

Abstract
One of the hallmarks of human immunodeficiency virus (HIV)-1 associated pathology in the CNS is deterioration of neuronal processes. Although there is mounting evidence of neuronal toxicity and cell death induced by the HIV-1 transactivating factor Tat, the molecular events linked directly to its detrimental effect on neuronal cells remain unclear. In this study, we used rat embryonic cortical neurons and demonstrated that Tat causes rapid degradation of microtubule-associated protein 2 (MAP2) and the collapse of cytoskeletal filaments. The mechanism of Tat action on MAP2 stability involved Tat-mediated translocation of the proteasome to the site of microtubule filaments. Immunohistochemical analysis of clinical samples from patients with HIV encephalopathy further revealed a significant decrease in MAP2 with predominant cytoplasmic 20S in cortical neurons near microglial nodules. These findings indicate a novel mechanism for the action of Tat on neuronal cells. It involves proteasome-mediated MAP2 degradation and may account for the loss of MAP2 and neuronal damage observed in the brain of AIDS patients with neurological dysfunctions.
Keywords: neuron, proteolysis, degeneration, cytoskeleton, apoptosis, recruitment
Introduction
The process of neurodegeneration in human immunodeficiency virus (HIV) encephalopathy (HIVE) is characterized by extensive synaptic and dendritic damage that can progress to neuronal loss (Del Valle et al., 2000; Masliah et al., 2004). Because neurons are rarely infected by HIV-1, indirect mechanisms, including release of viral proteins from infected cells, are thought to cause neuronal dysfunction and death. The viral regulatory protein Tat is among the critical factors that are believed to be involved in the onset and progression of autoimmunodeficiency syndrome (AIDS) and AIDS-associated neurological complications (Kaul et al., 2001; Nath, 2002; Peruzzi et al., 2005; Peruzzi, 2006). An intriguing aspect of Tat-induced neurotoxicity includes impairment of synaptic plasticity (Perry et al., 2005) and inhibition of neurite outgrowth (Bergonzini et al., 2004). Both extension and maintenance of neuronal processes are governed by the structural and functional integrity of the cytoskeleton. Tubulin is the major structural component of the microtubules that are required for neuronal outgrowth and plays essential roles in the regulation of cytoskeletal dynamics (Desai and Mitchison, 1997). Microtubule function is regulated by motor proteins such as kinesins and dyneins and structural microtubule-associated proteins (MAPs) (Hirokawa, 1994; Cassimeris and Spittle, 2001). The best studied MAPs in the nervous system are MAP2, mostly localized in the dendrites, and Tau, more abundant in axons (Binder et al., 1985; Riederer and Matus, 1985). MAP2 activity is critical for microtubule nucleation, polymerization, stability, and bundling (Chen et al., 1992; Ferralli et al., 1994), as well as microtubular transport (Lopez and Sheetz, 1995; Sato-Harada et al., 1996). In addition, both MAP2 and tubulin have been shown to possess a chaperone-like activity (Guha et al., 1998; Sarkar et al., 2004). In the brain, four isoforms of MAP2 are produced by alternative splicing: high molecular weight (HMW) MAP2 (a and b) and low molecular weight (LMW) MAP2 (c and d) isoforms. All variants consist of two major domains, the N-terminal projection domain and the C-terminal microtubule-binding domain. Importantly, loss of MAP2 in immunoreactive dendrites from pyramidal neurons of the frontoparietal cortex and hippocampus has been correlated with the severity of cognitive impairment in patients with HIVE (Masliah et al., 1997; Archibald et al., 2004). Downregulation of MAP2 expression has been demonstrated further by mRNA analysis in the frontal cortex of patients with HIVE (Masliah et al., 2004) and in primary human neurons exposed to Tat and methamphetamine (Langford et al., 2004).
Here, we investigated the effect of Tat on the extension and maintenance of neuronal processes of rat embryonic cortical neurons. In both instances, Tat promoted neuronal damage by a mechanism that involved recruitment of the proteasome to microtubules and MAP2 degradation. Immunohistochemical analysis of clinical samples further suggests the occurrence of a similar neurodegenerative mechanism in the setting of HIV encephalopathy.
Materials and Methods
Rat neuronal primary cultures.
Rat cortical neurons were obtained by enzymatic and mechanical treatment from Sprague Dawley rat embryonic day 17 (E17). Cortices were dissected out in dissecting medium (in mm: 1.6 sucrose, 2.2 glucose, 1 HEPES, 16 NaCl, 0.5 KCl, 0.1 Na2HPO4, and 0.022 KH2PO4) and placed in Hybernate E medium (BrainBits, Springfield, IL). After careful removal of the meninges, the intact tissue was incubated with TripleExpress enzyme (Invitrogen, Carlsbad, CA) at 37°C for 10 min, followed by three washes with Hybernate E medium. Tissue trituration was performed in culture medium (see below) using a fire-polished glass Pasteur pipette, and single cell suspension was diluted with culturing medium. Finally, cells were plated on poly-d-lysine-coated dishes at a density of 4.5 × 104/cm2 and cultured in Neurobasal medium containing B27 supplement, 0.25 mm Glutamax, and 0.25 mm l-glutamine (all from Invitrogen).
Mutagenesis.
pEYFP-Tat 1–86 served as template for the mutagenesis performed using the QuikChange II site-directed Mutagenesis kit (Stratagene, La Jolla, CA) following the instructions of the manufacturer. The mutagenic oligonucleotide primers were gttgctttcattgccaaaaagccttaggcatctcc and the complementary sequence on the opposite strand.
Nucleofection and transfection of neuronal cultures.
For nucleofection, freshly isolated cells (12 × 106/reaction) were centrifuged at 800 rpm for 5 min and resuspended in 100 μl of the nucleofection solution (rat neuron nucleofector kit; Amaxa, Gaithersburg, MD). The cell suspension was then mixed with 5 μg of the appropriate plasmid and transferred to a nucleofection cuvette. The nucleofection program was number O-03 as specified by the manufacturer. Cells were immediately plated in culture medium (as described above) at a concentration of 5 × 106/60 mm plate. For immunofluorescence experiments, nucleofected cells were plated at a concentration of 4 × 104/cm2 in poly-d-lysine/laminin-coated chamber slides. In all experiments, control untransfected cells were plated at half of the density. Efficiency of transfection was evaluated within 3 h from the nucleofection using a fluorescent microscope. When required, ALLN (N-acetyl-Leu-Leu-Nle-CHO; 0.25 μm) and MG-132 (carbobenzoxy-l-leucyl-l-leucyl-l-leucinal; 25 nm) inhibitors were added to the neuronal cultures 1 h after transfection. All of the inhibitors were from Calbiochem (La Jolla, CA) (EMD Biosciences, San Diego, CA).
Fully differentiated primary neurons were transfected 4 d after the original plating using the Lipofectamine 2000 system (Invitrogen), following the protocol of the manufacturer. If a fluorescent protein was used, expression of the positive cells was visualized under a fluorescent light microscope within 4 h from transfection.
Treatment of neuronal cultures with Tat protein and terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling assay.
Recombinant Tat1–72 protein was a kind gift from Dr. Avindra Nath (Johns Hopkins University, Baltimore, MD). After 8 d in culture, rat embryonic cortical neurons were exposed to recombinant Tat at a concentration of 100 nm or 500 nm. For Western blot analysis, cellular lysates were obtained 24 h after treatment with Tat1–72.
Western blots.
Rat primary cortical neurons were collected by scraping the plates in the presence of PBS, followed by centrifugation and lysis of the cell pellet in the appropriate volume of radioimmunoprecipitation assay buffer (50 mm Tris, pH 7.4, 1% NP-40, 0.25% sodium deoxycholate, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1 mm PMSF, 1 mm sodium orthovanadate, phosphatase inhibitors and protease inhibitor mixtures). Fifteen to 50 μg of the whole-cell lysate were separated on a 4–15% SDS-PAGE (Bio-Rad, Hercules, CA). MAP2 antibody was obtained from Cell Signaling Technology (Beverly, MA). Anti-living colors antibody was obtained from Transduction Laboratories (BD Biosciences, San Jose, CA). Actin, α-tubulin, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The antibody against Tat was the R705 polyclonal antibody obtained through the AIDS Research and Reference Reagent Program (Division of AIDS, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda MD): antiserum to HIV-1 Tat (from Dr. Bryan Cullen, Duke University Medical Center, Durham, NC). To detect the uptake of recombinant Tat1–72, we used anti-Tat polyclonal antibody (1:5000) kindly provided by Dr. Avindra Nath (Johns Hopkins University, Baltimore, MD).
Fluorescence microscopy.
For immunofluorescence analysis, rat primary neurons were grown in two- or eight-chamber slides (BD Falcon; BD Biosciences, Bedford, MA) coated with an aqueous solution containing poly-d-lysine and laminin (1:1) at concentrations of 0.01 and 0.005 mg/ml, respectively. Cells were washed once with PBS and fixed with 10% formaldehyde (methanol free) ultrapure EM grade (Polysciences, Warrington, PA) for 20 min at 4°C. After two washes with PBS, the slides were processed for immunofluorescence or stored at −20°C in 70% ethanol. For immunofluorescence, the chamber slides were washed three times with PBS, followed by 1 h of blocking at room temperature in PBS containing 10% Immunopure normal goat serum (Pierce Biotechnology, Rockford, IL). Primary antibody was diluted in PBS containing 2% goat serum and incubated over night at room temperature, followed by an additional incubation of 1 h at 37°C. Cells were rinsed three times with PBS and then incubated with the appropriate secondary antibody diluted in PBS containing 2% of goat serum for 1 h at 37°C. Finally, the chamber slides were rinsed three times with PBS and mounted with Vectashield mounting medium containing 4′6′-diamidino-2-phenylindole (DAPI; Vector Laboratories, Burlingame, CA). MAP2a/b (1:1000) antibody was obtained from Chemicon (Temecula, CA). Proteasome 20S (1:200) polyclonal antibody was obtained from Zymed (South San Francisco, CA).
Immunohistochemistry.
Samples from four cases of formalin-fixed, paraffin-embedded HIV-encephalopathy were collected from the HIV Manhattan Brain Bank at Mount Sinai Medical School. Four-micrometer-thick sections were cut using a microtome and placed on charged glass slides. Immunohistochemistry was then performed according to the instructions of the manufacturer (Vector Laboratories). Our protocol includes deparaffination, rehydration in alcohol up to water, antigen retrieval in citrate buffer, pH 6.0, endogenous peroxidase quenching, and blocking in normal horse (for mouse monoclonal antibodies) or normal goat (for rabbit polyclonal antibodies) serum for 2 h at room temperature. Primary antibodies were incubated overnight in a humidified chamber. After rinsing with PBS, secondary biotinylated antibodies were incubated for 1 h (1:200 dilution), followed by ABC incubation and developing with diaminobenzidine (Boehringer Mannheim, Mannheim, Germany), counterstained with hematoxylin and coverslipped.
Primary antibodies used in this study included a rabbit polyclonal anti-Tat (courtesy of Dr. Avindra Nath) used at a 1:1000 dilution, a mouse monoclonal anti-MAP-2 (clone MT-01; GeneTex, San Antonio, TX) used at a 1:100 dilution, and a mouse monoclonal antibody for Proteasome 20S alpha3-subunit (clone MCP257; Calbiochem) used at a 1:100 dilution.
Double-labeling immunofluorescence.
For double labeling, the first steps of the protocol are similar to the protocol described above. However, after incubation with the first primary antibody, a secondary fluorescein-tagged antibody was incubated for 1 h at room temperature, and after rinsing thoroughly, the second primary antibody was incubated overnight. Finally, a second secondary antibody tagged with rhodamin was incubated for 1 h. Sections were coverslipped with an aqueous-based mounting media containing DAPI and visualized under an inverted fluorescent microscope (see below).
Overexpression and purification of recombinant proteins.
GST and GST-Tat and its deletion mutant fusion proteins were prokaryotically expressed and purified as described previously (Amini et al., 2005). The integrity and purity of the GST fusion proteins were analyzed by SDS-PAGE followed by Coomassie blue staining. Known amounts of BSA were included as controls on the same gel. For the in vitro binding assays, 300 μg of neuronal lysates at day 19 postisolation were incubated with 5 μg of GST or appropriate GST-fusion proteins coupled to glutathione Sepharose. The bound proteins were eluted with Laemmli sample buffer and separated by SDS-PAGE.
Deconvolution analysis and quantification of colocalization between detected proteins.
Deconvolution of selected pictures was performed using the SlideBook 4 imaging software (Intelligent Imaging Innovations, Denver CO). In brief, a series of three-dimensional images of each individual picture were deconvoluted to one two-dimensional picture and resolved by adjusting the signal cutoff to near maximal intensity to increase resolution. Final pictures were prepared with Adobe Photoshop (Adobe Systems, San Jose, CA) to demonstrate subcellular localization and colocalization between detected proteins. The percentage of colocalization between tubulin and Tat or MAP2 and Tat was calculated from the entire volume of the cells by using SlideBook 4 software, according to the instructions of the manufacturer. Images were captured by inverted fluorescent microscope equipped with motorized z-axis.
Statistical analysis.
All results are represented as mean values ± SD. The significance of differences between means was performed using a two-tailed Student’s t test. p values ≤ 0.05 were considered to be statistically significant.
Results
Detrimental effects of Tat on neuronal processes
To determine the molecular events associated with Tat-mediated neuronal injury, primary cultures of E17 cortical neurons were transfected with a plasmid in which Tat (86 amino acids) was cloned in frame downstream of the enhanced yellow fluorescent protein (pEYFP-Tat). As a gene delivery system, we used nucleofection (see Materials and Methods), which has the advantage of giving a high efficiency of transfection (60–70%) and rapid expression of the transfected protein. We determined the levels of expression of EYFP and EYFP-Tat in neurons at 4, 10, and 32 h after transfection (Fig. 1A). Although the levels of the control EYFP increased with time, expression of EYFP-Tat was high at 4 h, decreased at 10 h, and was undetectable at 32 h. Analysis of EYFP and EYFP-Tat expressing cells by fluorescent microscopy demonstrated a similar efficiency of transfection for each plasmid at 4 h after nucleofection (Fig. 1B). However, at later time points, the number of EYFP-Tat-positive cells dramatically decreased compared with EYFP transfected cells. Quantitatively, 80 ± 5.8% decrease in the number of fluorescent cells was observed in the presence of EYFP-Tat between 4 and 24 h time points. In contrast, a slight increase (18 ± 3.7%) in EYFP-positive cells was observed at 24 h after transfection (data not shown). The apparent loss of Tat-expressing neurons was accompanied by the detection of nuclear GAPDH in these cells (data not shown), which has been shown previously to correlate with apoptosis (Ishitani and Chuang, 1996; Sawa et al., 1997; Saunders et al., 1999; Kusner et al., 2004; Hara et al., 2005).
Figure 1.

HIV-1 Tat expression and its damaging effects on rat embryonic cortical neurons. A, Representative Western blot showing levels of expression of fusion protein EYFP-Tat and control protein EYFP in rat cortical neurons at the indicated time points after nucleofection (see Materials and Methods). EYFP and EYFP-Tat proteins were detected using an antibody that recognizes YFP (top panel). The same blot was probed with anti-Tat antibody (middle panel) and with anti-actin antibody to verify equal loading (bottom panel). B, Expression of EYFP and EYFP-Tat detected by fluorescent light microscopy at 4 and 24 h (hrs) after nucleofection (original magnification, 100×). C, Fluorescent images showing differentiated neurons cotransfected either with pECFP (in blue) and the mitochondrial-targeted dsRed or with pEFP-Tat and dsRed-mito at 32 h after nucleofection (original magnification, 400×). The images were taken using an inverted fluorescent microscope equipped with motorized z-axis and deconvolution software. D, Representative pictures showing expression of EYFP and EYFP-Tat proteins in mature neurons at 4 and 24 h after transfection (original magnification, 200×).
To visualize Tat-mediated impairment of neurite outgrowth, cells were transfected with expression vector containing red fluorescent protein with mitochondrial localization signal (dsRed-mito) (Fig. 1C). Freshly isolated neurons were nucleofected with pECFP (enhanced cyan fluorescent protein) and dsRed-mito or pECFP-Tat and dsRed-mito. Expression of ECFP-Tat and ECFP proteins was visualized 24 h after transfection under cyan light emission using an inverted fluorescent microscope equipped with motorized z-axis and deconvolution software. Control cells (pECFP/dsRed-mito) efficiently extended neuronal processes as visualized by the distribution of blue fluorescent protein and mitochondrial dsRed-mito (Fig. 1C, top panel). In contrast, a Tat-expressing cell is unable to form neuronal processes and deteriorates as highlighted by condensed mitochondrial marker dsRed. In the same field, a Tat-negative (untransfected) cell shows normal distribution of the mitochondria throughout the neuronal processes (Fig. 1C, bottom panel, right side).
Tat-induced impairment of neuronal processes was further assessed in mature neurons. E17 cortical neurons were cultured for 5 d and then transfected by Lipofectamine 2000 system using pEYFP-Tat and pEYFP control vectors (Fig. 1D). Although the efficiency of transfection was lower compared with nucleofection, this method allowed gene delivery into neurons with well developed neuronal processes. Four h after transfection, both EYFP-Tat and the control EYFP are distributed along neuronal processes (Fig. 1D, top panels). However, 24 h later, neuronal processes in Tat-expressing cells showed clear signs of deterioration highlighted by a spotted localization of Tat along the dendrites (Fig. 1D, bottom panels) and similarly to the experiments performed in freshly isolated neurons (Fig. 1B), the number of Tat-expressing neurons decreased rapidly over a period of 24 h (data not shown).
These data demonstrate that Tat has detrimental effects on neuronal processes at two different stages of their maturation. It blocks neurite outgrowth in freshly isolated neurons, and it may induce damage to neuronal processes in mature cells. The end point in both instances is neuronal cell death, which occurs within 24 h of expression of this viral protein.
Tat-mediated MAP2 breakdown
To further study the inhibition of neurite outgrowth and neuronal damage by Tat, we investigated a possible interaction between Tat and microtubule structures in neuronal processes. MAP2 is a cytoskeletal protein that regulates microtubule dynamics and function (Hirokawa, 1994; Sanchez et al., 2000). Importantly, loss of MAP2 immunoreactivity is used as an indicator of neuronal damage after ischemia (Aoki et al., 1995; Dawson and Hallenbeck, 1996) and in patients with HIVE (Masliah et al., 1997; Archibald et al., 2004; Langford et al., 2004; Masliah et al., 2004).
We first determined expression levels of MAP2 proteins in cortical neurons in long-term cultures (Fig. 2A). Both high and low molecular weight MAP2s were present in rat embryonic neurons at 4 h after isolation. An elevated MAP2a/b expression was observed between days 2 and 19 after isolation. MAP2c expression also increased at day 2 in culture, but its decline started as early as 4 d after plating.
Figure 2.
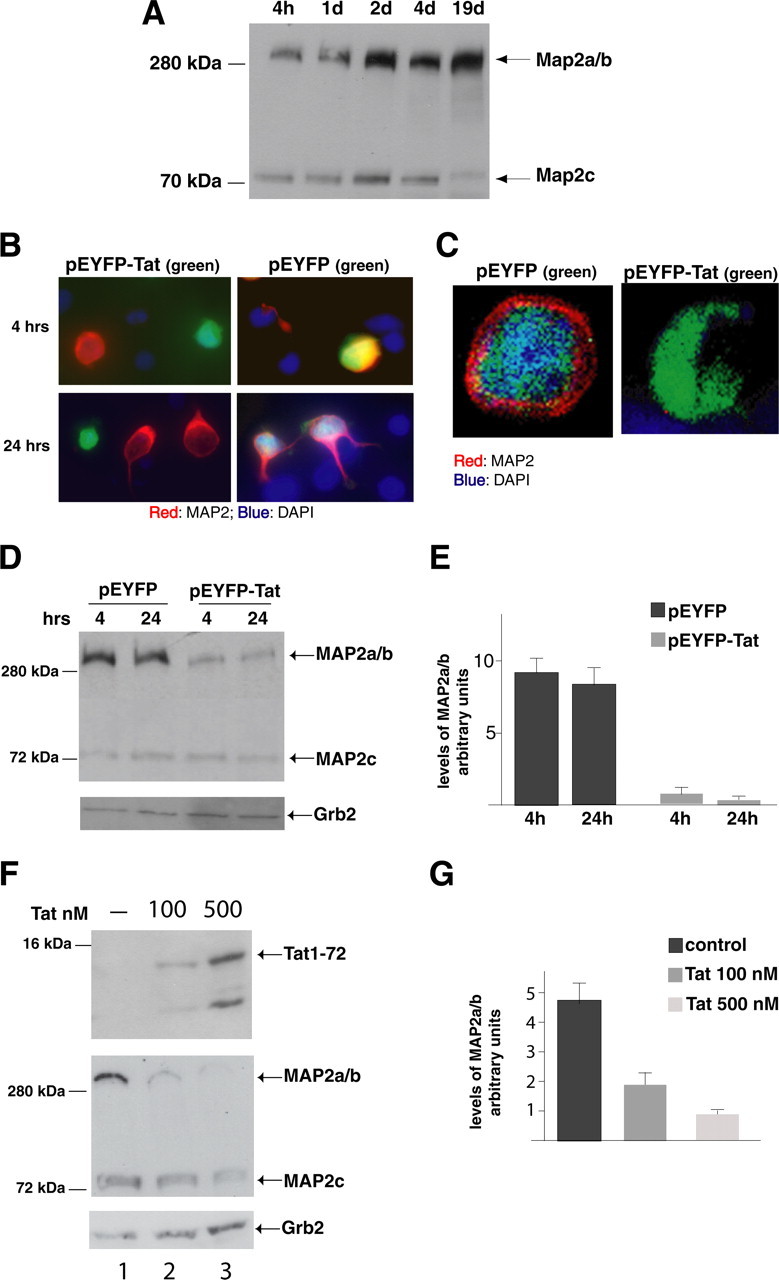
Tat-induced MAP2 degradation. A, Western blot analysis of protein extracts obtained from cultured rat cortical neurons at the indicated time points. Anti-MAP2 antibody recognizes MAP2 isoforms at 300 kDa (MAP2a/b) and 75 kDa (MAP2c). B, Immunofluorescence was performed using anti-MAP2 antibody (red) in pEYFP and pEYFP-Tat expressing neurons at 4 and 24 h (hrs). Colocalization of EYFP and MAP2 is indicated by yellow fluorescence. Tat-positive cells are indicated by green fluorescence (original magnification, 400×). C, Deconvolution evaluation of EYFP and EYFP-Tat cells fixed 24 h after transfection and immunolabeled with anti-MAP2 antibody. Note that Tat-expressing cell is MAP2 negative and is also characterized by loss of nuclear staining (DAPI) (original magnification, 400×). D, Western blot analysis showing expression of MAP2a/b isoforms in EYFP-Tat cells and pEYFP-positive cells at 4 and 24 h time points. Anti-Grb2 antibody was used to confirm equal loading. E, Densitometric analysis of MAP2a/b expression in EYFP and EYFP-Tat-positive cells normalized by Grb2 and by the efficiency of transfection. Values represent the average of three independent experiments. F, Western blot detecting intracellular Tat in control untreated neurons (lane 1, top panel) and in neurons exposed for 24 h to recombinant Tat1–72 protein at concentrations of 100 nm (lane 2) or 500 nm (lane 3). The same blot was reprobed with anti-MAP2 (middle panel) and anti-Grb2 (bottom panel) antibodies. G, Densitometric analysis relative to MAP2a/b expression after treatment with Tat1–72 evaluated from three independent experiments and normalization for Grb2 levels. Error bars represent SEM.
Next, we performed immunocytofluorescence analysis of MAP2a/b in cultures of cortical neurons at 4 and 24 h after transfection. We found that Tat-positive cells (green) are characterized by very low levels of MAP2 (red) (Fig. 2B, two left panels). In these two images, red cells (MAP2 staining) are Tat negative (lack of green). In contrast, neurons transfected with the control vector pEYFP are also positive for MAP2 (Fig. 2B, two right panels). Under higher magnification and by using deconvolution software, it was possible to demonstrate that in control EYFP cells, MAP2 was present in the entire perimeter of the cell, whereas Tat-positive cells were negative for MAP2 (Fig. 2C). Importantly, a dramatic difference in the nucleus staining (DAPI, blue fluorescence) between pEYFP and pEYFP-Tat indicates advanced apoptosis in Tat-expressing cells 24 h after transfection. Loss of MAP2 proteins in Tat-expressing neurons was additionally confirmed by Western blot analysis using an antibody that recognizes HMW as well as LMW MAP2 proteins (Fig. 2D). High levels of HMW MAP2 (MAP2a/b) are detected in cellular lysates obtained from control pEYFP-positive cells at 4 and 24 h after transfection. In contrast, neurons transfected with pEYFP-Tat vector showed lower levels of MAP2a/b at both 4 and 24 h after transfection. Interestingly, no evident changes were observed at the level of LMW MAP2 (MAP2c), suggesting that Tat action specifically targets MAP2 s a/b. Loss of MAP2a/b in Tat-positive neurons was quantified by densitometric analysis and normalized by efficiency of transfection and Grb2 levels (Fig. 2E). When compared with the values relative to the analysis of MAP2a/b expression in the control neurons (pEYFP), a reduction of ∼10-fold was observed for MAP2a/b levels in EYFP-Tat-positive neurons. In another experimental setting, 8-d-old cultures of rat embryonic cortical neurons were exposed to recombinant Tat (Tat1–72) at concentrations of 100 and 500 nm for 24 h. A dose-dependent intracellular accumulation of Tat was detected by Western blot analysis and is shown in Figure 2F (top panel). When tested for MAP2 expression, protein extracts obtained from the Tat-treated neuronal cultures showed a dramatic reduction in the MAP2a/b levels compared with untreated cells (Fig. 2F, middle panel). Quantitatively, a 2.6- and 6.7-fold decrease in MAP2 expression occurred in cells treated with 100 and 500 nm of recombinant Tat, respectively (Fig. 2G).
To further investigate Tat-induced loss of MAP2, we used GST-Tat to pull-down MAP2a/b from neuronal lysates (Fig. 3). Full-length 86 amino acids GST-Tat and three deletion mutants of Tat were tested: GST-TatΔ2–36 missing the amino acids 2–36, GST-TatΔ1–48 missing amino acids 1–48, and GST-Tat1–56, in which the sequence is truncated at the amino acid 56. The full-length GST-Tat1–86 and the mutant GST-Tat1–56 pulled down MAP2a/b isoforms from neuronal protein extracts. MAP2a/b was also pulled down by the mutant TatΔ2–36, although less efficiently, whereas the mutant GST-TatΔ1–48 did not (Fig. 3A). Therefore, the association between Tat and MAP2 seems to be mediated by the region between amino acids 36 and 48 of Tat, which has also been implicated in tubulin interactions in other studies (Chen et al., 2002). Next, we generated a mutant of Tat in which the tubulin-binding domain (amino acids 36–39) was deleted, and we used the new GST-TatΔ36–39 protein in GST pull-down experiments (Fig. 3B). First, we confirmed that TatΔ36–39 does not bind tubulin (Fig. 3B, lane 3). Among the GST-Tat proteins tested, only TatΔ2–36 and, to a lesser extent, the full-length Tat (1–86) pulled down tubulin, whereas both TatΔ36–39 and TatΔ1–48 completely failed to do so. When tested for binding to MAP2, TatΔ36–39 maintained, at least partially, its ability to bind MAP2 even in the absence of the tubulin-binding site (Fig. 3B, lane 3). The GST pull-down experiments suggest that the amino acids 36–48 in the Tat sequence likely mediate the association of Tat to the tubulin/MAP2a/b complex, with the strongest association observed within the tubulin-binding domain of Tat (amino acids 36–39).
Figure 3.

Pull-down reactions between GST-Tat fusion proteins and protein extracts isolated from differentiated cortical neurons. The Tat mutants were: TatΔ2–36 missing the amino acids 2–35; TatΔ1–48 lacks the first 48 amino acids, and Tat1–56 lacks amino acids 57–86. A, The results of the pull-downs are shown as a Western blot developed with anti MAP2a/b antibody. B, An additional GST-Tat pull-down using a mutant in which the tubulin-binding domain was deleted (Δ36–39). The same blot was probed with anti-MAP2a/b antibody (bottom panel).
In the next set of experiments, we evaluated the role of the tubulin-binding domain of Tat in Tat-induced MAP2a/b degradation and neuronal cell death. We generated a pEYFP-TatΔ36–39 expression vector and nucleofected it into freshly isolated rat embryonic cortical neurons. Levels of expression of TatΔ36–39 at 4 and 32 h after transfection were compared with the levels of cells transfected with the empty vector (pEYFP) and with wild-type Tat (pEYFP-Tat) (Fig. 4A). In contrast to the expression of full-length Tat, which is already undetectable after 32 h, substantial amounts of EYFP-TatΔ36–39 fusion protein are still present at the same time point. The slightly slower electrophoretic migration of the TatΔ36–39 mutant compared with the full-length Tat may reflect the removal of positively charged amino acids, in the TatΔ36–39 mutant, which changes the electrophoretic behavior of the mutated Tat (Creamer and Richardson, 1984; Armstrong and Roman, 1993). We next tested the effects of TatΔ36–39 on neuronal survival. Neurons transfected with pEYFP, pEYFP-Tat, or pEYFP-TatΔ36–39 plasmids were analyzed under fluorescent light, 24 h after transfection, when Tat-positive cells are heavily reduced in number (Fig. 4B). Interestingly, at this time point, the number of cells expressing TatΔ36–39 was comparable with the number of cells expressing the control vector. Quantitatively, an increase of ∼20% in cell number was observed when control EYFP and EYFP-TatΔ36–39 were expressed (Fig. 4C). At the same 24 h time point, Tat-expressing cells were almost completely eliminated. However, lack of toxicity of the TatΔ36–39 mutant is transient, because a substantial cell loss (50% ± 2.4%) was observed after 48 h from transfection (Fig. 4C). Importantly, the expression of MAP2a/b was reduced to a lesser extent in EYFP-TatΔ36–39 than in cells expressing the full-length Tat (Fig. 4D). Quantification analysis from three independent experiments showed a pattern of MAP2 expression in pEYFP-TatΔ36–39 neurons more similar to the one in control cells (pEYFP) than in Tat-expressing neurons (Fig. 4E). Altogether, these results indicate that Tat-mediated MAP2a/b degradation involves the tubulin-binding sequence of Tat and that a mutant of Tat lacking the tubulin binding domain partially rescues MAP2 expression and delays Tat-induced neuronal cell death.
Figure 4.
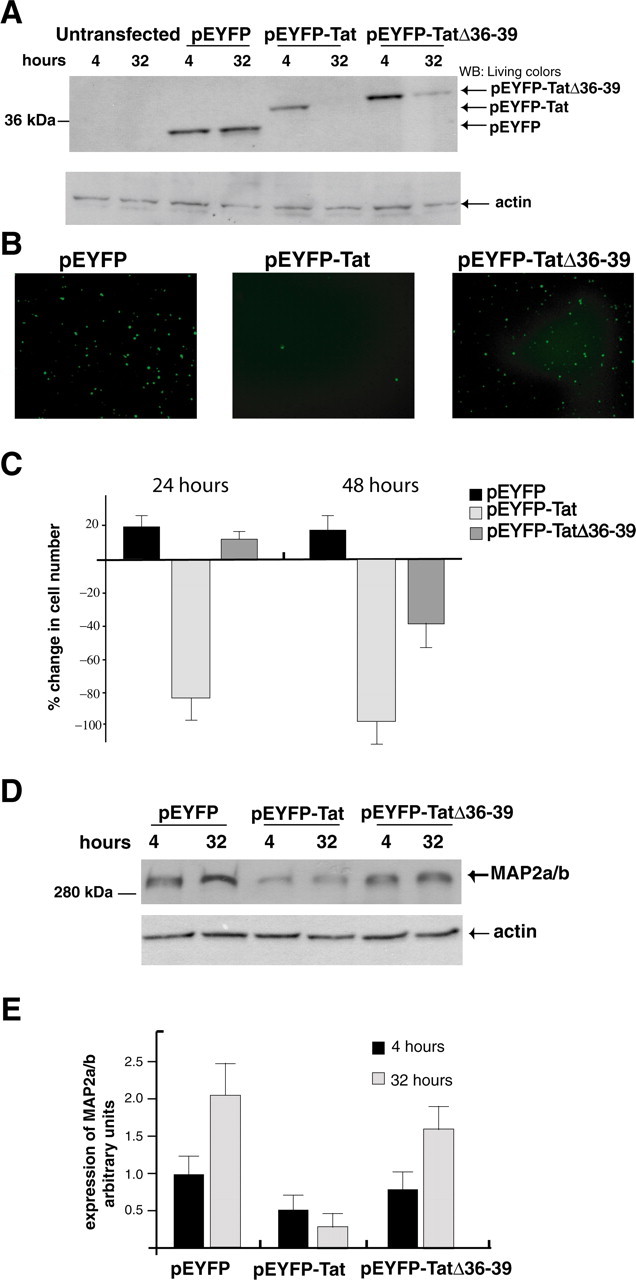
The tubulin-binding domain of Tat mediates MAP2a/b degradation. A, Western blots showing protein levels of EYFP, EYFP-Tat, and EYFP-Tat Δ36–39. The blot was developed with anti-living colors antibody that recognizes YFP. Actin was used to verify equal loading. B, Fluorescent images of EYFP, EYFP-Tat, and EYFP-TatΔ36–39-positive cells 24 h after transfection (original magnification, 100×). C, Quantitative analysis of the results from B at 24 and 48 h after transfection. Data represent the average of five independent experiments in triplicate (n = 15) with SD. D, Western blot analysis showing MAP2a/b protein level in cortical neurons at 4 and 24 h after transfection with pEYFP, pEYFP-Tat, and pEYFP-TatΔ36–39 expression vectors. E, Densitometry performed on three different Western blots obtained from independent experiments showing expression of MAP2a/b in the indicated samples. Error bars represent SEM.
Tat-mediated recruitment of the proteasome to the microtubules
To investigate the mechanisms of MAP2a/b breakdown following expression of Tat in neurons, we treated the pEYFP-Tat neurons with the calpain inhibitor (ALLN) and the proteasome inhibitor (MG-132). MAP2 proteins were detected by Western blot analysis 24 h after transfection (Fig. 5A). Among the two pathways blocked, only the treatment with proteasome inhibitor, MG-132, restored MAP2 protein levels in Tat-expressing cells. Quantitative analysis of MAP2 expression in the various experimental conditions from three independent experiments revealed a significant increase of MAP2a/b (threefold) in Tat-expressing cells treated with MG132 (Fig. 5B). Because Tat is known to bind several subunits of the 26S proteasome and enhance its proteolytic activity (Seeger et al., 1997; Apcher et al., 2003), we hypothesized that Tat-mediated proteasomal activity might be present in the proximity of neuronal microtubules. To examine this possibility, we performed immunofluorescence analysis in pEYFP-Tat transfected mature neurons (5 d after isolation) using an antibody against the 20S proteolytic core of the proteasome (Fig. 5C). Both untransfected and EYFP-expressing cells showed well-defined fluorescent particles of 20S (red) in the nucleus, and these did not overlap with cytoplasmic MAP2 (blue). In EYFP-Tat-positive cells, however, the 20S particles were detected equally well in the nucleus and in the neuronal processes where they colocalized with MAP2 (Fig. 5C). The predominant nuclear localization of the 20S in Tat-negative neurons was additionally confirmed by double labeling of the 20S proteasome and nuclear DAPI (Fig. 5D, insets). As mentioned previously, neurons transfected with pEYFP control plasmid showed homogenous distribution of the yellow fluorescent protein (green) along the processes, whereas the 20S staining was enhanced in the nuclear and perinuclear areas (highlighted in the inset picture). EYFP-Tat was also distributed through the neuronal processes but with a spotted pattern (middle panel), and the 20S particles seemed to be mainly dispersed away from the nuclear area. Of interest, EYFP-TatΔ36–39 had enhanced nuclear localization (third panel) and predominantly nuclear 20S. Quantitatively, 38 ± 7.1 and 49 ± 11.8% of the 20S proteasome colocalized with DAPI-stained nuclei in cells transfected with the control vector, EYFP, and the TatΔ36–39 mutant, respectively. However, only 9.8 ± 1.8% of 20S/DAPI colocalization was found in Tat-expressing neurons (Fig. 5D). These results strongly indicate that the tubulin-binding domain of Tat is important for the cytoplasmic redistribution of the 20S in Tat-bearing neurons.
Figure 5.
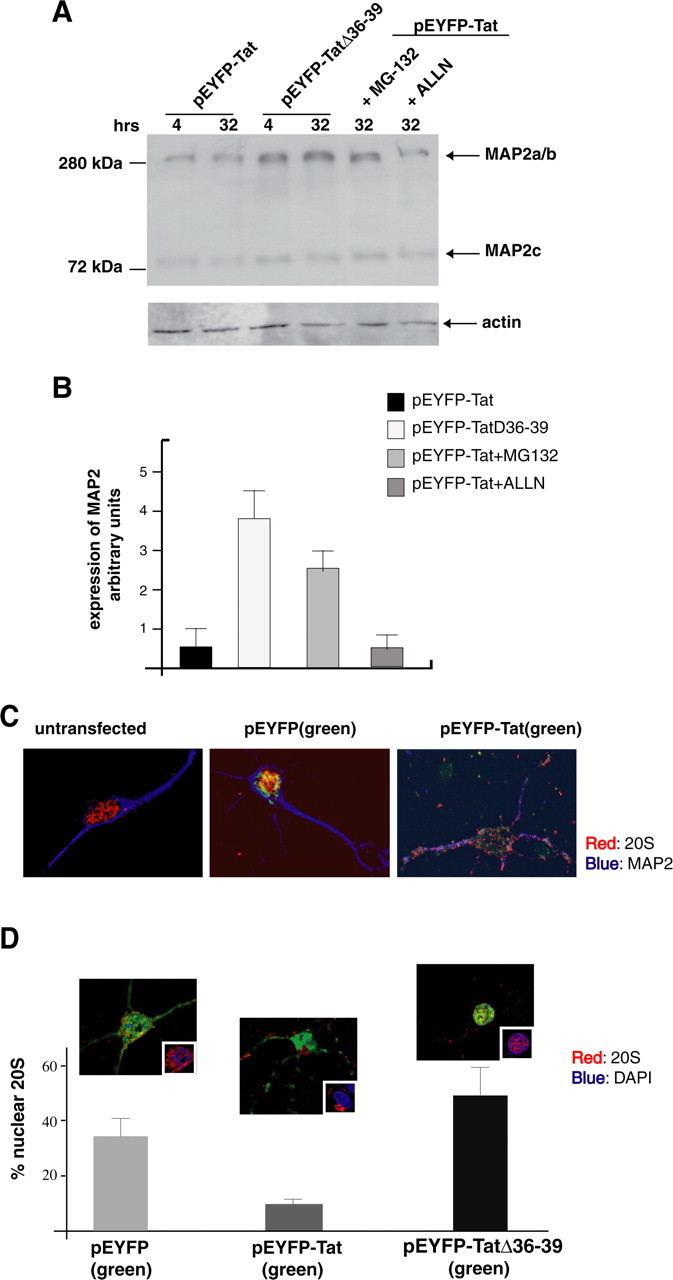
Tat-mediated recruitment of 20S particles and proteasomal degradation of MAP2a/b. A, Western blot of MAP2 expression in neurons transfected with pEYFP-Tat or pEYFP-TatΔ36–39 vectors. EYFP-Tat expressing cells were cultured in the presence or absence of protease inhibitors MG-132 (25 nm) or ALLN (0.25 μm) for the indicated time points. MG-132 as well as TatΔ36–39 preserves the stability of MAP2a/b. B, Densitometric analysis of MAP2a/b expression from three independent experiments. C, Immunofluorescent detection of the 20S proteasome (red) in differentiated neurons (cultured for 5 d, untransfected) and in EYFP or EYFP-Tat neurons 24 h after transfection (original magnification, 1000×). D, Diagram showing quantification of nuclear 20S in the indicated cell types as determined by deconvolution software (original magnification, 1000×). Representative images of neurons transfected with pEYFP, pEYFP-Tat, or pEYFP-TatΔ36–39 vectors are shown above each graph. The insets in the pictures represent the area in which colocalization between 20S (red) and DAPI (blue) was calculated. Error bars represent SEM.
Immunohistochemical analysis of MAP2, 20S, and Tat in clinical samples from HIVE cases
Next, we investigated subcellular distribution of Tat, MAP2, and 20S proteasome in brain tissue obtained from HIVE patients by immunohistochemistry. Figure 6A shows representative images of cortical sections from two HIVE cases and one control brain immunolabeled with Tat antibody. As expected, none of the neurons in the control tissue were positive for Tat, whereas in HIVE cases, Tat was detected in the cytoplasm of the neurons found in the proximity of microglial nodules (Fig. 6A, right panel). Interestingly, in surrounding reactive astrocytes, Tat was detected in both the nucleus and cytoplasm, suggesting cell type-dependent distribution of this viral protein. When labeled with the neuronal marker MAP2, a consecutive section of the same sample (control brain and the two HIVE cases) revealed a robust cytoplasmic staining in every neuron in the normal brain and significantly weaker MAP2 immunolabeling in the HIVE cases (Fig. 6A, middle panels). Quantitatively, only ∼30% of neurons in the HIVE cases showed a clear MAP2 labeling (Fig. 6B). Finally, we evaluated the subcellular localization of the 20S proteasome, and we found that the majority of control neurons were characterized by nuclear 20S immunolabeling. In contrast, a substantial number of neurons in the HIVE cases showed an apparent redistribution of the 20S particles into the cytoplasm, whereas it remained nuclear in surrounding reactive astrocytes (Fig. 6A, bottom panels). Quantitatively, the number of neurons in which 20S was detected in the cytoplasm increased from 12% in the control brain to 40% in the HIVE samples (Fig. 6C). Involvement of Tat in the cytoplasmic shift of 20S and MAP2 degradation in vivo was further investigated by immunofluorescence (Fig. 6D). HIVE brain samples were double labeled with 20S and Tat (top panels) or MAP2 and Tat (bottom panels). Similarly to the data obtained in our in vitro rat models, Tat colocalized with MAP2 or with 20S particles in the cytoplasm of human cortical neurons.
Figure 6.
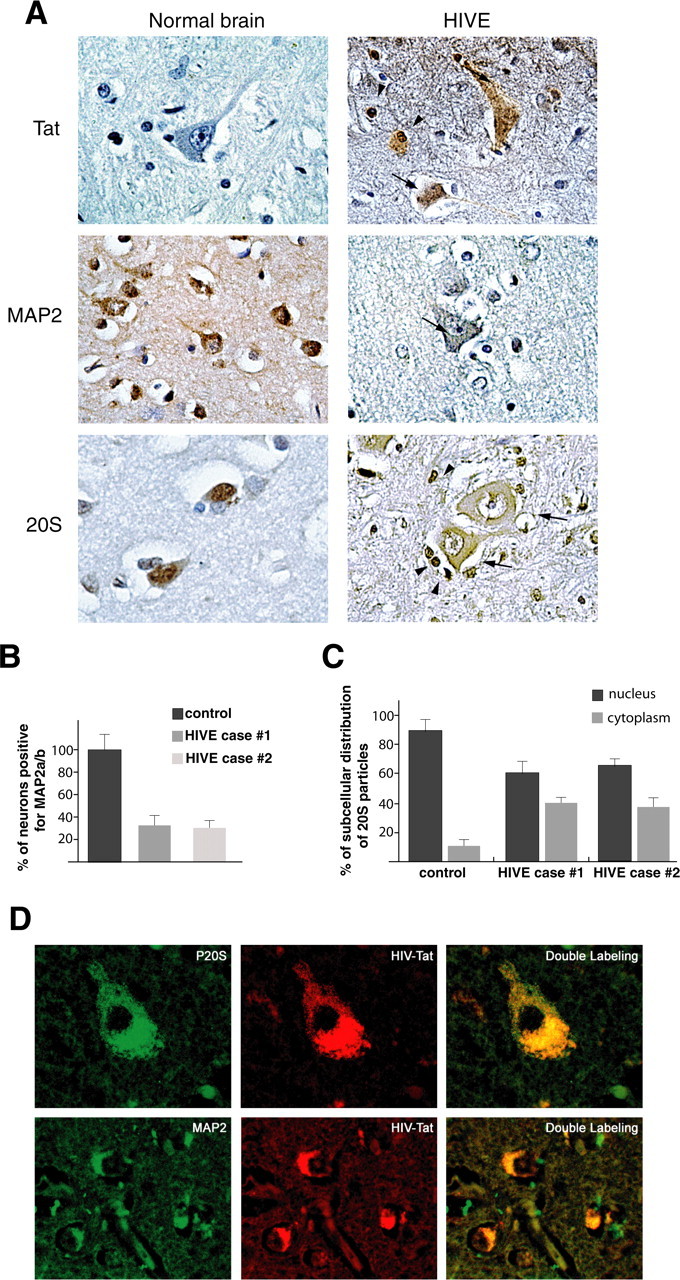
Detection of Tat, MAP2, and 20S in HIVE and control brains. A, Immunohistochemical analysis of cortical sections from a normal brain and one case of HIVE (see Materials and Methods). Black arrows indicate neurons; arrowheads indicate astrocytes (original magnification, 400×). B, Quantitative analysis showing percentage of neurons positive for MAP2 in the normal brain and in two cases of HIVE. C, Graph showing percentage of nuclear versus cytoplasmic distribution of 20S particles in normal brain and two cases of HIVE. D, Immunofluorescence analysis of HIVE brain tissues showing colocalization between 20S and Tat (top row; original magnification, 1000×) or MAP2 and Tat (bottom row; original magnification, 400×). Error bars represent SEM.
Together, our in vitro and in vivo data suggest a novel mechanism for Tat-induced neuronal damage (supplemental Fig. 7, available at www.jneurosci.org as supplemental material). It involves the binding of Tat to tubulin in the cytoplasmic compartment and recruitment of the 20S proteasome to the microtubule structure. Tubulin-mediated retention of Tat in the cytoplasm might displace MAP2 from the microtubule structure leading to its degradation via Tat-mediated 20S proteasome redistribution.
Discussion
Neuronal damage caused by HIV-1 infection is often manifested by compromised function of neurons and simplification of dendritic branches (Everall et al., 1994; Masliah et al., 1997; Gelman et al., 2004). The ability of HIV-1 Tat to be secreted by HIV-infected cells and to freely enter neighboring noninfected cells, including neurons, suggests a critical role for this viral protein in mediating neuronal damage. Considering that Tat can bind tubulin polymers (Chen et al., 2002) and block cellular differentiation and neurite outgrowth (Mondal and Agrawal, 1996; Bergonzini et al., 2004), we asked whether Tat-mediated neurotoxicity depends on its interaction with microtubules, which are the major structural and functional component of neuronal processes. Here, we used a model of rat primary neurons to investigate the effects of Tat in two different stages of neuronal maturation: an early stage in which the cells start to extend the neurites and a later stage in which neuronal processes are fully extended. Our data indicate that Tat has a dual effect in blocking neurite extension at the early stage and causing extensive damage to neuronal processes at the later stage. Experiments with a mutant Tat demonstrated that Tat-mediated inhibition of neurite outgrowth depends on the presence of the tubulin-binding site in the core region of Tat. This, in turn, induces degradation of the MAP2a/b proteins, which are critical for the stability of neuronal microtubules. The mechanism appears to involve Tat-mediated recruitment of the proteasome to the proximity of microtubules. This route of action of Tat also occurs in mature cells when neuronal processes are fully extended. Of interest, the extent of MAP2 degradation was comparable in neurons expressing Tat and in neurons treated with recombinant Tat.
Among the MAP2 isoforms, only the high molecular weight forms MAP2a and MAP2b appeared to be affected by Tat with deleterious consequences to the function and survival of the neurons. Because MAP2 protein levels decrease shortly after Tat expression (as early as 4 h after transfection), loss of MAP2 is an early event that precedes neuronal death. This finding is in line with several other reports showing a correlation between loss of MAP2 and synaptic damage to neuronal dendrites observed in patients with HIV encephalopathy (Masliah et al., 1997, 2004). Additionally, degradation of MAP2a/b occurred in a dose-dependent manner in neuronal cultures exposed to recombinant Tat1–72 (Fig. 2F,C).
The tubulin-binding domain of Tat is necessary for Tat-mediated MAP2 breakdown, because the Tat mutant lacking this particular sequence (TatΔ36–39) failed to promote MAP2 degradation. Previous reports indicate that the expression of MAP proteins can be regulated by activation of calpain proteolytic pathway (Buddle et al., 2003) and, to a lesser extent, by the proteasomal pathway (David et al., 2002; Feuillette et al., 2005). Therefore, to investigate possible mechanisms involved in Tat-mediated MAP2 breakdown, we treated Tat-expressing neurons with specific inhibitors of the calpain or proteasome pathways. Among the inhibitors used, only treatment with proteasome inhibitor, MG-132, preserved MAP2 degradation in Tat-positive neurons, suggesting a critical role for proteasomal activity in Tat-mediated MAP2 degradation.
With respect to apoptosis, the region of Tat containing the core and the basic domains (amino acids 31–61) has been often associated with neurotoxicity (Sabatier et al., 1991; Nath et al., 1996). In particular, the basic domain (amino acids 49–57) is the minimal neurotoxic region causing cell death (Sabatier et al., 1991). In line with these findings, expression of a mutant of Tat lacking the tubulin-binding region, TatΔ36–39, only delayed apoptotic events in rat embryonic cortical neurons. However, this tubulin-binding domain appeared necessary for Tat-mediated MAP2 degradation. Immunofluorescence studies performed on differentiated neurons further revealed Tat-dependent recruitment of 20S particles to microtubules (Fig. 5). In agreement with other studies showing that neurons from rat and human brains display mainly nuclear immunolabeling of the 20S proteasome (Adori et al., 2005), we also found predominant nuclear and perinuclear staining of the 20S in cultured rat embryonic cortical neurons and in control normal human brains. Importantly, a dramatic redistribution of the 20S particles from the nucleus to the cytoplasm was observed in Tat-expressing cell cultures, as well as in cortical neurons of HIVE brain tissue sections. It has also been reported that, during apoptosis, proteasomes are transported from the nucleus into the cytoplasm where they associate with cytoplasmic blebs (Pitzer et al., 1996). In this respect, the cytoplasmic shift of the 20S observed in Tat-positive cells might be an event that follows apoptotic stimuli induced by Tat. However, the intense nuclear staining of the 20S in TatΔ36–39, which is also toxic to the cells, suggests that the Tat-induced apoptosis does not require the shift of the 20S to the cytoplasm. In contrast, these data support a model in which Tat is retained in the cytoplasm via its tubulin binding domain, and that the tubulin/Tat complex is required for the Tat-mediated recruitment of the 20S to microtubules, where it promotes MAP2 degradation (supplemental Fig. 7, available at www.jneurosci.org as supplemental material). In this model, localization of Tat in the neuronal cytoplasm appears to play a critical role. In contrast to other cell types in which expression of Tat is detected exclusively in the nucleus/nucleolus, we observed cytoplasmatic redistribution of Tat protein in neurons both in vitro (Figs. 1B, 5B) and in vivo (Figs. 6, 7). Direct binding of Tat to proteins such as tubulin (Chen et al., 2002), Lis1 (Epie et al., 2005), and Hic (Gautier et al., 2005) that vary in abundance between different cell types could mediate the distribution of Tat within the cytoplasmic compartment. Retention of Tat in the neuronal cytoplasm may in turn trigger proteasome relocation to the cytoplasm causing MAP2 degradation. Indeed, a significant subcellular redistribution of 20S particles was observed in cortical neurons of HIVE clinical samples (Fig. 6), suggesting that the cytoplasmic shift of the 20S might play a role in HIV-associated neurological disorders. Although the nature of the 20S complexes remains to be elucidated, the Tat-dependent transfer of the 20S from the nucleus to the cytoplasm of neurons defines a new molecular mechanism by which viral proteins released in the brain could cause neuronal damage in HIV encephalopathy.
Footnotes
This work was supported by a grant from the National Institutes of Health (F.P.). We thank J. Otte and L. Hodge for technical support, Krzysztof Reiss for helpful discussion, and Martyn White for editorial assistance.
References
- Adori C, Low P, Moszkovkin G, Bagdy G, Laszlo L, Kovacs GG (2005). Subcellular distribution of components of the ubiquitin-proteasome-system in non-diseased human and rat brain. J Histochem Cytochem 54:263–267. [DOI] [PubMed] [Google Scholar]
- Amini S, Mameli G, Del Valle L, Skowronska A, Reiss K, Gelman BB, White MK, Khalili K, Sawaya BE (2005). p73 Interacts with human immunodeficiency virus type 1 Tat in astrocytic cells and prevents its acetylation on lysine 28. Mol Cell Biol 25:8126–8138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki M, Abe K, Yoshida T, Hattori A, Kogure K, Itoyama Y (1995). Early immunohistochemical changes of microtubule based motor proteins in gerbil hippocampus after transient ischemia. Brain Res 669:189–196. [DOI] [PubMed] [Google Scholar]
- Apcher GS, Heink S, Zantopf D, Kloetzel PM, Schmid HP, Mayer RJ, Kruger E (2003). Human immunodeficiency virus-1 Tat protein interacts with distinct proteasomal alpha and beta subunits. FEBS Lett 553:200–204. [DOI] [PubMed] [Google Scholar]
- Archibald SL, Masliah E, Fennema-Notestine C, Marcotte TD, Ellis RJ, McCutchan JA, Heaton RK, Grant I, Mallory M, Miller A, Jernigan TL (2004). Correlation of in vivo neuroimaging abnormalities with postmortem human immunodeficiency virus encephalitis and dendritic loss. Arch Neurol 61:369–376. [DOI] [PubMed] [Google Scholar]
- Armstrong DJ, Roman A (1993). The anomalous electrophoretic behavior of the human papillomavirus type 16 E7 protein is due to the high content of acidic amino acid residues. Biochem Biophys Res Commun 192:1380–1387. [DOI] [PubMed] [Google Scholar]
- Bergonzini V, Delbue S, Wang JY, Reiss K, Prisco M, Amini S, Khalili K, Peruzzi F (2004). HIV-Tat promotes cellular proliferation and inhibits NGF-induced differentiation through mechanisms involving Id1 regulation. Oncogene 23:7701–7711. [DOI] [PubMed] [Google Scholar]
- Binder LI, Frankfurter A, Rebhun LI (1985). The distribution of tau in the mammalian central nervous system. J Cell Biol 101:1371–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buddle M, Eberhardt E, Ciminello LH, Levin T, Wing R, DiPasquale K, Raley-Susman KM (2003). Microtubule-associated protein 2 (MAP2) associates with the NMDA receptor and is spatially redistributed within rat hippocampal neurons after oxygen-glucose deprivation. Brain Res 978:38–50. [DOI] [PubMed] [Google Scholar]
- Cassimeris L, Spittle C (2001). Regulation of microtubule-associated proteins. Int Rev Cytol 210:163–226. [DOI] [PubMed] [Google Scholar]
- Chen D, Wang M, Zhou S, Zhou Q (2002). HIV-1 Tat targets microtubules to induce apoptosis, a process promoted by the pro-apoptotic Bcl-2 relative Bim. EMBO J 21:6801–6810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Kanai Y, Cowan NJ, Hirokawa N (1992). Projection domains of MAP2 and tau determine spacings between microtubules in dendrites and axons. Nature 360:674–677. [DOI] [PubMed] [Google Scholar]
- Creamer LK, Richardson T (1984). Anomalous behavior of bovine alpha s1- and beta-caseins on gel electrophoresis in sodium dodecyl sulfate buffers. Arch Biochem Biophys 234:476–486. [DOI] [PubMed] [Google Scholar]
- David DC, Layfield R, Serpell L, Narain Y, Goedert M, Spillantini MG (2002). Proteasomal degradation of tau protein. J Neurochem 83:176–185. [DOI] [PubMed] [Google Scholar]
- Dawson DA, Hallenbeck JM (1996). Acute focal ischemia-induced alterations in MAP2 immunostaining: description of temporal changes and utilization as a marker for volumetric assessment of acute brain injury. J Cereb Blood Flow Metab 16:170–174. [DOI] [PubMed] [Google Scholar]
- Del Valle L, Croul S, Morgello S, Amini S, Rappaport J, Khalili K (2000). Detection of HIV-1 Tat and JCV capsid protein, VP1, in AIDS brain with progressive multifocal leukoencephalopathy. J Neurovirol 6:221–228. [DOI] [PubMed] [Google Scholar]
- Desai A, Mitchison TJ (1997). Microtubule polymerization dynamics. Annu Rev Cell Dev Biol 13:83–117. [DOI] [PubMed] [Google Scholar]
- Epie N, Ammosova T, Sapir T, Voloshin Y, Lane WS, Turner W, Reiner O, Nekhai S (2005). HIV-1 Tat interacts with LIS1 protein. Retrovirology 2:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everall IP, Glass JD, McArthur J, Spargo E, Lantos P (1994). Neuronal density in the superior frontal and temporal gyri does not correlate with the degree of human immunodeficiency virus-associated dementia. Acta Neuropathol (Berl) 88:538–544. [DOI] [PubMed] [Google Scholar]
- Ferralli J, Doll T, Matus A (1994). Sequence analysis of MAP2 function in living cells. J Cell Sci 107:3115–3125. [DOI] [PubMed] [Google Scholar]
- Feuillette S, Blard O, Lecourtois M, Frebourg T, Campion D, Dumanchin C (2005). Tau is not normally degraded by the proteasome. J Neurosci Res 80:400–405. [DOI] [PubMed] [Google Scholar]
- Gautier VW, Sheehy N, Duffy M, Hashimoto K, Hall WW (2005). Direct interaction of the human I-mfa domain-containing protein, HIC, with HIV-1 Tat results in cytoplasmic sequestration and control of Tat activity. Proc Natl Acad Sci USA 102:16362–16367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelman BB, Soukup VM, Schuenke KW, Keherly MJ, Holzer C III, Richey FJ, Lahart CJ (2004). Acquired neuronal channelopathies in HIV-associated dementia. J Neuroimmunol 157:111–119. [DOI] [PubMed] [Google Scholar]
- Guha S, Manna TK, Das KP, Bhattacharyya B (1998). Chaperone-like activity of tubulin. J Biol Chem 273:30077–30080. [DOI] [PubMed] [Google Scholar]
- Hara MR, Agrawal N, Kim SF, Cascio MB, Fujimuro M, Ozeki Y, Takahashi M, Cheah JH, Tankou SK, Hester LD, Ferris CD, Hayward SD, Snyder SH, Sawa A (2005). S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat Cell Biol 7:665–674. [DOI] [PubMed] [Google Scholar]
- Hirokawa N (1994). Microtubule organization and dynamics dependent on microtubule-associated proteins. Curr Opin Cell Biol 6:74–81. [DOI] [PubMed] [Google Scholar]
- Ishitani R, Chuang DM (1996). Glyceraldehyde-3-phosphate dehydrogenase antisense oligodeoxynucleotides protect against cytosine arabinonucleoside-induced apoptosis in cultured cerebellar neurons. Proc Natl Acad Sci USA 93:9937–9941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul M, Garden GA, Lipton SA (2001). Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature 410:988–994. [DOI] [PubMed] [Google Scholar]
- Kusner LL, Sarthy VP, Mohr S (2004). Nuclear translocation of glyceraldehyde-3-phosphate dehydrogenase: a role in high glucose-induced apoptosis in retinal Muller cells. Invest Ophthalmol Vis Sci 45:1553–1561. [PubMed] [Google Scholar]
- Langford D, Grigorian A, Hurford R, Adame A, Crews L, Masliah E (2004). The role of mitochondrial alterations in the combined toxic effects of human immunodeficiency virus Tat protein and methamphetamine on calbindin positive-neurons. J Neurovirol 10:327–337. [DOI] [PubMed] [Google Scholar]
- Lopez LA, Sheetz MP (1995). A microtubule-associated protein (MAP2) kinase restores microtubule motility in embryonic brain. J Biol Chem 270:12511–12517. [DOI] [PubMed] [Google Scholar]
- Masliah E, Heaton RK, Marcotte TD, Ellis RJ, Wiley CA, Mallory M, Achim CL, McCutchan JA, Nelson JA, Atkinson JH, Grant I (1997). Dendritic injury is a pathological substrate for human immunodeficiency virus-related cognitive disorders. HNRC Group. The HIV Neurobehavioral Research Center. Ann Neurol 42:963–972. [DOI] [PubMed] [Google Scholar]
- Masliah E, Roberts ES, Langford D, Everall I, Crews L, Adame A, Rockenstein E, Fox HS (2004). Patterns of gene dysregulation in the frontal cortex of patients with HIV encephalitis. J Neuroimmunol 157:163–175. [DOI] [PubMed] [Google Scholar]
- Mondal D, Agrawal KC (1996). Effect of HIV type 1 Tat protein on butyric acid-induced differentiation in a hematopoietic progenitor cell line. AIDS Res Hum Retroviruses 12:1529–1536. [DOI] [PubMed] [Google Scholar]
- Nath A (2002). Human immunodeficiency virus (HIV) proteins in neuropathogenesis of HIV dementia. J Infect Dis 186:[Suppl 2], S193–S198. [DOI] [PubMed] [Google Scholar]
- Nath A, Psooy K, Martin C, Knudsen B, Magnuson DS, Haughey N, Geiger JD (1996). Identification of a human immunodeficiency virus type 1 Tat epitope that is neuroexcitatory and neurotoxic. J Virol 70:1475–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry SW, Norman JP, Litzburg A, Zhang D, Dewhurst S, Gelbard HA (2005). HIV-1 transactivator of transcription protein induces mitochondrial hyperpolarization and synaptic stress leading to apoptosis. J Immunol 174:4333–4344. [DOI] [PubMed] [Google Scholar]
- Peruzzi F (2006). The multiple functions of HIV-1 Tat: proliferation versus apoptosis. Front Biosci 11:708–717. [DOI] [PubMed] [Google Scholar]
- Peruzzi F, Bergonzini V, Aprea S, Reiss K, Sawaya BE, Rappaport J, Amini S, Khalili K (2005). Cross talk between growth factors and viral and cellular factors alters neuronal signaling pathways: implication for HIV-associated dementia. Brain Res Brain Res Rev 50:114–125. [DOI] [PubMed] [Google Scholar]
- Pitzer F, Dantes A, Fuchs T, Baumeister W, Amsterdam A (1996). Removal of proteasomes from the nucleus and their accumulation in apoptotic blebs during programmed cell death. FEBS Lett 394:47–50. [DOI] [PubMed] [Google Scholar]
- Riederer B, Matus A (1985). Differential expression of distinct microtubule-associated proteins during brain development. Proc Natl Acad Sci USA 82:6006–6009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatier JM, Vives E, Mabrouk K, Benjouad A, Rochat H, Duval A, Hue B, Bahraoui E (1991). Evidence for neurotoxic activity of tat from human immunodeficiency virus type 1. J Virol 65:961–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez C, Diaz-Nido J, Avila J (2000). Phosphorylation of microtubule-associated protein 2 (MAP2) and its relevance for the regulation of the neuronal cytoskeleton function. Prog Neurobiol 61:133–168. [DOI] [PubMed] [Google Scholar]
- Sarkar T, Mitra G, Gupta S, Manna T, Poddar A, Panda D, Das KP, Bhattacharyya B (2004). MAP2 prevents protein aggregation and facilitates reactivation of unfolded enzymes. Eur J Biochem 271:1488–1496. [DOI] [PubMed] [Google Scholar]
- Sato-Harada R, Okabe S, Umeyama T, Kanai Y, Hirokawa N (1996). Microtubule-associated proteins regulate microtubule function as the track for intracellular membrane organelle transports. Cell Struct Funct 21:283–295. [DOI] [PubMed] [Google Scholar]
- Saunders PA, Chen RW, Chuang DM (1999). Nuclear translocation of glyceraldehyde-3-phosphate dehydrogenase isoforms during neuronal apoptosis. J Neurochem 72:925–932. [DOI] [PubMed] [Google Scholar]
- Sawa A, Khan AA, Hester LD, Snyder SH (1997). Glyceraldehyde-3-phosphate dehydrogenase: nuclear translocation participates in neuronal and nonneuronal cell death. Proc Natl Acad Sci USA 94:11669–11674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeger M, Ferrell K, Frank R, Dubiel W (1997). HIV-1 tat inhibits the 20 S proteasome and its 11 S regulator-mediated activation. J Biol Chem 272:8145–8148. [DOI] [PubMed] [Google Scholar]