

Abstract
The evolutionarily conserved Semaphorin family proteins are well known axon guidance ligands that mediate both attractive and repulsive responses in invertebrates and vertebrates. In this study, we show that the Drosophila Semaphorin-1a (Sema1a), a transmembrane Semaphorin, is required cell autonomously in adult photoreceptor (R-cell) axons for the establishment of an appropriate topographic termination pattern in the optic lobe. Loss of sema1a disrupts the association of neighboring R-cell growth cones leading to defects in local termination pattern, whereas overexpression of sema1a induces the hyper-fasciculation of R-cell axons. The function of Sema1a in R-cell axon guidance absolutely requires its cytoplasmic domain. We propose that Sema1a functions as a receptor in regulating R-cell axon guidance in the Drosophila visual system.
Keywords: Semaphorin-1a, axon guidance, axon guidance receptor, Drosophila visual system, neuronal connectivity, semaphorin
Introduction
The Semaphorin family proteins, including both secreted and membrane-associated forms, are well known axon guidance ligands (Pasterkamp and Kolodkin, 2003). In Drosophila, the transmembrane Semaphorin-1a (Sema1a) has been shown previously to mediate the defasciculation of motor axon bundles at specific choice points (Yu et al., 1998). Sema1a binds to its receptor Plexin A (PlexA) (Winberg et al., 1998), which in turn triggers downstream signaling events involving the receptor tyrosine kinase Otk (Winberg et al., 2001), the evolutionarily conserved flavoprotein monooxygenase molecule interacting with CasL (MICAL) (Terman et al., 2002), and the A kinase anchoring protein Nervy (Terman and Kolodkin, 2004), leading to a repulsive growth-cone response. Sema1a has also been shown to be involved in synaptic formation (Godenschwege et al., 2002). That overexpression of wild-type Sema1a, but not a truncated Sema1a mutant protein lacking the cytoplasmic domain, caused a gain-of-function phenotype (Godenschwege et al., 2002), raises the interesting possibility that Sema1a functions as a receptor in synaptic formation. However, because the cytoplasmic-domain-deleted Sema1a mutant still rescued the sema1a loss-of-function synaptic formation phenotype (Godenschwege et al., 2002), it remains unknown whether endogenous Sema1a truly functions as a receptor in the nervous system.
Our previous study implicates a role for Otk, a component of the PlexA receptor complex for Sema1a in mediating the defasciculation of embryonic motoneuron axons (Winberg et al., 2001), in layer-specific targeting of a subset of R-cell axons (i.e., R1–R6) in the adult visual system (Cafferty et al., 2004). The function of Otk in R-cell axons, however, appears to be independent of Sema1a because the R-cell projection pattern in sema1a null mutants was different from that in otk mutants (Cafferty et al., 2004). In this study, we show that Sema1a is required autonomously in R-cell axons for the establishment of appropriate topographic termination pattern in the optic lobe. The cytoplasmic domain of Sema1a is absolutely required for its function in R-cell axons. These results are consistent with a role for Sema1a to act as a receptor to regulate R-cell axon guidance.
Materials and Methods
Genetics.
Sema1a and Fasciclin II (Fas II) were overexpressed in R-cell axons by crossing UAS-sema1a and UAS-Fas II flies with the GMR-GAL4 driver line, respectively. Transgene rescue was performed by crossing elav-GAL4 (C155); Df(2)N22–5/Bc flies with UAS-sema1a, semaP1/Bc. The R-cell projection pattern in elav-GAL4 (C155)/+; UAS-sema1a, semaP1/Df(2)N22–5 was compared with that in UAS-sema1a, semaP1/Df(2)N22–5 or elav-GAL4 (C155)/+; UAS-sema1aΔcyt, semaP1/Df(2)N22–5. To generate single sema1a mutant R-cell axons, hsFLP, UAS-mCD8::GFP, elav-GAL4 (C155); semaP1, FRT40A/+ flies were crossed with Tub-GAL80, FRT40A flies. The progeny were heat-shocked at 37°C for 1 h at larval stage to induce mitotic recombination. To completely remove the MICAL gene in R-cells overexpressing Sema1a, genetic crosses were performed to generate the larvae with the genotype: eyFLP; GMR-GAL4, UAS-sema1a/+; FRT82B, Df(3R)swp2MICAL/FRT82B, w+M(3)RpS32.
Histology and immunohistochemistry.
Plastic sectioning of adult eyes was performed as described previously (Cafferty et al., 2004). Eye–brain complexes from third-instar larvae were dissected and stained as described previously (Ruan et al., 1999). Monoclonal antibody (MAb) 24B10 and anti-β-galactosidase antibodies were used at 1:200 and 1:1000 dilutions, respectively. The secondary antibodies (Jackson ImmunoResearch, West Grove, PA) were used at 1:200 dilution. Epifluorescent images were captured using a high-resolution fluorescence imaging system (Canberra Packard, Mississauga, Ontario, Canada) and analyzed by 2D Deconvolution using MetaMorph imaging software (Universal Imaging Corporation, Brandywine, PA). The severity of the R-cell hyper-fasciculation phenotype was quantified by counting the number of R-cell axon bundles that were located between lamina and medulla.
Results
Sema1a is expressed in R-cell axons and growth cones
To determine whether sema1a plays a role in R-cell axon guidance, we examined whether Sema1a is expressed in R-cell axons at the third-instar larval stage when the adult R-cell-to-optic-lobe connection pattern begins to form. The distribution of Semala in the developing visual system was examined using an affinity-purified anti-Sema1a antibody. At the third-instar larval stage, precursor cells in the eye-imaginal disc begin to differentiate into R-cells that project axons through the optic stalk into the optic lobe. The R-cell projection pattern at this stage can be visualized by staining using MAb 24B10 (Fig. 1A). Strong Sema1a staining was observed in R-cell bodies and their axons in the eye disc, the optic stalk, and the optic lobe (Fig. 1B,C). Within the lamina, the staining was present in R-cell axons as well as R1–R6 growth cones in the lamina plexus. Strong staining was also observed throughout the medulla neuropil comprising of R7 and R8 axons as well as non-R-cell axons. In sema1aP1 eye-specific mosaic individuals (Fig. 1D–F), the staining was missing in many regions of the eye disc, optic stalk, and optic lobe, confirming the specificity of anti-Sema1a antibody. We conclude that Sema1a is present in R-cell axons and their growth cones.
Figure 1.

Sema1a is present in R-cell axons and growth cones in the developing visual system. A, B, Wild-type (wt) third-instar larval eye–brain complexes were double stained with MAb 24B10 (green), which recognizes all R-cell axons, and anti-Sema1a (red). The merge is shown in C. Sema1a staining is present in R-cell bodies in the eye disc and their axonal trajectories from the eye disc through the optic stalk into the developing optic lobe. The lamina plexus, consisting mainly of R1–R6 growth cones, is strongly stained. D, E, sema1aP1 mosaic eye–brain complexes in which large patches of homozygous sema1aP1 mutant cells were generated in an otherwise heterozygous or wild-type eye and were double stained with MAb 24B10 (D) and anti-Sema1a (E). The merge is shown in F. Mosaic staining pattern is observed in the eye disc and optic stalk, confirming the specificity of this antibody. Scale bar: (in A) 20 μm.
sema1a is specifically required for the establishment of an appropriate topographic termination pattern in the optic lobe
We performed a detailed phenotypic analysis to determine the potential role of Sema1a in R-cell axon guidance. In wild type (Fig. 2A), the differentiating R-cells send out axons toward the posterior end of the eye disc where they converge and subsequently enter the optic stalk. After exiting the optic stalk, R-cell axons fan out to migrate over the superficial lamina. After reaching their appropriate topographic locations in between two layers of lamina glial cells (i.e., the intermediate target of R1–R6 axons), R1–R6 growth cones stop extension, expand significantly in size, and form close contacts with neighboring growth cones, resulting in the establishment of a continuous and dense terminal layer (Fig. 2A). During pupation, R1–R6 axons extend away from this region to seek appropriate lamina neurons for synaptic formation (Meinertzhagen and Hanson, 1993; Clandinin and Zipursky, 2002). R7 and R8 growth cones pass through the lamina into the medulla, where they also expand in size and elaborate a precise topographic termination pattern (Fig. 2A).
Figure 2.
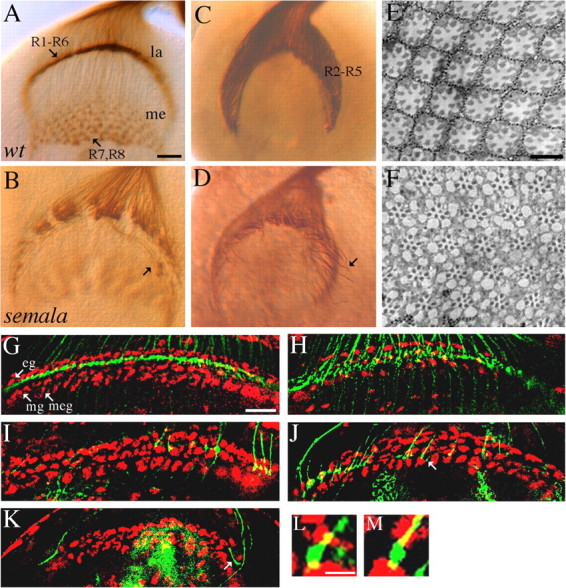
semala is specifically required for the establishment of an appropriate local retinotopic termination pattern. A, B, Third-instar larval eye–brain complexes were stained with MAb 24B10 to visualize all R-cell axons. In wild type (wt; A), expanded R1–R6 growth cones associate closely with each other and elaborate a smooth and dense terminal layer in the lamina (la). R7 and R8 axons project through the lamina into the medulla (me). In a sema1aP1 homozygote (B), R1–R6 growth cones associated loosely with neighboring growth cones. Some R1–R6 axons migrated laterally at the bottom of lamina into incorrect topographic locations (B, arrow). C, D, R2–R5 axons in wild type (C) and sema1aP1 homozygous mutants (D) were labeled with the ro-τ-lacZ marker. Like that in wild type (C), the vast majority of mutant R2–R5 axons still terminated in the lamina (D). Note that some R2-R5 axons (arrow) migrated abnormally at the bottom of lamina (D). E, F, Wild-type (E) and sema1aP1 mosaic (F) adult eyes were embedded in Epon and sectioned. G, H, R-cell axons (green) and glial nuclei (red) in wild-type (G) and sema1aP1 mosaic (H) third-instar larval brains were double-stained with MAb 24B10 and anti-Repo, respectively. In wild type (G), R1–R6 growth cones terminate in between epithelial (eg) and marginal (mg) glial layers. In a sema1aP1 mosaic individual (H) in which large clones of homozygous mutant eye tissues were generated (Newsome et al., 2000), R1–R6 growth cones were distributed in a much broader area between the lamina glial and the medulla glial cells (meg). I–K, Single wild-type (I) or sema1a mutant (J, K) axons were positively labeled. sema1a single-mutant axons frequently passed over the marginal glial layer (J, arrow). Note the three-layered glial structure at the edge of lamina termination site was distorted as a result of mounting. Axons at these regions were not included when the phenotype of stopping at incorrect glial layers was quantitated. Some (K, arrow) terminated at incorrect topographic locations. L, M, High-resolution view of expanded wild-type (L) and sema1aP1 mutant (M) R-cell growth cones (green) surrounded by glial cells (red) at lamina termination site. Scale bars: (in A) A–D, 20 μm; (in E) E, F, 10 μm; (in G) G–K, 20 mm; (in L) L, M, 4 μm.
To examine the potential role for Sema1a in R-cell axon guidance, we examined R-cell projection pattern in sema1a mutants at third-instar larval stage before synaptic formation. In homozygous sema1aP1 mutant larvae (Fig. 2B), the initial outgrowth of R-cell axons appeared normal. Mutant R-cell axons migrated correctly from the eye disc into the optic stalk, which was morphologically indistinguishable from that in wild type. After R-cell axons exited from the optic stalk en route to their termination region, however, severe defects were observed (Fig. 2B). R1–R6 growth cones failed to pack into a dense termination layer in all mutant hemispheres examined (n = 11). Instead, they scattered around the lamina terminal field and appeared to be unable to establish a close association in the target region. Some R1–R6 axons did not stop at their appropriate topographic termination region and instead migrated laterally into incorrect locations in the lamina (Fig. 2B). Similar expressivity of phenotype were observed in sema1aP1 hemizygotes (n = 30 hemispheres), consistent with the null nature of this allele (Yu et al., 1998).
Although severe defects in R-cell axon termination pattern within lamina were observed in sema1a mutants, the lamina-versus-medulla target selection appeared essentially normal. The majority of R2–R5 axons, a subset of R1–R6 axons labeled by the ro-τ-lacZ marker, still stop within the lamina layer in sema1a mutants (n = 15) (Fig. 2, compare D, C). Consistent with the phenotype observed with MAb 24B10 staining (Fig. 2B), we found that the organization of R2–R5 axons in the lamina plexus was disrupted in sema1a mutants (Fig. 2D).
To determine whether above defects were because of abnormal eye development, we examined R-cell differentiation and patterning in sema1a mutant eye disc and adult mosaic eye. Plastic sectioning of adult sema1a mosaic eye did not reveal any defect in either the number or the organization of R-cells within each mutant ommatidium (0 of 996 mutant ommatidia examined) (Fig. 2, compare F, E). The gross organization of mutant ommatidia in each clone also appeared normal. No defects in either the differentiation or the organization of R-cell clusters were observed in third-instar larval eye discs in sema1a mutants (data not shown). These data exclude the possibility that the R-cell axon guidance phenotype in sema1a mutants is secondary to abnormal R-cell development in the eye.
sema1a is required cell-autonomously in R-cells for axonal projections
To determine whether above defects reflect a role for sema1a in R-cells, we examined the projection pattern of sema1a mutant R-cell axons in an otherwise heterozygous or wild-type target region by using genetic mosaic analysis. In wild type, R1–R6 axons stop at their lamina intermediate target region in between two layers of glial cells (i.e., epithelial and marginal glia) (Fig. 2G). When large patches (>90% eye tissues) of sema1a mutant R-cells were generated by eye-specific mitotic recombination (Newsome et al., 2000), although the organization of glial cells in the lamina remained normal, R1–R6 axon termination pattern was disrupted (Fig. 2H). Many R1–R6 growth cones failed to stop at correct locations after reaching their intermediate target. This result indicates that sema1a is required in R-cells for correct axon termination.
To determine whether Sema1a is required in a cell-autonomous or non-cell-autonomous manner, we examined the projection of single mutant axons using the mosaic analysis with a repressible marker method (Lee and Luo, 1999). Single sema1a mutant cell mosaics in the eye were generated by expressing the FLP recombinase under the control of heat-inducible promoter at larval stage. In control (wild-type single mosaics), the vast majority (57 of 58 labeled single axons) of R1–R6 axons terminated normally (Fig. 2I). In contrast, many labeled single sema1a mutant R1–R6 axons (∼63%; n = 89) failed to stop at correct locations (Fig. 2J). Some mutant axons moved away from appropriate topographic locations and extended laterally within the lamina (∼10%; n = 89) (Fig. 2K), whereas in wild-type control, most labeled single axons stopped at correct topographic locations (57 of 58 labeled single R1–R6 axons). Like that in wild type (56 of 58 single R1–R6 axons) (Fig. 2L), R-cell growth cones still expanded normally in the lamina in sema1a mutants (87 of 89 single mutant axons) (Fig. 2M). These results indicate a cell autonomous role for Sema1a in R-cell axon guidance.
Overexpression of sema1a induced the hyper-fasciculation of R-cell axons
That R1–R6 growth cones failed to establish a highly condensed R1–R6 terminal layer in sema1a mutants raised the possibility that Sema1a is involved in promoting the local interaction between R1–R6 growth cones at their intermediate target region. To further address this possibility, we examined whether overexpression of Sema1a in R-cell axons would increase the association of R-cell axons.
Sema1a was overexpressed in R-cell axons by using the eye-specific GMR-GAL4 driver. When the GMR-GAL4 driver was used to overexpress Fas II, a well known homophilic cell adhesion molecule required for axonal fasciculation (Lin et al., 1994), we observed an axonal hyper-fasciculation phenotype (Fig. 3B; supplementary Table 1, available at www.jneurosci.org as supplemental material). If Sema1a, like Fas II, promotes R-cell axon interactions, one would predict that overexpression of Sema1a should produce a similar hyper-fasciculation phenotype. Overexpression of Sema1a was confirmed by staining R-cells with anti-Sema1a antibody (data not shown). Indeed, we found that overexpression of Sema1a caused a hyper-fasciculation phenotype similar to that in larvae overexpressing Fas II (Fig. 3, compare C, B; supplementary Table 1, available at www.jneurosci.org as supplemental material). Thicker axon bundles were formed in both lamina and medulla in all hemispheres examined (n = 50), coincident with the presence of large clumps of terminals in the lamina plexus. This phenotype is dosage dependant because an increase in the dosage of the sema1a transgene dramatically enhanced the phenotype (100%; n = 35) (Fig. 3D; supplementary Table 1, available at www.jneurosci.org as supplemental material). When both Sema1a and Fas II were overexpressed in R-cell axons, the hyper-fasciculation phenotype was dramatically enhanced (Fig. 3E; supplementary Table 1, available at www.jneurosci.org as supplemental material). This result is in marked contrast to the previous observation that sema1a counters the attractive action of Fas II in embryonic motor axons (Yu et al., 2000) and is consistent with a role for Sema1a to promote R-cell axon interactions.
Figure 3.
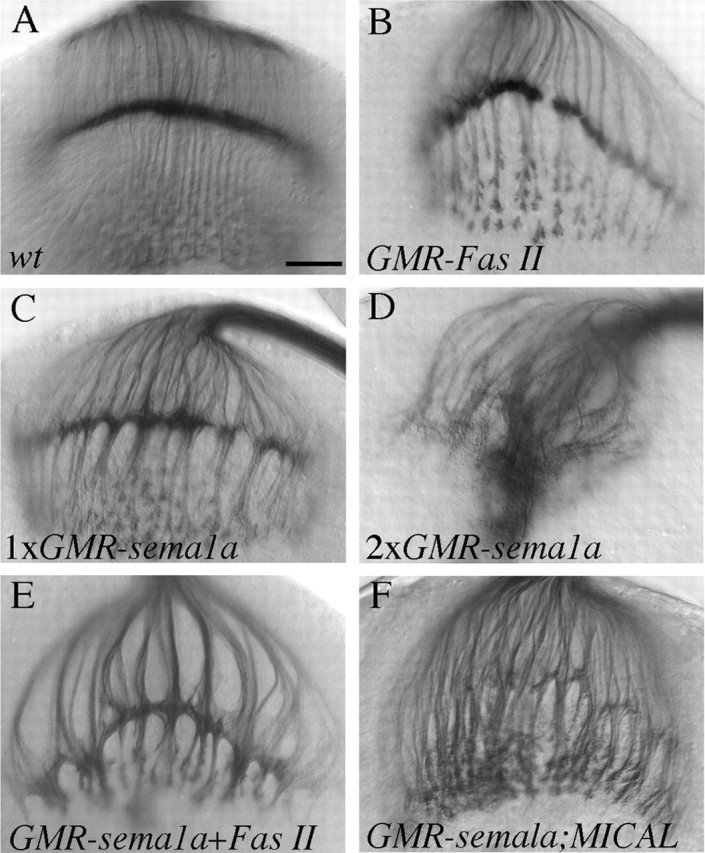
Overexpression of Sema1a induced the hyper-fasciculation of R-cell axons. A, Wild type (wt). B, Overexpression of Fas II in R-cell axons induced the formation of thicker bundles. C, Overexpression of Sema1a also caused a hyper-fasciculation phenotype. In larvae carrying two copies of the UAS-sema1a transgene (D), the phenotype becomes much more severe. R-cell axons form large clumps in the lamina and appeared to be unable to defasciculate and extend deeply into the medulla. In larvae carrying one copy of UAS-Fas II and UAS-sema1a (E), the phenotype is much stronger than that in larvae carrying a single copy of UAS-Fas II (B) or UAS-sema1a (C). The complete loss of the MICAL gene (F) did not suppress the Sema1a overexpression phenotype. Scale bar: (in A) 20 μm.
Sema1a has been shown to bind to its receptor PlexA to mediate repulsive interactions between motor axons (Winberg et al., 1998). To determine whether the above Sema1a-induced R-cell hyper-fasciculation phenotype is also dependent on the activation of the PlexA signaling pathway, we examined the potential epistatic interaction between sema1a and genes in the PlexA pathway. We found that the complete loss of the MICAL gene, which functions downstream of PlexA in both invertebrates and vertebrates (Terman et al., 2002), did not modify the sema1a overexpression phenotype (n = 19) (Fig. 3F; supplementary Table 1, available at www.jneurosci.org as supplemental material).
To further determine whether the classical PlexA signaling pathway plays a role in R-cell axon termination, we examined whether interfering with the PlexA signaling pathway would cause a sema1a-like phenotype. However, we found that R-cell axon termination pattern remained essentially normal in the absence of MICAL (supplementary Fig. 1C, available at www.jneurosci.org as supplemental material). The effect of interfering with PlexA signaling was also examined by overexpressing the Nervy gene encoding for a member of A kinase anchoring proteins (Terman and Kolodkin, 2004). Overexpression of Nervy in embryonic motor axons could effectively antagonize the PlexA signaling, causing a guidance phenotype identical to loss of PlexA or sema1a (Terman and Kolodkin, 2004). However, no obvious defect was observed when Nervy was overexpressed in R-cell axons (supplementary Fig. 1D, available at www.jneurosci.org as supplemental material) or glial cells in the optic lobe (supplementary Fig. 1E, available at www.jneurosci.org as supplemental material). These results suggest that the function of Sema1a in R-cell axons is independent of the classical PlexA signaling pathway.
The cytoplasmic domain of Sema1a is indispensable for its action in R-cell axon guidance
The above analyses support a specific role for Sema1a in R-cell axons. Sema1a may function as a ligand that activates its receptor on neighboring R-cell growth cones to promote their association. Alternatively, because Sema1a contains a cytoplasmic domain, it may function as a guidance receptor to mediate the interactions between R-cell axons, which is supported by that sema1a is required autonomously in single R1–R6 axons (Fig. 2). To address this, we examined whether the cytoplasmic domain of Sema1a is required for its action in R-cell axon guidance.
First, we tested whether deleting the cytoplasmic domain of Sema1a affects its ability to induce the hyper-fasciculation phenotype. The sema1aΔcyt transgene encoding a membrane-associated Semala mutant protein in which the C-terminal amino-acid sequence 695–899 of the cytoplasmic domain (amino acids 680–899) is deleted (Godenschwege et al., 2002) was overexpressed in R-cell axons. Surprisingly, we found that overexpression of sema1aΔcyt was unable to induce the hyper-fasciculation of R-cell axons (0 of 23 hemispheres) (Fig. 4B; supplementary Table 1, available at www.jneurosci.org as supplemental material), but instead caused a phenotype indistinguishable from that in sema1a mutants (∼80%; n = 23) (compare Figs. 4B, 2B). This result indicates that sema1aΔcyt acts as a dominant-negative form to interfere with the function of endogenous Sema1a.
Figure 4.

The cytoplasmic domain of Sema1a is required for its function in R-cell axon guidance. A, Overexpression of wild-type Sema1a under control of GMR-GAL4 caused the hyper-fasciculation of R-cell axons. B, Overexpression of sema1aΔcyt driven by GMR-GAL4 did not induce the formation of thicker bundles, but instead disrupted the organization of R1–R6 growth cones in the lamina plexus, which was indistinguishable from that in sema1aP1 mutants (Fig. 2B). C, Neuronal-specific expression of wild-type Sema1a under control of elav-GAL4 in a sema1aP1 hemizygote (i.e., sema1aP1/Df(2)N22–5) restored the normal R-cell projection pattern in 14 of 22 mutant hemispheres. The remaining eight mutant hemispheres displayed the axonal hyper-fasciculation phenotype (data not shown), likely caused by an above threshold expression level of the sema1a transgene in these individuals. D, No rescue was observed when the sema1aΔcyt transgene was expressed in a sema1aP1 hemizygote. Note the discontinuous lamina plexus and the aberrant projections of some R-cell axons (arrow) at the bottom of lamina. Scale bar: (in A) 20 μm.
We then tested whether the cytoplasmic domain is required for rescuing the sema1a phenotype. We chose the neuronal-specific driver elav-GAL4 for the rescue experiment. The expression of wild-type sema1a under control of this driver in wild-type larvae caused a weaker hyper-fasciculation phenotype (supplementary Fig. 2C, available at www.jneurosci.org as supplemental material), whereas no defect was observed when sema1aΔcyt was expressed under control of elav-GAL4 in wild-type larvae (supplementary Fig. 2D, available at www.jneurosci.org as supplemental material). Neuronal-specific expression of wild-type sema1a substantially rescued the phenotype in sema1a mutants (14 of 22 mutant hemispheres) (Fig. 4C). In contrast, all mutant hemispheres expressing sema1aΔcyt still displayed the sema1a phenotype (n = 15) (compare Figs. 4D, 2B), indicating that the cytoplasmic domain is essential for the function of Sema1a in R-cell axons. This data, together with that overexpression of sema1aΔcyt under control of GMR-GAL4 in wild-type larvae caused a sema1a-like loss-of-function phenotype (see above), suggests strongly that Sema1a functions as a receptor in R-cell axons.
Discussion
Semaphorin family members have been studied extensively for their role as axon guidance ligands in both invertebrates and vertebrates. Godenschwege et al. (2002) demonstrated previously that the cytoplasmic domain of Sema1a, the Drosophila transmembrane Semaphorin, is required for inducing a gain-of-function synaptic formation phenotype, raising the possibility that Sema1a functions as a receptor in synaptic formation. In this study, we provide several lines of evidence to support that Sema1a functions as a receptor in R-cell axons to regulate the formation of appropriate termination pattern in the optic lobe. First, we show that sema1a is required autonomously in single R-cell axons. Second, unlike overexpression of wild-type Sema1a, overexpression of the membrane-bound Sema1aΔcyt mutant lacking the cytoplasmic domain is incapable of inducing a R-cell hyper-fasciculation phenotype. Third, the cytoplasmic domain of Sema1a is required for rescuing the sema1a loss-of-function phenotype. And finally, the Sema1aΔcyt mutant lacking the cytoplasmic domain causes a dominant-negative effect when overexpressed in wild-type flies.
Sema1a may function as a guidance receptor in R1–R6 axons to promote the local interaction between neighboring growth cones when they reach their intermediate target region at the third-instar larval stage. The local interaction between R1–R6 growth cones has been shown to provide certain guidance information for R1–R6 axons to select their appropriate synaptic partners during pupation (Clandinin and Zipursky, 2000). Similarly, we speculate that a Sema1a-dependent interaction at third-instar larval stage may allow neighboring growth cones to communicate with each other, thus facilitating the formation of an appropriate retinotopic termination pattern in the lamina. An alternative model for the action of Sema1a is that Sema1a functions as a receptor in R1–R6 axons to mediate their interaction with cells in the lamina. Our current data does not allow us to distinguish among these possibilities. Although our data suggests strongly that Sema1a functions as a receptor in R-cell axons, the elucidation of the exact mechanism of Sema1a action awaits the identification of its upstream and downstream partners in the fly visual system.
It appears highly likely that transmembrane Semaphorins in vertebrates can also function as axon guidance receptors in the nervous system. Several vertebrate Semaphorins have been shown to be able to bind via their cytoplasmic domains to intracellular signaling proteins such as enabled/vasodilator-stimulated phosphoprotein-like protein (Klostermann et al., 2000) and c-Src (Eckhardt et al., 1997). While we were preparing this manuscript, Toyofuku et al. (2004) provided evidence to support that the chick transmembrane Semaphorin Sema6D can function as a receptor in cell migration during embryonic development. It will be of interest to determine whether these vertebrate transmembrane Semaphorins also function as axon guidance receptors in the nervous system.
Footnotes
*P.C. and L.Y. contributed equally to this work and are listed alphabetically.
This work was supported by an operating grant (MOP-14688) awarded to Y. R. from Canadian Institutes of Health Research. We thank Don van Meyel for critical reading of this manuscript; the Bloomington Stock Center for fly stocks; Developmental Studies Hybridoma Bank at University of Iowa for MAb 24B10; Corey Goodman for UAS-sema1a and UAS-Fas II; Alex Kolodkin for sema1aP1, UAS-Nervy, Df(3R)swp2MICAL, and anti-Sema1a antibody; and Rodney Murphey for sema1aΔcyt.
References
- Cafferty P, Yu L, Rao Y (2004). The receptor tyrosine kinase Off-track is required for layer-specific neuronal connectivity in Drosophila. Development 131:5287–5295. [DOI] [PubMed] [Google Scholar]
- Clandinin TR, Zipursky SL (2000). Afferent growth cone interactions control synaptic specificity in the Drosophila visual system. Neuron 28:427–436. [DOI] [PubMed] [Google Scholar]
- Clandinin TR, Zipursky SL (2002). Making connections in the fly visual system. Neuron 35:827–841. [DOI] [PubMed] [Google Scholar]
- Eckhardt F, Behar O, Calautti E, Yonezawa K, Nishimoto I, Fishman MC (1997). A novel transmembrane semaphorin can bind c-src. Mol Cell Neurosci 9:409–419. [DOI] [PubMed] [Google Scholar]
- Godenschwege TA, Hu H, Shan-Crofts X, Goodman CS, Murphey RK (2002). Bi-directional signaling by Semaphorin 1a during central synapse formation in Drosophila. Nat Neurosci 5:1294–1301. [DOI] [PubMed] [Google Scholar]
- Klostermann A, Lutz B, Gertler F, Behl C (2000). The orthologous human and murine semaphorin 6A-1 proteins (SEMA6A-1/Sema6A-1) bind to the enabled/vasodilator-stimulated phosphoprotein-like protein (EVL) via a novel carboxyl-terminal zyxin-like domain. J Biol Chem 275:39647–39653. [DOI] [PubMed] [Google Scholar]
- Lee T, Luo L (1999). Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron 22:451–461. [DOI] [PubMed] [Google Scholar]
- Lin DM, Fetter RD, Kopczynski C, Grenningloh G, Goodman CS (1994). Genetic analysis of Fasciclin II in Drosophila: defasciculation, refasciculation, and altered fasciculation. Neuron 13:1055–1069. [DOI] [PubMed] [Google Scholar]
- Meinertzhagen IA, Hanson TE (1993). The development of the optic lobe. In: The development of Drosophila melanogaster (Bate M, Martinez-Arias A, eds) pp. 1363–1491. Cold Spring Harbor, NY: Cold Spring Harbor.
- Newsome TP, Asling B, Dickson BJ (2000). Analysis of Drosophila photoreceptor axon guidance in eye-specific mosaics. Development 127:851–860. [DOI] [PubMed] [Google Scholar]
- Pasterkamp RJ, Kolodkin AL (2003). Semaphorin junction: making tracks toward neural connectivity. Curr Opin Neurobiol 13:79–89. [DOI] [PubMed] [Google Scholar]
- Ruan W, Pang P, Rao Y (1999). The SH2/SH3 adaptor protein dock interacts with the Ste20-like kinase misshapen in controlling growth cone motility. Neuron 24:595–605. [DOI] [PubMed] [Google Scholar]
- Terman JR, Kolodkin AL (2004). Nervy links protein kinase a to plexin-mediated semaphorin repulsion. Science 303:1204–1207. [DOI] [PubMed] [Google Scholar]
- Terman JR, Mao T, Pasterkamp RJ, Yu HH, Kolodkin AL (2002). MICALs, a family of conserved flavoprotein oxidoreductases, function in plexin-mediated axonal repulsion. Cell 109:887–900. [DOI] [PubMed] [Google Scholar]
- Toyofuku T, Zhang H, Kumanogoh A, Takegahara N, Yabuki M, Harada K, Hori M, Kikutani H (2004). Guidance of myocardial patterning in cardiac development by Sema6D reverse signalling. Nat Cell Biol 6:1204–1211. [DOI] [PubMed] [Google Scholar]
- Winberg ML, Noordermeer JN, Tamagnone L, Comoglio PM, Spriggs MK, Tessier-Lavigne M, Goodman CS (1998). Plexin A is a neuronal semaphorin receptor that controls axon guidance. Cell 95:903–916. [DOI] [PubMed] [Google Scholar]
- Winberg ML, Tamagnone L, Bai J, Comoglio PM, Montell D, Goodman CS (2001). The transmembrane protein Off-track associates with Plexins and functions downstream of Semaphorin signaling during axon guidance. Neuron 32:53–62. [DOI] [PubMed] [Google Scholar]
- Yu HH, Araj HH, Ralls SA, Kolodkin AL (1998). The transmembrane Semaphorin Sema I is required in Drosophila for embryonic motor and CNS axon guidance. Neuron 20:207–220. [DOI] [PubMed] [Google Scholar]
- Yu HH, Huang AS, Kolodkin AL (2000). Semaphorin-1a acts in concert with the cell adhesion molecules fasciclin II and connectin to regulate axon fasciculation in Drosophila. Genetics 156:723–731. [DOI] [PMC free article] [PubMed] [Google Scholar]