

Abstract
The cell adhesion molecule N-cadherin has been proposed to regulate synapse formation in mammalian central neurons. This is based on its synaptic localization enabling alignment of presynaptic and postsynaptic specializations by an adhesion mechanism. However, a potential role of N-cadherin in regulating synaptic transmission has remained elusive. In this paper, a functional analysis of N-cadherin knock-out synapses was enabled by in vitro neuronal differentiation of mouse embryonic stem cells circumventing the early embryonic lethality of mice genetically null for N-cadherin. In our in vitro system, initial synapse formation was not altered in the absence of N-cadherin, which might be attributable to compensatory mechanisms. Here, we demonstrate that N-cadherin is required for regulating presynaptic function at glutamatergic synapses. An impairment in the availability of vesicles for exocytosis became apparent selectively during high activity. Short-term plasticity was strongly altered with synaptic depression enhanced in the absence of N-cadherin. Most intriguingly, facilitation was converted to depression under specific stimulation conditions. This indicates an important role of N-cadherin in the control of short-term plasticity.
To analyze, whether N-cadherin regulates presynaptic function by a transsynaptic mechanism, we studied chimeric cultures consisting of wild-type neocortical neurons and ES cell-derived neurons. With N-cadherin absent only postsynaptically, we observed a similar increase in short-term synaptic depression as found in its complete absence. This indicates a retrograde control of short-term plasticity by N-cadherin. In summary, our results revealed an unexpected involvement of a synaptic adhesion molecule in the regulation of short-term plasticity at glutamatergic synapses.
Keywords: synaptic adhesion molecules, cadherins, synaptic depression, facilitation, retrograde regulation, ES cell-derived neurons
Introduction
Synaptic adhesion molecules are thought to control the formation and maintenance of presynaptic and postsynaptic structures by mediating transsynaptic, bidirectional signaling (Südhof, 2001; Garner et al., 2002; Scheiffele, 2003). However, their physiological role in modulating synaptic function has remained largely elusive. The cadherins constitute a superfamily of synaptic adhesion molecules, consisting of classical cadherins and protocadherins (Shapiro and Colman, 1999; Angst et al., 2001). Classical cadherins, e.g., N-cadherin, mediate cell adhesion by Ca2+-dependent, homophilic binding (Takeichi, 1990; Gumbiner, 2000). The intracellular domains interact with α- and β-catenins, resulting in regulation of the actin filament network (Yap and Kovacs, 2003). In the developing brain, cadherins have been implicated in growth cone guidance and target recognition (Matsunaga et al., 1988; Inoue and Sanes, 1997; Sanes and Yamagata, 1999; Redies, 2000; Clandinin and Zipursky, 2002; Poskanzer et al., 2003). Classical cadherins localize to CNS synapses at perisynaptic sites (Fannon and Colman, 1996; Uchida et al., 1996) in the vicinity of the active zone and the postsynaptic density (Uchida et al., 1996; Phillips et al., 2001).
N-cadherin is well known to be expressed at cortical glutamatergic synapses in mammals (Benson and Tanaka, 1998; Huntley and Benson, 1999). At the functional level, synaptic N-cadherin has been shown to dimerize and become protease resistant in response to synaptic activity, thus possibly enhancing adhesion (Tanaka et al., 2000). A role of N-cadherin in hippocampal long-term potentiation has been suggested by studies using function-blocking antibodies or peptides (Tang et al., 1998; Bozdagi et al., 2000). However, although these findings support a role for N-cadherin in glutamatergic synapse function, the direct analysis of its physiological significance by investigating N-cadherin knock-out synapses is still lacking, because N-cadherin knock-out mice die during early embryonic development because of a failure of heart formation (Radice et al., 1997). In an alternative approach, a functional inhibition of all classical cadherins by overexpression of a dominant-negative cadherin revealed a role of classical cadherins in dendritic spine morphogenesis (Togashi et al., 2002; Takeichi and Abe, 2005) and in synapse formation in general (Togashi et al., 2002; Bozdagi et al., 2004). However, because this type of inhibition affected all cadherins and no detailed analysis of synaptic function was done, the specific role of N-cadherin in regulating synaptic transmission remained unclear.
We have now achieved analysis of synaptic function in N-cadherin knock-out neurons: homozygous, N-cadherin-deficient embryonic stem (ES) cells (Moore et al., 1999) were differentiated in vitro, yielding neurons, which were purified by immunoisolation and cultured on glial microislands (Jüngling et al., 2003). N-cadherin has been shown to be restricted to glutamatergic synapses in cultured cortical neurons, whereas a different classical cadherin may be present at mature GABAergic synapses (Benson and Tanaka, 1998). Therefore, we focused on the analysis of glutamatergic synapses in our ES cell-derived synaptic networks. In this study, we report that the absence of N-cadherin leads to altered short-term plasticity at glutamatergic synapses. Furthermore, we present evidence that retrograde transsynaptic signaling mediated by N-cadherin might be involved in the regulation of short-term plasticity.
Materials and Methods
In vitro differentiation of ES cells
D3 wild-type, mouse ES cells were a kind gift from Dr. B. Fleischmann (Institute of Physiology I, University of Bonn, Bonn, Germany). Pluripotent mouse ES cells inheriting an N-cadherin deletion in homozygous (N-Cad−/−) or in heterozygous (N-Cad+/−) configuration had been derived previously from blastocysts (Moore et al., 1999) obtained from mice heterozygous for the N-cadherin mutation (Radice et al., 1997). All cell culture steps were performed at 37°C in 5% CO2 atmosphere. Proliferating ES cells were cultured on mitomycin C (Sigma, St. Louis, MO) inactivated mouse embryonic feeder cells in the presence of leukemia inhibitory factor (LIF; Chemicon, Temecula, CA) according to standard protocols (for details, see Jüngling et al., 2003). In vitro differentiation of ES cells was done using the embryoid body (EB) system (Strübing et al., 1995). Briefly, EB formation occurred in hanging drop culture with an initial cell density of 8000 cells/10 μl without LIF and in the presence of retinoic acid (20 nm). EBs were harvested after 4 d and were further cultured for another 3 d in the presence of retinoic acid in uncoated culture dishes (Falcon, Franklin Lakes, NJ). For additional in vitro differentiation, EBs were seeded on poly-l-ornithine (1 mg/ml; Sigma)-coated culture dishes and incubated for 2 weeks in the absence of retinoic acid (Jüngling et al., 2003).
Purification of ES cell-derived neurons
L1 immunoisolation of ES cell-derived neurons was performed as described previously (Jüngling et al., 2003). In brief, a plastic Petri dish (Falcon) was first coated with a goat anti-rat IgG (Jackson ImmunoResearch, West Grove, PA) and then, after incubation with bovine serum albumin, with supernatant from a rat anti-L1 antibody-producing hybridoma cell line. For L1 immunoisolation, EBs were dissociated mechanically after 50 min of papain (Worthington Biochemical, Lakewood, NJ) treatment. The cell suspension was added to the L1-coated Petri dish, and after 1.5 h at room temperature the nonadherent cells were washed off. The adherent cells were harvested from the Petri dish by trypsin treatment and were resuspended in PBS/25% fetal bovine serum.
In vitro synapse formation in microisland cultures
Purified, ES cell-derived neurons were cultured on glial cells in a microisland culture system (Lessmann and Heumann, 1997; Jüngling et al., 2003). Glial cells were obtained by dissociating the cortex of C57BL/6 mouse embryos (embryonic day 18–19) and culturing proliferating cells for several weeks in serum-containing medium [BME+ medium: Eagle’s basal medium with addition of insulin, l-glutamine, glucose, and 10% fetal bovine serum (Invitrogen, Carlsbad, CA)]. Confluent glial cells completely devoid of neurons were dissociated and seeded on uncoated glass coverslips in BME+ medium and cultured for 4–6 d in the presence of cytosine-β-d-arabinofuranoside (10 μm) to inhibit proliferation. ES cell-derived, L1-selected neurons were seeded on glial microislands at a density of 105 cells/35 mm2 dish and were cultured in Neurobasal medium (Invitrogen) with addition of B27 supplement, glutamax, and penicillin/streptomycin, leading to the formation of neuronal networks of three to eight neurons. Care was taken that the number of neurons per network was similar for the different genotypes. For chimeric cultures, enhanced green fluorescent protein (EGFP)-expressing wild-type neocortical neurons were obtained from transgenic EGFP-expressing mice (Hadjantonakis et al., 1998).
Immunocytochemistry
Synapsin I immunostaining was performed according to standard protocols (Mohrmann et al., 1999). A primary rabbit anti-synapsin I antibody (Chemicon) and a secondary Cy2-conjugated goat anti-rabbit antibody were used. Number of synapsin I-positive puncta on the dendrites of a cell were counted and then normalized to the dendritic length analyzed. Data are expressed as number per 30 μm dendritic length.
Electron microscopy
Cultured ES cell-derived neurons were washed once with washing buffer (0.1 m PIPES), pH 7.4, and fixed subsequently with 2% glutaraldehyde in washing buffer for 30 min. The cells were washed once in 0.1 m phosphate buffer (PB), pH 7.4, and treated with 1% OsO4 in PB for 1 h. Cells were washed twice with PB and dehydrated using series of ethanol dilutions, including a 70% ethanol dilution containing 0.5% uranyl acetate. Cells were embedded in Epon. Sections of 60 nm were examined using a Zeiss (Jena, Germany) EM10 electron microscope. Pictures were taken with a CCD camera at a final magnification of 40,000×. Synaptic profiles with at least 150 nm presynaptic membrane specialization and associated with at least 10 presynaptic vesicles were used for analysis. Single EM sections were used. In each section, the overall number of vesicles detectable within 500 nm distance from the active zone and the number of vesicles within two vesicle diameters (80 nm) from the presynaptic membrane were counted. To enable a comparison of vesicle pools between different genotypes, the number of vesicles counted was normalized to the length of the presynaptic membrane specialization. Numbers are given as mean ± SEM per 100 nm active zone length. Although this approach allows detection of drastic changes in vesicle pools, we cannot address changes in the three-dimensional structure of the synapses, because this would require three-dimensional reconstructions.
FM4-64 staining and destaining
To visualize presynaptic vesicle cycling, uptake of the styryl dye FM4-64 was used as described previously (Hartmann et al., 2001; Mohrmann et al., 2003). For staining of cycling vesicles, microisland cultures were incubated for 2 min in depolarizing extracellular solution (in mm: 40 KCl, 54 NaCl, 2 CaCl2, 1 MgCl2, 20 HEPES), pH 7.3, containing 10 μm FM4-64, and were subsequently washed three times in standard extracellular solution (with 1 mm Ca2+/10 mm Mg2+). FM4-64-labeled puncta were destained by electrical stimulation as described previously (Hartmann et al., 2001). A large-diameter (10 μm) glass pipette (filled with 3:1 standard extracellular solution:1 m NaCl) was used for extracellular stimulation (a 30 Hz pulse train lasting 1 s was repeated 10 times with 1 s intervals). Fluorescence imaging was done according to standard procedures at room temperature. In brief, digital fluorescence images of FM4-64 puncta (epifluorescence; excitation, 546 nm; emission, >590 nm) were acquired using a 40× oil-immersion objective (Olympus, Tokyo, Japan) in combination with a CCD camera system [CoolSNAPcf (Photometrics, Tucson, AZ); MetaView software (Universal Imaging, Downingtown, PA)]. To determine the fluorescence intensity associated with individual release sites, a region of interest was fitted to each FM4-64 punctum using MetaView software and the mean intensity (fluorescence/pixel) within this region was measured (after background subtraction). For analyzing destaining properties, all fluorescence intensity values obtained during destaining were normalized to the initial fluorescence intensity before stimulation. Destaining kinetics were fitted with a single exponential function.
Electrophysiology and data analysis
Somatic whole-cell patch-clamp recordings were obtained at 10–14 d in microisland culture [days in vitro (DIV)] from ES cell-derived neurons at room temperature using an EPC7 amplifier (HEKA Elektronik, Lambrecht/Pfalz, Germany) as described previously (Mohrmann et al., 1999, 2003). The standard patch-pipette solution contained the following (in mm): 110 KCl, 0.25 CaCl2, 10 EGTA, and 20 HEPES, pH 7.3. To record NMDA receptor-mediated postsynaptic currents (PSCs) at +40mV holding potential, an intracellular solution (in mm: 100 CsCl, 0.25 CaCl2, 20 TEACl, 10 EGTA, and 20 HEPES, pH 7.3) was used to reduce K+ channel activity. Patch pipettes had resistances of 3–8 MΩ. The standard extracellular solution contained the following (in mm): 130 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, and 20 HEPES, pH 7.3. PSCs were filtered at 3 kHz and were sampled at 10 kHz using pClamp6 software (Molecular Devices, Union City, CA).
Miniature PSCs.
For pharmacological isolation of AMPA receptor-mediated miniature EPSCs (mEPSCs), GABAA receptors were blocked with picrotoxin (100 μm; Tocris Bioscience, Ellisville, MO) and NMDA receptors with Mg2+ (1 mm). AMPA mEPSCs were recorded at −60 mV holding potential in the presence of TTX (1 μm) with increased extracellular K+ (30 mm) and increased extracellular Ca2+ (5 mm) to maximize spontaneous vesicle cycling. Analysis of mEPSC frequencies and amplitudes was performed using AUTESP software (custom-made by H. Zucker, Max Planck Institute of Neurobiology, Munich-Martinsried, Germany) as described previously (Mohrmann et al., 1999). Kinetics of individual mEPSCs (50 successive events per cell) were analyzed using pClamp software. Recording conditions and data analysis for GABAA receptor-mediated miniature PSCs (mPSCs) were similar except that AMPA receptors were blocked with DNQX (20 μm).
Sucrose-evoked EPSCs.
ES cell-derived neurons were completely superfused with hypertonic extracellular solution (500 mm sucrose, 1 μm TTX, 100 μm picrotoxin added) for 4 s using a one-barrel, local application system (Mohrmann et al., 2003). After complete removal of hypertonic solution, a second sucrose application was performed after a resting time of 1 min to study the refill properties of the depleted readily releasable pool. AMPA receptor-mediated EPSCs were recorded at a holding potential of −60 mV in the presence of picrotoxin and Mg2+ and were sampled at 2 kHz. The synaptic responses to both sucrose applications were integrated, and the ratio of charge influx (“refill”) was calculated.
Paired recordings.
Unitary EPSCs were recorded from pairs of synaptically connected ES cell-derived neurons on the same glial microisland. EPSCs were evoked by depolarizing current injection into the presynaptic cell leading to action potential (AP) generation. To achieve a more stable holding potential, in some experiments, action currents were elicited at the soma in voltage-clamp mode, whereas the axon was not voltage clamped. A train of four current pulses spaced by 50 ms was applied to the presynaptic neuron every 10 s. For tetanic activation, 20 action potentials at 50 Hz or 40 action potentials at 10 Hz were elicited. GABAA receptors were blocked by addition of picrotoxin (100 μm) to the standard extracellular solution. AMPA receptor-mediated EPSCs were recorded at −60 mV holding potential in the presence of Mg2+. Glycine (10 μm) was added to the standard extracellular solution for recording NMDA receptor-mediated PSC components at +40 mV holding potential. PSC amplitudes and failure (<2 SD noise) rates were determined with pClamp software.
All data are given as mean ± SEM. Statistical analysis was done using Student’s t test with Bonferroni correction, if applicable.
Results
Synapse formation in N-cadherin knock-out, ES cell-derived neurons
To investigate the functional role of N-cadherin at glutamatergic synapses, we first studied synapse formation in cultured N-cadherin knock-out, ES cell-derived neurons. We confirmed the absence of N-cadherin protein (Radice et al., 1997; Moore et al., 1999) by Western blots of protein extracts obtained from proliferating ES cells and from embryoid bodies after in vitro neuronal differentiation. Wild-type and heterozygous (N-Cad+/−) cells showed a clear expression of N-cadherin that was only slightly reduced in N-Cad+/− cells, whereas N-cadherin was undetectable in homozygous knock-out (N-Cad−/−) cells (Fig. 1B). After proliferation, we differentiated ES cells in vitro using an embryoid body system. ES cell-derived neurons were obtained from dissociated embryoid bodies by L1-immunoisolation and were then cultured on microislands of mouse cortical glia cells (Jüngling et al., 2003) (Fig. 1A,C). At 10 d in microisland culture (DIV), ES cell-derived neurons had formed small neuronal networks consisting of three to eight neurons. No gross differences in survival and outgrowth of neurites were observed between N-Cad−/−, N-Cad+/− and wild-type neurons (Fig. 1C).
Figure 1.

In vitro neuronal differentiation of N-cadherin knock-out, ES cell-derived neurons. A, Scheme of ES cell in vitro differentiation and purification of ES cell-derived neurons. EB, Embryoid body. B, Western blot analysis of N-cadherin expression in wild-type (+/+), heterozygous (+/−), and homozygous N-cadherin knock-out (−/−) ES cells. C, ES cell-derived neurons cultured on glial microislands at 10 DIV. Scale bars, 16 μm.
We analyzed the formation of synapses in N-cadherin knock-out, ES cell-derived neurons by immunocytochemical detection of presynaptic boutons using synapsin-I antibodies. For quantification, we calculated the density of synapsin-I-positive puncta per 30 μm dendrite length. At 10–12 DIV, the mean dendritic density of synapsin-I-positive puncta did not significantly differ between wild-type, N-Cad+/−, and N-Cad−/− neurons (Fig. 2A,B). This result indicates that the initial formation of synapses is not altered in the complete absence of N-cadherin. To further confirm at the ultrastructural level that the formation of excitatory glutamatergic synapses is not grossly altered in N-Cad−/− neurons, we studied asymmetric synapses in EM sections. In these and in the following experiments, N-Cad+/− neurons served as controls, because they have a genetic background identical to N-Cad−/− neurons, but express N-cadherin almost at the wild-type level (Fig. 1B). At 10–12 DIV, typical profiles of asymmetric synapses were present in cultures of both N-Cad+/− and N-Cad−/− neurons (Fig. 2C). No significant differences in the mean width of the synaptic cleft (N-Cad+/−: 25.8 ± 0.9 nm, n = 35 active zones; N-Cad−/−: 27.1 ± 0.6 nm, n = 46 active zones) were observed. Without stimulation, neither the overall number of vesicles detectable per 100 nm active zone length (N-Cad+/−: 10.1 ± 1.1, n = 18; N-Cad−/−: 8.5 ± 0.5, n = 29) nor the number of vesicles close (<80 nm) to the presynaptic membrane per 100 nm active zone length (N-Cad+/−: 2.4 ± 0.2, n = 18; N-Cad−/−: 2.4 ± 0.1, n = 29) differed significantly. However, we cannot exclude changes in the three-dimensional structure of asymmetric synapses in the absence of N-cadherin, because this would require three-dimensional reconstructions of the synapses at the EM level. In summary, our ultrastructural data revealed asymmetric synapses with basic properties similar to hippocampal neurons (Schikorski and Stevens, 1997, 2001).
Figure 2.
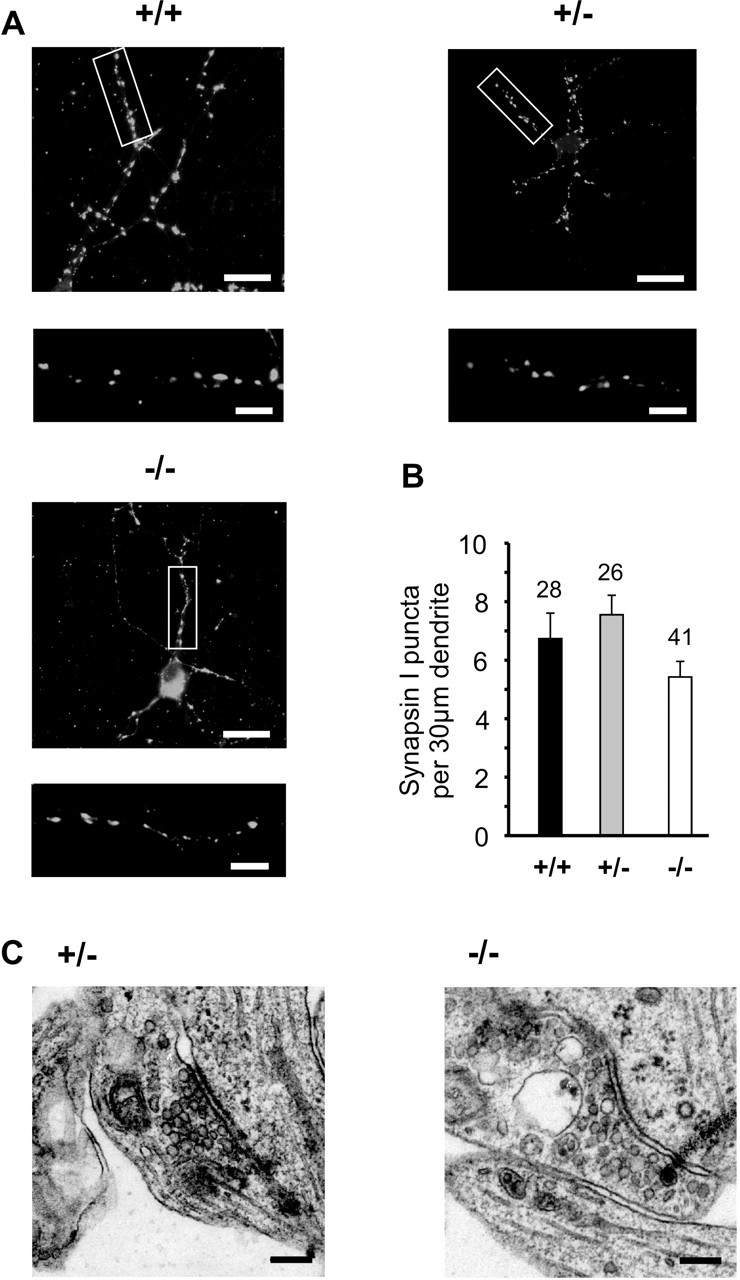
Initial synapse formation is not altered in N-cadherin knock-out, ES cell-derived neurons. A, Synapsin I immunostaining of presynaptic boutons at 10–12 DIV. The box in the top panel is shown enlarged in the bottom panel. Scale bars: top, 12 μm; bottom, 3 μm. B, Quantitative comparison of the dendritic density of synapsin I puncta. n, dendritic segments. C, Typical asymmetric synaptic profiles in cultures of N-Cad+/− and N-Cad−/− neurons. Scale bar, 100 nm. Ultrastructural characteristics were similar to those described for primary cultured cortical neurons. In this and all subsequent figures, genotypes are indicated by +/+, +/−, −/−; error bars represent SEM; and significant differences are indicated by asterisks (∗p < 0.05; ∗∗p < 0.01).
Functional impairment during enhanced synaptic activity as revealed by miniature postsynaptic currents
We next addressed the functional consequences of the absence of N-cadherin at glutamatergic synapses by performing whole-cell patch-clamp recordings of spontaneous AMPA receptor-mediated miniature postsynaptic currents (AMPA mEPSCs) at 10–12 DIV. AMPA mEPSCs were recorded at a holding potential of −60 mV in the presence of TTX and picrotoxin and were reversibly blocked by DNQX (20 μm). At 5 mm extracellular K+, no significant differences in the mean frequency (N-Cad+/−: 0.28 ± 0.04 Hz, n = 36; N-Cad−/−: 0.25 ± 0.04 Hz, n = 37) and in the mean amplitude (N-Cad+/−: 9.0 ± 0.2 pA, n = 36; N-Cad−/−: 8.7 ± 0.2 pA, n = 37) of AMPA mEPSCs were observed in the absence of N-cadherin (Fig. 3D). However, at elevated extracellular K+ (30 mm) to strongly stimulate vesicle exocytosis, a functional synaptic impairment was clearly indicated in N-Cad−/− neurons (Fig. 3A–C). The mean frequency of AMPA mEPSCs (2.0 ± 0.4 Hz) was markedly reduced (p < 0.01) compared with the threefold higher mean frequencies in N-Cad+/− (6.9 ± 1.4 Hz) and wild-type (6.7 ± 1.2 Hz) neurons. In contrast, no significant differences were observed in the mean amplitudes of AMPA mEPSCs between N-Cad−/−, N-Cad+/−, and wild-type neurons.
Figure 3.
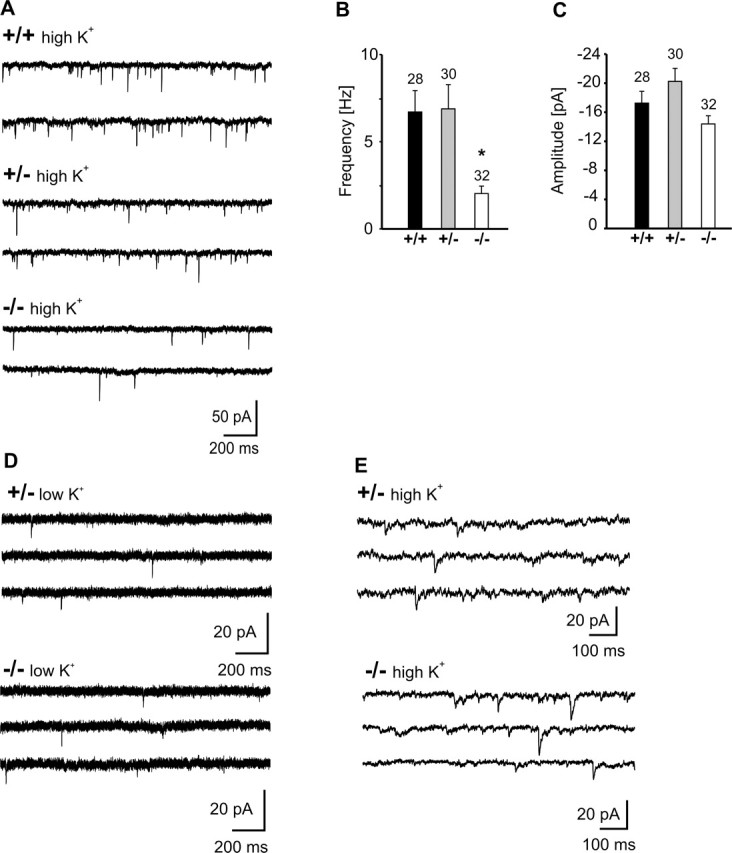
AMPA receptor-mediated miniature EPSCs revealed an impairment of presynaptic function during high activity. A, Example traces of AMPA mEPSCs evoked by elevated extracellular K+ (30 mm). B, C, Comparison of mean frequencies (B) and amplitudes (C) of AMPA mEPSCs at 30 mm K+. n (cells) is indicated. Note the significantly reduced frequency, indicating a presynaptic defect. D, Example traces of AMPA mEPSCs at 5 mm extracellular K+. E, Example traces of GABAA mPSCs evoked by elevated extracellular K+ (30 mm).
We also analyzed the kinetic properties of AMPA mEPSCs, because in particular the rise times of mEPSCs might indicate changes in glutamate concentration that might be caused by altered synaptic adhesion and thus altered width of the synaptic cleft. As expected from the ultrastructural data, we did not find any significant differences in the mean rise times of AMPA mPSCs between N-Cad−/−, N-Cad+/− and wild-type neurons (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Likewise, the mean decay time constant of AMPA mPSCs was not significantly different between N-Cad−/−, N-Cad+/− and wild-type neurons (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
In addition, we further studied whether the absence of N-cadherin affects miniature PSCs at GABAergic synapses. At 10–12 DIV, GABAA receptor-mediated mPSCs were recorded using a high K+ (30 mm) extracellular solution at a holding potential of −60 mV in the presence of TTX and DNQX. No significant differences in the mean frequency (N-Cad+/−: 0.85 ± 0.18 Hz, n = 18; N-Cad−/−: 1.09 ± 0.23 Hz, n = 20) and in the mean amplitude (N-Cad+/−: 13.1 ± 3.5 pA, n = 18; N-Cad−/−: 11.1 ± 1.6 pA, n = 20) of GABAA receptor-mediated mPSCs were detectable between N-Cad−/− and N-Cad+/− neurons (Fig. 3E). In summary, the analysis of miniature PSCs indicated a presynaptic functional impairment selectively at glutamatergic synapses in the absence of N-cadherin that became detectable under conditions of enhanced activity.
Functional defect in vesicle exocytosis as revealed by FM4-64 staining and destaining
We confirmed and further analyzed the presynaptic functional defect under conditions of high release activity in N-cadherin knock-out synapses by staining functional release sites with FM4-64. Presynaptic bouton staining was done in a depolarizing, high K+ solution, and destaining was performed by repeated (10×) high-frequency extracellular stimulation (30 Hz for 1 s) in physiological solution (2 mm Ca2+). At 10–12 DIV, the number of FM4-64 stained puncta per 30 μm dendrite length did not significantly differ between N-Cad+/− (6.8 ± 0.4, n = 52) and N-Cad−/− neurons (5.7 ± 0.5, n = 45). The mean initial fluorescence intensity of FM4-64 stained puncta was slightly but significantly (p < 0.01) reduced to 75.3% in N-Cad−/− neurons compared with N-Cad+/− controls (Fig. 4B).
Figure 4.
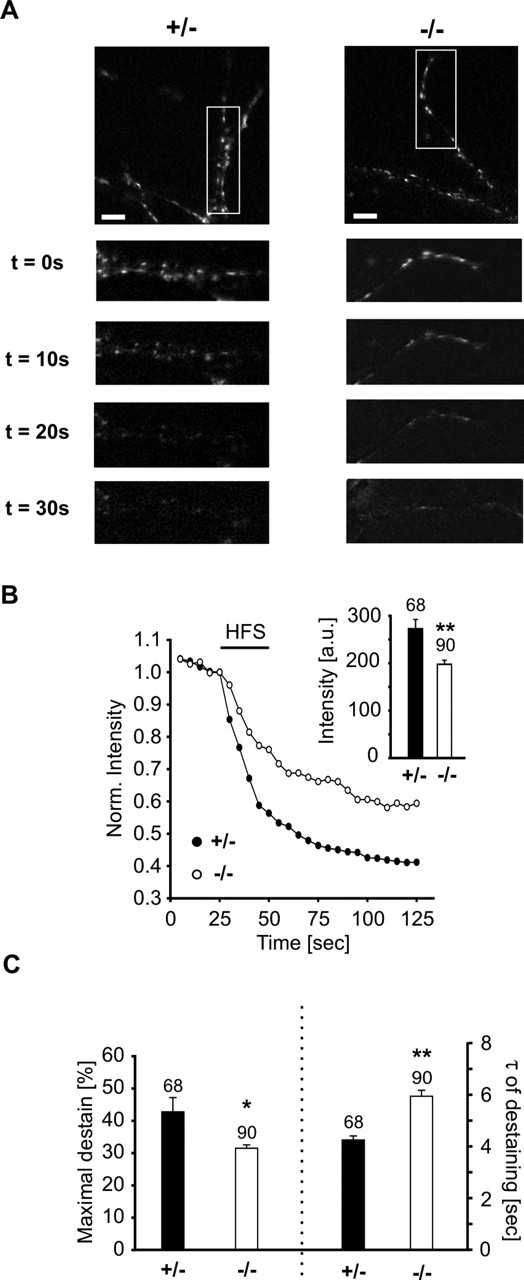
Reduced high-frequency stimulation-evoked vesicle exocytosis as revealed by FM4-64 staining/destaining of presynaptic release sites. A, Examples of high-frequency stimulation (HFS)-induced destaining of FM4-64-labeled presynaptic release sites. Dendritic segments in boxed areas in the top panels are shown enlarged below. Time after onset of HFS is indicated. Scale bars, 10 μm. B, Typical destaining kinetics of individual FM4-64 puncta in N-Cad+/− (filled dots) and N-Cad−/− (open dots) neurons. HFS is indicated by black bar. Inset, Initial mean fluorescence intensity without normalization. C, Quantitative analysis revealed a reduced amount of destaining (left part) and an increased mean time constant of destaining (right part) in N-Cad−/− neurons, indicating reduced vesicle exocytosis. Destaining kinetics were fitted monoexponentially. n indicates the number of FM4-64 puncta analyzed.
Intriguingly, using high-frequency stimulation we observed a substantial difference in destaining properties, indicating an impairment in evoked vesicle exocytosis (Fig. 4). The reduction in fluorescence intensity induced by high-frequency stimulation was significantly (p < 0.05) smaller in N-Cad−/− synapses (31.5 ± 1.0%) compared with N-Cad+/− controls (42.8 ± 4.4%). Destaining kinetics were fitted monoexponentially, and the mean time constant of destaining was significantly (p < 0.01) slower in N-Cad−/− neurons (5.9 ± 0.2 s) compared with N-Cad+/− controls (4.2 ± 0.2 s). In summary, these results demonstrate a presynaptic functional defect and support a role of N-cadherin in the regulation of exocytosis under conditions of high synaptic activity.
Impaired refill of the sucrose-depleted, readily releasable vesicle pool
To further characterize the presynaptic defect in N-Cad−/− neurons, we investigated the release properties of glutamatergic synapses by evoking Ca2+ influx-independent transmitter release with application of hypertonic sucrose solution. Application of sucrose (500 mm)-containing solution leads to a depletion of the readily releasable vesicle pool, allowing its refill properties to be studied (Rosenmund and Stevens, 1996). Sucrose-evoked AMPA receptor-mediated EPSCs were recorded in the whole-cell mode at a holding potential of −60 mV with GABAA and NMDA receptors blocked. Sucrose application was repeated after 1 min to study the refill of the readily releasable vesicle pool (Fig. 5). At 10–12 DIV, the mean postsynaptic charge influx evoked by the first sucrose application did not significantly differ between N-Cad+/− and N-Cad−/− neurons (Fig. 5B). This indicates, in line with the EM data given above, that the readily releasable pool is not affected by the absence of N-cadherin at low activity levels. In contrast, the refill of the readily releasable pool as tested by the second sucrose application was significantly (p < 0.05) reduced in N-Cad−/− neurons (59 ± 7%) compared with N-Cad+/− controls (96 ± 13%) (Fig. 5B). No change in the time course of charge influx was observed. This difference in refill properties strongly supports the idea that the replenishment of the readily releasable vesicle pool is impaired in the absence of N-cadherin.
Figure 5.
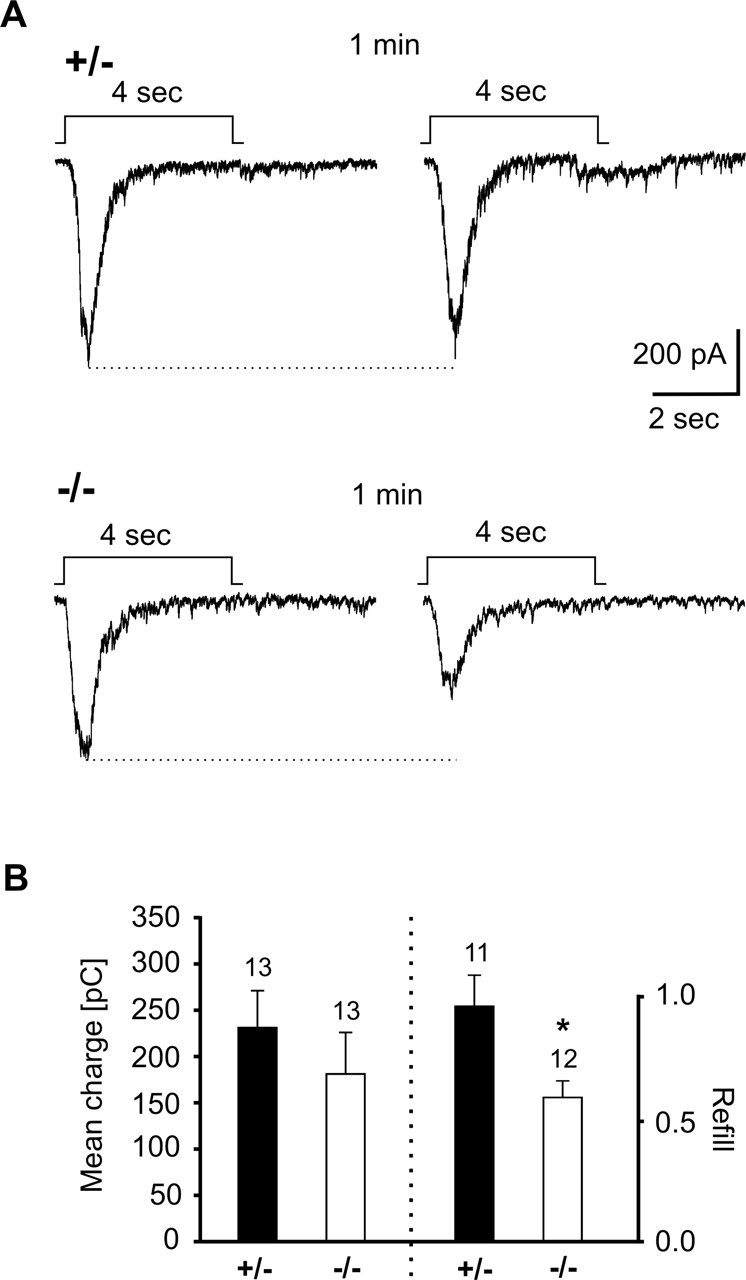
Impaired refill of the readily releasable vesicle pool in N-cadherin knock-out synapses. A, Example traces of AMPA EPSC bursts elicited by two successive hypertonic sucrose applications in N-Cad+/− and N-Cad−/− neurons. The application protocol is indicated above the EPSC traces. B, Quantitative comparison of the mean charge influx during the first sucrose application (left part) and the mean refill (ratio of charge influx during second and first sucrose response) of the readily releasable pool (right part). Note the substantially reduced refill in N-cadherin knock-out synapses.
To further study whether ultrastructural changes in presynaptic vesicle pools can be observed under conditions of high synaptic activity in the absence of N-cadherin, we analyzed EM sections obtained from cultures that had been exposed to a high K+ (30 mm) extracellular solution for 15 min immediately before fixation. We observed a significantly (p < 0.01) reduced number of synaptic vesicles close (within 80 nm distance) to the presynaptic membrane specialization in stimulated N-Cad−/− synapses (1.4 ± 0.1 vesicles per 100 nm active zone length, n = 26 active zones from 5 independent experiments) compared with stimulated N-Cad+/− controls (2.5 ± 0.1 vesicles per 100 nm active zone, n = 23 active zones from 5 independent experiments). No significant change in the overall number of vesicles was detectable (N-Cad+/−: 6.8 ± 0.5 vesicles per 100 nm active zone length, n = 26; N-Cad−/−: 7.7 ± 0.5 vesicles per 100 nm active zone length, n = 23). In line with the impaired refill of the readily releasable pool in sucrose application experiments, these ultrastructural data indicate that the availability of vesicles for release is strongly reduced in N-cadherin knock-out synapses during high activity levels.
Increased depression of evoked synaptic responses indicates a defect in the regulation of short-term plasticity
For an in-depth analysis of a presynaptic defect in the classical action potential-evoked transmission, we performed paired whole-cell recordings at glutamatergic synapses. Current injection into the presynaptic cell was used to elicit an action potential. To test for potential changes in excitability in N-Cad−/− neurons, APs were elicited by depolarizing current pulses in current-clamp mode (Fig. 6A). No significant differences in AP threshold, in AP amplitude and shape, and in firing patterns were found (data not shown). Action potential-evoked AMPA EPSCs were recorded using a physiological extracellular Ca2+ concentration (2 mm) at a holding potential of −60 mV in the presence of Mg2+ and picrotoxin (Fig. 6B). Addition of DNQX (10 μm) or the AMPA receptor-specific antagonist SYM2206 [(±)-4-(4-amino-phenyl)-1,2-dihydro-1-methyl-2-propylcarbamoyl-6,7-methylene-dioxyphthalazine] (100 μm) completely blocked AMPA EPSCs. At 10–12 DIV, the mean amplitudes (including failures) and the mean failure rates of AMPA EPSCs evoked by a single action potential did not differ significantly between N-Cad+/− and N-Cad−/− synapses (Fig. 7C,D), demonstrating that there is no general impairment in vesicle exocytosis under low-frequency stimulation. To further examine whether the glutamate concentration reached in the synaptic cleft at the postsynaptic AMPA receptors is changed in the absence of N-cadherin, we analyzed the blockade of AMPA EPSCs by the low-affinity glutamate receptor antagonist γ-d-glutamylglycine (γ-DGG) (Diamond and Jahr, 1997; Wadiche and Jahr, 2001; Silver et al., 2003). In the presence of 1 mm γ-DGG, the fractional block of AMPA EPSCs was similar in N-Cad−/− (35.5 ± 10.9%, n = 7) compared with N-Cad+/− synapses (48.6 ± 8.9, n = 8), suggesting that the cleft glutamate concentration is not affected (Fig. 6C).
Figure 6.

Properties of glutamatergic synaptic transmission evoked by a single stimulus are not altered in N-cadherin knock-out synapses. A, Typical action potentials evoked by a depolarizing current pulse (bottom) in N-Cad+/− and N-Cad−/− neurons. B, AMPA EPSCs (post) elicited by action currents in the presynaptic cell soma (pre) in a paired recording. Asterisks indicate autaptic PSCs. ampl, Amplitude. C, Partial block of evoked AMPA EPSCs by 1 mm γ-DGG. D, MK-801 (40 μm) block of evoked NMDA PSC component at +40 mV holding potential (measured 40 ms after onset of PSC, as indicated by arrows in D). The control response before and the fourth response after addition of MK-801 are superimposed.
Figure 7.

Increased synaptic depression in N-cadherin knock-out synapses as revealed by paired-pulse analysis. A, Typical paired-pulse behavior of action potential-evoked AMPA EPSCs in paired recordings in N-Cad+/− and N-Cad−/− neurons. Presynaptic action potentials elicited by a 5 ms depolarizing twin pulse (pre) and the corresponding AMPA EPSCs (post) are shown. B, Mean amplitudes and mean half-widths of action potentials did not differ between N-Cad+/− and N-Cad−/− neurons. The ratio of the AP amplitudes and the ratio of the AP half widths of APs elicited by the second and the first pulse in individual presynaptic cells are shown. Filled circles: N-Cad+/−; open circles: N-Cad−/−. C, D, Mean amplitudes (including failures) (C) and mean failure rates (D) of AMPA EPSCs. Responses to the first action potential were not different, whereas responses to the second action potential showed a reduced amplitude and an increased failure rate in N-Cad−/− zneurons. E, Quantitative comparison of paired-pulse ratio of AMPA EPSCs. Note the markedly enhanced paired-pulse depression in N-cadherin knock-out synapses. n (cells) is indicated.
Similarly to AMPA EPSCs, the NMDA receptor-mediated PSC component recorded at +40 mV holding potential (measured 40 ms after PSC onset) (Fig. 6D) did not significantly differ between N-Cad−/− synapses (25.4 ± 6.4 pA, n = 21) and N-Cad+/− controls (34.1 ± 6.2 pA, n = 21). Consequently, the NMDA/AMPA ratio, which was calculated as the ratio of the NMDA component of the PSC at +40 mV and the AMPA EPSC at −60 mV, did not significantly differ in the absence of N-cadherin (N-Cad+/−: 0.39 ± 0.06, n = 20; N-Cad−/−: 0.34 ± 0.08, n = 21). Although NMDA PSC components showed only small amplitudes at 10–12 DIV, we studied their use-dependent block by (+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleate (MK-801) (40 μm) (Rosenmund et al., 1993) (Fig. 6D). The mean time constant of the progressive block of the NMDA PSC component (40 ms after PSC onset) by MK-801 did not significantly differ between N-Cad+/− (2.9 ± 1.1 stimuli, n = 5) and N-Cad−/− synapses (2.4 ± 1.1 stimuli, n = 6). These data suggest that the probability of vesicle release in response to a single action potential was not altered in the absence of N-cadherin.
We further analyzed the paired-pulse behavior of the above described evoked AMPA EPSCs with stimulations spaced at an interval of 50 ms (20 Hz). In these experiments, no differences in presynaptic action potentials were observed in the absence of N-cadherin (Fig. 7A,B). Intriguingly, in N-Cad−/− synapses, we observed a pronounced paired-pulse depression of the AMPA EPSC amplitude (paired-pulse ratio, 0.34 ± 0.10), whereas in N-Cad+/− controls the mean paired-pulse ratio was 1.00 ± 0.25. The significantly (p < 0.05) increased paired-pulse depression was accompanied by a significant (p < 0.05) increase in the incidence of failures in response to the second action potential (Fig. 7C–E), strongly indicating a presynaptic defect in the absence of N-cadherin.
In addition to paired-pulse behavior, we also studied synaptic responses elicited by tetanic stimulation in paired recordings. Two different stimulation frequencies were used to enable analysis of different forms of short-term plasticity. Using a 50 Hz stimulation protocol (20 pulses), we observed a moderate depression of evoked AMPA EPSCs during the stimulation protocol in N-Cad+/− synapses. Strikingly, in N-Cad−/− synapses, this synaptic depression was significantly [Kolmogorov–Smirnov (KS) test, p < 0.001] stronger (Fig. 8A,B), suggesting that the availability of vesicles for exocytosis during high-frequency stimulation is reduced. No significant differences in the amplitudes of the first AMPA EPSC in the train were observed (N-Cad+/−, 75.7 ± 23.6 pA; N-Cad−/−, 58.3 ± 18.6 pA). We also studied the time course of recovery from multiple-pulse depression by eliciting AMPA EPSCs at different time intervals after the end of the 50 Hz stimulation protocol. In N-Cad−/− synapses, the recovery of the AMPA EPSC amplitude was significantly (p < 0.05) smaller at intervals of up to 20 s, and the full recovery was reached much later compared with control N-Cad+/− synapses (Fig. 8C). This finding indicates, in line with the activity-dependent depletion of vesicles close to the presynaptic membrane in EM sections, that the delivery of vesicles to the active zone is impaired in the absence of N-cadherin. Using a 10 Hz stimulation protocol, we observed an initial facilitation of evoked AMPA EPSCs that was in some cells followed by a slight depression during the stimulation protocol in N-Cad+/− synapses. In contrast, in N-Cad−/− synapses, we found a strong synaptic depression (Fig. 8D,E). Again, no significant differences in the amplitudes of the first AMPA EPSC in the train were observed (N-Cad+/−, 93.8 ± 26.2 pA; N-Cad−/−, 89.5 ± 45.0 pA). Our data from paired recordings indicate an impairment in the regulation of short-term plasticity. This seems to be caused by a reduction in the availability of vesicles for release (Südhof, 1995) during enhanced activity.
Figure 8.
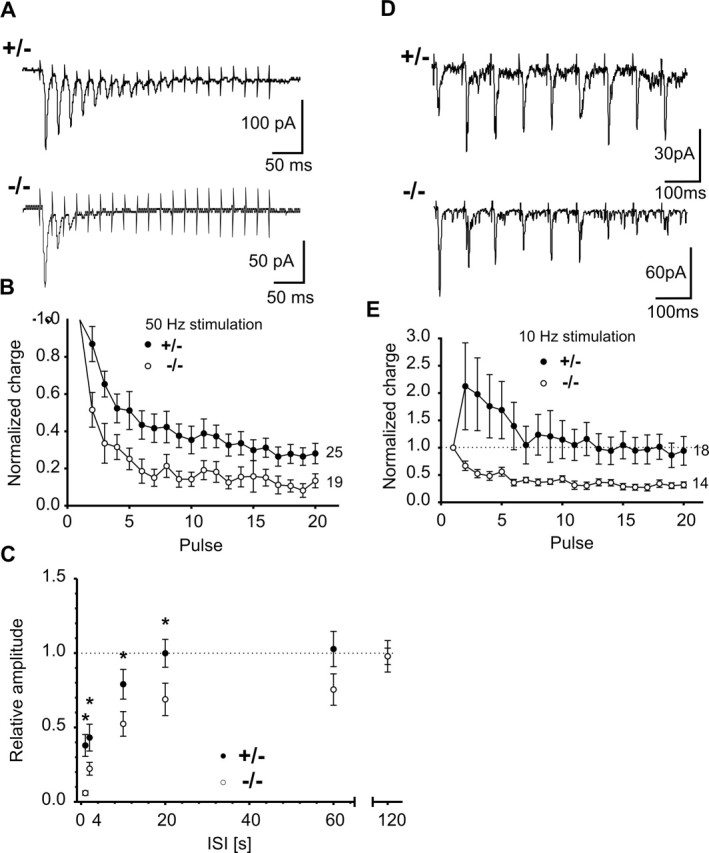
Altered short-term plasticity in N-cadherin knock-out synapses as revealed by tetanic stimulation. A, Typical responses to high-frequency stimulation in N-Cad+/− and N-Cad−/− neurons. AMPA EPSCs were elicited by 20 action potentials at 50 Hz. B, Quantitative comparison of responses to high-frequency stimulation in N-Cad+/− (filled circles) and N-Cad−/− (open circles) synapses revealed strongly increased synaptic depression. C, Recovery from depression elicited by 20 action potentials at 50 Hz. The normalized AMPA EPSC amplitude evoked by a test pulse (N-Cad+/−, n = 8; N-Cad−/−, n = 11) at different time intervals [interstimulus interval (ISI): 1, 2, 10, 20, 60, and 120 s] after the end of the conditioning high-frequency pulse train is shown. Note the significantly slower recovery in N-Cad−/− synapses. D, Typical AMPA EPSCs evoked by 10 Hz stimulation in N-Cad+/− and N-Cad−/− neurons. E, Quantitative comparison of responses to 10 Hz stimulation in N-Cad+/− and N-Cad−/− synapses revealed a switch from facilitation to depression. n (cells) is indicated.
Transsynaptic retrograde regulation of short-term plasticity by N-cadherin
Classical cadherins are thought to bridge the synaptic cleft by adhesive interactions. Thus, the postsynaptic target cell might regulate presynaptic short-term plasticity via an N-cadherin-mediated homophilic interaction. To test for such a transsynaptic regulation, we studied glutamatergic synapses in chimeric microisland cultures consisting of EGFP-expressing neocortical neurons (wild-type) and N-Cad−/− or N-Cad+/− neurons derived from ES cells (Fig. 9A). We performed paired recordings from a green fluorescent wild-type neuron and an N-Cad−/− or an N-Cad+/− neuron, respectively. Evoked AMPA EPSCs were elicited as described above. Recordings from synapses between presynaptic N-Cad−/− neurons and postsynaptic neocortical wild-type neurons showed only very small amplitudes (17.5 ± 5.2 pA, n = 11) of the AMPA EPSCs evoked by a single action potential compared with presynaptic N-Cad+/− neurons (68.3 ± 22.5, n = 12). In addition, the mean failure rate was strongly increased (>25%), suggesting that functional synapse formation is affected in this chimeric configuration. This result indicates that N-cadherin is, in addition to regulating short-term plasticity, involved in a different process controlling synapse formation or maturation (Togashi et al., 2002; Bozdagi et al., 2004), which deserves further investigation.
Figure 9.
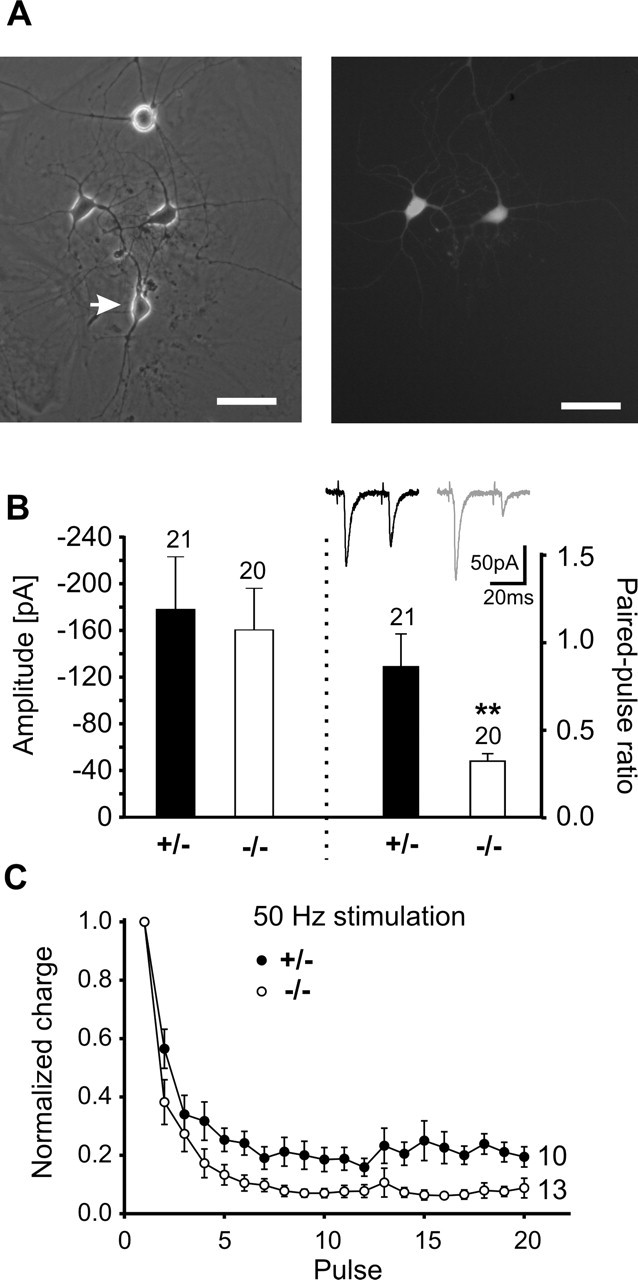
Transsynaptic retrograde regulation of short-term plasticity by N-cadherin. A, Chimeric microisland culture (left) consisting of EGFP-expressing, wild-type neocortical neurons (right) and unlabeled ES cell-derived neurons (arrow) at 12 DIV. Paired recordings were obtained from a presynaptic wild-type neuron and a postsynaptic N-Cad+/− or N-Cad−/− neuron. B, Right, Mean amplitudes of AMPA EPSCs elicited by the first stimulus of a paired-pulse protocol. Left, Paired-pulse analysis of evoked AMPA EPSCs revealed an increased paired-pulse depression in the postsynaptic absence of N-cadherin. Insets, Original recordings. C, High-frequency stimulation of synapses between presynaptic wild-type neurons and postsynaptic N-Cad+/− or N-Cad−/− neurons revealed an increased depression of AMPA EPSCs in the postsynaptic absence of N-cadherin. n (cells) is indicated.
Our major aim in this paper was to analyze synapses between presynaptic neocortical and postsynaptic ES cell-derived neurons to study transsynaptic regulation of short-term plasticity. The mean amplitudes of the AMPA EPSCs evoked by the first stimulation of a paired-pulse protocol did not significantly differ between postsynaptic N-Cad−/− and postsynaptic N-Cad+/− neurons (Fig. 9B). Intriguingly, synapses with postsynaptic N-Cad−/− neurons showed a significantly (p < 0.05) increased paired-pulse depression (0.34 ± 0.06) compared with synapses with postsynaptic N-Cad+/− neurons (0.84 ± 0.2) (Fig. 9B). In addition, synaptic depression in response to 50 Hz stimulation was significantly (KS test, p < 0.01) stronger in synapses, in which N-cadherin was postsynaptically absent (Fig. 9C). In summary, our results indicate that N-cadherin transsynaptically regulates short-term plasticity at glutamatergic synapses.
Discussion
N-cadherin is a synaptic adhesion molecule well known to be localized perisynaptically in the vicinity of the active zone and the PSD of glutamatergic synapses. In contrast to its synaptic localization, the specific physiological importance in CNS synapse function has remained unclear. Using in vitro differentiation of ES cells, we were able to circumvent the early embryonic lethality of N-cadherin knock-out mice (Radice et al., 1997) and investigated the properties of synapses formed in vitro between N-cadherin knock-out neurons. Our analysis revealed a presynaptic defect in the availability of vesicles for exocytosis during high activity and, consequently, in short-term plasticity at glutamatergic synapses devoid of N-cadherin. We further found evidence for a role in the transsynaptic regulation of the short-term modulation of vesicle release. Initial synapse formation, however, appeared to be unaffected by the specific lack of N-cadherin.
Initial synapse formation, as indicated by the density of Synapsin I puncta, the density of FM4-64 puncta, the basic ultrastructure of asymmetric synapses, and the presence of postsynaptic AMPA and NMDA receptor-mediated currents, was not affected in N-cadherin knock-out neurons, suggesting that glutamatergic synapse formation can occur in the complete absence of N-cadherin. However, our experiments cannot address a crucial role of N-cadherin in specific synapse formation in vivo, because synapse formation in our in vitro system may be altered. In addition, because other cadherins might have compensated for N-cadherin, a general role in initial synapse formation might have been obscured. A functional “knock-down” approach of classical cadherin function using expression of a dominant-negative mutant cadherin had resulted in a partial loss of synaptic sites (Togashi et al., 2002; Bozdagi et al., 2004). This indicates a general role of cadherins in synapse formation that does not specifically require N-cadherin. In addition, our data show that the absence of N-cadherin-mediated synaptic adhesion did not lead to changes in the synaptic cleft as suggested by ultrastructural data and by the normal cleft glutamate concentration. However, N-cadherin may nevertheless cooperate with other adhesion molecules in regulating synaptic adhesion.
Despite the lack of a gross effect on synapse number, we observed drastic defects in synaptic function selectively under conditions of high synaptic activity in N-cadherin knock-out neurons. A presynaptic defect was strongly indicated by a reduction in the frequency of K+ depolarization-stimulated AMPA receptor-mediated miniature EPSCs. A reduced efficiency of vesicle exocytosis during high synaptic activity was confirmed by high-frequency stimulation-evoked FM4-64 destaining of presynaptic release sites. Moreover, repeated sucrose stimulations revealed a reduced refill of the readily releasable vesicle pool in the absence of N-cadherin, indicating that after depletion, the replenishment of the readily releasable pool is impaired. A defect in the activity-dependent recruitment of vesicles to the active zone was further supported by a reduction in the number of vesicles close (<80 nm) to the presynaptic membrane in EM sections from N-cadherin-deficient synapses that had been K+-depolarized to strongly stimulate vesicle release. Together, these results indicate a reduced availability of vesicles during high synaptic activity. Both the reduction in miniature EPSC frequency and the impaired FM4-64 destaining are in line with such a reduced availability of vesicles.
The detailed functional analysis of N-cadherin knock-out synapses using paired recordings again revealed an impairment in high-frequency vesicle exocytosis leading to alterations in short-term plasticity properties. Synaptic responses elicited by a single action potential did not differ in respect to amplitude, failure rate (AMPA EPSCs), and blockade by MK-801 (NMDA PSCs), strongly suggesting that the release probability in response to a single action potential is not altered in the absence of N-cadherin. Paired recordings further revealed a markedly increased paired-pulse depression that was accompanied by an increased failure rate in response to the second stimulus. Moreover, synaptic depression in response to tetanic stimulation was substantially increased. Most intriguingly, synaptic facilitation was converted to depression under appropriate stimulation conditions. Thus, our functional analysis of N-cadherin knock-out synapses revealed an unexpected involvement of N-cadherin in the regulation of short-term plasticity at glutamatergic synapses. An analogous phenotype consisting of enhanced synaptic depression during repetitive stimulation was described in neural cell adhesion molecule (NCAM)-deficient mouse neuromuscular synapses (Rafuse et al., 2000).
Our findings could be mechanistically explained by a reduced availability of vesicles for release during enhanced synaptic activity (Regehr and Stevens, 2001; Schneggenburger et al., 2002; Xu-Friedmann and Regehr, 2004). In particular, the slower recovery from multipulse synaptic depression and the reduced refill during repeated sucrose applications in N-cadherin-deficient synapses indicate that the replenishment of the readily releasable vesicle pool is impaired. As suggested by our EM data from stimulated synapses, a defect in vesicle recruitment to the active zone might lead to an enhanced depletion of docked vesicles in the absence of N-cadherin. In line with this, deletion of β-catenin has been described to perturb presynaptic vesicle localization, suggesting a role of cadherin/catenin complexes in organizing vesicle pools (Bamji et al., 2003). Because it occurs on a very short time scale, the increased paired-pulse depression might in addition indicate that the priming reaction of already docked vesicles and thus the release competence of vesicles (Südhof, 1995, 2004) is impaired during high synaptic activity.
N-cadherin might regulate the availability of vesicles for release indirectly by organizing or modulating the actin cytoskeleton (Kovacs et al., 2002; Goodwin and Yap, 2004). The underlying molecular mechanisms might use the classical cadherin signal transduction pathway. Cadherins associate with β-catenin, which interacts with α-catenin, leading to a regulation of the actin cytoskeleton. In addition, the cytoplasmic domain of cadherins also interacts with proteins of the p120 catenin family, which regulate Rho GTPases and might thus influence action organization (Goodwin and Yap, 2004). The state of the actin cytoskeleton is thought to control vesicle pools and vesicle recruitment to the active zone at glutamatergic synapses (Zhang and Benson, 2001; Sakaba and Neher, 2003). However, the role of actin in vesicle pool organization and vesicle recruitment might be rather indirect. Recent evidence suggests that actin filaments might provide a scaffold for other regulatory molecules that more directly control vesicle cycling (Sankaranarayanan et al., 2003). In addition to regulating the actin cytoskeleton, synaptic cadherin–catenin complexes interact with other signaling pathways including presynaptic PDZ proteins such as Veli, which complexes with CASK (Bamji et al., 2003; Takeichi and Abe, 2005). This might provide a signaling pathway more directly regulating active zone processes such as docking and priming of synaptic vesicles. In line with this idea, the synaptic vesicle clustering activity of β-catenin was proposed to be mediated by actin-independent mechanisms involving presynaptic PDZ proteins (Bamji et al., 2003).
To study transsynaptic regulatory effects of N-cadherin, we analyzed glutamatergic synapses in heterogeneous cultures consisting of wild-type neocortical neurons and ES cell-derived neurons. With N-cadherin absent selectively on the presynaptic side, a reduction in synaptic responses to a single action potential was observed, whereas in the selective postsynaptic absence of N-cadherin, AMPA EPSCs evoked by a single action potential were unaltered. A comparable asymmetry in synaptic strength has been described in heterogenotypic cultures of control and NCAM−/− neurons (Dityatev et al., 2000). The molecular mechanisms underlying such an asymmetry in synaptic strength are currently unclear and need additional detailed investigation. Regarding transsynaptic regulation of short-term plasticity, we found that the selective postsynaptic absence of N-cadherin was sufficient to induce the same alterations in short-term plasticity, i.e., increased synaptic depression, as were observed in homogeneous cultures with N-cadherin absent presynaptically and postsynaptically. This suggests that postsynaptic N-cadherin is able to retrogradely regulate release properties of the presynaptic site, presumably by a homophilic interaction with presynaptic N-cadherin. A similar regulation of short-term plasticity by the postsynaptic target neuron has been described at glutamatergic synapses onto different types of inhibitory interneurons in the mammalian neocortex (Reyes et al., 1998). Excitatory synapses formed by an individual pyramidal neuron can exhibit either facilitation or depression, depending on the cell type of the target neurons (Markram et al., 1998). The transsynaptic regulation of short-term plasticity by N-cadherin might represent a molecular mechanism involved in such a target-dependent regulation of presynaptic functional properties.
In addition to its specific regulatory role in presynaptic vesicle exocytosis as analyzed and discussed above, N-cadherin might have additional postsynaptic functions in more mature synapses, which were not addressed in this paper. In hippocampal neurons, a role of cadherin–catenin complexes in spine formation (Murase et al., 2002; Togashi et al., 2002; Abe et al., 2004; Okamura et al., 2004) and dendritic arborization (Yu and Malenka, 2003) has been demonstrated. Furthermore, N-cadherin has been proposed to play a stabilizing role at newly formed synapses underlying the long-term maintenance of synaptic plasticity (Tang et al., 1998; Bozdagi et al., 2000; Murase and Schuman, 1999).
Our functional analysis of mammalian glutamatergic synapses specifically devoid of N-cadherin revealed an important role of N-cadherin in regulating synapse function during high activity. Based on our findings, we propose that N-cadherin is a transsynaptic retrograde regulator of presynaptic short-term plasticity. Furthermore, N-cadherin might interact with other synaptic adhesion molecules to regulate additional aspects of synapse formation, function, and plasticity. However, such additional roles may be obscured by compensatory mechanisms involving other adhesion molecules.
Footnotes
This work was supported by grants from the European Molecular Biology Organization to R.M. and from the Deutsche Forschungsgemeinschaft to K.J. (GRK736), V.L. (SFB509 and SFB553), and K.G. (SFB509 and Go710/3). We thank A.Copi for establishing ES cell culture, F. W. Pfrieger and D. Mauch for L1 antibodies, C. Walz for help with fluorescence imaging, W. Hofer for help with ultrastructural analysis, and M. Bohndorf, J. Gerkrath, and H. Bartel for excellent technical assistance. We also thank H. Hatt and N. Brose for generous support.
References
- Abe K, Chisaka O, van Roy F, Takeichi M (2004). Stability of dendritic spines and synaptic contacts is controlled by αN-catenin. Nat Neurosci 7:357–363. [DOI] [PubMed] [Google Scholar]
- Angst BD, Marcozzi C, Magee AI (2001). The cadherin superfamily: diversity in form and function. J Cell Sci 114:629–641. [DOI] [PubMed] [Google Scholar]
- Bamji SX, Shimazu K, Kimes N, Huelsken J, Birchmeier W, Lu B, Reichardt LF (2003). Role of β-catenin in synaptic vesicle localization and presynaptic assembly. Neuron 40:719–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson DL, Tanaka H (1998). N-cadherin redistribution during synaptogenesis in hippocampal neurons. J Neurosci 18:6892–6904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozdagi O, Shan W, Tanaka H, Benson DL, Huntley GW (2000). Increasing numbers of synaptic puncta during late-phase LTP: N-cadherin is synthesized, recruited to synaptic sites, and required for potentiation. Neuron 28:245–259. [DOI] [PubMed] [Google Scholar]
- Bozdagi O, Valcin M, Poskanzer K, Tanaka H, Benson DL (2004). Temporally distinct demands for classic cadherins in synapse formation and maturation. Mol Cell Neurosci 27:509–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clandinin TR, Zipursky SL (2002). Making connections in the fly visual system. Neuron 35:827–841. [DOI] [PubMed] [Google Scholar]
- Diamond JS, Jahr CE (1997). Transporters buffer synaptically released glutamate on a submillisecond time scale. J Neurosci 17:4672–4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dityatev A, Dityateva G, Schachner M (2000). Synaptic strength as a function of post- versus presynaptic expression of the neural cell adhesion molecule NCAM. Neuron 26:207–217. [DOI] [PubMed] [Google Scholar]
- Fannon AM, Colman DR (1996). A model for central synaptic junctional complex formation based on the differential adhesive specificities of the cadherins. Neuron 17:423–434. [DOI] [PubMed] [Google Scholar]
- Garner CC, Zhai RG, Gundelfinger ED, Ziv NE (2002). Molecular mechanisms of CNS synaptogenesis. Trends Neurosci 25:243–251. [DOI] [PubMed] [Google Scholar]
- Goodwin M, Yap AS (2004). Classical cadherin adhesion molecules: coordinating cell adhesion, signaling and the cytoskeleton. J Mol Histol 35:839–844. [DOI] [PubMed] [Google Scholar]
- Gumbiner BM (2000). Regulation of cadherin adhesive activity. J Cell Biol 148:399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjantonakis AK, Gerstenstein M, Ikawa M, Okabe M, Nagy A (1998). Generating green fluorescent mice by germline transmission of green fluorescent ES cells. Mech Dev 76:79–90. [DOI] [PubMed] [Google Scholar]
- Hartmann M, Heumann R, Lessmann V (2001). Synaptic secretion of BDNF after high-frequency stimulation of glutamatergic synapses. EMBO J 20:5887–5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntley GW, Benson DL (1999). Neural (N)-cadherin at developing thalamocortical synapses provides an adhesion mechanism for the formation of somatopically organized connections. J Comp Neurol 407:453–471. [PubMed] [Google Scholar]
- Inoue A, Sanes JR (1997). Lamina-specific connectivity in the brain: regulation by N-cadherin, neurotrophins, and glycoconjugates. Science 276:1428–1431. [DOI] [PubMed] [Google Scholar]
- Jüngling K, Nägler K, Pfrieger FW, Gottmann K (2003). Purification of embryonic stem cell-derived neurons by immunoisolation. FASEB J 17:2100–2102. [DOI] [PubMed] [Google Scholar]
- Kovacs EM, Goodwin M, Ali RG, Paterson AD, Yap AS (2002). Cadherin-directed actin assembly: E-cadherin physically associates with the Arp2/3 complex to direct actin assembly in nascent adhesive contacts. Curr Biol 12:379–382. [DOI] [PubMed] [Google Scholar]
- Lessmann V, Heumann R (1997). Cyclic AMP endogenously enhances synaptic strength of developing glutamatergic synapses in serum-free microcultures of rat hippocampal neurons. Brain Res 763:111–122. [DOI] [PubMed] [Google Scholar]
- Markram H, Wang Y, Tsodyks M (1998). Differential signaling via the same axon of neocortical pyramidal neurons. Proc Natl Acad Sci USA 95:5323–5328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunaga M, Hatta K, Nagafuchi A, Takeichi M (1988). Guidance of optic nerve fibres by N-cadherin adhesion molecules. Nature 334:62–64. [DOI] [PubMed] [Google Scholar]
- Mohrmann R, Werner M, Hatt H, Gottmann K (1999). Target-specific factors regulate the formation of glutamatergic transmitter release sites in cultured neocortical neurons. J Neurosci 19:10004–10013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohrmann R, Lessmann V, Gottmann K (2003). Developmental maturation of synaptic vesicle cycling as a distinctive feature of central glutamatergic synapses. Neuroscience 117:7–18. [DOI] [PubMed] [Google Scholar]
- Moore R, Radice GL, Dominis M, Kemler R (1999). The generation and in vivo differentiation of murine embryonal stem cells genetically null for either N-cadherin or N- and P-cadherin. Int J Dev Biol 43:831–834. [PubMed] [Google Scholar]
- Murase S, Schuman EM (1999). The role of cell adhesion molecules in synaptic plasticity and memory. Curr Opin Cell Biol 11:549–553. [DOI] [PubMed] [Google Scholar]
- Murase S, Mosser E, Schuman EM (2002). Depolarization drives β-catenin into neuronal spines promoting changes in synaptic structure and function. Neuron 35:91–105. [DOI] [PubMed] [Google Scholar]
- Okamura K, Tanaka H, Yagita Y, Saeki Y, Taguchi A, Hiraoka Y, Zeng LH, Colman DR, Miki N (2004). Cadherin activity is required for activity-induced spine remodeling. J Cell Biol 167:961–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips GR, Huang JK, Wang Y, Tanaka H, Shapiro L, Zhang W, Shan WS, Arndt K, Frank M, Gordon RE, Gawinowicz MA, Zhao Y, Colman DR (2001). The presynaptic particle web: ultrastructure, composition, dissolution, and reconstitution. Neuron 32:63–77. [DOI] [PubMed] [Google Scholar]
- Poskanzer K, Needleman LA, Bozdagi O, Huntley GW (2003). N-cadherin regulates ingrowth and laminar targeting of thalamocortical axons. J Neurosci 23:2294–2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radice GL, Rayburn H, Matsunami H, Knudsen KA, Takeichi M, Hynes RO (1997). Developmental defects in mouse embryos lacking N-cadherin. Dev Biol 181:64–78. [DOI] [PubMed] [Google Scholar]
- Rafuse VF, Polo-Parada L, Landmesser LT (2000). Structural and functional alterations of neuromuscular junctions in NCAM-deficient mice. J Neurosci 20:6529–6539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redies C (2000). Cadherins in the central nervous system. Prog Neurobiol 61:611–648. [DOI] [PubMed] [Google Scholar]
- Regehr WG, Stevens CF (2001). Physiology of synaptic transmission and short-term plasticity. In: Synapses (Cowan WM, Südhof TC, Stevens CF, eds) pp. 135–175. Baltimore: The Johns Hopkins UP.
- Reyes A, Lujan R, Rozov A, Burnashev N, Somogyi P, Sakmann B (1998). Target-cell-specific facilitation and depression in neocortical circuits. Nat Neurosci 1:279–285. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF (1996). Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron 16:1197–1207. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Clements JD, Westbrook GL (1993). Nonuniform probability of glutamate release at a hippocampal synapse. Science 262:754–757. [DOI] [PubMed] [Google Scholar]
- Sakaba T, Neher E (2003). Involvement of actin polymerization in vesicle recruitment at the calyx of Held synapse. J Neurosci 23:837–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes JR, Yamagata M (1999). Formation of lamina-specific synaptic connections. Curr Opin Neurobiol 9:79–87. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan S, Atluri PP, Ryan TA (2003). Actin has a molecular scaffolding, not propulsive, role in presynaptic function. Nat Neurosci 6:127–135. [DOI] [PubMed] [Google Scholar]
- Scheiffele P (2003). Cell-cell signaling during synapse formation in the CNS. Annu Rev Neurosci 26:485–508. [DOI] [PubMed] [Google Scholar]
- Schikorski T, Stevens CF (1997). Quantitative ultrastructural analysis of hippocampal excitatory synapses. J Neurosci 17:5858–5867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schikorski T, Stevens CF (2001). Morphological correlates of functionally defined synaptic vesicle populations. Nat Neurosci 4:391–395. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Sakaba T, Neher E (2002). Vesicle pools and short-term synaptic depression: lessons from a large synapse. Trends Neurosci 25:206–212. [DOI] [PubMed] [Google Scholar]
- Shapiro L, Colman DR (1999). The diversity of cadherins and implications for a synaptic adhesive code in the CNS. Neuron 23:427–430. [DOI] [PubMed] [Google Scholar]
- Silver RA, Lübke J, Sakmann B, Feldmeyer D (2003). High-probability uniquantal transmission at excitatory synapses in barrel cortex. Science 302:1981–1984. [DOI] [PubMed] [Google Scholar]
- Strübing C, Ahnert-Hilger G, Shan J, Wiedenmann B, Hescheler J, Wobus AM (1995). Differentiation of pluripotent embryonic stem cells into the neuronal lineage in vitro gives rise to mature inhibitory and excitatory neurons. Mech Dev 53:275–278. [DOI] [PubMed] [Google Scholar]
- Südhof TC (1995). The synaptic vesicle cycle: a cascade of protein-protein interactions. Nature 375:645–653. [DOI] [PubMed] [Google Scholar]
- Südhof TC (2001). The synaptic cleft and synaptic cell adhesion. In: Synapses (Cowan WM, Südhof TC, Stevens CF, eds) pp. 275–313. Baltimore: The Johns Hopkins UP.
- Südhof TC (2004). The synaptic vesicle cycle. Annu Rev Neurosci 27:509–547. [DOI] [PubMed] [Google Scholar]
- Takeichi M (1990). Cadherins: a molecular family important in selective cell-cell adhesion. Annu Rev Biochem 59:237–252. [DOI] [PubMed] [Google Scholar]
- Takeichi M, Abe K (2005). Synaptic contact dynamics controlled by cadherin and catenins. Trends Cell Biol 15:216–221. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Shan W, Phillips GR, Arndt K, Bozdagi O, Shapiro L, Huntley GW, Benson DL, Colman DR (2000). Molecular modification of N-cadherin in response to synaptic activity. Neuron 25:93–107. [DOI] [PubMed] [Google Scholar]
- Tang L, Hung CP, Schuman EM (1998). A role for the cadherin family of cell adhesion molecules in hippocampal long-term potentiation. Neuron 20:1165–1175. [DOI] [PubMed] [Google Scholar]
- Togashi H, Abe K, Mizoguchi A, Takaoka K, Chisaka O, Takeichi M (2002). Cadherin regulates dendritic spine morphogenesis. Neuron 35:77–89. [DOI] [PubMed] [Google Scholar]
- Uchida N, Honjo Y, Johnson KR, Wheeklock MJ, Takeichi M (1996). The catenin/cadherin adhesion system is localized in synaptic junctions bordering transmitter release zones. J Cell Biol 135:767–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadiche JI, Jahr CE (2001). Multivesicular release at climbing fiber-Purkinje cell synapses. Neuron 32:301–313. [DOI] [PubMed] [Google Scholar]
- Xu-Friedmann MA, Regehr WG (2004). Structural contributions to short-term synaptic plasticity. Physiol Rev 84:69–85. [DOI] [PubMed] [Google Scholar]
- Yap AS, Kovacs EM (2003). Direct cadherin-activated cell signaling: a view from the plasma membrane. J Cell Biol 160:11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Malenka RC (2003). β-catenin is critical for dendritic morphogenesis. Nat Neurosci 6:1169–1177. [DOI] [PubMed] [Google Scholar]
- Zhang W, Benson DL (2001). Stages of synapse development defined by dependence on F-actin. J Neurosci 21:5169–5181. [DOI] [PMC free article] [PubMed] [Google Scholar]