the rat: an overview based on an alternative nomenclature (Ch1–Ch6). Neuroscience 10:1185–1201. [DOI] [PubMed] [Google Scholar]
- Mitani A, Ito K, Hallanger AE, Wainer BH, Kataoka K, McCarley RW (1988). Cholinergic projections from the laterodrosal and pedunculopontine tegmental nuclei to the pontine gigantocellular tegmental field in the cat. Brain Res 451:397–402. [DOI] [PubMed] [Google Scholar]
- Nuñez A, Buño W (1999). In vitro electrophysiological properties of rat dorsal column nuclei neurons. Eur J Neurosci 11:1865–1876. [DOI] [PubMed] [Google Scholar]
- Nuñez A, Buño W (2001). Properties and plasticity of synaptic inputs to rat dorsal column neurones recorded in vitro. J Physiol (Lond) 535:483–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuñez A, De la Roza C, Rodrigo-Angulo M, Buño W, Reinoso-Suarez F (1997). Electrophysiological properties and cholinergic responses of rat ventral oral pontine reticular neurons in vitro. Brain Res 754:1–11. [DOI] [PubMed] [Google Scholar]
- Nuñez A, Panetsos F, Avendano C (2000). Rhythmic neuronal interactions and synchronization in the rat dorsal column nuclei. Neuroscience 100:599–609. [DOI] [PubMed] [Google Scholar]
- Nyberg G, Blomqvist A (1982). The termination of forelimb nerves in the feline cuneate nucleus demonstrated by the transganglionic transport method. Brain Res 248:209–222. [DOI] [PubMed] [Google Scholar]
- Oh JD, Woolf NJ, Roghani A, Edwards RH, Butcher LL (1992). Cholinergic neurons in the rat central nervous system demonstrated by in situ hybridization of choline acetyltransferase mRNA. Neuroscience 47:807–822. [DOI] [PubMed] [Google Scholar]
- Palkovits M, Jacobowitz DM (1974). Topographic atlas of catecholamine and acetylcholinesterase-containing neurons in the rat brain. II. Hindbrain (mesencephalon, rombencephalon). J Comp Neurol 157:29–42. [DOI] [PubMed] [Google Scholar]
- Panetsos F, Nuñez A, Avendano C (1998). Sensory information processing in the dorsal column nuclei by neuronal oscillators. Neuroscience 84:635–639. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C (1986). In: The rat brain in stereotaxic coordinates New York: Academic.
- Popratiloff A, Rustioni A, Weinberg RJ (1997). Heterogeneity of AMPA receptors in the dorsal column nuclei of the rat. Brain Res 754:333–339. [DOI] [PubMed] [Google Scholar]
- Qian J, Saggau P (1997). Presynaptic inhibition of synaptic transmission in the rat hippocampus by activation of muscarinic receptors: involvement of presynaptic calcium influx. Br J Pharmacol 122:511–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmusson DD, Dykes RW (1988). Long-term enhancement of evoked potentials in cat somatosensory cortex produced by coactivation of the basal forebrain and cutaneous receptor. Exp Brain Res 70:276–286. [DOI] [PubMed] [Google Scholar]
- Reboreda A, Sanchez E, Romero M, Lamas JA (2003). Intrinsic spontaneous activity and subthreshold oscillations in neurones of the rat dorsal column nuclei in culture. J Physiol (Lond) 551:191–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rustioni A, Weinberg RJ (1989). The somatosensory system. In: Handbook of chemical neuroanatomy (Björklund A, Hökfelt T, Swanson LW, eds) pp. 219–321. Amsterdam: Elsevier.
- Satoh K, Fibiger HC (1986). Cholinergic neurons of the laterodorsal tegmental nucleus: efferent and afferent connections. J Comp Neurol 253:277–302. [DOI] [PubMed] [Google Scholar]
- Schäfer MK-H, Eiden LE, Weihe E (1998). Cholinergic neurons and terminal fields revealed by immunohistochemistry for the vesicular acetylcholine transporter. I. Central nervous system. Neuroscience 84:331–359. [DOI] [PubMed] [Google Scholar]
- Scheibel AB (1980). Anatomical and physiological substrates of arousal: a view from the bridge. In: The reticular formation revisited (Hobson JA, Brazier MAB, eds) pp. 55–66. New York: Raven.
- Semba K, Reiner PB, Fibiger HC (1990). Single cholinergic mesopontine tegmental neurons project to both the pontine reticular formation and the thalamus in the rat. Neuroscience 38:643–654. [DOI] [PubMed] [Google Scholar]
- Shiromani PJ, Armstrong DM, Gillin JC (1988). Cholinergic neurons from the dorsolateral pons project to the medial pons: a WGA-HRP and choline acetyltransferase immunohistochemical study. Neurosci Lett 95:19–23. [DOI] [PubMed] [Google Scholar]
- Shulz DE, Ego-Stengel V, Ahissar E (2003). Acetylcholine-dependent potentiation of temporal frequency representation in the barrel cortex does not depend on response magnitude during conditioning. J Physiol (Paris) 97:431–439. [DOI] [PubMed] [Google Scholar]
- Simon JR, Oderfeld-Nowak B, Felten DL, Aprison MH (1981). Distribution of choline acetyltransferase, acetylcholinesterase, muscarinic receptor binding, and choline uptake in discrete areas of the rat medulla oblongata. Neurochem Res 6:497–505. [DOI] [PubMed] [Google Scholar]
- Spann BM, Grofova I (1989). Origin of ascending and spinal pathways from the nucleus tegmenti pedunculopontinus in the rat. J Comp Neurol 283:13–27. [DOI] [PubMed] [Google Scholar]
- Thomson AM (2000). Facilitation, augmentation and potentiation at central synapses. Trends Neurosci 23:305–312. [DOI] [PubMed] [Google Scholar]
- Valentino RJ, Dingledine R (1981). Presynaptic inhibitory effect of acetylcholine in the hippocampus. J Neurosci 1:784–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valverde F (1966). The pyramidal tract in rodents. A study of its relations with the posterior column nuclei, dorsolateral reticular formation of the medulla, and cervical spinal cord (Golgi and electron microscopic observations). Z Zellforsch Mikrosk Anat 71:297–363. [PubMed] [Google Scholar]
- Vertes RB (1977). Selective firing of rat pontine gigantocellular neurons during movement and REM sleep. Brain Res 128:146–152. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Mishina M, Inoue Y (1994). Distinct distributions of five NMDA receptor channel subunit mRNAs in the brainstem. J Comp Neurol 343:520–531. [DOI] [PubMed] [Google Scholar]
- Yajeya J, De la Fuente J, Bajo VM, Rinolobos AS, Heredia M, Criado JM (1999). Muscarinic activation of a non-selective cationic conductance in pyramidal neurons in rat basolateral amygdala. Neuroscience 88:159–167. [DOI] [PubMed] [Google Scholar]