

Abstract
Ischemic strokes are often associated with late-onset epilepsy, but the underlying mechanisms are poorly understood. In the hippocampus, which is one of the regions most sensitive to ischemic challenge, global ischemia induces a complete loss of CA1 pyramidal neurons, whereas the resistant CA3 pyramidal neurons display a long-term hyperexcitability several months after the insult. The mechanisms of this long-term hyperexcitability remain unknown despite its clinical implication. Using chronic in vivo EEG recordings and in vitro field recordings in slices, we now report spontaneous interictal epileptiform discharges in the CA3 area of the hippocampus from post-ischemic rats several months after the insult. Whole-cell recordings from CA3 pyramidal neurons, revealed a permanent reduction in the frequency of spontaneous and miniature GABAergic IPSCs and a parallel increase in the frequency of spontaneous and miniature glutamatergic postsynaptic currents. Global ischemia also induced a dramatic loss of GABAergic interneurons and terminals together with an increase in glutamatergic terminals in the CA3 area of the hippocampus. Altogether, our results show a morpho-functional reorganization in the CA3 network several months after global ischemia, resulting in a net shift in the excitatory–inhibitory balance toward excitation that may constitute a substrate for the generation of epileptiform discharges in the post-ischemic hippocampus.
Keywords: GABA, glutamate, interneuron, ischemia, interictal, stroke
Introduction
Seizures are among the most common neurological sequelae of stroke (McNamara, 1979; Cocito et al., 1982; Silverman et al., 2002), and ischemic episodes are often associated with late-onset epilepsy (Bladin et al., 2000; Afsar et al., 2003; Lossius et al., 2005). Studies in animal models of global ischemia report a specific neuronal loss (Pulsinelli, 1985; Schmidt-Kastner and Freund, 1991) associated with a long-term hyperexcitability in the most vulnerable regions, notably the hippocampus (Mittmann et al., 1998; Congar et al., 2000; Crépel et al., 2003). The mechanisms of ischemia-induced neuronal death have been studied extensively; however, despite their clinical implications, the mechanisms of long-term network hyperexcitability are still poorly understood.
One major issue raised by brain lesions is the fate of surviving neurons and synapses. Indeed, functional reorganization often occurs after central or peripheral lesions and involves modifications in the excitatory–inhibitory balance in resistant cells. In animal models of the temporal lobe of epilepsy (TLE), the death of specific types of GABAergic interneurons (Obenaus et al., 1993; Buckmaster and Jongen-Relo, 1999; Cossart et al., 2001; Dinocourt et al., 2003; Kobayashi and Buckmaster, 2003) is associated with an aberrant sprouting of glutamatergic fibers (Represa et al., 1987; Sutula et al., 1988; Okazaki et al., 1995; Buckmaster and Dudek, 1999; Esclapez et al., 1999; Epsztein et al., 2005) in different hippocampal fields. These changes result in an enhanced synaptic excitation (Esclapez et al., 1999; Wuarin and Dudek, 2001; Epsztein et al., 2005) together with a decreased synaptic inhibition (Cossart et al., 2001; Kobayashi and Buckmaster, 2003; Shao and Dudek, 2005).
After global ischemia (as occurs in cardiac arrest, coronary artery occlusion, or heart bypass surgery in humans), a specific pattern of neuronal degeneration is observed in the hippocampus both in patients (Brierley and Cooper, 1962; Siesjo, 1981; Zola-Morgan et al., 1986) and animal models (Pulsinelli and Brierley, 1979; Pulsinelli, 1985; Crépel et al., 1989; Schmidt-Kastner and Freund, 1991; Arabadzisz and Freund, 1999) with the degeneration of CA1 pyramidal cells, whereas CA3 pyramidal cells are preserved. A loss of subsets of GABAergic interneurons have also been reported in the CA3 area (Hsu and Buzsaki, 1993; Arabadzisz and Freund, 1999) together with a long-term hyperexcitability (Congar et al., 2000). The aim of the present study was to analyze the long-term morpho-functional consequences of global ischemia on glutamatergic and GABAergic systems in the CA3 area of the hippocampus that could account for the hyperexcitability of resistant CA3 pyramidal cells (Congar et al., 2000).
Using in vivo and in vitro recordings, we report spontaneous epileptiform discharges in the hippocampus several months after ischemia. This enhancement of excitability is associated with an important reduction in GABAergic spontaneous and miniature synaptic transmission in CA3 pyramidal neurons together with a massive loss of GABAergic interneurons in the CA3 region of the hippocampus. We also report an increase in spontaneous and miniature glutamatergic synaptic transmission in these cells associated with an enhancement of glutamatergic terminals in the CA3 area. We conclude that a morpho-functional reorganization occurs in the CA3 network several months after global ischemia that may be a substrate for the occurrence of synchronized epileptiform discharges in the hippocampus of post-ischemic rats.
Materials and Methods
Forebrain ischemia.
Experiments involving animals were approved by the French ethical science committee. Rats had access to food and water ad libitum and were housed under a 12 h light/dark cycle at 22–24°C. Male Wistar rats (180–200 g; Janvier Breeding Center, Le Genest-Saint-Isle, France) were used in these experiments. The global ischemic episode was achieved by four-vessel occlusion, according to a previously described method (Pulsinelli and Brierley, 1979; Congar et al., 2000). The vertebral arteries were first electrocauterized under anesthesia (7% chloral hydrate). The alar foramina of the first cervical vertebrae was identified trough a dorsal incision. Direct observation of the bony tunnel of the first vertebra (through which the vertebral arteries travel) was accomplished by pivoting a dissecting microscope (10×) so that the viewing angle of the microscope was in line with the axis of the tunnel. A small (<1 mm) electrocautery needle was inserted vertically through the alar foramina and down into the bony tunnel of the first vertebra. Brief intermittent application of current to the needle tip cauterized the vertebral arteries and averted bleeding in the majority of rats. Complete vertebral artery occlusion was assured in all rats by direct visualization of the bony perimeter of the tunnel in which the vertebral arteries travel. If this tunnel is empty and if its perimeter can be visualized, the vertebral arteries are well cauterized. An atraumatic arterial occlusion device was then placed around each common carotid artery and exteriorized through a ventral neck incision. The next day, when the animals had recovered from the procedure, carotid arteries were clamped in the unanesthetized rat for 20–25 min. Only 10% of the rats died within 2–3 min from respiratory failures. Seventy percent of the rats became unresponsive and lost their righting reflex within 30 s of carotid artery ligation while spontaneously breathing, thanks to the relatively good perfusion of the brainstem via the anterior spinal artery (Pulsinelli and Brierley, 1979). Twenty percent of the rats became only lethargic after artery occlusion and were not included in the study as described previously (Pulsinelli and Brierley, 1979). The carotid artery clamps from unresponsive rats were then released and recovered a locomotor behavior within the next half-hour. Among these rats, 10% developed spontaneous seizures within the first 72 h after the ischemic insult and subsequently died or were removed from the study. Thus, all in all, ∼60% of the operated rats could be used in the present study (n = 25 post-ischemic rats). These rats were allowed to survive until morphological or electrophysiological experiments were performed (2–6 months later). Rats with only electrocauterized vertebral arteries (sham-operated rats) and controls rats of the same age were used as controls (n = 23).
In vivo recordings. Post-ischemic adult Wistar rats (n = 3) were anesthetized with a mixture of ketamine (1 mg/kg) and xylazine (0.5 mg/kg). One bipolar steel (200 μm in diameter) hippocampal electrode was stereotaxically implanted in the CA3 area (3.5 mm posteriorly, 2.5 mm laterally, and 3.7 mm deeply from the bregma) and anchored to a micro-connector on the skull with dental cement. After a 3 d recovery, animals were connected and recorded continuously with a video-EEG system (Deltamed, Paris, France) for 7 d, 24 h per day. The animals were freely moving, and food and water were given ad libitum. EEG traces represented the difference of potential between the two hippocampal electrodes. Signals were amplified (1000×), filtered at 0.16–97 Hz pass, acquired at 256 Hz using a time constant of 0.3 s and a notch at 50 Hz, and analyzed using the Coherence 3NT program (Deltamed). Tracings were visually inspected for interictal epileptiform discharges (IEDs), and electrographic seizure activity as defined previously (Pedley, 1980). IED frequency was determined during 60 s epochs for 24 h (1440 contiguous epochs per rat). Animals that did not experience ischemia (n = 3) were implanted using the same protocol, as control rats. At the end of the recording period, animals were killed and the position of the bipolar electrode in the hippocampus was verified histologically (data not shown).
Hippocampal slices preparation.
Experiments were performed in CA3 hippocampal neurons in slices obtained from 15 rats (350–500 g) 2–6 months after ischemia and from 15 control rats. The animals were perfused intracardially, under chloral hydrate (350 mg/kg) anesthesia, with an ice-cold and oxygenated modified artificial CSF (mACSF; 0–3°C) containing the following (in mm): 2 KCl, 0.5 CaCl2, 7 MgCl2, 1.2 NaH2PO4, 26 NaHCO3, 11 glucose, and 250 sucrose equilibrated with 95% O2--5% CO2, pH 7.4. After decapitation, the brain was quickly removed from the skull, hippocampi were dissected free, and 400-μm-thick transverse slices were prepared using a Leica (Nussloch, Germany) VT 1000E tissue slicer in the same 0–3°C ice-cold mACSF equilibrated with 95% O2–5% CO2, pH 7.4. The slices were then incubated at room temperature in an oxygenated ACSF containing (in mm) 126 NaCl, 3.5 KCl, 2 CaCl2, 1.3 MgCl2, 1.2 NaH2PO4, 25 NaHCO3, and 11 glucose, equilibrated with 95% O2–5% CO2, pH 7.4. After a 2 h recovery period, hippocampal slices were transferred one at a time to a submerged recording chamber and superfused continuously (2.5–3 ml/min at 33–34°C or 30–32°C depending on the experiment) with ACSF. The study was performed using hippocampal slices from the medial pole to have a homogenous extent of the neuronal lesion (Crépel et al., 1989) (see below, Histology and immunocytochemistry).
Extracellular and patch-clamp recordings.
Extracellular recordings of spontaneous activity in the CA3 pyramidal layer were performed with glass electrodes (2–3 MΩ, filled with ACSF) using a DAM-80 amplifier (World Precision Instruments, Sarasota, FL). Signals were filtered with a 0.1 Hz to 3 kHz bandpass. Signals were digitized with a Labmaster interface card (Molecular Devices, Union City, CA) at 10 kHz. Ripple oscillations were determined from 400 to 500 Hz low-pass filtered raw data (8-pole Bessel filter). Fast ripple oscillations were determined from 200 Hz high-pass-filtered raw data (8-pole Bessel filter). Population IED-like activities were determined from 1 to 100 Hz bandpass-filtered raw data (8-pole Bessel filter). Filtering was performed using Clampfit 8.2 (Molecular Devices). Power spectrum analysis was performed on raw data by applying a Kaiser function (α = 1) for each population burst or ripple event using Clampfit 8.2 and averaged across all our recordings.
Whole-cell patch-clamp recordings of spontaneous and miniature synaptic currents were obtained using tight-seal electrodes (5–7 MΩ) filled with a Cs-gluconate-based internal solution that contained the following (in mm): 120 Cs-gluconate, 10 KCl, 10 NaCl, 0.1 CaCl2, 1 EGTA, 10 HEPES, 2 MgATP, and 0.5 GTP, pH 7.25. Biocytin (0.5%) was added to the patch pipette solution passively diffused into neurons for post hoc analysis. morphological analysis (see below, Histology and Immunocytochemistry). Signals were fed to an Axopatch 2B amplifier (Molecular Devices), filtered (2 kHz), digitized (10 kHz) with a Labmaster interface card (Molecular Devices) to a personal computer, and analyzed with the MiniAnalysis 6.0.1 program (Synaptosoft, Decatur, GA). Access resistance ranged between 10 and 20 MΩ, and the results were discarded if it changed by >20%. The baseline noise ranged between 3 and 5 pA, and an amplitude threshold of 2 pA more than the baseline noise was used for event detection. The false-positive and false-negative events represented <5% of the total events. Each single event in each recorded cell was fully characterized by the following parameters: rise time (10–90%), amplitude, and decay time constants, calculated using MiniAnalysis 6.0.1. Glutamatergic events were discriminated based on their kinetic characteristics as described previously (Cossart et al., 2002). All of the experimental values are given as means ± SEMs. The Student’s t test was used for statistical comparisons, except when indicated otherwise (p < 0.05 was considered significant).
Histology and immunocytochemistry.
The extent and the specificity of the lesion was determined using cresyl violet on slices used for electrophysiological recordings (Crépel et al., 1989). Previous data have shown that a 20–25 min occlusion is necessary to reproducibly obtain the specific histopathological pattern observed in the hippocampus of patients after transient global ischemia (Brierley and Cooper, 1962; Siesjo, 1981; Zola-Morgan et al., 1986) (i.e., a near complete loss of CA1 pyramidal cells with a good preservation of CA3 pyramidal and dentate granule cells). Furthermore, this pattern of neuronal degeneration show a clear septotemporal gradient: (1) in the septal and medial pole, an almost complete pyramidal cell loss is observed in the CA1 area, but CA3 pyramidal cells and dentate granule cells are preserved, while (2) in the most temporal pole, a loss of CA3 pyramidal and granule neurons is also observed (Crépel et al., 1989). Because we were interested in the long-term consequences of global ischemia on surviving CA3 pyramidal cells and to limit heterogeneity in the results, only slices from the medial pole were used in our electrophysiological and immunochemical experiments. Using these slices, we observed a near complete loss of CA1 pyramidal cells with a good preservation of CA3 pyramidal cells (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) (Pulsinelli and Brierley, 1979; Crépel et al., 1989; for review, see Schmidt-Kastner and Freund, 1991). Pyramidal cells recorded in patch-clamp configuration were filled with biocytin and morphologically identified post hoc accordingly to the protocol used previously (Cossart et al., 2002). Only well defined CA3 pyramidal cells were considered in the present study.
For immunocytochemical analysis of interneurons, control (n = 5) and ischemic (n = 7) animals were perfused 2–6 months after the insult with 4% paraformaldehyde and 0.2% glutaraldehyde. Vibratome sections (60 μm thick) were obtained and kept in a cold PBS–sodium azide mix until use. Slices from control and ischemic animals were processed in parallel, with sections from each brain in different wells, but using the same solutions. Each experiment including both control and ischemic animals was repeated three to five times, to minimize differences related to manipulations. The following antibodies were used: rabbit anti-calretinin (1:5000; Swant, Bellinzona, Switzerland), rabbit anti-cholecystokinin (CCK; 1:500; Peninsula Laboratories, San Carlos, CA), rabbit anti-GABA (1:1000; Sigma, St. Louis, MO), mouse anti-parvalbumin (PV; 1:5000; Swant), rabbit anti-somatostatin (1:4000; Peninsula Laboratories), guinea pig anti-vesicular GABA transporter (vGAT; 1:2000; Synaptic Systems, Goettingen, Germany), rabbit anti-vesicular glutamate tranporter 1 (vGLUT1; 1:1000; Synaptic Systems), and vGLUT3 (1:1000; a generous gift from Dr. S. El Mestikawy, Institut National de la Santé et de la Recherche Médicale Unité 513, Creteil, France). They were revealed with Cy3- or FITC-conjugated secondary antibodies (The Jackson Laboratory, Bar Harbor, ME). In all cases, no labeling was detected when specific antibodies were omitted or replaced by normal rabbit or mouse serums. Preparations (eight different sections per animal per antibody) were observed in a confocal microscope (LSM 510; Zeiss, Thornwood, NY) using for each antibody the same scanning conditions for control and ischemic sections. For stereological analysis of cell densities, stacks of six optical sections (5 μm thick) from each preparation were collected and the analysis software ImageJ 1.31g (Wayne Rasband, National Institutes of Health, Bethesda, MD) was used. Values were calculated and expressed as a percentage of control values/mm3± SE. Puncta stainings in the pyramidal cell layer obtained with GABA, vGAT, vGLUT1, vGLUT3, CCK, and PV antibodies were evaluated measuring the optical fluorescence density in the pyramidal cell layer and the stratum oriens of control and ischemic animals, excluding from the analysis identified dendrites and cell bodies. Individual particle counting was not feasible for vGLUT1 (namely within the stratum lucidum) or PV stainings because many immunoreactive elements were in contact. We, however, did particle counting for vGAT and CCK stainings using the automatic tools provided by the ImageJ software and calculated the total number of particles in the selected fields. The results obtained were not significantly different from that obtained after fluorescence intensity analysis (particle numbers after ischemia decreased 25 ± 5% for vGAT and 40 ± 7% for CCK compared with controls; n = 7 for each). For homogeneity reasons only, values obtained using the first protocol were kept in this report. Fluorescence values from the corpus callosum were used as a reference value for background. Puncta staining for vGLUT1 and vGAT likely represents terminal boutons. Statistical analyses were performed using SigmaStat 3.1 (Systat Software, Richmond, CA).
Chemicals.
Bicuculline, d-APV, NBQX (2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonoamide), CNQX, and biocytin were obtained from Sigma, GYKI 52466 was obtained from Tocris (Ellisville, MO), and TTX was obtained from Latoxan (Valence, France).
Results
Post-ischemic rats (n = 25) experienced a transitory global ischemic episode using the four-vessel occlusion model (4VO; see Materials and Methods). This model has been widely used to simulate cerebral global ischemia such as appear in the brain of a patient during cardiac arrest, coronary artery occlusion, or heart bypass surgery (Pulsinelli and Brierley, 1979; Ginsberg and Busto, 1989; Schmidt-Kastner and Freund, 1991). Transient global ischemia was achieved by the transitory occlusion of the carotid arteries during 20–25 min after a permanent occlusion of the vertebral arteries. Only rats that were unresponsive and have lost their righting reflex during the occlusion were used in this study. These rats were then allowed to survive until experiments were performed (2–6 months later). Nissl staining of hippocampal slices from these animals showed a dramatic loss of CA1 pyramidal neurons with a good preservation of CA3 pyramidal and dentate granule cells, as observed in patients after transient global ischemia (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) (Congar et al., 2000). Rats with only vertebral arteries occlusion (sham-operated rats) and control rats of the same age were used as controls (n = 23).
The post-ischemic hippocampus displays spontaneous interictal burst discharges in vivo and in vitro
To test whether the hippocampus from long-term post-ischemic rats displayed epileptiform burst discharges months after the insult, depth in vivo, electroencephalographic recordings were performed. Rats were implanted unilaterally in the CA3 area of the hippocampus, and the spontaneous activity was recorded 4 months after global ischemia (together with 24 h video monitoring) during 7 d. None of the recorded post-ischemic rats (n = 3) showed behavioral or electrographic seizures in the hippocampus during the recording period. However, EEGs from all post-ischemic rats showed IEDs at a frequency of 0.40 ± 0.03 events/min (n = 3) (Fig. 1A) when recorded during periods of quiet wakefulness. No IED was recorded in the hippocampus of age-matched control rats (n = 3) (Fig. 1A). We next performed extracellular recordings in hippocampal slices obtained from post-ischemic animals. Extracellular recordings were performed at 34–35°C with a glass electrode in the CA3 pyramidal cell layer. In keeping with in vivo recordings, spontaneous bursts of activity were recorded in all slices from post-ischemic animals at a frequency of 0.14 ± 0.02 Hz (n = 791 bursts recorded in seven slices) (Fig. 1B–E) but not control rats (n = 10 slices). These activities were very similar to IEDs recorded in vivo when identically filtered in the EEG bandpass (1–100 Hz) (Fig. 1E). Fast Fourier filtering of these activities in higher bandpass (>200 Hz) revealed high-frequency population spikes (Fig. 1C) that were reminiscent of fast ripples (FRs) recorded in animal models of the TLE and in patients with TLE (Bragin et al., 1999, 2002). Power spectral analysis (Fig. 1D) of all recorded IED-like activities (n = 791 in seven slices) showed a peak between 200 and 350 Hz (mean, 240 Hz) (Fig. 1D). No IED-like activities or FRs were recorded in the CA3 area in hippocampal slices from control rats (n = 10) but only lower-frequency oscillations of smaller amplitude (ripple) (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material) as described previously (Buzsaki et al., 1992; Draguhn et al., 1998). Power spectral analysis of these activities revealed a peak between 100 and 200 Hz (mean, 144 Hz; n = 176 events recorded in seven slices) (supplemental Fig. 2B, available at www.jneurosci.org as supplemental material). Similar ripple oscillations were also recorded in hippocampal slices from post-ischemic rats (mean, 171 Hz; n = 107 events recorded in four slices) (supplemental Fig. 2A,B, available at www.jneurosci.org as supplemental material) in addition to IED-like activities.
Figure 1.
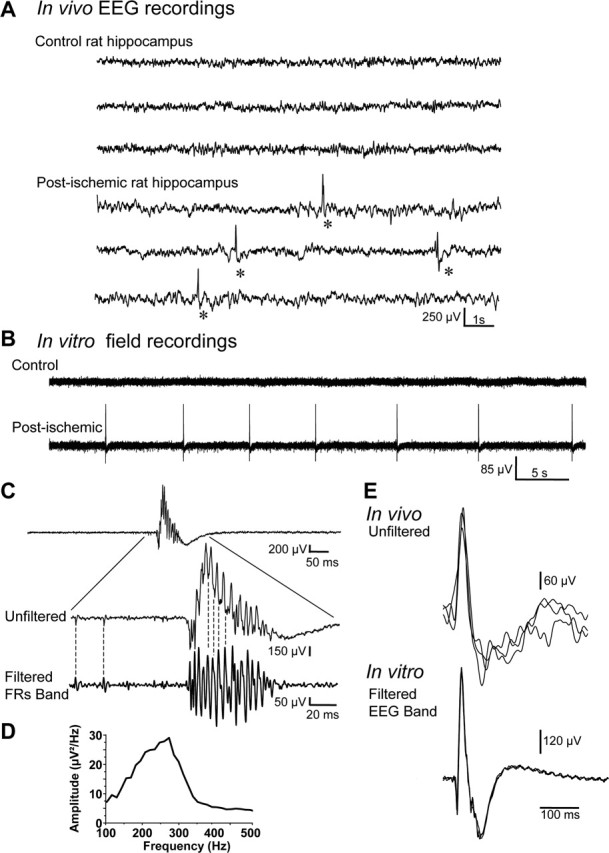
In vivo and in vitro epileptiform discharges in the hippocampus from a control and post-ischemic rat. A, Depth electroencephalographic (EEG) recordings of the hippocampus from a control and a post-ischemic rat (4 months after the insult) in the awake state. Recordings from the post-ischemic rat showed recurrent IEDs (asterisks; bottom). No IED was recorded in the hippocampus from the age-matched control rat (top). B, In vitro extracellular recording in the CA3 pyramidal layer in hippocampal slices from a control (top) and a post-ischemic (same animal shown in A, bottom) rat. Note that in the hippocampal slice from the post-ischemic rat, but not from the control rat, spontaneous epileptiform burst discharges are recorded that have the same shape as in vivo recorded IEDs (see also E). C, Example of a population burst extracellularly recorded in the CA3 pyramidal cell layer in vitro (top) before (Unfiltered) and after filtering in the FR band (Filtered FRs Band; high-pass, 200 Hz). D, Power spectrum of in vitro recorded IED-like activities (n = 791 in 7 slices) reveal high-frequency oscillations in the FR band (between 150 and 350 Hz; mean, 240 Hz). E, Comparison of three superimposed spontaneous events recorded in vivo (Unfiltered, top) and in vitro (Filtered EEG Band; bandpass, 1–100 Hz; bottom). Note that spontaneous bursts recorded in vitro were very similar to IEDs recorded in vivo when identically filtered in the EEG bandpass.
Therefore, the hippocampus displays spontaneous IEDs both in vivo and in vitro several months after a global ischemic insult.
The inhibitory drive is strongly reduced in CA3 pyramidal cells from post-ischemic rats
GABAergic synaptic transmission plays a key role in the control of neuronal excitability. To test a possible change of inhibitory drive in the post-ischemic CA3 pyramidal cells, GABAergic synaptic events were recorded in CA3 pyramidal neurons in vitro in hippocampal slices several months after ischemia and compared with control cells. IPSCs were recorded in whole-cell configuration in the voltage-clamp mode at the reversal potential for glutamatergic currents (Vh = +10 mV). Recordings were performed at 30–32°C to prevent spontaneous burst discharges that could preclude the analysis of ongoing activity. In these conditions, we observed that the frequency of spontaneous IPSCs was significantly reduced in post-ischemic CA3 pyramidal cells by 31% compared with control neurons (30.7 ± 2.1 Hz in control, n = 12; 21.2 ± 1.8 Hz in post-ischemic CA3 pyramidal cells, n = 13; p < 0.01) (Fig. 2A,C). We next considered the analysis of miniature IPSCs (mIPSCs) in the presence of the sodium channel blocker TTX (1 μm). We first observed a dramatic reduction in the frequency of mIPSCs by 68% in post-ischemic CA3 pyramidal cells (3.0 ± 0.6 Hz in control; 0.96 ± 0.46 Hz in post-ischemic CA3 pyramidal cells; n = 10 for each condition; p < 0.01) (Fig. 2B,C). Interestingly, we also observed that the mean decay time constant of mIPSCs was significantly slower in post-ischemic CA3 pyramidal cells compared with control cells (Table 1) as shown by cumulative probability plots (n = 6; χ2, p < 0.0001) (Fig. 2D) without modification of the mean rise time and amplitude (Table 1, Fig. 2D). A similar result was obtained focusing on focally generated mIPSCs with a fast rise time (<1.5 ms): their mean decay time constant was increased from 20 ± 0.97 ms in control cells to 29.3 ± 2.6 ms in post-ischemic CA3 pyramidal cells (n = 10 for each condition; p < 0.001) without a significant modification of their rise time (0.9 ± 0.03 ms in control; 1.0 ± 0.06 ms in post-ischemic CA3 pyramidal cells; n = 10 for each condition; p < 0.001).
Figure 2.

The GABAergic inhibitory drive is decreased in CA3 post-ischemic neurons. A, B, Spontaneous (A) and miniature (B) IPSCs, respectively, recorded in the absence and the presence of TTX (1 μm) in a control and post-ischemic CA3 pyramidal cell. Vh = +10 mV. C, Bar graph of the averaged frequency value of spontaneous IPSCs (sIPSC; left) and mIPSCs (right) in control (open bar; n = 12 and 10 for spontaneous and miniature events, respectively) and post-ischemic (filled bar; n = 13 and 10 for spontaneous and miniature events, respectively) CA3 pyramidal cells (∗∗p < 0.01). Note that the frequency of spontaneous and miniature IPSCs is decreased in post-ischemic cells. D, Cumulative probability distribution of 10–90% rise time (left) and decay time constants (right) of mIPSCs in control (2643 events from 6 cells; solid line) and post-ischemic (1323 events from 6 cells; dotted line) cells. Note that the cumulative plot of decay time constants is significantly shifted toward slower values in CA3 pyramidal cells from post-ischemic rats (χ2, p < 0.0001), whereas no significant difference was observed for rise times (χ2, p > 0.05). Post-isch., Post-ischemic. Error bars correspond to SEM.
Table 1.
Kinetic properties of miniature EPSCAMPA, EPSCKA, EPSCAMPA/KA, and IPSCs
| Event type | Control | Post-ischemic |
|---|---|---|
| EPSCAMPA | ||
| Rise time (ms) | 3.1 ± 0.4 | 3.4 ± 0.6 |
| Tau (ms) | 21.3 ± 1.6 | 24.3 ± 2.9 |
| Amplitude (pA) | 11.2 ± 1.7 (n = 9) | 12.9 ± 1 (n = 9) |
| EPSCKA | ||
| Rise time (ms) | 15.1 ± 0.8 | 14.3 ± 1.5 |
| Tau (ms) | 167.9 ± 39.2 | 137.82 ± 31.3 |
| Amplitude (pA) | 11.91 ± 1.7 (n = 9) | 9.8 ± 1.2 (n = 9) |
| EPSCAMPA/KA | ||
| Rise time (ms) | 3.7 ± 0.4 | 3.6 ± 0.8 |
| Tau1 (ms) | 18.4 ± 1.2 | 19.6 ± 2.9 |
| Amplitude 1 (pA) | 14.6 ± 3.1 | 13.7 ± 2.4 |
| Tau2 (ms) | 125.5 ± 14.9 | 141 ± 18.9 |
| Amplitude 2 (pA) | 6.6 ± 0.9 (n = 9) | 5.2 ± 0.6 (n = 9) |
| IPSC | ||
| Rise time (ms) | 2.35 ± 0.17 | 2.9 ± 0.29 |
| Tau (ms) | 19.7 ± 1.35 | 30.3 ± 2.25* |
| Amplitude (pA) | 13.4 ± 1.36 (n = 10) | 13.8 ± 2.1 (n = 10) |
*p < 0.05.
Altogether, these results show a strong reduction in the frequency of both spontaneous and mIPSCs several months after ischemia.
Global ischemia induces a dramatic loss of interneurons in the CA3 field of the hippocampus
The reduction in the frequency of mIPSCs with no change in their amplitude could be attributable to a loss of GABAergic terminals consecutive to the degeneration of interneurons after global ischemia. We thus investigated whether GABAergic terminals were affected several months after ischemia by analyzing the distribution of vGAT and GABA immunostainings in CA3 (Fig. 3A–D). In control sections, vGAT and GABA puncta immunoreactivity was quite intense around pyramidal cell bodies and few neurons in neuropile layers (Fig. 3A,C). This staining was significantly reduced after ischemia (Fig. 3B,D), notably in the pyramidal cell layer (by 25%, p < 0.001 for vGAT; by 31%, p < 0.005 for GABA) (Fig. 3K). In agreement with this, the number of GABA-positive cell bodies in CA3 was decreased by 37% (p < 0.05) (Fig. 3D,K). These data suggest that the perisomatic innervation of pyramidal cells is significantly reduced after ischemia and that this reduction may result from a loss of inhibitory interneurons, presumably basket cells.
Figure 3.
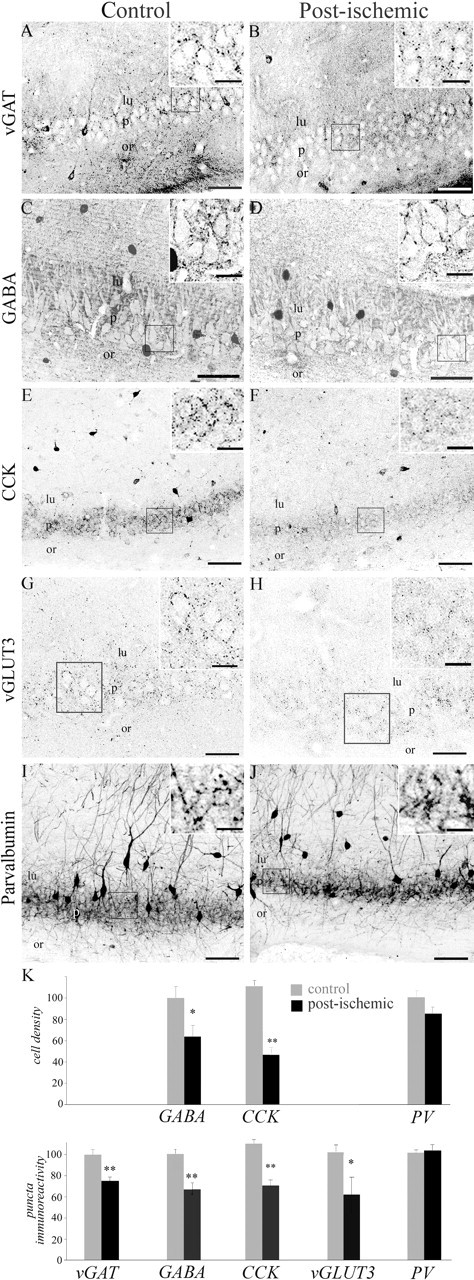
Ischemia induces a loss of GABAergic, CCK-positive, and parvalbumin-positive interneurons in the CA3 area of the hippocampus. Left column, Representative sections from control animals stained with vGAT (A), GABA (C), CCK (E), vGLUT3 (G), and parvalbumin (I). Right column, Representative sections from ischemic animals stained with the same antibodies (B, D, F, H, J). The insets are enlarged magnifications of the corresponding squares. A, B, Ischemia resulted in a dramatic reduction in vGAT staining in the stratum pyramidale of CA3 compared with control animals, suggesting a loss of perisomatic inhibition. C, D, Immunostainings with GABA antibodies revealed a significant decrease in GABA-positive cells and perisomatic puncta in ischemic animals. E, F, Immunostaining with CCK antibodies reveals a significant loss of immunopositive cell bodies in CA3 associated with a decrease in immunoreactive puncta around pyramidal cell bodies. G, H, Immunostainings with vGLUT3 antibodies depicting a significant decrease in bouton immunoreactivity in the pyramidal cell layer of ischemic animals. I, J, Immunostaining with parvalbumin antibodies reveals no major changes after ischemia. K, Quantitative analysis of the number of immunopositive cell bodies in the CA3 area (cell density, expressed as a percentage of control; values ± SEM) and fluorescence intensity in the CA3 stratum pyramidale (puncta immunoreactivity, expressed as a percentage of control; values ± SEM). ∗p < 0.05; ∗∗p < 0.001. Scale bars: A, B, E–G, H–J, 100 μm; C, D, 50 μm; insets, 25 μm. lu, Stratum lucidum; or, stratum oriens; p, stratum pyramidale.
Two populations of basket cells can be differentiated using antibodies against CCK and PV (for review, see Freund and Buzsaki, 1996; Somogyi and Klausberger, 2005). To see whether ischemia differentially affected these populations of basket cells, we investigated changes in the number of CCK- and PV-immunopositive cell bodies in the CA3 region of the hippocampus and changes in the density of CCK and PV puncta immunoreactivity in the CA3 stratum pyramidale months after global ischemia (Fig. 3E–J). In control sections, antibodies against CCK labeled numerous cell bodies mostly in the stratum radiatum, whereas antibodies against PV labeled cell bodies mostly in the stratum pyramidale, as reported previously (Freund and Buzsaki, 1996) (Fig. 3E,I). In sections from ischemic animals, CCK-immunopositive cells were significantly reduced by 58% (p < 0.0001) (Fig. 3F,K) and the density of CCK-immunopositive puncta was significantly reduced by 36% in the CA3 pyramidal cell layer (p < 0.0001) (Fig. 3F,K). To further test for the disappearance of terminals from CCK basket cells, we investigated the distribution of vGLUT3 immunoreactivity, because this vesicular glutamate transporter is present in axon terminals of CCK basket cells (Somogyi et al., 2004). In agreement with this report, in control animals, the highest density of vGLUT3-positive boutons was observed in the CA3 pyramidal cell layer, outlining the soma of pyramidal cells (Fig. 1G). In post-ischemic rats, a 39% decrease in this immunoreactivity was observed (p < 0.005) (Fig. 1H,K), confirming the loss of CCK terminals in the CA3 stratum pyramidale from the post-ischemic rat. In contrast to CCK interneurons, examination of PV-immunopositive cell bodies revealed that this subpopulation remained unaffected (p > 0.05) (Fig. 3J,K). Accordingly, no significant change was detected for PV-positive puncta immunostaining in the CA3 stratum pyramidale (Fig. 3J,K). These data strongly suggest that global ischemia induces a selective loss of CCK-positive basket cells and terminals while preserving PV-positive cells (i.e., PV basket and PV axo-axonic cells). We then investigated whether dendritic targeting interneurons were also affected months after ischemia. For this purpose, the density of cells immunoreactive to somatostatin that mainly stains dendritic targetting interneurons (for review, see Freund and Buzsaki, 1996; Somogyi and Klausberger, 2005) was analyzed. The density of somatostatin-positive cells was dramatically reduced after ischemia (by 60%; p < 0.001) (Fig. 4A,B,E). Thus, global ischemia also resulted in a massive loss of dendritic targeting CA3 interneurons.
Figure 4.
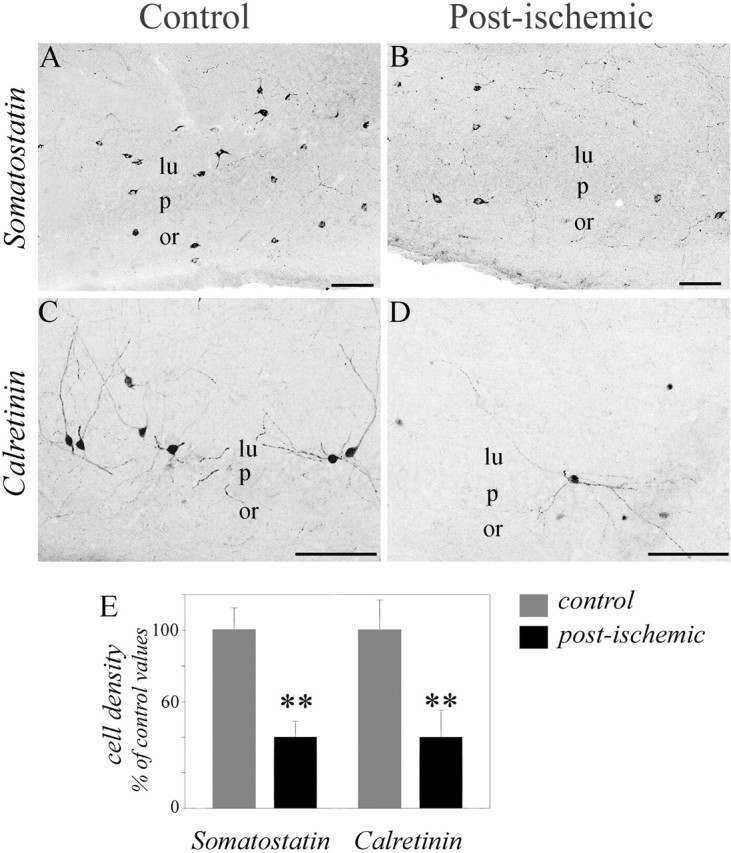
Ischemia induces a loss of somatostatin and calretinin interneurons in the CA3 area of the hippocampus. A–D, Representative sections from control (left) and ischemic (right) animals stained with somatostatin (A, B) and calretinin (C, D). A, B, Somatostatin-immunopositive neurons are significantly lost in the CA3 field of ischemic (B) compared with control (A) animals. C, D, Calretinin-immunopositive neurons are significantly lost in the CA3 field of ischemic (D) compared with control (C) animals; the decrease is observed both in the stratum radiatum and the stratum oriens. E, Histograms depicting the mean values of somatostatin- and calretinin-immunopositive cell bodies in the CA3 area (expressed as a percentage of control; values ± SEM). ∗∗p < 0.001. Scale bars, 100 μm. lu, Stratum lucidum; or, stratum oriens; p, stratum pyramidale.
Finally, we tested whether global ischemia also affected interneurons that express the calcium-binding protein calretinin. We found that the densities of calretinin-positive cell bodies was significantly reduced after ischemia (by 68%; p < 0.0001) (Fig. 4C–E) as described previously (Freund and Magloczky, 1993). Because most of these neurons innervate other interneurons that are also affected by ischemia (i.e., other calretinin, somatostatin, and CCK interneurons) (Gulyas et al., 1996), the functional implications of their loss are not easy to predict.
In conclusion, global ischemia induces an important loss of GABAergic interneurons that presumably innervate both the soma and the dendrites of CA3 pyramidal cells. These changes may explain the drastic reduction in GABAergic synaptic transmission observed in post-ischemic CA3 cells.
The glutamatergic excitatory drive is strongly enhanced in CA3 pyramidal cells from post-ischemic rats
The excitability of principal cells is controlled both by excitatory and inhibitory inputs. Therefore, in parallel to the study of the GABAergic system, we also examined the alterations of the glutamatergic synaptic system in the post-ischemic hippocampus. In a first attempt to evaluate a possible change in the glutamatergic synaptic system, we examined glutamatergic boutons using vGLUT1 antibodies in the CA3 area in hippocampal sections from control and post-ischemic rats. In control rats, vGLUT1-positive boutons distributed mainly within neuropile layers and were scarcely present within the pyramidal cell layer (Fig. 5A). In post-ischemic animals, a significant increase in vGLUT1 staining was observed both in the stratum oriens (by 32.3 ± 9.30%; p < 0.01) and in the stratum lucidum (by 25.2 ± 7.90%; p < 0.01), the terminal zone of the mossy fibers (Fig. 5B,C). No significant change in the density of staining was observed in the pyramidal cell layer and radiatum (p > 0.5) (Fig. 5C). The increased vGLUT1 staining in the stratum oriens could correspond to a sprouting of mossy fibers in the CA3 infrapyramidal band as observed in animal models of the TLE (Represa et al., 1987). To test this hypothesis, we selectively labeled the mossy fibers using the Timm staining. No change was observed in the trajectory of the mossy fiber pathway in the CA3 area from post-ischemic rats (data not shown) as described previously (Arabadzisz and Freund, 1999). Thus, the increased vGLUT1 staining in the stratum oriens after ischemia does not correspond to the sprouting of mossy fibers in the CA3 infrapyramidal band.
Figure 5.

Ischemia induces an increase in vGLUT1 immunostaining in the CA3 area of the hippocampus. A, B, Representative sections from control (A) and ischemic (B) animals stained with vGLUT1 antibodies. Note that vGLUT1 immunoreactivity is significantly increased in the CA3 strata lucidum and oriens of post-ischemic rats. Scale bars: A, B, 100 μm; insets, 25 μm. C, Bar graphs of averaged values of fluorescence intensity in the stratum oriens (left), the stratum lucidum (middle), and the stratum radiatum (right) in the CA3 area (expressed as a percentage of control; values ± SEM). ∗p < 0.05. lu, Stratum lucidum; or, stratum oriens; py, stratum pyramidale; ra, stratum radiatum.
Altogether, these data are compatible with an increased density of glutamatergic terminals in the CA3 region after ischemia.
In a second set of experiments, glutamatergic synaptic currents received by post-ischemic CA3 pyramidal cells were recorded several months after ischemia and compared with controls. Recordings were performed in hippocampal slices in the whole-cell configuration in the voltage-clamp mode at Vh = −70mV (corresponding to the reversal potential for GABAergic currents and to a potential at which NMDA receptor-mediated currents are nearly abolished by the Mg2+ block) (Ascher and Nowak, 1988). The temperature was kept at 30–32°C to prevent spontaneous burst discharges. In these conditions, we observed that the frequency of spontaneous non-NMDA receptor-mediated EPSCs was significantly increased to 243% of control in post-ischemic CA3 pyramidal cells (10.7 ± 1.6 Hz in control and 26.0 ± 5.2 Hz in post-ischemic CA3 pyramidal cells; n = 11 for each; p < 0.05) (Fig. 6A,C). This increase in the frequency of spontaneous EPSCs might result from an enhanced network activity after the loss of inhibitory interneurons and/or to an increase in the number of glutamatergic terminals as suggested by the vGLUT1 staining. To discriminate between these hypotheses, we measured the frequency of miniature EPSCs (mEPSCs) in the continuous presence of TTX (1 μm) in both control and post-ischemic CA3 pyramidal cells. We found that the frequency of mEPSCs was increased to 278% of control in post-ischemic CA3 pyramidal cells (0.27 ± 0.05 Hz and 0.75 ± 0.2 Hz in the control and post-ischemic CA3 pyramidal cells, respectively; n = 13 for each; p < 0.05) (Fig. 6B,C). In a previous study, we have shown that, relying on their pharmacological profile and kinetic properties, three types of non-NMDA mEPSCs can be isolated in control CA3 pyramidal cells: (1) the fast pure AMPA receptor-mediated events (mEPSCAMPA), (2) the mixed AMPA/kainate receptor-mediated events (mEPSCAMPA/KA), and (3) the slow pure kainate receptor-mediated events (mEPSCKA) (Cossart et al., 2002). We next examined whether the frequency of these three types of events was differentially affected after ischemia. Using their kinetics and pharmacological profile (see Materials and Methods), these three types of events were isolated in post-ischemic CA3 pyramidal cells. As reported previously (Cossart et al., 2002), EPSCKA recorded in those cells were resistant to NMDA (50 μm d-APV) and AMPA (100 μm GYKI 52466) receptor antagonists and fully blocked by the nonselective AMPA/KA antagonist CNQX (50 μm) (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). We found no significant differences between the kinetics of mEPSCAMPA, mEPSCAMPA/KA, and mEPSCKA recorded in CA3 pyramidal cells from control and post-ischemic rats (Table 1, Fig. 6B). We then estimated the total contribution of AMPA and KA receptor-mediated events to glutamatergic miniature activity in control and post-ischemic CA3 pyramidal neurons, relying on their kinetics characteristics (see Table 1 and Materials and Methods) (cf. Cossart et al., 2002). We found that the frequency of mEPSCAMPA was strongly increased to 323% of control in post-ischemic CA3 pyramidal cells (0.17 ± 0.036 Hz in control and 0.55 ± 0.16 Hz in post-ischemic CA3 pyramidal cells; n = 12 for each condition; p < 0.05) (Fig. 6C). Regarding the change in KA receptor-mediated responses, we found that the frequency of mEPSCKA was increased, but not significantly (0.02 ± 0.009 Hz in control and 0.046 ± 0.09 Hz in post-ischemic CA3 pyramidal cells; n = 12 cells for each condition; p > 0.05) (Fig. 6C). Moreover, we observed that the frequency of mEPSCAMPA/KA was significantly enhanced to 422% of control in post-ischemic CA3 pyramidal cells (0.045 ± 0.02 Hz in control and 0.19 ± 0.06 Hz in post-ischemic CA3 pyramidal cells; n = 12 cells for each condition; p < 0.05) (Fig. 6C). Thus, both the AMPA and KA receptor-mediated synaptic transmission is strongly enhanced in post-ischemic CA3 pyramidal cells. Interestingly, these changes in the frequency of AMPA and KA receptor-mediated responses were not associated with a change in their amplitude (as shown by averaged values in Table 1), in accordance with a change at the presynaptic side. Therefore, there is a strong enhancement of the glutamatergic excitatory drive in CA3 pyramidal cells several months after the ischemic insult that may result from an increased number of glutamatergic terminals.
Figure 6.

Glutamatergic excitatory drive is increased in CA3 post-ischemic neurons. A, B, Spontaneous EPSCs (A) and mEPSCs (B), respectively, recorded in the absence and the presence of TTX (1 μm), d-APV (50 μm), and bicuculline (10 μm) in a control and post-ischemic CA3 pyramidal cell. Vh = −70 mV. In miniature activity, three types of mEPSCs can be distinguished based on their decay time: the fast AMPA receptor-mediated EPSC (star), the slow KA receptor-mediated EPSC (circle), and the mixed AMPA/KA receptor-mediated EPSC (with a double decaying phase; triangle). C, Bar graphs (top) of the averaged frequency values of spontaneous EPSCs (left) and mEPSCs (right) in control (open bar; n = 11 and 13 for spontaneous and miniature events, respectively) and post-ischemic (filled bar; n = 11 and 13 for spontaneous and miniature events, respectively) CA3 pyramidal cells. Note that the frequency of both spontaneous and miniature events is significantly increased several months after global ischemia. Bar graphs (bottom) of averaged frequency values of miniature EPSCAMPA, EPSCAMPA/KA, and EPSCKA in control (open bar; n = 12) and post-ischemic (filled bar; n = 12) CA3 pyramidal (∗p < 0.05; ∗∗p < 0.01) cells. Note that the frequency of EPSCAMPA and EPSCAMPA/KA is significantly increased in post-ischemic CA3 pyramidal cells. D, Bar graphs of the averaged values of frequency (top) or charge transfer (bottom) ratio of EPSCs versus IPSCs for both spontaneous (left) and miniature (right) synaptic activity in control (open bar; n = 8 and 7 for spontaneous and miniature events, respectively) and post-ischemic (filled bar; n = 8 and 7 for spontaneous and miniature events, respectively) CA3 pyramidal cells (Mann–Whitney test; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001). sEPSCs, Spontaneous EPSCs; Post-isch., post-ischemic; Spont., spontaneous; Mini., miniature. Error bars correspond to SEM.
Finally, we assessed the ratio of excitatory versus inhibitory drive in control and post-ischemic CA3 pyramidal cells by calculating the frequency ratio of non-NMDA EPSCs versus IPSCs for spontaneous and miniature events. We found an increase in the frequency ratio for both spontaneous (from 0.4 ± 0.07 to 1.0 ± 0.1 in eight control and eight post-ischemic neurons, respectively; Mann–Whitney test; p < 0.01) (Fig. 6D) and miniature (from 0.2 ± 0.12 to 4.1 ± 1.6 in seven control and seven post-ischemic neurons, respectively; Mann–Whitney test; p < 0.05) events (Fig. 6D). The prolongation of IPSCs kinetics that we observed several months after ischemia might partly compensate the shift toward excitation observed in CA3 pyramidal cells several months after ischemia. To test this hypothesis, we calculated the charge transfer ratio of non-NMDA excitatory versus inhibitory synaptic transmission. We found that, despite the kinetic changes, this ratio was significantly shifted toward excitation in post-ischemic rats for both spontaneous (from 0.34 ± 0.07 to 1.28 ± 0.43 in eight control and eight post-ischemic neurons, respectively; Mann–Whitney test; p < 0.05) (Fig. 6D) and miniature (from 0.39 ± 0.06 to 1.75 ± 0.19 in seven control and seven post-ischemic neurons, respectively; Mann–Whitney test; p < 0.01) (Fig. 6D) synaptic transmission. In conclusion, we show that in post-ischemic CA3 pyramidal cells, the balance between excitatory and inhibitory drive is strongly shifted toward excitation.
Discussion
The main findings of the present study are that the CA3 pyramidal cells surviving several months after an ischemic insult receive less inhibitory GABAergic influx and more excitatory glutamatergic drive. In keeping with these observations, we show a dramatic loss of GABAergic interneurons and terminals together with an increase in glutamatergic terminals in the CA3 area of the hippocampus from post-ischemic rats. The consequence is a net shift in the excitatory–inhibitory balance toward excitation. This might explain the spontaneous epileptiform discharges observed both in vivo and in vitro in the hippocampus from post-ischemic rats.
Decreased inhibitory drive and loss of GABAergic interneurons in the CA3 area from post-ischemic rats
The study of GABAergic synaptic transmission shows that the frequency of spontaneous and miniature IPSCs is significantly reduced in CA3 pyramidal cells several months after global ischemia. This decrease is associated with a reduction in the number of GABAergic interneurons together with a loss of GABAergic terminals. The decrease in GABAergic terminals was assessed using puncta immunoreactivity for vGAT because this transporter accumulates within the synaptic boutons (Chaudhry et al., 1998) apposed to gephyrin-positive synapses (McIntire et al., 1997). Although puncta staining changes might well represent changes on the protein content in each terminal, the decrease in vGAT puncta immunoreactivity associated with the loss of GABAergic interneurons and to the decreased frequency of mIPSCs with no change in their amplitude suggest a loss of GABAergic synapses in the CA3 pyramidal cell from post-ischemic rats. This loss was significant within the stratum pyramidale, suggesting that perisomatic inhibition is impaired in the CA3 area months after ischemia. In keeping with this observation, we found a reduction in the number of CCK interneurons and CCK puncta immunoreactivity within the stratum pyramidale in the CA3 area from post-ischemic rats. In contrast, CA3 PV-positive basket cells are preserved months after ischemia as reported previously (Mudrick and Baimbridge, 1989; Freund et al., 1991; Arabadzisz and Freund, 1999). Basket cells play a pivotal role in the generation of fast network oscillations in the hippocampus (Cobb et al., 1995; Fisahn et al., 1998; Csicsvari et al., 2003; Hajos et al., 2004). Recently, it has been proposed that this process mainly involves PV basket cells (Freund, 2003), whereas CCK basket cells are involved in long-lasting inhibition of principal neurons (Hefft and Jonas, 2005). Thus, the preferential loss of CCK but not PV basket cells after ischemia may result in a selective impairment of long-lasting somatic inhibition with a good preservation of high-frequency oscillations. We also observed, as described previously (Represa et al., 1991; Arabadzisz and Freund, 1999), a dramatic decrease in somatostatin-positive interneurons in the CA3 area that are known to preferentially innervate pyramidal cell dendrites (Freund and Buzsaki, 1996; Somogyi and Klausberger, 2005). Altogether, these data show that an ischemic episode induces a dramatic loss of GABAergic interneurons innervating both the perisomatic and dendritic domains of pyramidal cells in the CA3 area. The loss of interneurons is likely to be responsible for the strong reduction in the inhibitory drive observed in CA3 pyramidal cells months after ischemia, although we cannot exclude an additional change in the quantal release properties of GABAergic synapses (Hirsch et al., 1999).
The reduction in inhibitory drive in the post-ischemic CA3 pyramidal cells is associated with an enhancement of the decay time constant of mIPSCs without any significant modification of their rise times and amplitudes. The absence of change of the rise time is in accordance with the immunohistochemical data showing a decrease in both somatic and dendritic targeting interneurons, as reported in the dentate gyrus of chronic epileptic rats (Kobayashi and Buckmaster, 2003). The prolonged decay time constant of remaining mIPSCs in the post-ischemic CA3 pyramidal cell is more surprising because only PV basket cells that are known to generate fast decaying IPSCs in CA3 pyramidal cells (Bartos et al., 2002) are preserved. A similar result was obtained focusing on focally generated mIPSCs with a fast rise time (<1.5 ms). Therefore, mIPSCs with an increased decay time constant in post-ischemic CA3 pyramidal cells do not correspond to distally generated events. This kinetics change might rather correspond to other changes as modifications in the subunit composition of GABAA receptors (Brooks-Kayal et al., 1998; Goldstein et al., 2002; for review, see Fritschy and Brunig, 2003; Cossart et al., 2005) or in the properties of GABAergic transporters (Calcagnotto et al., 2005).
Increased excitatory drive and enhancement of glutamatergic terminals in the CA3 area from post-ischemic rats
The study of the fate of the ongoing glutamatergic influx in CA3 pyramidal cells from post-ischemic rats reveals an important increase in the frequency of spontaneous and miniature glutamatergic synaptic events. These changes are associated with an increase in glutamatergic terminals both in the stratum oriens and in the stratum lucidum of CA3, which is compatible with the sprouting of glutamatergic fibers as observed in animal models of the TLE (Esclapez et al., 1999; Wuarin and Dudek, 2001; Epsztein et al., 2005). In the CA3 stratum lucidum, the glutamatergic mossy fibers contact CA3 pyramidal cells through the large mossy fiber boutons and stratum lucidum interneurons through small en passant boutons or filopodia extensions (Acsady et al., 1998). It has been shown previously that the number of mossy fiber filopodia extensions is decreased several months after ischemia (Arabadzisz and Freund, 1999). Thus, the increased vGLUT1 staining in the stratum lucidum might correspond to glutamatergic terminals contacting CA3 pyramidal cells. In keeping with this, we observed an increase in the frequency of mEPSCAMPA/KA, which are specifically generated at mossy fiber synapses (Castillo et al., 1997; Cossart et al., 2002), in CA3 pyramidal cells from post-ischemic rats. These results are also compatible with the postlesional plasticity of KA receptor-operated synapses after mossy fiber sprouting in animal models of epilepsy (Epsztein et al., 2005). The increased vGLUT1 staining in the stratum oriens does not result from the aberrant sprouting of the mossy fibers in the CA3 infrapyramidal band, because we did not observe a change in the trajectory of the mossy fiber pathway in post-ischemic animals as reported previously (Arabadzisz and Freund, 1999). Therefore, this enhancement might be attributable to the sprouting of other fibers such as associational/commissural glutamatergic fibers that massively project in this area (Sik et al., 1993).
Spontaneous IEDs in the hippocampus from post-ischemic rats
Spontaneous IEDs were recorded in the hippocampus from all post-ischemic rats but not control rats. Although IEDs can occasionally be observed in the absence of seizure, they are often associated with an increased probability for seizure generation in patients with TLE (Gloor, 1991; Sundaram et al., 1999; for review, see Staley et al., 2005) and in animal models of the TLE (Leite et al., 1996; Bragin et al., 2004). Furthermore, in focal onset epilepsy, IEDs are localized to the epileptic brain region and frequently disappear after resection of the epileptogenic brain tissue or spontaneous resolution of epilepsy (Wennberg et al., 1998; Salanova et al., 1999). However, none of the in vivo recorded post-ischemic rats showed electrophysiological or behavioral seizures during our in vivo EEG recordings and video monitoring. The reasons for the lack of observable seizure in this model might be the death of CA1 pyramidal cells, the main output pathway of the hippocampal formation, a lower seizure frequency than in status epilepticus models of epilepsy, or a longer latent period between the initial insult and seizure onset. Spontaneous IED-like activities (i.e., similar in shape and duration to the in vivo recorded IEDs) were also recorded in vitro in all hippocampal slices from post-ischemic animals. Filtering of these IEDs revealed the presence of high-frequency oscillations (200–300 Hz) termed FRs, whereas only ripple events were observed in control rats. Similar FR activities were specifically observed in the hippocampus from patients with TLE and from chronic epileptic rats (Bragin et al., 1999) but not from control rats, where only ripples were observed (Buzsaki et al., 1992). In animal models of TLE, FRs are specifically generated in regions capable of generating seizures (Bragin et al., 1999), and their observation is correlated to the subsequent development of spontaneous seizures (Bragin et al., 2004). Altogether, these studies suggest that FRs may reflect neuronal mechanisms responsible for epilepsy in TLE. The CA3 recurrent network form a powerful excitatory circuit that can easily generate IEDs and FRs in conditions of reduced inhibition (Miles and Wong, 1987) or enhanced excitation (Dzhala and Staley, 2003, 2004). Recent in vitro work show that they are generated through an increase in the firing rate of CA3 pyramidal cells, a buildup of excitatory events, and synchronization through the recurrent CA3 excitatory network (Dzhala and Staley, 2004; Menendez et al., 2006). Thus, both the increased synaptic excitation and decreased synaptic inhibition in CA3 pyramidal cells described in this study, together with the previously described changes in their intrinsic membrane properties (Congar et al., 2000), can explain the generation of spontaneous IEDs in the CA3 network of the post-ischemic rat hippocampus. Because IED- and FR-generating areas are associated with an enhanced susceptibility for seizure generation, these modifications could have profound implications for the occurrence of post-stroke epilepsy (Staley et al., 2005).
Footnotes
This work was supported by Institut National de la Santé et de la Recherche Médicale. J.E. was supported by the French Ministère de la Recherche and the Fondation pour la Recherche Médicale. We thank Drs. R. Cossart and R. Khazipov for helpful comments.
References
- Acsady L, Kamondi A, Sik A, Freund T, Buzsaki G (1998). GABAergic cells are the major postsynaptic targets of mossy fibers in the rat hippocampus. J Neurosci 18:3386–3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afsar N, Kaya D, Aktan S, Sykut-Bingol C (2003). Stroke and status epilepticus: stroke type, type of status epilepticus, and prognosis. Seizure 12:23–27. [DOI] [PubMed] [Google Scholar]
- Arabadzisz D, Freund TF (1999). Changes in excitatory and inhibitory circuits of the rat hippocampus 12–14 months after complete forebrain ischemia. Neuroscience 92:27–45. [DOI] [PubMed] [Google Scholar]
- Ascher P, Nowak L (1988). The role of divalent-cations in the N-methyl-D-aspartate responses of mouse central neurons in culture. J Physiol (Lond) 399:247–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartos M, Vida I, Frotscher M, Meyer A, Monyer H, Geiger JR, Jonas P (2002). Fast synaptic inhibition promotes synchronized gamma oscillations in hippocampal interneuron networks. Proc Natl Acad Sci USA 99:13222–13227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bladin CF, Alexandrov AV, Bellavance A, Bornstein N, Chambers B, Cote R, Lebrun L, Pirisi A, Norris JW (2000). Seizures after stroke: a prospective multicenter study. Arch Neurol 57:1617–1622. [DOI] [PubMed] [Google Scholar]
- Bragin A, Engel J Jr, Wilson CL, Fried I, Mathern GW (1999). Hippocampal and entorhinal cortex high-frequency oscillations (100–500 Hz) in human epileptic brain and in kainic acid–treated rats with chronic seizures. Epilepsia 40:127–137. [DOI] [PubMed] [Google Scholar]
- Bragin A, Mody I, Wilson CL, Engel J Jr (2002). Local generation of fast ripples in epileptic brain. J Neurosci 22:2012–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bragin A, Wilson CL, Almajano J, Mody I, Engel J Jr (2004). High-frequency oscillations after status epilepticus: epileptogenesis and seizure genesis. Epilepsia 45:1017–1023. [DOI] [PubMed] [Google Scholar]
- Brierley JB, Cooper JE (1962). Cerebral complications of hypotensive anaesthesia in a healthy adult. J Neurol Neurosurg Psychiatry 25:24–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks-Kayal AR, Shumate MD, Jin H, Rikhter TY, Coulter DA (1998). Selective changes in single cell GABA(A) receptor subunit expression and function in temporal lobe epilepsy. Nat Med 4:1166–1172. [DOI] [PubMed] [Google Scholar]
- Buckmaster PS, Dudek FE (1999). In vivo intracellular analysis of granule cell axon reorganization in epileptic rats. J Neurophysiol 81:712–721. [DOI] [PubMed] [Google Scholar]
- Buckmaster PS, Jongen-Relo AL (1999). Highly specific neuron loss preserves lateral inhibitory circuits in the dentate gyrus of kainate-induced epileptic rats. J Neurosci 19:9519–9529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsaki G, Horvath Z, Urioste R, Hetke J, Wise K (1992). High-frequency network oscillation in the hippocampus. Science 256:1025–1027. [DOI] [PubMed] [Google Scholar]
- Calcagnotto ME, Paredes MF, Tihan T, Barbaro NM, Baraban SC (2005). Dysfunction of synaptic inhibition in epilepsy associated with focal cortical dysplasia. J Neurosci 25:9649–9657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo PE, Malenka RC, Nicoll RA (1997). Kainate receptors mediate a slow postsynaptic current in hippocampal CA3 neurons. Nature 388:182–186. [DOI] [PubMed] [Google Scholar]
- Chaudhry FA, Reimer RJ, Bellocchio EE, Danbolt NC, Osen KK, Edwards RH, Storm-Mathisen J (1998). The vesicular GABA transporter, VGAT, localizes to synaptic vesicles in sets of glycinergic as well as GABAergic neurons. J Neurosci 18:9733–9750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb SR, Buhl EH, Halasy K, Paulsen O, Somogyi P (1995). Synchronization of neuronal activity in hippocampus by individual GABAergic interneurons. Nature 378:75–78. [DOI] [PubMed] [Google Scholar]
- Cocito L, Favale E, Reni L (1982). Epileptic seizures in cerebral arterial occlusive disease. Stroke 13:189–195. [DOI] [PubMed] [Google Scholar]
- Congar P, Gaiarsa JL, Popovici T, Ben Ari Y, Crépel V (2000). Permanent reduction of seizure threshold in post-ischemic CA3 pyramidal neurons. J Neurophysiol 83:2040–2046. [DOI] [PubMed] [Google Scholar]
- Cossart R, Dinocourt C, Hirsch JC, Merchan-Perez A, De Felipe J, Esclapez M, Bernard C, Ben Ari Y (2001). Dendritic but not somatic GABAergic inhibition is decreased in experimental epilepsy. Nat Neurosci 4:52–62. [DOI] [PubMed] [Google Scholar]
- Cossart R, Epsztein J, Tyzio R, Becq H, Hirsch J, Ben Ari Y, Crépel V (2002). Quantal release of glutamate generates pure kainate and mixed AMPA/kainate EPSCs in hippocampal neurons. Neuron 35:147–159. [DOI] [PubMed] [Google Scholar]
- Cossart R, Bernard C, Ben Ari Y (2005). Multiple facets of GABAergic neurons and synapses: multiple fates of GABA signalling in epilepsies. Trends Neurosci 28:108–115. [DOI] [PubMed] [Google Scholar]
- Crépel V, Represa A, Beaun M, Ben-Ari Y (1989). Hippocampal damage induced by ischemia and intra-amygdaloid kainate injection: effects on N-methyl-D-aspartate, (N-(1-[2-thienyl]cyclohexyl)piperidine and glycine binding sites. Neuroscience 31:605–612. [DOI] [PubMed] [Google Scholar]
- Crépel V, Epsztein J, Ben Ari Y (2003). Ischemia induces short- and long-term remodeling of synaptic activity in the hippocampus. J Cell Mol Med 7:401–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csicsvari J, Jamieson B, Wise KD, Buzsaki G (2003). Mechanisms of gamma oscillations in the hippocampus of the behaving rat. Neuron 37:311–322. [DOI] [PubMed] [Google Scholar]
- Dinocourt C, Petanjek Z, Freund TF, Ben Ari Y, Esclapez M (2003). Loss of interneurons innervating pyramidal cell dendrites and axon initial segments in the CA1 region of the hippocampus following pilocarpine-induced seizures. J Comp Neurol 459:407–425. [DOI] [PubMed] [Google Scholar]
- Draguhn A, Traub RD, Schmitz D, Jefferys JG (1998). Electrical coupling underlies high-frequency oscillations in the hippocampus in vitro Nature 394:189–192. [DOI] [PubMed] [Google Scholar]
- Dzhala VI, Staley KJ (2003). Transition from interictal to ictal activity in limbic networks in vitro J Neurosci 23:7873–7880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzhala VI, Staley KJ (2004). Mechanisms of fast ripples in the hippocampus. J Neurosci 24:8896–8906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epsztein J, Represa A, Jorquera I, Ben Ari Y, Crépel V (2005). Recurrent mossy fibers establish aberrant kainate receptor-operated synapses on granule cells from epileptic rats. J Neurosci 25:8229–8239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esclapez M, Hirsch JC, Ben Ari Y, Bernard C (1999). Newly formed excitatory pathways provide a substrate for hyperexcitability in experimental temporal lobe epilepsy. J Comp Neurol 408:449–460. [DOI] [PubMed] [Google Scholar]
- Fisahn A, Pike FG, Buhl EH, Paulsen O (1998). Cholinergic induction of network oscillations at 40 Hz in the hippocampus in vitro Nature 394:186–189. [DOI] [PubMed] [Google Scholar]
- Freund TF (2003). Interneuron Diversity series: Rhythm and mood in perisomatic inhibition. Trends Neurosci 26:489–495. [DOI] [PubMed] [Google Scholar]
- Freund TF, Buzsaki G (1996). Interneurons of the hippocampus. Hippocampus 6:347–470. [DOI] [PubMed] [Google Scholar]
- Freund TF, Magloczky Z (1993). Early degeneration of calretinin-containing neurons in the rat hippocampus after ischemia. Neuroscience 56:581–596. [DOI] [PubMed] [Google Scholar]
- Freund TF, Buzsaki G, Leon A, Baimbridge KG, Somogyi P (1991). Relationship of neuronal vulnerability and calcium-binding protein immunoreactivity in ischemia. Exp Brain Res 83:55–66. [DOI] [PubMed] [Google Scholar]
- Fritschy JM, Brunig I (2003). Formation and plasticity of GABAergic synapses: physiological mechanisms and pathophysiological implications. Pharmacol Ther 98:299–323. [DOI] [PubMed] [Google Scholar]
- Ginsberg MD, Busto R (1989). Rodent models of cerebral ischemia. Stroke 20:1627–1642. [DOI] [PubMed] [Google Scholar]
- Gloor P (1991). Preoperative electroencephalographic investigation in temporal lobe epilepsy: extracranial and intracranial recordings. Can J Neurol Sci 18:554–558. [DOI] [PubMed] [Google Scholar]
- Goldstein PA, Elsen FP, Ying SW, Ferguson C, Homanics GE, Harrison NL (2002). Prolongation of hippocampal miniature inhibitory postsynaptic currents in mice lacking the GABA(A) receptor alpha1 subunit. J Neurophysiol 88:3208–3217. [DOI] [PubMed] [Google Scholar]
- Gulyas AI, Hajos N, Freund TF (1996). Interneurons containing calretinin are specialized to control other interneurons in the rat hippocampus. J Neurosci 16:3397–3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajos N, Palhalmi J, Mann EO, Nemeth B, Paulsen O, Freund TF (2004). Spike timing of distinct types of GABAergic interneuron during hippocampal gamma oscillations in vitro J Neurosci 24:9127–9137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefft S, Jonas P (2005). Asynchronous GABA release generates long-lasting inhibition at a hippocampal interneuron-principal neuron synapse. Nat Neurosci 8:1319–1328. [DOI] [PubMed] [Google Scholar]
- Hirsch JC, Agassandian C, Merchan-Perez A, Ben Ari Y, DeFelipe J, Esclapez M, Bernard C (1999). Deficit of quantal release of GABA in experimental models of temporal lobe epilepsy. Nat Neurosci 2:499–500. [DOI] [PubMed] [Google Scholar]
- Hsu M, Buzsaki G (1993). Vulnerability of mossy fiber targets in the rat hippocampus to forebrain ischemia. J Neurosci 13:3964–3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Buckmaster PS (2003). Reduced inhibition of dentate granule cells in a model of temporal lobe epilepsy. J Neurosci 23:2440–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leite JP, Babb TL, Pretorius JK, Kuhlman PA, Yeoman KM, Mathern GW (1996). Neuron loss, mossy fiber sprouting, and interictal spikes after intrahippocampal kainate in developing rats. Epilepsy Res 26:219–231. [DOI] [PubMed] [Google Scholar]
- Lossius MI, Ronning OM, Slapo GD, Mowinckel P, Gjerstad L (2005). Poststroke epilepsy: occurrence and predictors—a long-term prospective controlled study (Akershus Stroke Study). Epilepsia 46:1246–1251. [DOI] [PubMed] [Google Scholar]
- McIntire SL, Reimer RJ, Schuske K, Edwards RH, Jorgensen EM (1997). Identification and characterization of the vesicular GABA transporter. Nature 389:870–876. [DOI] [PubMed] [Google Scholar]
- McNamara JO (1979). Human hypoxia and seizures: effects and interactions. Adv Neurol 26:137–143. [PubMed] [Google Scholar]
- Menendez dlP, Huberfeld G, Cohen I, Miles R (2006). Threshold behavior in the initiation of hippocampal population bursts. Neuron 49:131–142. [DOI] [PubMed] [Google Scholar]
- Miles R, Wong RKS (1987). Inhibitory control of local excitatory circuits in the guinea-pig hippocampus. J Physiol (Lond) 388:611–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittmann T, Qu M, Zilles K, Luhmann HJ (1998). Long-term cellular dysfunction after focal cerebral ischemia: in vitro analyses. Neuroscience 85:15–27. [DOI] [PubMed] [Google Scholar]
- Mudrick LA, Baimbridge KG (1989). Long-term structural changes in the rat hippocampal formation following cerebral ischemia. Brain Res 493:179–184. [DOI] [PubMed] [Google Scholar]
- Obenaus A, Esclapez M, Houser CR (1993). Loss of glutamate-decarboxylase messenger-RNA containing neurons in the rat dentate gyrus following pilocarpine-induced seizures. J Neurosci 13:4470–4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki MM, Evenson DA, Nadler JV (1995). Hippocampal mossy fiber sprouting and synapse formation after status epilepticus in rats—visualization after retrograde transport of biocytin. J Comp Neurol 352:515–534. [DOI] [PubMed] [Google Scholar]
- Pedley TA (1980). Interictal epileptiform discharges: discriminating characteristics and clinical correlations. Am J EEG Technol 20:101–119. [Google Scholar]
- Pulsinelli WA (1985). Selective neuronal vulnerability: morphological and molecular characteristics. Prog Brain Res 63:29–37. [DOI] [PubMed] [Google Scholar]
- Pulsinelli WA, Brierley JB (1979). A new bilateral hemispheric ischemia in the unanesthetised rat. Stroke 10:267–272. [DOI] [PubMed] [Google Scholar]
- Represa A, Tremblay E, Ben-Ari Y (1987). Kainate binding sites in the hippocampal mossy fibers: localization and plasticity. Neuroscience 20:739–748. [DOI] [PubMed] [Google Scholar]
- Represa A, Crépel V, Ben Ari Y (1991). Transient cerebral ischemia induces changes in SRIF mRNA in the fascia dentata. Brain Res Mol Brain Res 10:337–342. [DOI] [PubMed] [Google Scholar]
- Salanova V, Markand O, Worth R (1999). Longitudinal follow-up in 145 patients with medically refractory temporal lobe epilepsy treated surgically between 1984 and 1995. Epilepsia 40:1417–1423. [DOI] [PubMed] [Google Scholar]
- Schmidt-Kastner R, Freund TF (1991). Selective vulnerability of the hippocampus in brain ischemia. Neuroscience 40:599–636. [DOI] [PubMed] [Google Scholar]
- Shao LR, Dudek FE (2005). Changes in mIPSCs and sIPSCs after kainate treatment: evidence for loss of inhibitory input to dentate granule cells and possible compensatory responses. J Neurophysiol 94:952–960. [DOI] [PubMed] [Google Scholar]
- Siesjo BK (1981). Cell damage in the brain: a speculative synthesis. J Cereb Blood Flow Metab 1:155–185. [DOI] [PubMed] [Google Scholar]
- Sik A, Tamamaki N, Freund TF (1993). Complete axon arborization of a single CA3 pyramidal cell in the rat hippocampus, and its relationship with postsynaptic parvalbumin-containing interneurons. Eur J Neurosci 5:1719–1728. [DOI] [PubMed] [Google Scholar]
- Silverman IE, Restrepo L, Mathews GC (2002). Poststroke seizures. Arch Neurol 59:195–201. [DOI] [PubMed] [Google Scholar]
- Somogyi P, Klausberger T (2005). Defined types of cortical interneurone structure space and spike timing in the hippocampus. J Physiol (Lond) 562:9–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somogyi J, Baude A, Omori Y, Shimizu H, El Mestikawy S, Fukaya M, Shigemoto R, Watanabe M, Somogyi P (2004). GABAergic basket cells expressing cholecystokinin contain vesicular glutarnate transporter type 3 (VGLUT3) in their synaptic terminals in hippocampus and isocortex of the rat. Eur J Neurosci 19:552–569. [DOI] [PubMed] [Google Scholar]
- Staley K, Hellier JL, Dudek FE (2005). Do interictal spikes drive epileptogenesis? Neuroscientist 11:272–276. [DOI] [PubMed] [Google Scholar]
- Sundaram M, Sadler RM, Young GB, Pillay N (1999). EEG in epilepsy: current perspectives. Can J Neurol Sci 26:255–262. [DOI] [PubMed] [Google Scholar]
- Sutula T, He XX, Cavazos J, Scott G (1988). Synaptic reorganization in the hippocampus induced by abnormal functional activity. Science 239:1147–1150. [DOI] [PubMed] [Google Scholar]
- Wennberg R, Quesney F, Olivier A, Rasmussen T (1998). Electrocorticography and outcome in frontal lobe epilepsy. Electroencephalogr Clin Neurophysiol 106:357–368. [DOI] [PubMed] [Google Scholar]
- Wuarin JP, Dudek FE (2001). Excitatory synaptic input to granule cells increases with time after kainate treatment. J Neurophysiol 85:1067–1077. [DOI] [PubMed] [Google Scholar]
- Zola-Morgan S, Squire LR, Amaral DG (1986). Human amnesia and the medial temporal region: enduring memory impairment following a bilateral lesion limited to field CA1 of the hippocampus. J Neurosci 6:2950–2967. [DOI] [PMC free article] [PubMed] [Google Scholar]